

Abstract
Background
Multiple sclerosis is an acquired demyelinating disease of the central nervous system. It is the second most common cause of disability in adults in United States after head trauma.
Discussion
The etiology of MS is probably multifactorial, related to genetic, environmental, and several other factors. The pathogenesis is not fully understood but is believed to involve T-cell-mediated inflammation directed against myelin and other related proteins with a possible role for B cells. The McDonald criteria have been proposed and revised over the years to guide the diagnosis of MS and are based on clinical presentation and magnetic resonance imaging (MRI) of the brain and spinal cord to establish dissemination in time and space. The treatment of MS includes disease modification with immunomodulator drugs and symptom management to address the specific symptoms such as fatigue, spasticity, and pain.
Conclusion
An update on etiology, pathogenesis, diagnosis, and immunomodulatory treatment of MS is presented.
Keywords: Demyelination, diagnosis, etiology, immunomodulator, multiple sclerosis, pathogenesis, treatment
Introduction
Multiple sclerosis is an autoimmune inflammatory disorder of CNS of unknown etiology, characterized by demyelination and variable degrees of axonal loss. The disease affects mostly young women (between ages 20 and 40 years) and is one of leading causes of disability in young adults in United States (Orton et al. 2006; Kister et al. 2013). Its prevalence in the United States is about 400,000 and over two million people are affected worldwide with an expected increase in the number of cases in future (Weinshenker 1996; Mayr et al. 2003). The disease appears to be more common in northern hemisphere and there is some genetic susceptibility as well in individuals of Scandinavian or northern European ancestry (Williams et al. 1995; Compston and Coles 2008). The etiology although unknown presumably involves interaction between genetic, environmental, and other factors triggering an aberrant autoimmune attack resulting in damage to myelin and axons (Bruck and Stadelmann 2005). The course of MS can be variable with a significant proportion of patients experiencing some progression following the initial relapsing remitting phase leading to significant disability (Weinshenker et al. 1989; Lublin and Reingold 1996). Much progress has been made in the past two decades in treating MS with the advent of effective immunomodulatory therapies which can potentially slow down the progression and alter the disease course.
Etiology
The etiology of MS remains unknown; however, it is believed to be caused by immune dysregulation triggered by genetic and environmental factors (Ascherio and Munger 2007a,b). Although MS is not an inherited disease, there is a strong genetic component to its etiology as evidenced by clustering of MS cases within families. The risk of MS among first-degree relatives of MS patients is 10–50 times higher than the general population (absolute risk 2–5%); the concordance rate in monzygotic twins is about one-third (Weinshenker 1996; Kantarci 2008). Linkage analysis studies have revealed several gene loci as risk factors, with the major histocompatibility complex (MHC) HLA DR15/DQ6 allele being the strongest one (Barcellos et al. 2003; Sawcer et al. 2011). More recently, alleles of interleukin-2 receptor alpha gene (IL2RA) and interleukin-7 receptor alpha gene (IL7RA) have also been identified as inheritable risk factors (Hafler et al. 2007; Sawcer et al. 2011). However, most of the genetic factors underlying susceptibility still remain to be defined. Furthermore, genetic susceptibility does not fully explain the changes in MS risk that occur with migration suggesting a likely role for environmental factors.
Among environmental factors, Epstein-Barr virus (EBV) infection and vitamin D deficiency have been extensively studied and strongly linked to MS risk (Ascherio and Munger 2007a,b) MS is prevalent in geographic areas farther away from the equator (Simpson et al. 2011). Low vitamin D levels from reduced sun exposure may be a factor contributing to increased susceptibility to MS in these regions (Ascherio and Munger 2007b; Correale et al. 2009; Ascherio et al. 2010). Studies have suggested that higher levels of vitamin D have a possible protective role in certain susceptible patient populations (Munger et al. 2004, 2006). The risk of developing MS is approximately 15-fold higher among individuals with a history of EBV infection in childhood and about 30-fold higher among those infected with EBV in adolescence or later in life (Ascherio 2013). However, the difference in the risk of MS among migrants from high to low MS prevalence areas suggests that other infectious or noninfectious factors in addition to EBV may be involved (Ascherio and Munger 2007a,b). The “hygiene hypothesis,” supported by many epidemiological observations, suggests that improved sanitation and reduced childhood infections in developed countries may account for the increased rates of autoimmune diseases (T-helper 1 mediated) and allergy (T-helper 2 mediated) (Conradi et al. 2011). However, this hypothesis does not explain the higher MS prevalence in rural compared to urban areas (with expected improved sanitation) reported in some studies (Sotgiu et al. 2003).
Cigarette smoking has also been proposed at a potential environmental risk factor with several studies reporting an association between smoking and MS risk and disease activity (Wingerchuk 2012). The odds ratio for developing MS is approximately 1.5 for smokers compared with nonsmokers (Wingerchuk 2012; Fragoso 2014). As with other risk factors, smoking appears to influence the MS susceptibility in conjunction with the genetic and other environmental factors.
There is no specific diet associated with increased risk of MS. The role of dietary factors appears to be complex and related to the influence of multiple dietary components including vitamin A and D, salt, omega-3-unsaturated fatty acid, and polyphenol on immune regulation. Some recent reports have suggested that salt modulates the differentiation of human and mouse Th17 cells (Kleinewietfeld et al. 2013; Wu et al. 2013). A more aggressive course of experimental autoimmune encephalomyelitis (EAE) was observed in mice fed a high sodium diet. In a small observational study, higher sodium intake was associated with increased clinical and radiological disease activity in patients with MS (Farez et al. 2015).
The potential risk factors for MS are listed in Table .
Table 1.
Potential risk factors for multiple sclerosis
| Female gender |
| Caucasian race |
| Genetic |
| HLA DR15/DQ6, IL2RA and IL7RA alleles |
| Infections |
| Epstein–Barr virus (EBV) infection |
| Temperate climate |
| Low vitamin D level |
| Lack of sunlight exposure |
| Cigarette smoking |
Immunopathogenes
The pathogenesis of MS involves immune attack against CNS antigens mediated through activated CD4+ myelin-reactive T cells with a possible contribution by B cells. Much of our understanding of immunopathogenesis of MS is derived from the study of experimental autoimmune encephalomyelitis (EAE), an animal model of CNS inflammatory demyelination that can be induced by peripheral immunization with myelin protein components. EAE shares many of the histologic features of MS including active demyelination, oligodendrocyte and axonal loss, all of which are presumably mediated by myelin specific T cells (Yong 2004; Gold et al. 2006). The immunopathogenesis of MS is thought to involve a breach of self-tolerance toward myelin and other CNS antigens resulting in persistent peripheral activation of autoreactive T cells (Hafler et al. 2005; Selter and Hemmer 2013). In a genetically susceptible individual, this loss of self-tolerance may be triggered by an environmental antigen, presumably an infectious agent such as a virus. The infection could cause bystander activation of T cells or result in release of autoantigens due to cellular damage, which can then lead to activation of T cells by cross reactivity between an endogenous protein (e.g., myelin basic protein) and the pathogenic exogenous protein (viral or bacterial antigen), a process known as molecular mimicry (Fujinami and Oldstone 1985; Wucherpfennig and Strominger 1995; Aichele et al. 1996; Gran et al. 1999; O’Connor et al. 2001).
