

Abstract
Noonan syndrome (NS) is an autosomal dominant genetic disorder characterized by short stature, craniofacial dysmorphism, and congenital heart defects. A significant fraction of NS-patients also develop myeloproliferative disorders. The penetrance of these defects varies considerably among patients. In this study, we have examined the effect of 2 genetic backgrounds (C57BL/6J.OlaHsd and 129S2/SvPasCrl) on the phenotypes displayed by a mouse model of NS induced by germline expression of the mutated K-RasV14I allele, one of the most frequent NS-KRAS mutations. Our results suggest the presence of genetic modifiers associated to the genetic background that are essential for heart development and function at early stages of postnatal life as well as in the severity of the haematopoietic alterations.
Keywords: developmental disorders, genotype-phenotype correlation, genetic backgrounds, mouse models, modifiers, Noonan Syndrome, Rasopathies, Ras oncogenes
Introduction
Noonan syndrome (NS) is a relatively frequent developmental disorder (incidence of about 1/1000–1/2500 newborns) characterized by a broad spectrum of clinical symptoms including craniofacial dysmorphism, short stature, cardiovascular and skeletal defects, delayed puberty, and learning impairment.1,2 It belongs to a group of clinically related developmental disorders known as RASopathies caused by germline mutations in genes that encode components or regulators of the RAS/MAPK signaling pathway. Genes mutated in NS include PTPN11, SOS1, KRAS, NRAS, RAF1, BRAF, MEK1, SHOC2, CBL, RIT1 and RRAS.1,3–5 Among these genes, KRAS has been found mutated in around 5% of patients.6
Although there are some genotype-phenotype trends, none of the features of NS patients appear to exclusively correlate with a particular genotype. For instance, PTPN11-associated NS is linked to a high incidence of pulmonary stenosis and atrial septal defects, bleeding diathesis, short stature, and juvenile myelomonocytic leukemia (JMML). In contrast, the incidence of hypertrophic cardiomyopathy (HCM) and coarctation of the aorta is lower in patients with these mutations.2,7 On the other hand, SOS1-associated NS patients are mostly characterized by a higher prevalence of ectodermal abnormalities that are generally associated with a lower incidence of intellectual disability, short stature, atrial septal defects, and cryptorchidism.2,8 NS patients with RAF1 mutations are associated with HCM and a predisposition to hyperpigmented cutaneous lesions,9 whereas those carrying mutations in the highly related BRAF locus display neonatal growth failure, feeding problems, hypotonia, and a higher prevalence of multiple nevi and dark colored lentigines.10 Finally, patients with KRAS mutations present a severe medical and cognitive impairment. For this reason, vertical familial transmission in KRAS-associated NS patients is less frequent.2 The low number of patients carrying a KRAS-associated NS mutation (less than 5%) and the different KRAS-associated mutations described make very difficult to establish a solid genotype-phenotype correlation, although these patients display a global developmental delay and their mental retardation is more prevalent than in patients with PTPN11 and SOS1 mutations.9,11
Moreover, the phenotypic variability of NS patients has been observed even between members of the same family.12-14 Interestingly, there have been cases of apparently unaffected relatives identified after the diagnosis of their affected children.12,13 The lack of a robust genotype-phenotype correlation suggests that the penetrance of this syndrome cannot be completely attributed to the mutational spectrum, suggesting that genetic modifiers and different environmental factors must influence the type and grade of alterations suffered by these patients. The genetic variability of NS makes the establishment of a reliable genotype-phenotype correlation essential for the management of these patients.
During the last decade, scientists have generated genetically-engineered mouse (GEM) models of NS carrying germline mutations in their Ptpn11 and Raf1 loci. These mice have helped to illustrate the impact of the genetic background in some of their phenotypic alterations.15,16 For instance, backcross studies showed that while Ptpn11+/D61G mice on a 129S6/SvEv genetic background display nearly normal viability, mice carrying this mutation on a C57BL6/J background were no longer viable due to severe cardiac defects.15 Likewise, the NS-associated Raf1+/L613V mice were viable on a mixed genetic background but not when backcrossed to C57BL6/J mice.16 The incomplete penetrance of the growth, facial dysmorphia, and viability phenotypes of Raf1D486N/D486N mice on a mixed 129Sv/C57BL/6 background, as well as the identification of a 129Sv locus on mouse chromosome 8 that is strongly linked to the mutation, suggest the existence of modifier alleles in the 129Sv strain that could affect the disease phenotype.17,18
We have recently reported the generation and characterization of a GEM model that carries the K-RasV14I mutation. This strain recapitulates most of the alterations described in NS patients, including short stature, facial dysmorphism, cardiac defects, and a myeloproliferative disease (MPD) resembling JMML.19 Here, we report the phenotypic characterization of this GEM model in the context of the C57BL/6J.OlaHsd and 129S2/SvPasCrl genetic backgrounds. Our results support the concept that the limited genotype-phenotype correlation observed in NS patients likely results from genetic modifiers. Moreover, our observations highlight the need to identify such modifiers, since they could play important roles for the treatment of these patients.
