

Abstract
Slow growing Mycobacterium avium subsp. paratuberculosis (MAP) causes a deadly condition in cattle known as Johne's disease where asymptomatic carriers are the major source of disease transmission. MAP was also shown to be associated with chronic Crohn's disease in humans. Mycobacterium smegmatis is a model mycobacterium that can cause opportunistic infections in a number of human tissues and, rarely, a respiratory disease. Currently, there are no rapid, culture-independent, reliable and inexpensive tests for the diagnostics of MAP or M. smegmatis infections. Bacteriophages are viruses producing a number of proteins that effectively and specifically recognize the cell envelopes of their bacterial hosts. We demonstrate that the mycobacterial phage L5 minor tail protein Gp6 and lysin Gp10 are useful tools for the rapid capture of mycobacteria. Immobilized Gp10 was able to bind both MAP and M. smegmatis cells whereas Gp6 was M. smegmatis specific. Neither of the 2 proteins was able to capture E. coli, salmonella, campylobacter or Mycobacterium marinum cells. Gp6 was detected previously as a component of the phage particle and shows no homology to proteins with known function. Therefore, electrospray ionization mass spectrometry was used to determine whether recombinant Gp6 could bind to a number of chemically synthesized fragments of mycobacterial surface glycans. These findings demonstrate that mycobacteriophage proteins could be used as a pathogen capturing platform that can potentially improve the effectiveness of existing diagnostic methods.
Keywords: bacteriophage, Johne's disease, lysin, mycobacterium, receptor binding protein, Crohn's disease
Abbreviations
- BSA
bovine serum albumin
- MAP
Mycobacterium avium subsp. paratuberculosis
- PBS
phosphate buffered saline
- RBP
receptor binding protein
- RT-PCR
real time polymerase chain reaction
- SDS-PAGE
polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate, ESI-MS, electrospray ionization mass spectrometry
- IMAC
immobilized metal affinity chromatography
- FM
fluorescent microscopy
- SEM
scanning electron microscopy
Introduction
The importance of non-tuberculous mycobacteria for human and animal health is often underestimated. Many non-tuberculous mycobacteria are pathogenic to humans and animals. “Slow growing” Mycobacterium avium subsp. paratuberculosis (MAP) can cause paratuberculosis or Johne's disease in cattle. The infection occurs in the first few months of life but the symptoms usually develop after several years and can lead to significant damage of the gastrointestinal tract, severe diarrhea and eventual death of the animal.1 Johne's disease has a major impact on the ruminant industry.2 At least 1 in 5 dairy herds in Ontario has MAP infected cows according to the Ontario Johne's Education and Management Assistance Program (www.johnes.ca) and asymptomatic animals are the major source of disease transmission. MAP is also associated with human Crohn's disease although the literature describing the role of MAP in Crohn's disease pathogenesis remains controversial.3 “Fast growing” M. smegmatis is a model mycobacterium that can cause opportunistic infections in a number of human tissues and, rarely, respiratory disease.4 Modern techniques used to detect bacterial pathogens are expensive and have substantial drawbacks. Enzyme-linked immunosorbent assays (ELISA) can be impeded by antibody degradation whereas polymerase chain reactions (PCR) can be inhibited by many naturally abundant agents, e.g. fatty acids and calcium ions. Thus, rapid, culture-independent, reliable and inexpensive tests are urgently needed for effective prophylactics and therapeutics against mycobacterial infections both in humans and industrial animals.