As depicted in Figure1, once activated in the periphery, myelin-reactive T cells are able to migrate across the blood–brain barrier (BBB). The transmigration process involves interaction between very late antigen-4 (VLA-4) present on T lymphocytes and the vascular cell adhesion molecule-1 (VCAM-1) expressed on capillary endothelial cells; this process is facilitated by expression and upregulation of various adhesion molecules, chemokines, and matrix metalloproteinases (MMPs) (Yong 2004; Gold and Wolinsky 2011). After entering the CNS, the autoreactive peripherally activated T cells can be reactivated upon encountering the autoantigenic peptides within the brain parenchyma in the context of MHC class II molecules expressed by local antigen presenting cells (dendritic cells, macrophages, and B cells) triggering an inflammatory cascade leading to release of cytokines and chemokines, recruitment of additional inflammatory cells including T cells, monocytes, and B cells and persistent activation of microglia and macrophages resulting in myelin damage (Hemmer et al. 2002; Frohman et al. 2006; Inglese 2006). The local inflammation and demyelination results in exposure of sequestered myelin autoantigens providing an additional target for self-reactive T cells, a phenomenon called “epitope spreading” (Miller et al. 1997). Activation of resident CNS glial cells (such as microglia) results in persistent inflammation even in absence of further infiltration of exogenous inflammatory cells (O’Connor et al. 2001). The evidence based on animal studies suggests that CD4+ T-helper 1 (TH1) cells which release proinflammatory cytokines such as interferon-gamma, interleukin-2 (IL-2), and tumor necrosis factor-α (TNF-α) are the key players in mediating inflammation in MS with some role for the novel CD4+ T-helper-17 (TH17) cell subset which secretes IL-17 (O’Connor et al. 2001; Selter and Hemmer 2013). The CD4+ T-helper 2 (TH2) cells, which secret interleukins 4, 5, and 10 are believed to have a counter regulatory role limiting the TH1-cell-mediated injury (Tzartos et al. 2008). The TH1/TH2 paradigm is more apparent in EAE; in MS, indirect evidence exists for a predominant role of Th1 cells based on the success of therapies that shift the cytokine profile away from Th1 toward Th2. CD8+ T cells are believed to be involved as well and can induce axonal pathology by direct injury to MHC I/antigen expressing cells such as neurons and oligodendrocytes (Batoulis et al. 2010). The contribution of B cells to MS pathogenesis (possibly through autoantibody secretion and antigen presentation to T cells) has recently been recognized and is supported by observed pathologic heterogeneity of MS lesions, the presence of meningeal inflammation and B-cell follicle-like structures adjacent to subpial cortical lesions, and the success of B-cell-based immunotherapies (O’Connor et al. 2001; Batoulis et al. 2010; Naismith et al. 2010).
Figure 1.
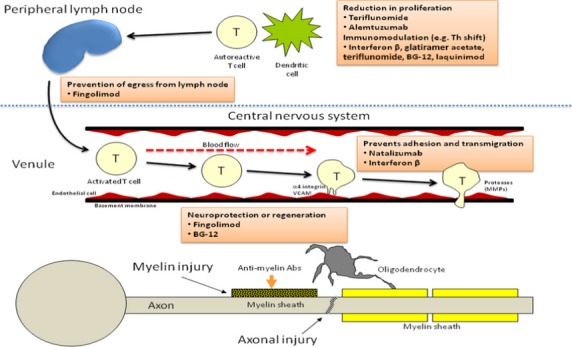
Immunopathogenic mechanisms in MS and proposed targets of different disease modifying therapies.
Although demyelination is the hallmark of MS pathology, early axonal injury and axonal loss also occur and may drive disability progression (Trapp et al. 1998). The exact mechanism(s) of both myelin and axonal injury are not completely understood, but are likely to include both direct injury to myelin and oligodendrocytes, and axons by CD4+ and CD8+ T lymphocytes, activated microglia/macrophages, and/or antibody and complement as well as the indirect effects of proinflammatory cytokines such as IL-1beta, TNF-α, nitric oxide, and MMPs (Lucchinetti et al. 2000; Hemmer et al. 2002; Gold and Wolinsky 2011). Meningeal inflammatory infiltrates reported in association with subpial cortical lesions may contribute to cortical inflammation and disability in some cases (Howell et al. 2011; Lucchinetti et al. 2011).
Pathology
The MS plaques or lesions are focal areas of demyelination associated with variable inflammation and axonal loss that predominantly affect the white matter of the brain, spinal cord, and optic nerves but can also involve the cerebral cortex including subpial regions (Sobel and Moore 2008; Popescu and Lucchinetti 2012). The inflammatory infiltrates associated with plaques consist of activated T cells (predominantly CD8+ with variable presence of CD4+ cells), activated macrophages/microglia, plasma cells, and B cells (Hauser et al. 1986; O’Connor et al. 2001). MS plaques can be further classified histologically as active, chronic, and remyelinated. Active lesions are common in relapsing remitting MS and are characterized by myelin degradation (with relative axonal preservation), macrophage infiltration, reactive astrocytes, and perivascular and parenchymal inflammation (Bruck et al. 1995; Frischer et al. 2009). Chronic or inactive plaques are more often seen in patients with progressive disease and are associated with more extensive demyelination, often with marked axonal depletion, loss of oligodendrocytes, and relative absence of active inflammation (Prineas et al. 2001; Sobel and Moore 2008; Popescu and Lucchinetti 2012). Remyelinated plaques are seen within or more often at the margins of active plaques and contain thinly myelinated axons and often increased numbers of oligodendrocyte precursor cells (Bruck et al. 1995; Popescu and Lucchinetti 2012). “Shadow plaques” are lesions that show more diffuse (but still incomplete) remyelination and are seen in patients with relapsing and progressive disease (Barkhof et al. 2003). The presence of cortical demyelination and axonal loss has been increasingly recognized in early MS (Trapp et al. 1998; Cifelli et al. 2002). Lucchinetti and colleagues have described four distinct immmopathological patterns (pattern I with macrophage and T-cell predominance, II with additional immunoglobulin and complement deposition, III with apoptotic oligodendrocyte loss, and the rare type IV pattern with nonapoptotic death of oligodendrocytes) in active MS lesions, suggesting that there may be pathological heterogeneity among MS patients (Lucchinetti et al. 2000). However, the observed pathologic heterogeneity may not be exclusive to a subset of MS patients and is probably related to the stage of disease in a given patient (Barnett and Prineas 2004). Cortical involvement can occur in MS and may reflect either the presence of cortical demyelination or actual neuronal loss. Within the cortex, three distinct lesion types have been described based on the location of the plaques: subpial, intracortical, and leukocortical (Bogdan et al. 2013). Cortical lesions seen in early MS are usually highly inflammatory and correlate with cognitive impairment (Geurts and Barkhof 2008; Lucchinetti et al. 2011).
Clinical Presentation and Diagnosis
The clinical symptoms and signs of MS are variable and may result from involvement of sensory, motor, visual, and brainstem pathways. The majority of patients with MS initially present with relapsing remitting episodes of new or recurrent neurological symptoms. The first clinical event in these patients, termed clinically isolated syndrome (CIS), can be optic neuritis, incomplete myelitis, or brainstem syndrome (Miller et al. 2005). The presence of classic demyelination lesions on baseline brain or spinal cord MRI is the most important predictor of having a second relapse in CIS patients (Filippi et al. 1994). The presence of cerebrospinal fluid (CSF) abnormalities (positive oligoclonal bands) may have additional prognostic value in patients with CIS and positive brain MRI (Miller et al. 2005; Awad et al. 2010). A variable proportion of patients with relapsing remitting MS (25–40%) develop secondary progressive disease over time with progressive accumulation of disability with infrequent or no relapses (Lublin and Reingold 1996). Primary progressive MS (seen in approximately 10–15% patients) is defined by progressive accumulation of disability from the onset with no or minor relapses and typically presents with a progressive myelopathy with an older age of onset and involving a higher proportion of men (Miller and Leary 2007). Both primary and secondary progressive MS share some clinical and imaging features and are now considered to be part of the progressive disease spectrum (Lublin and Reingold 1996; Ingle et al. 2005; Lublin et al. 2014). The progressive relapsing form of MS with worsening disability from onset and clear acute relapses with or without full recovery is now considered to be progressive disease with disease activity (Lublin et al. 2014).