Results
Genetic background modulates perinatal viability and life span of K-RasV14I “Noonan” mice
We have recently generated a mouse model for NS carrying KRAS mutation by genetically modifying codon 14 of the endogenou mouse K-Ras locus in embryonic stem (ES) cells.19 These ES cells were derived from 129S2/Sv mice due to their higher levels of germline transmission. This modification resulted in a K-Ras allele that encodes an isoleucine residue at position 14 of the K-Ras protein instead of the valine residue present in the wild-type protein. These engineered K-Ras+/V14I ES cells were used to generate chimeric mice that were subsequently crossed to the most widely used inbred strain, C57BL/6J.OlaHsd (from now B6), according to standard procedures. As a consequence, the resulting K-Ras+/V14I strain had a mixed B6/129 genetic background. These mice were born at the expected Mendelian ratios, indicating that the V14I mutation did not affect embryonic development. However, when we crossed these mice among themselves, the resulting homozygous K-RasV14I/V14I (from now K-RasV14I) animals displayed high perinatal lethality (only 10% of homozygous mice, obtained from crosses between heterozygous mice, were alive after birth), mainly due to cardiovascular defects.19
Next, we backcrossed the B6/129 K-Ras+/V14I mice to 129S2/SvPasCrl mice (from now on 129) or B6 wild-type animals for at least 7 generations. All heterozygous mice were viable regardless of genetic background. However, homozygous K-RasV14I mice backcrossed to B6 mice displayed complete perinatal lethality. Although analysis of E13.5 and E18.5 embryos revealed expected Mendelian ratios (data not shown), no homozygous mice were found at postnatal day 0 (P0) (Table 1). Thus, suggesting that, in the B6 genetic background, the cardiac defects responsible for the lethality of mice in the mixed B6/129 background become exacerbated and homozygous mice die of cardiac failure either during the late stages of embryonic development or during birth. Finally, homozygous K-RasV14I mice backcrossed to 129 mice showed high perinatal lethality similar to that observed in homozygous mice in the mixed B6/129 genetic background.19
Table 1.
Progeny at birth (postnatal day P0) from crosses between B6 K-Ras+/V14I mice. Pregnant females were either no treated or treated with the MEK inhibitor PD0325901
| Treatment | n | K-Ras+/+ | K-Ras+/V14I | K-RasV14I |
|---|---|---|---|---|
| Untreated | 14 | 3(21%) | 11(79%) | 0(0%) |
| PD0325901 | 6 | 2(33%) | 1(17%) | 3(50%) |
Interestingly, exposure of pregnant mothers to the MEK inhibitor PD0325901 (1 mg/kg of body weight, daily intraperitoneal injection) from E7.5 until P9, followed by treatment of the pups (1 mg/kg of body weight, every other day by intraperitoneal injection) until P21, rescued the lethality observed for homozygous K-RasV14I mice in the B6 background (Table 1), as previously reported in the mixed B6/129 K-RasV14I model19 and in mice expressing the NS-associated Sos1 mutation, E846K.20 K-RasV14I mice exposed to the MEK inhibitor until weaning survived for at least 6 months, time when the animals were sacrificed. These mice displayed normal hearts although they had enlarged spleens affected by MPD (data not shown). These results strengthen the idea that the perinatal lethality observed in B6 K-RasV14I mice is due to increased K-Ras signaling in heart tissue, a defect that could be blocked by exposure to the MEK inhibitor.
K-Ras+/V14I and K-RasV14I mice of a mixed B6/129 background presented reduced life span.19 Now we investigated whether the lifespan of these mice could also be affected by the genetic background. Interestingly, the lifespan of B6 K-Ras+/V14I mice was similar to that of B6/129 heterozygous mice (half-life of 64 weeks vs. 62 weeks). However, backcrossing K-Ras+/V14I mice into the 129 genetic background resulted in increased life span with a half-life of 82 weeks (a 32% increase in the life span compared to B6/129 heterozygous mice). Similar results were obtained for homozygous K-RasV14I mice, which displayed a half-life of 57 weeks (a 58% increase in the life span compared to B6/129 homozygous mice, which had a life span of 36 weeks) (Fig. 1).
Figure 1.
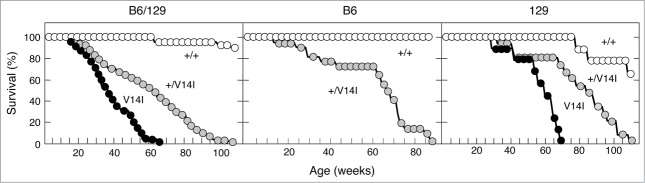
Phenotypic consequences of the genetic backgrounds on the survival rate. (Left) Survival curve of wild-type (n = 25) (+/+, open circles), K-Ras+/V14I (n = 68) (+/V14I, gray circles), and K-RasV14I (n = 30) (V14I, solid circles) B6/129 mice. (Middle) Survival curve of wild-type (n = 5) (+/+, open circles) and K-Ras+/V14I (n = 22) (+/V14I, gray circles) B6 mice. (Right) Survival curve of wild-type (n = 9) (+/+, open circles), K-Ras+/V14I (n = 15) (+/V14, gray circles) and K-RasV14I(n = 9) (V14I, solid circles) 129 mice. Note, the left panel was previously published,19 included here for comparison.
Phenotypic consequences of the genetic background on the body size
Next, we analyzed the effect of the different genetic backgrounds on body weight and body size. As illustrated in Figure 2, the body weight and body size of heterozygous K-Ras+/V14I mice were similar to that of wild-type littermates in the 3 genetic backgrounds, B6/129, B6 and 129. However, the genetic background played a role in homozygous K-RasV14I mice. For instance, 129 homozygous mice (n = 8) displayed significantly reduced body size and body weight compared to their wild-type littermates. This reduction in size was not apparent at birth. However, they were significantly smaller at weaning (Fig. 2). Moreover, at 4 weeks of age, K-RasV14I males weighed only 69% [9.29 ± 3.02 g (n = 8) vs. 13.62 ± 2.06 g (n = 14)] of their wild-type littermates. In addition, they were 22% shorter [(6.6 ± 5.98 cm (n = 8) vs. 7.5 ± 4.97 cm (n = 14)] (Fig. 2). These results are reminiscent of those previously observed in homozygous mice in the mixed B6/129 background.19 However, whereas these differences were maintained in older 129 K-RasV14I mice (Fig. 2), they became ameliorated with time in a mixed B6/129 background.19
Figure 2.