Bacteriophages are viruses that bind to the cell surface of their host bacteria and initiate infection resulting in the amplification of phage DNA and production of new phage particles. This recognition is highly specific and is used widely for bacterial typing, i.e. the identification of bacterial species on the basis of their ability to be lysed by particular subsets of phages. Importantly, bacteriophages are usually resilient to conditions responsible for antibody degradation, such as the presence of proteases in the natural samples. Immobilized bacteriophage particles have been suggested to be potential platforms for culture-independent diagnosis of bacterial infections.5,6 This approach is especially promising for the diagnosis of slow growing and/or fastidious bacteria. Phage–host specificity is mediated by proteins located at their tails called receptor binding proteins – RBPs.7-10 The use of RBPs presents additional advantages over the use of whole phages as probes for bacterial infections.11 The substantially smaller size of RBPs leads to a more uniform surface coverage of biosensor elements used in different diagnostic platforms. RBPs can be easily engineered for increased affinity and/or specificity compared to often cumbersome phage genome engineering. Also, unlike whole phage particles, RBPs recognize and bind to the host bacteria without inducing the lytic cycle. The latter event may preclude the effective detection of a captured pathogen because of the destruction of bacterial DNA and/or specific marker antigens. We have recently employed genetically engineered RBPs for the reliable and specific capture of Salmonella, Shigella and Campylobacter cells.11-14 We demonstrated the use of RBP-based magnetic separation with real time PCR for the rapid, sensitive and specific detection of Campylobacter jejuni in artificially contaminated milk and chicken broth. Recovery rates, assessed by real time PCR, were greater than 80% for the samples spiked with as low as 100 cfu/mL of C. jejuni cells. The total sample preparation and analysis time in the proposed protocol was less than 3 hours.15
Lysins represent another class of cell envelope binding phage proteins. Lysins are peptidoglycan hydrolases that degrade the bacterial cell wall facilitating the release of newly formed virions.16 They have been proposed to act as a new class of antimicrobials capable of targeting antibiotic resistant pathogens.17-19 Interestingly, catalytically inactive recombinant lysins were recently engineered as a novel tool for the detection of Listeria strains.20,21
The current study represents an attempt to develop culture-independent diagnostic probes for mycobacteria using mycobacteriophage L5 host envelope binding proteins. Phage L5 of the Siphoviridae family is one of the best studied mycobacterial phages. Its genome sequence was the first obtained for a temperate non-Escherichia coli phage.22 L5 virions have a long non-contractile tail and contain 52 297 bp of double-stranded linear DNA.22 Luciferase-expressing L5 phage was also proposed to be useful for the detection of live mycobacteria.23,24 L5 phage was initially described as having a broad host range, including M. avium,22 although it is much more effective in infecting fast growing Mycobacterium smegmatis and requires special conditions to infect slow growing mycobacteria.24 Our study suggests that the minor tail protein Gp6 and lysin Gp10 recognize and bind to the host cell surface. Immobilized Gp10 was able to bind both MAP and M. smegmatis cells whereas Gp6 was M. smegmatis specific. Neither of the 2 proteins was able to capture E. coli, salmonella, campylobacter or M. marinum cells. Our approach demonstrates that mycobacteriophage proteins are capable of pathogen capture and can potentially be used to improve the effectiveness of existing MAP diagnostic platforms.
Results and Discussion
Selection and identification of mycobacteriophage cell binding proteins
It was shown that phages L5 and D29 infect M. smegmatis via mechanism(s) different from that used by the related phages Bxb1 and TM4.33,34 Yet, neither the RBP(s) nor the nature of the phage receptor has been described for any of these phages. Proteins reported as minor tail proteins of phage L5 were chosen first as RBP candidates. Comparison of the L5 genome sequence with that of the D29, Bxb1 and TM4 phages was performed to reveal the possible RBP genes among the genes encoding tail proteins. Notably, the homolog of the gene encoding the L5 minor tail protein Gp6 (NCBI gene ID 2942962, protein ID NP_039673.1) was found in the genome of phage D29, but not in the genomes of Bxb1 and TM4. These facts suggest a possible involvement of Gp6 in adsorption of L5 phage to the host cells although no known functional domains were revealed by BLAST analysis or the Phyre2 protein structure prediction engine. The native Gp6 has 313 amino acid residues, a predicted molecular weight of 34 kDa and a pI of 4.52 according to the ProtParam tool (www.expasy.org). All 20 types of amino acid residues are present within its primary sequence. The Phyre2 software predicted Gp6 to have at least 17 % α helical structure, 49 % β strands and 15 % disordered regions.