There is no single diagnostic test for MS and the diagnosis is usually based on the clinical presentation, supported by neuroimaging and in some cases by CSF analysis (to look for inflammatory markers oligoclonal bands and/or elevated IgG index) and evoked potential studies (to look for clinically silent lesion in visual, brainstem, or spinal cord pathways). CSF inflammatory markers are present in up to 85% patients with MS (Link and Huang 2006); IgG index is less sensitive and specific than oligoclonal bands Awad et al. 2010). There have been several proposed diagnostic criteria incorporating the clinical and ancillary data, the most commonly used one is the McDonald criteria initially proposed in 2001 and revised in 2005 and most recently in 2011 (Polman et al. 2011). The basic concept behind these criteria is demonstration of dissemination in time (DIT) and space using the clinical and/or MRI data. A detailed discussion of McDonald criteria is beyond the scope of this review; in summary, the definitive diagnosis of MS requires ≥2 attacks or objective clinical evidence of ≥ 2 lesions or objective clinical evidence of 1 lesion with historical evidence of a prior attack. With one clinical attack, DIT can be demonstrated by presence of asymptomatic gadolinium-enhancing and nonenhancing lesions at any time or by presence of new lesions on a follow-up scan obtained anytime after the initial symptom onset or the simultaneous (see Table 1996). Dissemination in space (DIS) in a patient with two clinical attacks but objective evidence of one lesion can be demonstrated by using the MRI criteria detailed in Table 2013. The criteria for primary progressive MS include 1 year of disease progression plus two of the following criteria: a. evidence of DIS in brain, b. DIS in spinal cord (≥ 2 T2 lesions in the cord), c. positive CSF oligoclonal bands and/or elevated IgG index.
Table 2.
McDonald MRI criteria for demonstration of DIT (Polman et al. 2011)
| DIT Can be Demonstrated by |
|---|
| 1. A new T2 and/or gadolinium-enhancing lesion(s) on follow-up MRI, with reference to a baseline scan, irrespective of the timing of the baseline MRI |
| 2. Simultaneous presence of asymptomatic gadolinium-enhancing and nonenhancing lesions at any time |
Table 3.
McDonald MRI criteria for demonstration of DIS (Polman et al. 2011)
| DIS can be demonstrated by ≥ 1 T2 lesions1 in at least 2 of the 4 area of the CNS |
|---|
| Periventricular |
| Juxtacortical |
| Infratentorial |
| Spinal cord2 |
MRI, magnetic resonance imaging; DIS, lesion dissemination in space; CNS, central nervous system.
Adapted from Polman et al. (2011).
Gadolinium enhancement of lesions is not required for DIS.
If a subject has a brainstem or spinal cord syndrome, the symptomatic lesions are excluded from the Criteria and do not contribute to lesion count.
In patients presenting with typical relapsing remitting symptoms and classic demyelination lesions (e.g., shown in Fig.2) on neuroimaging meeting the radiological criteria, the differential diagnosis is limited and often no further diagnostic testing is indicated in these cases.
Figure 2.
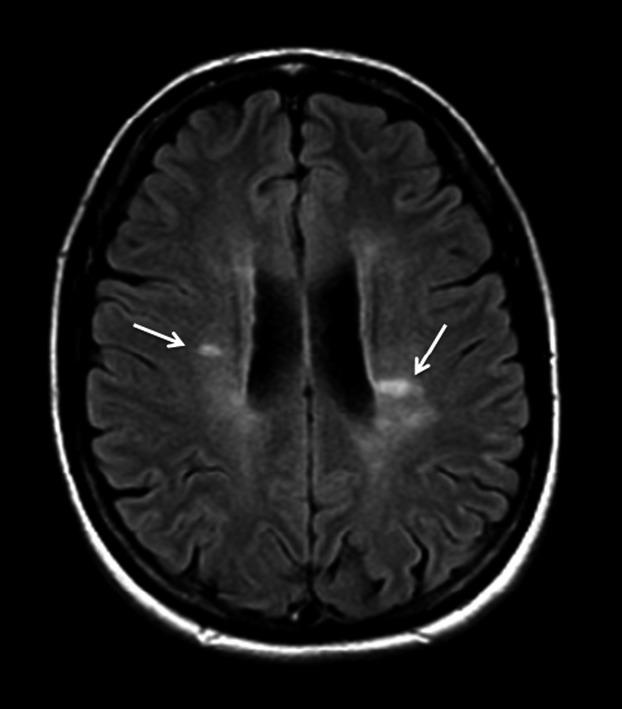
Brain MRI axial fluid attenuated inversion recovery (FLAIR) image shows the characteristic periventricular areas of increased signal intensity (arrows) that are oriented perpendicular to and often contiguous with the lateral ventricles.
The differential diagnosis in other cases depends on the clinical presentation and is outlined in Table 2007a.
Table 4.
Differential diagnosis of multiple sclerosis
| Optic neuritis/neuropathy |
| Inflammatory, neuromyelitis optica (NMO) spectrum disorder, genetic, ischemic |
| Myelitis/myelopathy— |
| Inflammatory demyelination—idiopathic, postviral, postvaccinialNMO spectrum disorder, Autoimmune–systemic lupus erythematosus, antiphospholipid antibody syndrome, other systemic autoimmune disorders |
| Infectious (Lyme disease, HIV, viral, others) |
| Ischemic/vascular |
| Others–compressive, nutritional |
| Brainstem syndrome |
| Stroke, tumor, vasculitis (lupus, Sjögren’s syndrome, Behçet’s disease) |
| Cerebral white matter lesions |
| Small vessel disease (Leukoaraiosis) |
| Migraine |
| Primary CNS vasculitis |
| Sarcoidosis |
| CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) |
Atypical presentation or variants of MS
There are some less common clinical variants of MS which present with atypical clinical and radiological features, these include tumefactive MS, Balo’s concentric sclerosis, and Marburg disease. The radiological hallmark of tumefactive MS is a large solitary >2 cm lesion associated with mass effect, edema and/or ring enhancement (hence the name tumefactive). The clinical symptoms depend on the size and location of the lesion and often include aphasia, agnosia, seizures and visual field defects, not typically seen in CIS or RRMS patients (Lucchinetti et al. 2008). Marburg’s disease and Balo’s concentric sclerosis are characterized by a rapidly evolving fulminant clinical course and poor prognosis. The Marburg variant has the distinct radiological feature of large tumor-like multifocal demyelinating lesions in deep white matter; the pathological changes are similar to those of classicMS but may appear more destructive and have more inflammatory infiltrates (Karussis 2014). The pathological changes seen in Balo’s concentric sclerosis are quite unique and consist of alternating bands of normally myelinated or remyelinated, and demyelinated white matter; this pattern has been described as resembling hypoxia-induced injury (Stadelmann et al. 2005). The MRI may show alternating isointense and hypointense concentric rings with partial enhancement on T1-weighted images (Zettl et al. 2012; Karussis 2014).