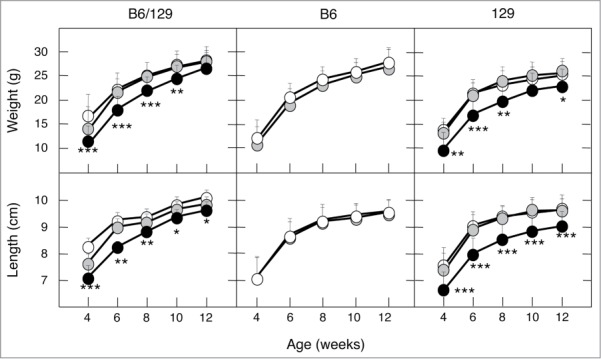
Phenotypic consequences of the genetic background on body size. Growth curves of male mice in mixed B6/129, B6 and 129 (F5). (Left) Body weight and body length of wild-type (n = 24 and 6, respectively), K-Ras+/V14I (n = 18 and 14, respectively) and K-RasV14I (n = 18 and 7, respectively) B6/129 male mice. (Middle) Body weight and body length of wild-type (n = 30) and K-Ras+/V14I (n = 25) B6 male mice. (Right) Body weight and body length of wild-type (n = 14), K-Ras+/V14I (n = 21) and K-RasV14I (n = 8) 129 male mice. Error bars indicate SD. *P < 0.05; **P < 0.01; ***P < 0.001. Note, the left panel was previously published,19 included here for comparison.
Phenotypic consequences of the genetic background on the heart
To determine the effect of modifier alleles on the cardiac phenotype, we sacrificed 4 month-old mutant and control mice after the fifth-generation backcross onto B6 or 129 backgrounds. Heart weight and heart/body weight ratio of 4 month-old B6 and 129 heterozygous K-Ras+/V14I mice were higher compared to wild-type littermates. This increase in weight was similar in both B6 and 129 genetic backgrounds and comparable to mice of the mixed B6/129 background. Hearts of 4 month-old K-Ras+/V14I mice were 24% heavier in the B6 background (n = 14), 26% heavier in the 129 background (n = 10), and 16% heavier in the B6/129 background (n = 30) than wild-type littermates (Fig. 3). The heart weight of homozygous K-RasV14I animals was also larger than in wild-type mice, regardless of the genetic background. However, the heart weight in the 129 mice was smaller than in animals of mix background. As illustrated in Figure 3, whereas the hearts of homozygous 129 K-RasV14I animals (n = 5) were 32% heavier than that of their wild-type littermates, the hearts of homozygous animals in the mixed B6/129 genetic background (n = 13) were 57% heavier than the hearts of the corresponding wild-type littermates.
Figure 3.
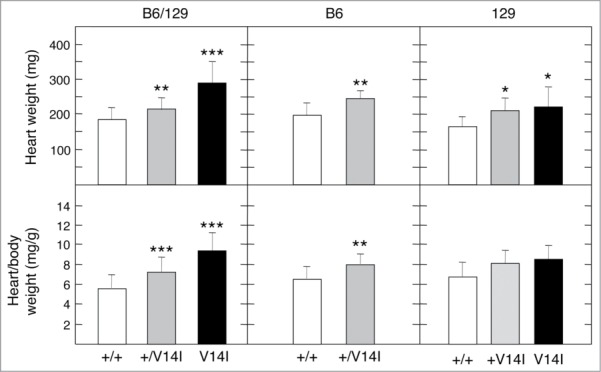
Phenotypic consequences of the genetic background on the heart. Heart weight and heart/body weight ratio of mixed B6/129, B6 and 129 (F5) 4 month-old male mice. (Left) Heart weight and heart/body weight ratio of wild-type (n = 22) (+/+, open bars), K-Ras+/V14I (n = 30) (+/V14I, gray bars) and K-RasV14I (n = 13) (V14I, solid bars) B6/129 male mice. (Middle) Heart weight and heart/body weight ratio of wild-type (n = 9) (+/+, open bars) and K-Ras+/V14I (n = 14) (+/V14I, gray bars) B6 male mice. (Rigth) Heart weight and heart/body weight ratio of wild-type (n = 10) (+/+, open bars), K-Ras+/V14I (n = 10) (+/V14I, gray bars) and K-RasV14I (n = 5) (V14I, solid bars) B6 male mice. Error bars indicate SD. *P < 0.05; **P < 0.01; ***P < 0.001. Note, the left panel was previously published,19 included here for comparison.
Phenotypic consequences of the genetic background on haematopoietic alterations
Heterozygous K-Ras+/V14I mice displayed haematopoietic alterations regardless of their genetic backgrounds. Yet, the phenotypic defects appeared to be more pronounced in mice backcrossed onto the B6 background. For instance, 4 month-old B6 K-Ras+/V14I mice displayed an increase of leukocytes in their peripheral blood, mainly due to expansion of the neutrophil, eosinophil, and basophil populations (n = 14). These populations were significantly increased in B6 K-Ras+/V14I mice compared to the control littermates [2.71 ± 1.56 109/l (n = 11) vs. 1.17 ± 0.41 109/l (n = 6)] (Table 2). In contrast, no significant differences were found in the B6/129 K-Ras+/V14I mice when compared to wild-type littermates [2.28 ± 2.14 109/l (n = 15) vs. 0.81 ± 0.39 109/l (n = 10)] (Table 2).19 B6 K-Ras+/V14I animals were also anemic and suffered from thrombocytopenia, displaying 79% less red blood cells (RBC) and 24% less platelets compared to their wild-type littermates (Table 2). However, in the B6/129 K-Ras+/V14I mice, these parameters were almost similar to those observed in the wild-type littermates (Table 2).19 Indeed, their spleens were, on average, 3 times larger than those of wild-type mice (n = 9) [368.1 ± 0.10 mg (n = 14) vs. 118.6 ± 0.03 mg (n = 9)]. Heterozygous K-Ras+/V14I mice of B6/129 (n = 22) and 129 (n = 10) backgrounds also displayed splenomegaly. However, the size of their spleens, on average, was only 2-fold larger than that of their corresponding wild-type littermates [B6/129: 186.9 ± 0.12 mg (n = 22) vs. 94.9 ± 0.03 mg (n = 13); 129: 109.1 ± 0.04 mg (n = 10) vs. 63.4 ± 0.01 mg (n = 10)]. Similar results were obtained with the spleen/body weight ratios [B6/129: 5.96 ± 0.0031 mg (n = 22) vs. 2.78 ± 0.0007 mg (n = 13); B6: 3.92 ± 0.0033 mg (n = 14) vs. 1.20 ± 0.0010 mg (n = 9); 129: 4.22 ± 0.0017 mg (n = 10) vs. 2.57 ± 0.0004 mg (n = 10)] (Fig. 4). Hence, the presence of modifiers in the B6 background seems to have an important impact on the haematopoietic alterations. Yet, as mentioned before, these quantitative differences did not have a significant influence on the life span of B6 heterozygous mice. In contrast, 129 K-Ras+/V14I mice survived significantly longer than the B6/129 K-Ras+/V14I mice.