It was shown recently that gene 10 (NCBI gene ID 2942936, protein ID NP_039674.1) encodes a putative lysin with an N-terminal peptidase domain and C-terminal cell wall binding domain.35 Interestingly, while gene 10 is located apart from the gene cluster encoding most of the minor tail proteins in the L5 phage genome, it is close to gene 6 separated only by the small genes 7–9 encoding tRNA.22 Thus, Gp6 and Gp10 were chosen as cell binding proteins to be recombinantly produced and tested for use as diagnostics for mycobacteria.
Production of putative mycobacteriophage cell binding proteins
Comparison of the nucleotide sequences of cloned genes 6 and 10 with that available in the NCBI database revealed the presence of point mutations that resulted in the corresponding amino acid substitutions. Gene 6 had a T44C mutation that led to the Leu15Pro amino acid substitution in the recombinant protein. Gene 10 had 2 point mutations C95T and A670G, which resulted in the amino acid substitutions Thr32Ile and Ile224Val. Use of the non-proofreading Taq DNA polymerase was the probable reason for the appearance of these mutations, which were limited to pyrimidine/pyrimidine and purine/purine substitutions36 although one cannot exclude the existence of different gene variants within the phage population.
E. coli cells producing either Gp6 or Gp10 demonstrated similar growth characteristics and no inhibition of growth was revealed after induction of gene 6 or gene 10 expression. Similar amounts of recombinant protein was produced in each case – about 5 mg per 1 L of culture. Recombinant Gp6 and Gp10 proteins, however, differed greatly in their solubility. While production of Gp6 led to high amounts of soluble protein, the yield of soluble Gp10 was markedly lower (Fig. 1). Recombinant Gp10 was mostly confined to the insoluble cell pellet (not shown). Both proteins did not form SDS-resistant oligomers, which were shown to be characteristic of various phage RBPs including ones from campylobacter and salmonella phages.37-41 SDS PAGE analysis of the soluble Gp10 revealed a contaminant band at 60 kDa, i.e., about 20 kDa higher than the 40 kDa band corresponding to the His6-Gp10 polypeptide that has the predicted molecular weight of 38.1 kDa (Fig. 1). A similar band was absent in the preparation of Gp6, which contained, almost exclusively, a 40 kDa protein in agreement with the predicted mass of 39.7 kDa for His6-Gp6. We did not investigate further the identity of the 60 kDa band, which could represent a subunit of the chaperonin GroEL that is often observed in a complex with recombinant proteins.42
Figure 1.
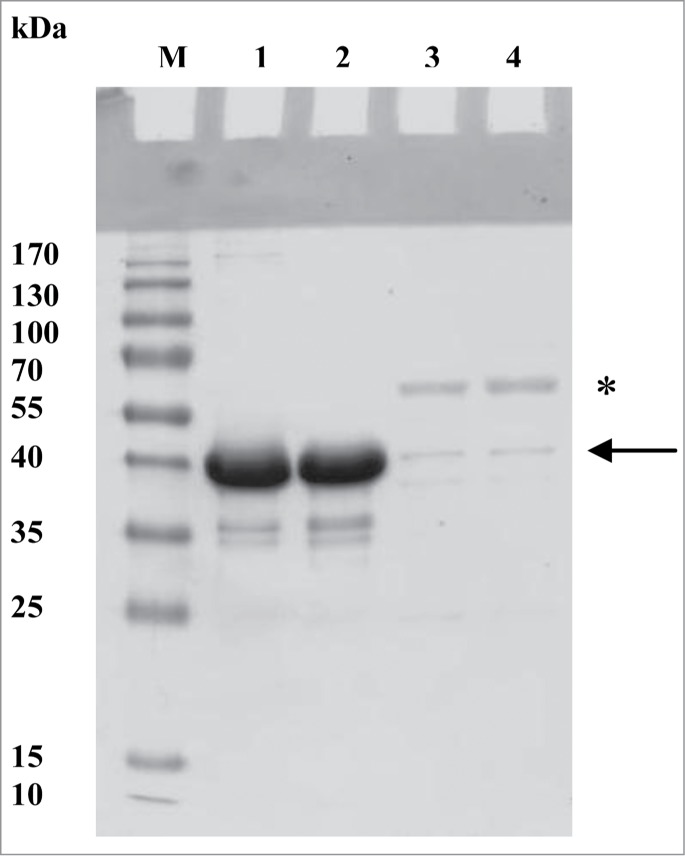
SDS-PAGE analysis of recombinant Gp6 and Gp10. (A) Comparison of the yield and properties of soluble Gp6 and Gp10. Both samples were obtained from the same volumes of expression cultures using the same IMAC protocol. M, molecular weight markers; lane 1, Gp6 sample incubated in SDS-PAGE sample buffer with 2% SDS for 10 min at room temperature; lane 2, same as lane 1, but sample was preheated at 95°C for 10 min; lanes 3 and 4 are similar to lanes 1 and 2 but Gp10 samples were used. The arrow indicates the expected position of recombinant Gp10; the asterisk labels the ca. 60 kDa contaminant.