Therapeutic Options
The management of MS includes treatment with immunomodulatory agents that help alter the course of the disease, symptomatic management focusing on relieving specific symptoms such as fatigue, spasticity, bladder dysfunction, and pain (not discussed in this review). Corticosteroids (methylprednisolone) and adrenocorticotropic hormone (ACTH) have anti-inflammatory and immunomodulatory effects and are typically used to treat acute relapse to hasten the recovery (Berkovich 2013). The immunomodulatory therapies (IMT) used in long-term disease modification are discussed in the next section.
Immunomodulatory Therapies
The most significant progress in the treatment of MS in the last two decades has been the development of IMT. Since the introduction of first immunomodulating medication, interferon beta-1b in 1993, several other medications with different mechanism of action (figure1), mode and frequency of administration have become available. Currently, there are 12 medications approved for treatment of MS, including six self-injectable, three infusion based, and three oral medications as listed in Table 2007b.
Table 5.
Approved therapies for multiple sclerosis
| Medication | Dose | Route | Frequency | Major side effects |
|---|---|---|---|---|
| First-line therapies | ||||
| Beta-interferon-1b (Betaseron®, Bayer HealthCare Pharmaceuticals Inc., Whippany, NJ) | 250 µg | SC | Every other day | Flu-like symptoms, injection site reactions, liver enzyme elevation, thyroid abnormalities, leukopenia or anemia, and depression |
| Beta-interferon-1a (Avonex®, Biogen Idec, Cambridge, MA ) | 30 µg | IM | Once a week | |
| Peginterferon beta-1a (Plegridy®, Biogen Idec, Cambridge, MA) | 125 µg | SC | Every 14 days | |
| Beta-interferon-1a (Rebif®, EMD Serono, Inc. Rockland, MA ) | 44 µg | SC | Three times weekly | |
| Beta-interferon-1b (Extavia®, Bayer HealthCare Pharmaceuticals Inc., Montville, NJ) | 250 µg | SC | Every other day | |
| Glatiramer acetate (Copaxone®, TEVA Neuroscience, Inc., Overland Park, KS ) | 20 mg | SC | Daily | Local injection site reactions, postinjection reaction (flushing, chest tightness, palpitation, and dyspnea) and rare lipoatrophy with prolonged use |
| 40 mg | SC | Three times weekly | ||
| Second line therapies | ||||
| Natalizumab (Tysabri®, Biogen Idec, Cambridge, MA) | 300 mg | IV | Every 4 weeks | Hepatotoxicity, infusion reactions, progressive multifocal encephalopathy (PML) |
| Mitoxantrone (Novantrone®, EMD Serono, Inc. Rockland, MA) | Weight-based dose | IV | Every 3 months | Cardiotoxicity, secondary leukemia |
| Fingolimod (Gilenya®, Novartis Pharma Stein AG Stein, Switzerland) | 0.5 mg | Oral | Once daily | First dose bradycardia, atrioventricular block, herpes virus infection, macular edema, elevated blood pressure, rare risk of PML |
| Teriflunomide (Aubagio®, Genzyme Corporation, Cambridge, MA) | 7 and 14 mg | Oral | Once daily | Hair loss, headache, diarrhea, hepatotoxicity, teratogenicity, increased risk of infections due to lymphopenia |
| Dimethyl fumarate (Tecfidera®, Biogen Idec., Cambridge, MA ) | 240 mg | Oral | Twice daily | GI effects-nausea, diarrhea, abdominal pain, flushing, pruritus, liver enzyme elevation, lymphopenia, rare cases of PML |
| Alemtuzumab (Lemtrada®, Genzyme Corporation, Cambridge, MA ) | 12 mg or 24 mg | IV | Per day (5 days in year 1, 3 days in year 2) | Serious infusion reactions, secondary autoimmune diseases-thryoiditis, thrombocytopenia, anti-glomerual basement membrane disease, increased risk of malignancies—thyroid cancer, melanoma, lymphoproliferative disorders |
The mechanism of action of IMT used for treatment of MS is broad suppression of the immune response mediated by autoreactive lymphocytes; most of these are effective in relapsing remitting MS where inflammatory demyelination is the primary process (Weinstock-Guttman et al. 1995; Rudick et al. 1997; Damal et al. 2013). The goal of these therapies is to reduce the frequency of relapses and number of MRI lesions (new, enlarging and/or enhancing T2 lesions), and slow the disability progression. Most of these agents have shown good efficacy in patients with relapsing remitting MS and clinically isolated syndrome, however, their benefit in patients with progressive disease has been questionable (Filippini et al. 2013). The mechanism of action and side-effect profile of different IMTs are briefly discussed here with the exception of alemtuzumab, the latest medication to be approved for treatment of MS.
Beta-Interferon
Interferons (IFNs) are endogenous proteins that are involved in immune response against viral and bacterial agents and were the first class of disease modifying agents developed for treatment of MS. The beta-interferons (IFN-β) have multiple actions including stabilizing the BBB thereby limiting the entry of T cells into the CNS, modulating T- and B-cell function, and altering the expression of cytokines (Yong et al. 1998; Weber et al. 1999; Dhib-Jalbut 2002). Several different preparations of IFN- β are available and are listed in Table 2007b. Both IFN- β1a and IFN-β1b have shown similar efficacy and are considered first line agents for treating patients with relapsing MS and CIS (Rudick et al. 1997). Although two different trials with IFN- β1b showed conflicting results in secondary progressive MS, it may be indicated in patients with continued relapses (Kappos et al. 2004). The side effects of beta-interferon include flu-like symptoms, depression, liver enzyme elevation, thyroid abnormalities, leukopenia or anemia, and injection site reactions (Rudick et al. 1997).
Glatiramer Acetate
Glatiramer acetate (GA) or Copolymer 1 is a synthetic complex of four amino acids that mimics myelin basic protein (MBP), one of the autoantigens targeted by the T cells. Due to its structural similarity to MBP, GA blocks the formation of myelin reactive T cells and induces GA-specific regulatory T-cell expression and Th2 anti-inflammatory cytokine production (bystander suppression) (Wolinsky 1995; Rudick et al. 1997; Gran et al. 2000; Dhib-Jalbut 2002; Ruggieri et al. 2007). The clinical efficacy of GA in terms of reducing relapse rate and MRI lesions is similar to IFN-β, however, GA has somewhat limited effect on disability progression (Rudick et al. 1997; La Mantia et al. 2010). The side effect profile of GA is, however, more favorable and includes local injection site reactions, post injection reaction (flushing, chest tightness, palpitation, and dyspnea within minutes of injection with spontaneous resolution) and rare lipoatrophy with prolonged use.
Natalizumab
Natalizumab is a humanized monoclonal antibody that binds α4β1-integrin on lymphocytes blocking their interaction with VCAM-1 on endothelial cells thereby preventing the transmigration of lymphocytes across the BBB (Ransohoff 2007). Its superior efficacy has been demonstrated in two phase 3 studies with a robust effect on relapse rate reduction and disability progression (Miller et al. 2003; Polman et al. 2006). The major safety concern with natalizumab is progressive multifocal leukoencephalopathy (PML), a serious potentially fatal opportunistic brain infection caused by reactivation of JC virus (Yousry et al. 2006). As of December 2014, 514 cases of PML have been reported worldwide with postmarketing use of natalizumab (TYSABRI [natalizumab] 2014). The overall risk of PML in MS patients with natalizumab use is 3.78 per 1000 with a much higher risk (13/1000) among patients with prolonged duration of therapy (≥24 months), history of prior immunosuppressive therapy, and positive JC virus antibody status (Bloomgren et al. 2012; Information M, 2014). Due to the risk of PML, natalizumab now has a more limited use as a second line drug in patients with breakthrough disease or intolerable side effects with first line therapies.