Table 2.
Blood cells counts of 4 month-old B6/129, B6 and 129 mice
| B6/129 mice |
|||||||
|---|---|---|---|---|---|---|---|
| K-Ras+/+ (n = 10) | K-Ras+/V14I (n = 15) | K-RasV14I (n = 10) | p value +/+ vs +/V14I | p value +/+ vs V14I | |||
| WBC (109/l) | 9.76 ± 3.12 | 12.16 ± 2.15 | 16.77 ± 5.57 | 0.0825 | NS | 0.0100 | ** |
| LYM (109/l) | 8.43 ± 2.91 | 8.53 ± 1.86 | 7.37 ± 2.94 | 0.9363 | NS | 0.5148 | NS |
| MID (109/l) | 0.43 ± 0.26 | 0.53 ± 0.31 | 0.83 ± 0.87 | 0.5316 | NS | 0.3306 | NS |
| GRA (109/l) | 0.81 ± 0.39 | 2.28 ± 2.14 | 8.87 ± 4.97 | 0.0747 | NS | 0.0122 | * |
| RBC (1012/l) | 9.36 ± 0.33 | 9.07 ± 1.47 | 6.21 ± 2.08 | 0.1441 | NS | 0.0010 | *** |
| PLT (109/l) | 714 ± 265 | 547.7 ± 276 | 344.5 ± 129 | 0.2557 | NS | 0.1009 | NS |
|
B6 mice (F5) |
|||||||
| |
K-Ras+/+ (n = 6) |
K-Ras+/V14I (n = 11) |
p value +/+ vs +/V14I |
||||
| WBC (109/l) | 9.96 ± 0.14 | 13.80 ± 5.56 | 0.1413 | NS | |||
| LYM (109/l) | 8.42 ± 2.96 | 10.71 ± 5.36 | 0.3505 | NS | |||
| MID (109/l) | 0.38 ± 0.44 | 0.38 ± 0.18 | 0.9969 | NS | |||
| GRA (109/l) | 1.17 ± 0.41 | 2.71 ± 1.56 | 0.0089 | ** | |||
| RBC (1012/l) | 10.62 ± 1.03 | 8.38 ± 1.26 | 0.0022 | ** | |||
| PLT (109/l) | 959.6 ± 425.1 | 230.0 ± 163.3 | 0.0142 | * | |||
|
129 mice (F5) |
|||||||
| |
K-Ras+/+ (n = 9) |
K-Ras+/V14I (n = 10) |
K-RasV14I (n = 6) |
p value +/+ vs +/V14I |
p value+/+ vs V14I |
||
| WBC (109/l) | 7.33 ± 2.19 | 9.07 ± 2.76 | 15.64 ± 7.04 | 0.1510 | NS | 0.0332 | * |
| LYM (109/l) | 6.13 ± 1.73 | 7.57 ± 2.78 | 8.84 ± 3.83 | 0.1995 | NS | 0.1530 | NS |
| MID (109/l) | 0.19 ± 0.12 | 0.29 ± 0.15 | 0.90 ± 0.74 | 0.1369 | NS | 0.0981 | NS |
| GRA (109/l) | 1.02 ± 0.69 | 1.21 ± 0.59 | 6.04 ± 5.17 | 0.5173 | NS | 0.0630 | NS |
| RBC (1012/l) | 10.43 ± 1.68 | 10.85 ± 0.88 | 10.36 ± 0.67 | 0.9241 | NS | 0.4952 | NS |
| PLT (109/l) | 305.0 ± 153.7 | 363.89 ± 190.7 | 182.67 ± 147 | 0.3721 | NS | 0.2247 | NS |
WBC: white blood cells; LYM: lymphocytes; MID: monocytes and some eosinophiles; GRA: granulocytes; RBD: red blood cells; PLT: platelets. Numbers shown are the media ± SD. Statistical significance was assessed by 2-tailed Student's t-test.
*P < 0.05; **P < 0.01;
***P <0.001; NS, not significant.
Note, the B6/129 data was previously published,19 included here for comparison.
Figure 4.