Assessment of immobilized Gp6 and Gp10 as probes for MAP and M. smegmatis
To assess mycobacteriophage proteins as probes for mycobacteria, preparations of Gp6 and Gp10 were immobilized onto activated gold surfaces and these surfaces were used to assess the specificity of bacterial capture. Bacterial capture was monitored by fluorescent microscopy (FM) and scanning electron microscopy (SEM). A negative control was run in parallel where a similar gold surface was treated with all the reagents, but the surface was not exposed to the recombinant phage proteins. No significant bacterial capture was observed in the absence of phage proteins.
Capture efficiency of M. smegmatis cells was 31.3 ± 1.5 × 10−3 cells/μm2 and 28.7 ± 1.18 × 10−3 cells/μm2 by Gp6 and Gp10, respectively when a 20 μg/ml solution of either protein preparation was used for immobilization onto the gold chip (Figs. 2 and 3). Gold chips covered with Gp10 demonstrated a capture efficiency of 12.3 ± 1.3 × 10−3 MAP cells/μm2 under similar conditions (Fig. 3). Remarkably, the immobilization procedure with either 20 or 40 μg/ml of Gp 6 did not lead to capture of any MAP cells (Fig. 2). Neither Gp6 or Gp10 was able to serve as a capturing agent for M. marinum cells (Figs. 2 and 3). The specificity of recognition was confirmed by exposing Gp6 or Gp10 covered surfaces to suspensions of E. coli K12, S. Typhimurium or C. jejuni cells at 109 cfu/ml in PBS. These negative control experiments demonstrated no bacterial capture (not shown). It can be concluded that Gp10 preparations can specifically bind both M. smegmatis and MAP cells whereas Gp6 can only bind M. smegmatis cells under the conditions used. Thus, Gp6 may be applied for the specific detection of M. smegmatis whereas Gp10 appears to be a promising candidate for the development of a capturing element for a high throughput MAP diagnostic platform.
Figure 2.

Images of M. smegmatis, MAP and M. marinum cells captured by the recombinant Gp6 protein immobilized on gold surfaces (both SEM and FM images are shown for the same experiment). (A) Gold surface treated with 20 μg/ml of Gp6 and incubated with M. smegmatis cells; (B) same as A but MAP cells were used; (C) same as A but with M. marinum cells; (D) the surface was treated in a same way as in (A), except that it was not exposed to the recombinant protein prior to incubation with M. smegmatis cells.
Figure 3.

Images of M. smegmatis, MAP and M. marinum cells captured by the recombinant Gp10 protein immobilized on gold surfaces (both SEM and FM images are shown for the same experiment). (A) Gold surface treated with 20 μg/ml of recombinant protein Gp10 and incubated with M. smegmatis cells; (B) same as (A), but MAP cells were used; (C) same as (A), but with M. marinum cells; (D) the surface was treated in a same way as in (A), except that it was not exposed to the recombinant protein prior to incubation with M. smegmatis cells.