Mitoxantrone
Mitoxantrone is a synthetic anthracendione antineoplastic agent; its immunomodulatory effects include suppression of T and B lymphocytes and macrophage proliferation. Mitoxantrone is indicated for reducing disability and relapse frequency in patients with worsening relapsing remitting and secondary-progressive MS, however, its use has been limited due to risk of dose-related cardiotoxicity and treatment-related leukemia (Fox 2006).
Oral Therapies
Three new oral medications have recently become available for treatment of relapsing MS: fingolimod, teriflunomide, and dimethylfumarate. The efficacy of these medications has been established in several phase 3 studies with comparable or somewhat better effect (compared to some injectable therapies) on relapse rate reduction, MRI lesions, and disability progression.
Fingolimod is a sphingosine-1-phosphate receptor (S1P1) modulator and was the first oral drug approved for treatment of MS. By binding to S1P1 receptor on the T cells, it prevents emigration of activated T cells from lymph nodes thereby limiting their entry into the CNS (Chiba et al. 1998; Pelletier and Hafler 2012). The potential side effects of fingolimod include first dose bradycardia, atrioventricular block, herpes virus infection, macular edema, elevated blood pressure, and a reported cases of PML (Cohen et al. 2010; Kappos et al. 2010; Samson 2013; Calic et al. 2015).
There have been a total of three reported cases of PML associated with use of fingolimod, two of these occurred in context of prior immunosuppressive therapy, the third most recent case, however, was reported in a patient with no prior immunosuppressive therapy after more than 4 years of fingolimod use (Calic et al. 2015; Case of PML reported in patient treated with Gilenya, 2015). A single case of sudden suspected cardiac death within 24 h of taking first dose of fingolimod was reported in December 2011 (Lindsey et al. 2012). Even though a direct association with the medication could not be established, the US Food and Drug Administration and European regulatory agency released new monitoring guidelines for first dose monitoring and the drug is now contraindicated in patients with history of cardiac disease or stroke and patients on antiarrhythmic medications.
Teriflunomide is an active metabolite of leflunomide (a drug used to treat rheumatoid arthritis) and is an inhibitor of enzyme dihyrdro-orotate dehydrogenase (DHODH) which interferes with denovo synthesis of pyrimidine in rapidly dividing cells (Oh and O’Connor 2013). Its anti-inflammatory effect in MS is believed to be mediated by reducing the activity of proliferating T and B lymphocytes. Teriflunomide does not affect the resting or slowly dividing hematopoietic cells which use the alternate DHODH independent “salvage pathway” for pyrimidine synthesis, therefore, preserving the basic homeostatic functions of these cells and immune surveillance (Oh and O’Connor 2013). Leflunomide is converted almost entirely into teriflunomide after absorption and taken at the recommended doses, both drugs result in similar plasma concentration of teriflunomide (AUBAGIO® (teriflunomide) Prescibing information. 2014). The short-term side effects of teriflunomide are relatively mild and include hair loss, headache, diarrhea, and elevated liver enzymes (O’Connor et al. 2011). Reduction in lymphocyte and neutrophil counts, elevated blood pressure, and a single case of latent tuberculosis are some of the other side effects reported (O’Connor et al. 2011; Confavreux et al. 2014). The potential teratogenecity of teriflunomide remains a major concern although several pregnancies reported during its clinical trial did not have any adverse outcome. Nevertheless, strict contraception is recommended to avoid pregnancy and a rapid elimination process is undertaken in women who become pregnant while taking teriflunomide as the drug can remain in the systems for 8 months to 2 years (Confavreux et al. 2014).
Dimethylfumarate (DMF) or BG12 is the latest oral therapy to be approved for treatment of MS. Related to fumaric acid ester which has been used for treatment of psoriasis in Germany since 1990s; BG12 is as an enteric coated formulation of DMF with improved GI tolerability. It is hydrolyzed to monomethyl fumarate soon after oral absorption. The mechanism of action of DMF involves inhibition of proinflammatory pathways via activation of nuclear factor erythroid 2-related factor 2 (Nrf-2) antioxidant response pathway (Linker et al. 2011). The most common side effect with DMF include nausea, diarrhea, abdominal pain which can be minimized by taking the medication with food and flushing which can be reduced by aspirin (Gold et al. 2012; Fox et al. 2014). Lymphopenia may occur although no increased infection risk was observed in phase 3 studies (Fox et al. 2012; Gold et al. 2012). There have been a few cases of PML reported with use of fumaric acid ester formulations in patients with psoriasis with pronounced prolonged lymphopenia being the major risk factor (Ermis et al. 2013; van Oosten et al. 2013; Sweetser et al. 2013). There has been a recent report of a fatal case of PML in a patient with multiple sclerosis treated with dimethyl fumarate (FDA Drug Safety Communication, 2014). In response to these cases of PML, JC virus antibody testing and close monitoring for lymphopenia has been suggested in patients initiating DMF therapy to improve surveillance.
The oral medications are currently considered second line due to a greater risk of serious side effects making their safety profile less favorable in patients with early and mild disease. The main safety concern with oral medications is the potential for prolonged immunosuppression increasing the risk of serious infections and also malignancies due to altered immune surveillance. A simplified treatment algorithm for patients with RRMS and CIS is outlined in Figure3.
Figure 3.
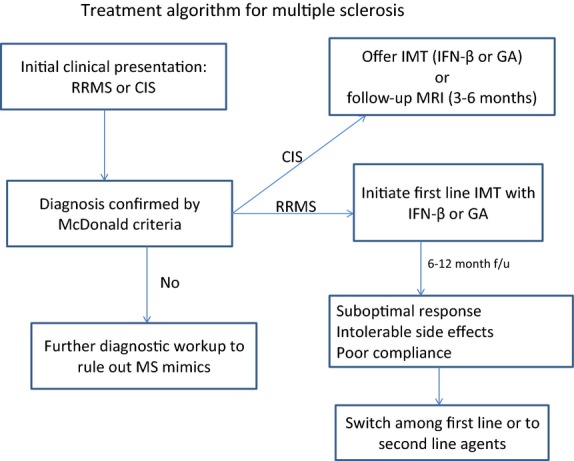
Treatment approach to patients with RRMS and CIS.
In summary, there has been significant progress in the field of MS with better understanding of immunopathogenesis, wider availability and use of MRI, and availability of disease modifying therapies. There are currently a wide range of therapeutic options to treat MS including the recently approved oral drugs. However, balancing the safety and efficacy of these drugs remains a challenge due to serious side effects associated with more effective therapies. Given the heterogeneity of MS, personalized treatment by recognizing specific genetic markers in individual patients has been proposed. Also, there is an urgent need for novel therapeutic agents that can prevent or minimize the neuronal and/or axonal degeneration occurring in patients with progressive MS.
Conflict of Interest
None declared.