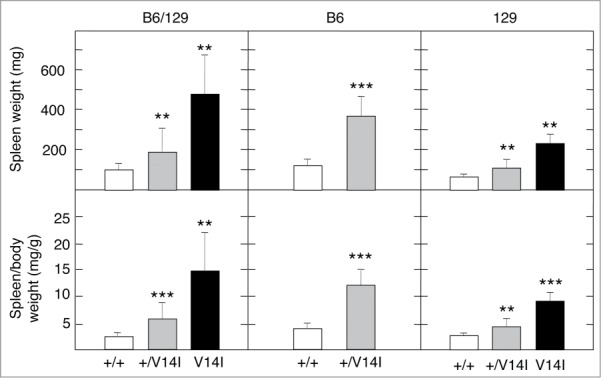
Phenotypic consequences of the genetic background on haematopoietic alterations. Spleen weight and spleen/body weight ratio of mixed B6/129, B6 and 129 (F5) 4 month-old male mice. (Left) Spleen weight and spleen/body weight ratio of wild-type (n = 13) (+/+, open bars), K-Ras+/V14I (n = 22) (+/V14I, gray bars) and K-RasV14I (n = 12) (V14I, solid bars) B6/129 male mice. (Middle) Spleen weight and spleen/body weight ratio of wild-type (n = 9) (+/+, open bars) and K-Ras+/V14I (n = 14) (+/V14I, gray bars) B6 male mice. (Rigth) Spleen weight and spleen/body weight ratio of wild-type (n = 10) (+/+, open bars), K-Ras+/V14I (n = 10) (+/V14I, gray bars) and K-RasV14I (n = 5) (V14I, solid bars) B6 male mice. Error bars indicate SD. *P < 0.05; **P < 0.01; ***P < 0.001. Note, the left panel was previously published,19 included here for comparison.
Homozygous K-RasV14I mice displayed an exacerbated phenotype. For instance, the spleen of mixed B6/129 homozygous mice (n = 12) was 5 times bigger than the spleen of control mice (n = 13) [475.5 ± 0.26 mg (n = 12) vs. 94.9 ± 0.03 mg (n = 13)]. Interestingly, the splenomegaly seemed to be ameliorated in the 129 genetic background (n = 5), since their spleens were only 3.6 times larger than those of their wild-type littermates (n = 10) [230.0 ± 0.05 mg (n = 5) vs. 63.4 ± 0.01 mg (n = 10)]. Similar results were obtained with the spleen/body weight ratios [B6/129: 15.78 ± 0.0090 mg (n = 12) vs. 2.78 ± 0.0007 mg (n = 13); 129: 9.06 ± 0.0017 mg (n = 10) vs. 2.57 ± 0.0004 mg (n = 10)] (Fig. 4). Blood count analyses in 4 month-old mice confirmed that the MPD is ameliorated in the 129 background, at least in homozygous mice. Significant differences were only found in 129 homozygous mice in leukocytosis in comparison to their wild-type littermates (Table 2). There were no significant differences in neutrophils, eosinophil, and basophil populations, either in RBC or in platelets (Table 2). Interestingly, these parameters significantly changed in B6/129 mutant mice19 (Table 2). These observations might explain the longer life span of 129 homozygous mice compared to B6/129 mice (Fig. 1).
Discussion
NS is a dominant disorder that clinically overlaps with other syndromes, such as Costello syndrome (CS), Cardio-Facio-Cutaneous (CFC) syndrome, Noonan-like syndrome with loose anagen hair (NS/LAH), and Neurofibromatosis type 1 (NF1), all known as RASopathies.9,21 Each of these syndromes exhibits unique characteristic phenotypes likely to be mediated by the nature of the mutated components of the RAS-MAPK pathway. However, there is an important phenotypic variability between patients with the same genotype that affects the nature and penetrance of their clinical features. This phenotypic variability can only be explained by the existence of genetic modifiers in the highly outbred human population. In this regard, NS has shown a large genotype-phenotype variation, a fact complicated by the diverse array of mutations assigned to this syndrome.
The best-studied example within the RASopathies is NF1, a syndrome characterized by marked inter– and intra–familial variation.22 Since different affected members of the same NF1 family often have quite different disease phenotypes, it is clear that variation in the mutant NF1 allele itself does not account for all the disease variability.23–26 Although challenging, identifying the genetic modifiers is of great interest to improve treatment and genetic counseling. In spite of these efforts, the mechanisms underlying NF1 clinical variability remain still poorly understood. Some of the modifier loci that influence resistance to peripheral nerve sheath tumors and astrocytoma, some of the typical features of NF1 patients, have been recently identified.27,28
CS patients also display significant genotype-phenotype variability. For instance, although all CS patients carry alterations in the HRAS locus, and around 84% of the patients carry the G12S mutation, HCM (the most frequent cardiac phenotype) was only present in 64% of them.29 Just recently, the phenotypic variability in 2 patients affected by the NS/LAH syndrome has been reported.30 This rare condition is caused by a missense A4G (Ser2Gly) change in SHOC2, which encodes a regulatory protein that participates in RAS signaling. These two patients, with the same alteration in SHOC2, displayed extremely different phenotypic alterations, in particular concerning the severity of the cardiac phenotype and neurocognitive profile.30 Around 50% of NS patients display mutations in PTPN11, being the most commonly reported mutation the substitution N308D. Interestingly, not all patients carrying this mutation display the same clinical signs.7,31-33 For instance, it was recently reported that from 47 patients carrying the PTPN11N308D mutation, 14 patients did not display pulmonary valve stenosis, the most prevalent cardiac phenotype in NS-PTPN11 patients.34 Hence, the fact that the alterations are different between patients with the same mutation, even in members of the same family carrying the same mutation, strongly indicates the existence of genetic modifiers.
Studies with GEM models have illustrated that modifiers in the genetic background affect the severity of the disease. Since different inbred B6 and 129 substrains have been used in different laboratories, caution must been taken comparing the results due to the substantial genetic variation between substrains.35 This may be the case for the variability described in 2 knock-in mouse models of CS with the same H-RasG12V mutation. These mouse models displayed different phenotypes that can only be attributed to the use of different inbred substrains. Interestingly, while one study showed cranial alteration, papillomas, and angiosarcomas,36 the other GEM models displayed cranial alterations, cardiac hypertrophy, and hypertension.37 Genetic background differences have also been studied in a B-Raf knock-in mouse model for CFC.38 Mice on B6 background displayed shorter survival than mice with increased 129 or CD1 genetic background contribution. Moreover, although the alterations (growth retardation, craniofacial dysmorphism, cataracts, heart defects, and seizures) were present in all the studied genotypes, they were more prevalent in B6 mice.38
In the context of NS, the previous studies describing mouse models expressing mutations in Ptpn1115 and Raf1,16 as well as the results obtained in the context of the K-RasV14I mutation presented here, have illustrated clear differences in disease spectrum and severity of the same mutation on different strain backgrounds. We previously observed that B6/129 K-RasV14I mice displayed perinatal lethality with incomplete penetrance, suggesting the presence of modifier alleles, in one or the other strain, that modulate this phenotype.19 This incomplete penetrance disappeared when mice were backcrossed to B6 background, since no B6 homozygous K-RasV14I mice were obtained just after birth, likely due to the severity of cardiac defects. Similar results were described in mice that express the Ptpn1D61G and Raf1L613V mutations, which display complete lethality after the fifth and the third backcross generation to B6, respectively, due to the severity of the cardiac defects.15,16 In contrast, no change in the perinatal lethality was found when K-RasV14I mice were backcrossed to 129 background, suggesting that there is a small contribution of the 129 background to the perinatal lethality of the K-RasV14I mice. However, unlike our results, Ptpn11+/D16G mice backcrossed to 129S6/SvEv displayed nearly normal viability.15
Our results illustrate that while genetic modifiers associated with the B6 background seem to be essential for the heart function at early stages of postnatal life, that is not the case in the adulthood since the adult cardiac phenotype does not show significant variability. The studied genetic backgrounds did not have an important impact in either growth alterations or facial dysmorphism.19 Heterozygous mice, regardless of the genetic background, displayed similar facial dysmorphism, body weight and length compared to the wild-type littermates.19 However, 129 homozygous K-RasV14I mice displayed bigger reduction in body weight and length compared to B6/129 mixed background. Similar to growth alterations, 129 homozygous mice displayed higher craniofacial alterations than the mixed background.19
In contrast, the progression of the MPD was affected by differences in the genetic background. The haematopoietic alterations (splenomegaly, leukocytosis, and anemia) were more severe in B6 K-Ras+/V14I mice. The increase of spleen weight and spleen/body weight ratio was bigger in B6 K-Ras+/V14I mice compared to the mixed B6/129 K-Ras+/V14I mice. However, the MPD was ameliorated in 129 K-RasV14I mice, resulting in a longer survival rate. It still remains to be addressed whether this is due to a slower progression of the MPD and/or to a longer period of latency.
The lack of a perfect genotype-phenotype correlation in human patients leads to speculation on phenotypes influenced by genetic modifiers. All of these data suggest that incomplete penetrance reflects strain-specific modifiers. Future studies need to focus on the identification of genetic modifiers that may explain the wide phenotypic variability observed in these patients. Genomic scans using single nucleotide polymorphism (SNP) panels should help to determine the presence of modifiers responsible for the variable penetrance. Moreover, the study of mutant mouse models under different genetic backgrounds will help to elucidate the existence of potential genetic modifiers.
A better understanding of an individual's risk for a given phenotype will lead to a more focused prevention and better and more specific treatments. Furthermore, identification of genetic modifiers will improve the treatment of patients and, in some cases, may improve survival.
Materials and Methods
Mice
Animal use was in accordance with the animal care standards established by the European Union. All the experiments were reviewed and approved by the Animal Care Committee of the Institute of Health Carlos III. The inbred strains were purchased from Harlan, the Netherlands (C57BL/6J.OlaHsd) and from Charles River, Germany (129S2/SvPasCrl). The K-RasV14I strain has previously been described.19
Blood analysis
Blood collection from the renal vein was used as a terminal method of blood extraction during the necropsy. The blood was obtained using EDTA-tubes and blood populations were quantified using a blood analyzer (Abacus Junior Vet).
MEK inhibitor treatment
The MEK inhibitor PD0325901 (Wuhan Sunrise Technology Development Company, Ltd) was prepared and administrated as previously described.19
Statistical Analyses
Results were expressed as mean ± SD. K-Ras+/V14I and K-RasV14I were compared with K-Ras+/+ mice using a 2-tailed Student's test. A P value less than 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
We thank M. Barbacid for his advice and support and I. Agudo, I. Aragón, N. Cabrera, M.C. González, M. Lamparero, P. Nogales, M. San Román, and R. Villar for technical assistance.
Funding
Work was supported by grants from Fondo de Investigación Sanitaria (PI042124, PI08–1623, PI11–02529), Autonomous Community of Madrid (GR/SAL/0349/2004), and Fundación Ramón Areces (FRA 01–09–001) to C.G. I.H.-P. was supported by PFIS grant from the Instituto de Salud Carlos III and A.J.S. by a FPU fellowship from the Spanish Ministry of Economy and Competitiveness and by the COFUND scheme of the Seventh Framework Program of the European Union (grant agreement 291820).