References
- Aichele P, Bachmann MF, Hengartner H. Zinkernagel RM. Immunopathology or organ-specific autoimmunity as a consequence of virus infection. Immunol. Rev. 1996;152:21–45. doi: 10.1111/j.1600-065x.1996.tb00909.x. [DOI] [PubMed] [Google Scholar]
- Ascherio A. Environmental factors in multiple sclerosis. Expert Rev. Neurother. 2013;13:3–9. doi: 10.1586/14737175.2013.865866. [DOI] [PubMed] [Google Scholar]
- Ascherio A. Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann. Neurol. 2007a;61:288–299. doi: 10.1002/ana.21117. [DOI] [PubMed] [Google Scholar]
- Ascherio A. Munger KL. Environmental risk factors for multiple sclerosis. Part II: noninfectious factors. Ann. Neurol. 2007b;61:504–513. doi: 10.1002/ana.21141. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Munger KL. Simon KC. Vitamin D and multiple sclerosis. Lancet Neurol. 2010;9:599–612. doi: 10.1016/S1474-4422(10)70086-7. [DOI] [PubMed] [Google Scholar]
- AUBAGIO®. 2014. (teriflunomide) Prescibing information. Genzyme Corp. (Accessed July 23, 2014)
- Awad A, Hemmer B, Hartung HP, Kieseier B, Bennett JL. Stuve O. Analyses of cerebrospinal fluid in the diagnosis and monitoring of multiple sclerosis. J. Neuroimmunol. 2010;219:1–7. doi: 10.1016/j.jneuroim.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Barcellos LF, Oksenberg JR, Begovich AB, Martin ER, Schmidt S, Vittinghoff E, et al. HLA-DR2 dose effect on susceptibility to multiple sclerosis and influence on disease course. Am. J. Hum. Genet. 2003;72:710–716. doi: 10.1086/367781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkhof F, Bruck W, De Groot CJ, Bergers E, Hulshof S, Geurts J, et al. Remyelinated lesions in multiple sclerosis: magnetic resonance image appearance. Arch. Neurol. 2003;60:1073–1081. doi: 10.1001/archneur.60.8.1073. [DOI] [PubMed] [Google Scholar]
- Barnett MH. Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann. Neurol. 2004;55:458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Batoulis H, Addicks K. Kuerten S. Emerging concepts in autoimmune encephalomyelitis beyond the CD4/T(H)1 paradigm. Annals Anat. 2010;192:179–193. doi: 10.1016/j.aanat.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Berkovich R. Treatment of acute relapses in multiple sclerosis. Neurotherapeutics. 2013;10:97–105. doi: 10.1007/s13311-012-0160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N. Engl. J. Med. 2012;366:1870–1880. doi: 10.1056/NEJMoa1107829. [DOI] [PubMed] [Google Scholar]
- Bogdan FP, Pirko I, Lucchinetti FC. Pathology of multiple sclerosis: Where do we stand? Cont. Lifelong Learn. Neurol. 2013;19:901–921. doi: 10.1212/01.CON.0000433291.23091.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck W. Stadelmann C. The spectrum of multiple sclerosis: new lessons from pathology. Curr. Opin. Neurol. 2005;18:221–224. doi: 10.1097/01.wco.0000169736.60922.20. [DOI] [PubMed] [Google Scholar]
- Bruck W, Porada P, Poser S, Rieckmann P, Hanefeld F, Kretzschmar HA, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann. Neurol. 1995;38:788–796. doi: 10.1002/ana.410380514. [DOI] [PubMed] [Google Scholar]
- Calic Z, Cappelen-Smith C, Hodgkinson SJ, McDougall A, Cuganesan R. Brew BJ. Treatment of progressive multifocal leukoencephalopathy-immune reconstitution inflammatory syndrome with intravenous immunoglobulin in a patient with multiple sclerosis treated with fingolimod after discontinuation of natalizumab. J. Clin. Neurosci. 2015;22:598–600. doi: 10.1016/j.jocn.2014.08.016. [DOI] [PubMed] [Google Scholar]
- Case of PML reported in patient treated with Gilenya. 2015. ®. https://beta.mssociety.ca/research-news/article/case-of-pml-reported-in-patient-treated-with-gilenya.
- Chiba K, Yanagawa Y, Masubuchi Y, Kataoka H, Kawaguchi T, Ohtsuki M, et al. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. I. FTY720 selectively decreases the number of circulating mature lymphocytes by acceleration of lymphocyte homing. J. Immunol. 1998;160:5037–5044. [PubMed] [Google Scholar]
- Cifelli A, Arridge M, Jezzard P, Esiri MM, Palace J. Matthews PM. Thalamic neurodegeneration in multiple sclerosis. Ann. Neurol. 2002;52:650–653. doi: 10.1002/ana.10326. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- Compston A. Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Confavreux C, O’Connor P, Comi G, Freedman MS, Miller AE, Olsson TP, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13:247–256. doi: 10.1016/S1474-4422(13)70308-9. [DOI] [PubMed] [Google Scholar]
- Conradi S, Malzahn U, Schroter F, Paul F, Quill S, Spruth E, et al. Environmental factors in early childhood are associated with multiple sclerosis: a case-control study. BMC Neurol. 2011;11:123. doi: 10.1186/1471-2377-11-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correale J, Ysrraelit MC. Gaitan MI. Immunomodulatory effects of Vitamin D in multiple sclerosis. Brain. 2009;132:1146–1160. doi: 10.1093/brain/awp033. [DOI] [PubMed] [Google Scholar]
- Damal K, Stoker E. Foley JF. Optimizing therapeutics in the management of patients with multiple sclerosis: a review of drug efficacy, dosing, and mechanisms of action. Biologics. 2013;7:247–258. doi: 10.2147/BTT.S53007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhib-Jalbut S. Mechanisms of action of interferons and glatiramer acetate in multiple sclerosis. Neurology. 2002;58:S3–S9. doi: 10.1212/wnl.58.8_suppl_4.s3. [DOI] [PubMed] [Google Scholar]
- Ermis U, Weis J. Schulz JB. PML in a patient treated with fumaric acid. N. Engl. J. Med. 2013;368:1657–1658. doi: 10.1056/NEJMc1211805. [DOI] [PubMed] [Google Scholar]
- Farez MF, Fiol MP, Gaitan MI, Quintana FJ. Correale J. Sodium intake is associated with increased disease activity in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry. 2015;86:26–31. doi: 10.1136/jnnp-2014-307928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA Drug Safety Communication. 2014. FDA warns about case of rare brain infection PML with MS drug Tecfidera (dimethyl fumarate) http://www.fda.gov/Drugs/DrugSafety/ucm424625.htm. (Accessed January 8, 2015)
- Filippi M, Horsfield MA, Morrissey SP, MacManus DG, Rudge P, McDonald WI, et al. Quantitative brain MRI lesion load predicts the course of clinically isolated syndromes suggestive of multiple sclerosis. Neurology. 1994;44:635–641. doi: 10.1212/wnl.44.4.635. [DOI] [PubMed] [Google Scholar]
- Filippini G, Del Giovane C, Vacchi L, D’Amico R, Di Pietrantonj C, Beecher D, et al. Immunomodulators and immunosuppressants for multiple sclerosis: a network meta-analysis. Cochrane Database Syst. Rev. 2013;6:CD008933. doi: 10.1002/14651858.CD008933.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox EJ. Management of worsening multiple sclerosis with mitoxantrone: a review. Clin. Ther. 2006;28:461–474. doi: 10.1016/j.clinthera.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012;367:1087–1097. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- Fox RJ, Kita M, Cohan SL, Henson LJ, Zambrano J, Scannevin RH, et al. BG-12 (dimethyl fumarate): a review of mechanism of action, efficacy, and safety. Curr. Med. Res. Opin. 2014;30:251–262. doi: 10.1185/03007995.2013.849236. [DOI] [PubMed] [Google Scholar]