References
- 1.Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet 2013; 14:355-69; PMID:23875798; http://dx.doi.org/ 10.1146/annurev-genom-091212-153523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet 2013; 381:333-42; PMID:23312968; http://dx.doi.org/ 10.1016/S0140-6736(12)61023-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K, Otaga T, Takada F, Yano M, Ando T, et al.. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet 2013; 93:173-80; PMID:23791108; http://dx.doi.org/ 10.1016/j.ajhg.2013.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flex E, Jaiswal M, Pantaleoni F, Martinelli S, Strullu M, Fansa EK, Caye A, De Luca A, Lepri F, Dvorsky R, et al.. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet 2014; 23:4315-27; PMID:24705357; http://dx.doi.org/ 10.1093/hmg/ddu148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab 2011; 25:161-79; PMID:21396583; http://dx.doi.org/ 10.1016/j.beem.2010.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 2007; 7:295-308; PMID:17384584; http://dx.doi.org/ 10.1038/nrc2109 [DOI] [PubMed] [Google Scholar]
- 7.Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van der Burgt I, Brunner HG, Bertola DR, Crosby A, Ion A, et al.. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 2002; 70:1555-63; PMID:11992261; http://dx.doi.org/ 10.1086/340847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zenker M, Horn D, Wieczorek D, Allanson J, Pauli S, van der Burgt I, Doerr HG, Gaspar H, Hofbeck M, Gillessen-Kaesbach G, et al.. SOS1 is the second most common Noonan gene but plays no major role in cardio-facio-cutaneous syndrome. J Med Genet 2007; 44:651-6; PMID:17586837; http://dx.doi.org/ 10.1136/jmg.2007.051276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tartaglia M, Gelb BD. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann N Y Acad Sci 2010; 1214:99-121; PMID:20958325; http://dx.doi.org/ 10.1111/j.1749-6632.2010.05790.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC, Pantaleoni F, Scioletti AP, Esposito G, Cordeddu V, Lepri F, et al.. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum. Hum Mutat 2009; 30:695-702; PMID:19206169; http://dx.doi.org/ 10.1002/humu.20955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee BH, Kim JM, Jin HY, Kim GH, Choi JH, Yoo HW. Spectrum of mutations in Noonan syndrome and their correlation with phenotypes. J Pediatr 2011; 159:1029-35; PMID:21784453; http://dx.doi.org/ 10.1016/j.jpeds.2011.05.024 [DOI] [PubMed] [Google Scholar]
- 12.Bertola DR, Pereira AC, de Oliveira PS, Kim CA, Krieger JE. Clinical variability in a Noonan syndrome family with a new PTPN11 gene mutation. Am J Med Genet A 2004; 130A:378-83; PMID:15384080; http://dx.doi.org/ 10.1002/ajmg.a.30270 [DOI] [PubMed] [Google Scholar]
- 13.Zenker M, Voss E, Reis A. Mild variable Noonan syndrome in a family with a novel PTPN11 mutation. Eur J Med Genet 2007; 50:43-7; PMID:17052965; http://dx.doi.org/ 10.1016/j.ejmg.2006.08.003 [DOI] [PubMed] [Google Scholar]
- 14.Bentires-Alj M, Kontaridis MI, Neel BG. Stops along the RAS pathway in human genetic disease. Nat Med 2006; 12:283-5; PMID:16520774; http://dx.doi.org/ 10.1038/nm0306-283 [DOI] [PubMed] [Google Scholar]
- 15.Araki T, Chan G, Newbigging S, Morikawa L, Bronson RT, Neel BG. Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc Natl Acad Sci U S A 2009; 106:4736-41; PMID:19251646; http://dx.doi.org/ 10.1073/pnas.0810053106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu X, Simpson J, Hong JH, Kim KH, Thavarajah NK, Backx PH, Neel BG, Araki T. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J Clin Invest 2011; 121:1009-25; PMID:21339642; http://dx.doi.org/ 10.1172/JCI44929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu X, Yin J, Simpson J, Kim KH, Gu S, Hong JH, Bayliss P, Backx PH, Neel BG, Araki T. Increased BRAF heterodimerization is the common pathogenic mechanism for noonan syndrome-associated RAF1 mutants. Mol Cell Biol 2012; 32: 3872-90; PMID:22826437; http://dx.doi.org/ 10.1128/MCB.00751-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritchard CA, Bolin L, Slattery R, Murray R, McMahon M. Post-natal lethality and neurological and gastrointestinal defects in mice with targeted disruption of the A-Raf protein kinase gene Curr Biol 1996; 6: 614-7; PMID:8805280; http://dx.doi.org/ 10.1016/S0960-9822(02)00548-1 [DOI] [PubMed] [Google Scholar]
- 19.Hernandez-Porras I, Fabbiano S, Schuhmacher AJ, Aicher A, Canamero M, Camara JA, Cusso L, Desco M, Heeschen C, Mulero F, et al.. K-RasV14I recapitulates Noonan syndrome in mice Proc Natl Acad Sci U S A 2014; 111:16395-16400; PMID:25359213; http://dx.doi.org/ 10.1073/pnas.1418126111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen PC, Wakimoto H, Conner D, Araki T, Yuan T, Roberts A, Seidman CE, Bronson R, Neel BG, Seidman JG et al.. Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome-associated Sos1 mutation. J Clin Invest 2010; 120: 4353-65; PMID:21041952; http://dx.doi.org/ 10.1172/JCI43910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tidyman WE, Rauen KA. Noonan, Costello and cardio-facio-cutaneous syndromes: dysregulation of the Ras-MAPK pathway. Expert Rev Mol Med 2008; 10:e37; PMID:19063751; http://dx.doi.org/ 10.1017/S1462399408000902 [DOI] [PubMed] [Google Scholar]