- Fragoso YD. Modifiable environmental factors in multiple sclerosis. Arq. Neuropsiquiatr. 2014;72:889–894. doi: 10.1590/0004-282x20140159. [DOI] [PubMed] [Google Scholar]
- Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132:1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman EM, Racke MK. Raine CS. Multiple sclerosis–the plaque and its pathogenesis. N. Engl. J. Med. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- Fujinami RS. Oldstone MB. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043–1045. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- Geurts JJ. Barkhof F. Grey matter pathology in multiple sclerosis. Lancet Neurol. 2008;7:841–851. doi: 10.1016/S1474-4422(08)70191-1. [DOI] [PubMed] [Google Scholar]
- Gold R. Wolinsky JS. Pathophysiology of multiple sclerosis and the place of teriflunomide. Acta Neurol. Scand. 2011;124:75–84. doi: 10.1111/j.1600-0404.2010.01444.x. [DOI] [PubMed] [Google Scholar]
- Gold R, Linington C. Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- Gran B, Hemmer B, Vergelli M, McFarland HF. Martin R. Molecular mimicry and multiple sclerosis: degenerate T-cell recognition and the induction of autoimmunity. Ann. Neurol. 1999;45:559–567. doi: 10.1002/1531-8249(199905)45:5<559::AID-ANA3>3.0.CO;2-Q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gran B, Tranquill LR, Chen M, Bielekova B, Zhou W, Dhib-Jalbut S, et al. Mechanisms of immunomodulation by glatiramer acetate. Neurology. 2000;55:1704–1714. doi: 10.1212/wnl.55.11.1704. [DOI] [PubMed] [Google Scholar]
- Hafler DA, Slavik JM, Anderson DE, O’Connor KC, De Jager P. Baecher-Allan C. Multiple sclerosis. Immunol. Rev. 2005;204:208–231. doi: 10.1111/j.0105-2896.2005.00240.x. [DOI] [PubMed] [Google Scholar]
- Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, et al. Risk alleles for multiple sclerosis identified by a genome wide study. N. Engl. J. Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C. Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann. Neurol. 1986;19:578–587. doi: 10.1002/ana.410190610. [DOI] [PubMed] [Google Scholar]
- Hemmer B, Archelos JJ. Hartung HP. New concepts in the immunopathogenesis of multiple sclerosis. Nat. Rev. Neurosci. 2002;3:291–301. doi: 10.1038/nrn784. [DOI] [PubMed] [Google Scholar]
- Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134:2755–2771. doi: 10.1093/brain/awr182. [DOI] [PubMed] [Google Scholar]
- Ingle GT, Sastre-Garriga J, Miller DH. Thompson AJ. Is inflammation important in early PPMS? a longitudinal MRI study. J. Neurol. Neurosurg. Psychiatry. 2005;76:1255–1258. doi: 10.1136/jnnp.2004.036590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese M. Multiple sclerosis: new insights and trends. AJNR Am. J. Neuroradiol. 2006;27:954–957. [PMC free article] [PubMed] [Google Scholar]
- Kantarci OH. Genetics and natural history of multiple sclerosis. Semin. Neurol. 2008;28:7–16. doi: 10.1055/s-2007-1019125. [DOI] [PubMed] [Google Scholar]
- Kappos L, Weinshenker B, Pozzilli C, Thompson AJ, Dahlke F, Beckmann K, et al. Interferon beta-1b in secondary progressive MS: a combined analysis of the two trials. Neurology. 2004;63:1779–1787. doi: 10.1212/01.wnl.0000145561.08973.4f. [DOI] [PubMed] [Google Scholar]
- Kappos L, Radue EW, O’Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- Karussis D. The diagnosis of multiple sclerosis and the various related demyelinating syndromes: a critical review. J. Autoimmun. 2014;48–49:134–142. doi: 10.1016/j.jaut.2014.01.022. [DOI] [PubMed] [Google Scholar]
- Kister I, Chamot E, Salter AR, Cutter GR, Bacon TE. Herbert J. Disability in multiple sclerosis: a reference for patients and clinicians. Neurology. 2013;80:1018–1024. doi: 10.1212/WNL.0b013e3182872855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–522. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Mantia L, Munari LM. Lovati R. Glatiramer acetate for multiple sclerosis. Cochrane Database Syst. Rev. 2010:CD004678. doi: 10.1002/14651858.CD004678.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey JW, Haden-Pinneri K, Memon NB. Buja LM. Sudden unexpected death on fingolimod. Mult. Scler. 2012;18:1507–1508. doi: 10.1177/1352458512438456. [DOI] [PubMed] [Google Scholar]
- Link H. Huang YM. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. J. Neuroimmunol. 2006;180:17–28. doi: 10.1016/j.jneuroim.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- Lublin FD. Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46:907–911. doi: 10.1212/wnl.46.4.907. [DOI] [PubMed] [Google Scholar]
- Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83:278–286. doi: 10.1212/WNL.0000000000000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M. Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Lucchinetti CF, Gavrilova RH, Metz I, Parisi JE, Scheithauer BW, Weigand S, et al. Clinical and radiographic spectrum of pathologically confirmed tumefactive multiple sclerosis. Brain. 2008;131:1759–1775. doi: 10.1093/brain/awn098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, et al. Inflammatory cortical demyelination in early multiple sclerosis. N. Engl. J. Med. 2011;365:2188–2197. doi: 10.1056/NEJMoa1100648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr WT, Pittock SJ, McClelland RL, Jorgensen NW, Noseworthy JH. Rodriguez M. Incidence and prevalence of multiple sclerosis in Olmsted County, Minnesota, 1985–2000. Neurology. 2003;61:1373–1377. doi: 10.1212/01.wnl.0000094316.90240.eb. [DOI] [PubMed] [Google Scholar]
- Miller DH. Leary SM. Primary-progressive multiple sclerosis. Lancet Neurol. 2007;6:903–912. doi: 10.1016/S1474-4422(07)70243-0. [DOI] [PubMed] [Google Scholar]
- Miller SD, Vanderlugt CL, Begolka WS, Pao W, Yauch RL, Neville KL, et al. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 1997;3:1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GPA, Libonati MA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2003;348:15–23. doi: 10.1056/NEJMoa020696. [DOI] [PubMed] [Google Scholar]
- Miller D, Barkhof F, Montalban X, Thompson A. Filippi M. Clinically isolated syndromes suggestive of multiple sclerosis, part I: natural history, pathogenesis, diagnosis, and prognosis. Lancet Neurol. 2005;4:281–288. doi: 10.1016/S1474-4422(05)70071-5. [DOI] [PubMed] [Google Scholar]
- Montalban X, Tintore M, Swanton J, Barkhof F, Fazekas F, Filippi M, et al. MRI criteria for MS in patients with clinically isolated syndromes. Neurology. 2010;74:427–434. doi: 10.1212/WNL.0b013e3181cec45c. [DOI] [PubMed] [Google Scholar]
- Munger KL, Zhang SM, O’Reilly E, Hernán MA, Olek MJ, Willett WC, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology. 2004;62:60–65. doi: 10.1212/01.wnl.0000101723.79681.38. [DOI] [PubMed] [Google Scholar]
- Munger KL, Levin LI, Hollis BW, Howard NS. Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006;296:2832–2838. doi: 10.1001/jama.296.23.2832. [DOI] [PubMed] [Google Scholar]
- Naismith RT, Piccio L, Lyons JA, Lauber J, Tutlam NT, Parks BJ, et al. Rituximab add-on therapy for breakthrough relapsing multiple sclerosis: a 52-week phase II trial. Neurology. 2010;74:1860–1867. doi: 10.1212/WNL.0b013e3181e24373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor KC, Bar-Or A. Hafler DA. The neuroimmunology of multiple sclerosis: possible roles of T and B lymphocytes in immunopathogenesis. J. Clin. Immunol. 2001;21:81–92. doi: 10.1023/a:1011064007686. [DOI] [PubMed] [Google Scholar]