- 22.Friedman JM, Gutmann DH, MacCollin M, Riccardi VM. Neurofibromatosis: phenotype, natural history, and pathogenesis. Taylor & Francis, Baltimore: 1999. [Google Scholar]
- 23.Easton DF, Ponder MA, Huson SM, Ponder BA. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. Am J Hum Genet 1993; 53:305-13; PMID:8328449 [PMC free article] [PubMed] [Google Scholar]
- 24.Szudek J, Joe H, Friedman JM. Analysis of intrafamilial phenotypic variation in neurofibromatosis 1 (NF1). Genet Epidemiol 2002; 23:150-64; PMID:12214308; http://dx.doi.org/ 10.1002/gepi.1129 [DOI] [PubMed] [Google Scholar]
- 25.Sabbagh A, Pasmant E, Laurendeau I, Parfait B, Barbarot S, Guillot B, Combemale P, Ferkal S, Vidaud M, Aubourg P, et al.. Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum Mol Genet 2009; 18:2768-78; PMID:19417008; http://dx.doi.org/ 10.1093/hmg/ddp212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pemov A, Sung H, Hyland PL, Sloan JL, Ruppert SL, Baldwin AM, Boland JF, Bass SE, Lee HJ, Jones KM, et al.. Genetic Modifiers of Neurofibromatosis Type 1-Associated Cafe-au-Lait Macule Count Identified Using Multi-platform Analysis. PLoS Genet 2014; 10:e1004575; PMID:25329635; http://dx.doi.org/ 10.1371/journal.pgen.1004575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reilly KM, Broman KW, Bronson RT, Tsang S, Loisel DA, Christy ES, Sun Z, Diehl J, Munroe DJ, Tuskan RG. An imprinted locus epistatically influences Nstr1 and Nstr2 to control resistance to nerve sheath tumors in a neurofibromatosis type 1 mouse model. Cancer Res 2006; 66:62-8; PMID:16397217; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walrath JC, Fox K, Truffer E, Gregory Alvord W, Quinones OA, Reilly KM. Chr 19(A/J) modifies tumor resistance in a sex- and parent-of-origin-specific manner. Mamm Genome 2009; 20:214-23; PMID:19347398; http://dx.doi.org/ 10.1007/s00335-009-9179-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin AE, Alexander ME, Colan SD, Kerr B, Rauen KA, Noonan J, Baffa J, Hopkins E, Sol-Church K, Limongelli G, et al.. Clinical, pathological, and molecular analyses of cardiovascular abnormalities in Costello syndrome: a Ras/MAPK pathway syndrome. Am J Med Genet A 2011; 155A:486-507; PMID:21344638; http://dx.doi.org/ 10.1002/ajmg.a.33857 [DOI] [PubMed] [Google Scholar]
- 30.Baldassarre G, Mussa A, Banaudi E, Rossi C, Tartaglia M, Silengo M, Ferrero GB. Phenotypic variability associated with the invariant SHOC2 c.4A>G (p.Ser2Gly) missense mutation. Am J Med Genet A 2014; 164A:3120-5; PMID:25331583; http://dx.doi.org/ 10.1002/ajmg.a.36697 [DOI] [PubMed] [Google Scholar]
- 31.Jongmans M, Sistermans EA, Rikken A, Nillesen WM, Tamminga R, Patton M, Maier EM, Tartaglia M, Noordam K, van der Burgt I. Genotypic and phenotypic characterization of Noonan syndrome: new data and review of the literature. Am J Med Genet A 2005; 134A:165-70; PMID:15723289; http://dx.doi.org/ 10.1002/ajmg.a.30598 [DOI] [PubMed] [Google Scholar]
- 32.Musante L, Kehl HG, Majewski F, Meinecke P, Schweiger S, Gillessen-Kaesbach G, Wieczorek D, Hinkel GK, Tinschert S, Hoeltzenbein M, et al.. Spectrum of mutations in PTPN11 and genotype-phenotype correlation in 96 patients with Noonan syndrome and five patients with cardio-facio-cutaneous syndrome. Eur J Hum Genet 2003; 11:201-6; PMID:12634870; http://dx.doi.org/ 10.1038/sj.ejhg.5200935 [DOI] [PubMed] [Google Scholar]
- 33.Zenker M, Buheitel G, Rauch R, Koenig R, Bosse K, Kress W, Tietze HU, Doerr HG, Hofbeck M, Singer H, et al.. Genotype-phenotype correlations in Noonan syndrome. J Pediatr 2004; 144:368-74; PMID:15001945; http://dx.doi.org/ 10.1016/j.jpeds.2003.11.032 [DOI] [PubMed] [Google Scholar]
- 34.Ezquieta B, Santome JL, Carcavilla A, Guillen-Navarro E, Perez-Aytes A, Sanchez del Pozo J, Garcia-Miñuar S, Castillo E, Alonso M, Vendrell T, et al.. Alterations in RAS-MAPK genes in 200 Spanish patients with Noonan and other neuro-cardio-facio-cutaneous syndromes. Genotype and cardiopathy. Rev Esp Cardiol (Engl Ed) 2012; 65:447-55; PMID:22465605; http://dx.doi.org/ 10.1016/j.recesp.2011.12.016 [DOI] [PubMed] [Google Scholar]
- 35.Simpson EM, Linder CC, Sargent EE, Davisson MT, Mobraaten LE, Sharp JJ. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat Genet 1997; 16:19-27; PMID:9140391; http://dx.doi.org/ 10.1038/ng0597-19 [DOI] [PubMed] [Google Scholar]
- 36.Chen X, Mitsutake N, LaPerle K, Akeno N, Zanzonico P, Longo VA, Mitsutake S, Kimura ET, Geiger H, Santos E, et al.. Endogenous expression of Hras(G12V) induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc Natl Acad Sci U S A 2009; 106:7979-84; PMID:19416908; http://dx.doi.org/ 10.1073/pnas.0900343106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuhmacher AJ, Guerra C, Sauzeau V, Canamero M, Bustelo XR, Barbacid M. A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J Clin Invest 2008; 118:2169-79; PMID:18483625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urosevic J, Sauzeau V, Soto-Montenegro ML, Reig S, Desco M, Wright EM, Cañamero M, Mulero F, Ortega S, Bustelo XR, et al.. Constitutive activation of B-Raf in the mouse germline provides a model for human cardio-facio-cutaneous syndrome. Proc Natl Acad Sci U S A 2011; 108:5015-20; PMID:21383153; http://dx.doi.org/ 10.1073/pnas.1016933108 [DOI] [PMC free article] [PubMed] [Google Scholar]