- O’Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N. Engl. J. Med. 2011;365:1293–1303. doi: 10.1056/NEJMoa1014656. [DOI] [PubMed] [Google Scholar]
- Oh J. O’Connor PW. An update of teriflunomide for treatment of multiple sclerosis. Ther. Clin. Risk Manag. 2013;9:177–190. doi: 10.2147/TCRM.S30947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosten BW, Killestein J, Barkhof F, Polman CH. Wattjes MP. PML in a patient treated with dimethyl fumarate from a compounding pharmacy. N. Engl. J. Med. 2013;368:1658–1659. doi: 10.1056/NEJMc1215357. [DOI] [PubMed] [Google Scholar]
- Orton SM, Herrera BM, Yee IM, Valdar W, Ramagopalan SV, Sadovnick AD, et al. Sex ratio of multiple sclerosis in Canada: a longitudinal study. Lancet Neurol. 2006;5:932–936. doi: 10.1016/S1474-4422(06)70581-6. [DOI] [PubMed] [Google Scholar]
- Pelletier D. Hafler DA. Fingolimod for multiple sclerosis. N. Engl. J. Med. 2012;366:339–347. doi: 10.1056/NEJMct1101691. [DOI] [PubMed] [Google Scholar]
- Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu BF. Lucchinetti CF. Pathology of demyelinating diseases. Annu. Rev. Pathol. 2012;7:185–217. doi: 10.1146/annurev-pathol-011811-132443. [DOI] [PubMed] [Google Scholar]
- Prineas JW, Kwon EE, Cho ES, Sharer LR, Barnett MH, Oleszak EL, et al. Immunopathology of secondary-progressive multiple sclerosis. Ann. Neurol. 2001;50:646–657. doi: 10.1002/ana.1255. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. Natalizumab for multiple sclerosis. N. Engl. J. Med. 2007;356:2622–2629. doi: 10.1056/NEJMct071462. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Cohen JA, Weinstock-Guttman B, Kinkel RP. Ransohoff RM. Management of multiple sclerosis. N. Engl. J. Med. 1997;337:1604–1611. doi: 10.1056/NEJM199711273372207. [DOI] [PubMed] [Google Scholar]
- Ruggieri M, Avolio C, Livrea P. Trojano M. Glatiramer acetate in multiple sclerosis: a review. CNS Drug Rev. 2007;13:178–191. doi: 10.1111/j.1527-3458.2007.00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson K. PML in suspected MS patient taking Fingolimod Raises FDA concerns. Neurology Today. 2013;13:37. [Google Scholar]
- Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selter CR. Hemmer B. Update on immunopathogenesis and immunotherapy in multiple sclerosis. Immuno. Targets Therapy. 2013;2:21–30. doi: 10.2147/ITT.S31813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson S, Jr, Blizzard L, Otahal P, Van der Mei I. Taylor B. Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. J. Neurol. Neurosurg. Psychiatry. 2011;82:1132–1141. doi: 10.1136/jnnp.2011.240432. [DOI] [PubMed] [Google Scholar]
- Sobel R. Moore W. Vol. 2. London: UK Oxford Univ. Press; 2008. pp. 1513–1608. Demyelinating diseases. Greenfield’s neuropathology, [Google Scholar]
- Sotgiu S, Pugliatti M, Sotgiu A, Sanna A. Rosati G. Does the “hygiene hypothesis” provide an explanation for the high prevalence of multiple sclerosis in Sardinia? Autoimmunity. 2003;36:257–260. doi: 10.1080/0891693031000151607. [DOI] [PubMed] [Google Scholar]
- Stadelmann C, Ludwin S, Tabira T, Guseo A, Lucchinetti CF, Leel-Ossy L, et al. Tissue preconditioning may explain concentric lesions in Balo’s type of multiple sclerosis. Brain. 2005;128:979–987. doi: 10.1093/brain/awh457. [DOI] [PubMed] [Google Scholar]
- Swanton JK, Fernando K, Dalton CM, Miszkiel KA, Thompson AJ, Plant GT, et al. Modification of MRI criteria for multiple sclerosis in patients with clinically isolated syndromes. J. Neurol. Neurosurg. Psychiatry. 2006;77:830–833. doi: 10.1136/jnnp.2005.073247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton JK, Rovira A, Tintore M, Altman DR, Barkhof F, Filippi M, et al. MRI criteria for multiple sclerosis in patients presenting with clinically isolated syndromes: a multicentre retrospective study. Lancet Neurol. 2007;6:677–686. doi: 10.1016/S1474-4422(07)70176-X. [DOI] [PubMed] [Google Scholar]
- Sweetser MT, Dawson KT. Bozic C. Case reports of PML in patients treated for psoriasis. N. Engl. J. Med. 2013;369:1082. doi: 10.1056/NEJMc1307680. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S. Bo L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- TYSABRI®. 2014. medinfo@biogenidec.com(natalizumab): PML Incidence in Patients Receiving TYSABRI ( December ). Medical Information, http://medinfo.biogenidec.com.
- Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 2008;172:146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Janovskaja J, Polak T, Poser S. Rieckmann P. Effect of interferon beta on human myelin basic protein-specific T-cell lines: comparison of IFNbeta-1a and IFNbeta-1b. Neurology. 1999;52:1069–1071. doi: 10.1212/wnl.52.5.1069. [DOI] [PubMed] [Google Scholar]
- Weinshenker BG. Epidemiology of multiple sclerosis. Neurol. Clin. 1996;14:291–308. doi: 10.1016/s0733-8619(05)70257-7. [DOI] [PubMed] [Google Scholar]
- Weinshenker BG, Bass B, Rice GP, Noseworthy J, Carriere W, Baskerville J, et al. The natural history of multiple sclerosis: a geographically based study. I. Clinical course and disability. Brain. 1989;112(Pt 1):133–146. doi: 10.1093/brain/112.1.133. [DOI] [PubMed] [Google Scholar]
- Weinstock-Guttman B, Ransohoff RM, Kinkel RP. Rudick RA. The interferons: biological effects, mechanisms of action, and use in multiple sclerosis. Ann. Neurol. 1995;37:7–15. doi: 10.1002/ana.410370105. [DOI] [PubMed] [Google Scholar]
- Williams R, Rigby AS, Airey M, Robinson M. Ford H. Multiple sclerosis: it epidemiological, genetic, and health care impact. J. Epidemiol. Community Health. 1995;49:563–569. doi: 10.1136/jech.49.6.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingerchuk DM. Smoking: effects on multiple sclerosis susceptibility and disease progression. Therapeut. Advan. Neurol. Dis. 2012;5:13–22. doi: 10.1177/1756285611425694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolinsky JS. Copolymer 1: a most reasonable alternative therapy for early relapsing-remitting multiple sclerosis with mild disability. Neurology. 1995;45:1245–1247. doi: 10.1212/wnl.45.7.1245. [DOI] [PubMed] [Google Scholar]
- Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig KW. Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong VW. Immunopathogenesis of multiple sclerosis. Contin. Lifelong Learn. Neurol. 2004;10:11–27. [Google Scholar]
- Yong VW, Chabot S, Stuve O. Williams G. Interferon beta in the treatment of multiple sclerosis: mechanisms of action. Neurology. 1998;51:682–689. doi: 10.1212/wnl.51.3.682. [DOI] [PubMed] [Google Scholar]
- Yousry TA, Major EO, Ryschkewitsch C, Fahle G, Fischer S, Hou J, et al. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. N. Engl. J. Med. 2006;354:924–933. doi: 10.1056/NEJMoa054693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zettl UK, Stuve O. Patejdl R. Immune-mediated CNS diseases: a review on nosological classification and clinical features. Autoimmun. Rev. 2012;11:167–173. doi: 10.1016/j.autrev.2011.05.008. [DOI] [PubMed] [Google Scholar]