Our results with Gp10 are unexpected and contradictory to the recent survey of mycobacterial endolysins where the expression of the L5 gene 10 in M. smegmatis caused a dramatic rise in ATP-release 3 hours after induction and cell lysis appeared to be complete 7 hours after the induction of expression.35 Also, no overproduction of Gp10 could be detected in either soluble or insoluble fractions of M. smegmatis cells.35 We were able to successfully produce a small amount of soluble Gp10 using the conventional E. coli expression strain BL21(DE3) and observed most of the recombinant protein in the insoluble fraction in agreement with what was observed for other mycobacterial lysins.35 Successful heterologous expression of gene 10 in E. coli may be explained by the lack of toxic effects of Gp10 on a phylogenetically non-related bacterium as well as by the fact that the overproduction was performed at room temperature, not at 37°C as described in the previous study.35 The ability of the L5 lysin Gp10 to bind effectively both M. smegmatis and MAP cells could be explained. First, the enzymatic activity of Gp10 could be impaired under the conditions used in the cell binding assays. For example, it was shown that the catalytic activities of M. tuberculosis peptidoglycan hydrolases RipA and RipB are optimal at acidic pH whereas tight binding to the peptidoglycan is still observed at pH 7 when the hydrolysis is quite slow.43 Another possible reason for the impaired Gp10 lysin activity may be the presence of the non-conserved Thr32Ile substitution in the cloned gene 10 that was used in this current study. This substitution is located fairly close to the putative catalytic Cys41 of the N-terminal NlpC/p60-like peptidase domain of Gp10.35,43 Also, proper orientation of the immobilized RBPs was demonstrated to be an important factor influencing the efficiency of cell capture.12,13 The “random,“ un-oriented immobilization via primary amino groups may leave the N-terminal catalytic domain inaccessible in many Gp10 molecules where only the cell binding C-terminal region would be exposed. This may essentially lead to the situation similar to that described previously where catalytically inactive cell binding domains of listeriaphages were used for the detection of Listeria cells.20,21 In addition, our binding experiment requires considerably shorter exposures of the cell suspensions to the immobilized Gp10 (1 hour at room temperature) compared to several hours of endogenous production at 37°C described earlier.35 In any case, our study demonstrates the successful application of phage lysins for the rapid capture of mycobacteria.
Identification of possible Gp6 carbohydrate ligands
Complex surface carbohydrates were shown to be putative receptors of mycobacteriophages.44,45 Bioinformatic analysis did not reveal any conserved domains that could be assigned to Gp6 so the protein was tested for its ability to bind mycobacterial complex carbohydrates using ESI-MS and a set of chemically synthesized fragments of known mycobacterial surface glycans. ESI mass spectrometry analysis suggests that recombinant His6-Gp6 exists as a monomer of 39760 ± 10 Da which is close to the predicted mass of 39 729 Da. ESI-MS also demonstrated that Gp6 has a modest affinity toward chemically synthesized mycobacterial arabinan fragments (Table 1). The oligosaccharide/Gp6 interaction was sensitive to minor changes in the oligosaccharide structure similar to the interaction of monoclonal antibody CS-35 with the oligosaccharide fragments of mycobacterial arabinan where Ka values were ranging from 102 to 105 M−1.46 The presence of α-(1→3) glycosidic bonds between the second and the third arabinose residues enables the binding of tetrasaccharide 7 in contrast to isomeric tetrasaccharide 4 where only β-(1→2) and α-(1→5) bonds are present. The addition of the mannose residue to tetrasaccharide 4 (compound 1), removing the terminal β-arabinose residue (compound 5) or changing the terminal arabinose residue to an α-configuration (compound 8) also promotes binding to Gp6. It should be mentioned that the presence of additional mannose residues (compounds 2 and 9) or the branching structure (compounds 3 and 6) did not increase Gp6 affinity for the oligosaccharides.
Table 1.
Association constants (Ka) for Gp6 and mycobacterial surface oligosaccharides measured at 25°C and pH 7.2 using the direct ESI-MS assay.1
| # | Bacterial oligosaccharide structures | MW, Da | Ka, M-1 × 103 |
|---|---|---|---|
| 1 | α-Manp-(1→5)-β-Araf-(1→2)-α-Araf-(1→5)-α-Araf-(1→5)-α-Araf-octyl-NHCOCF3 | 931 | 3.9 ± 1.1 |
| 2 | α-Manp-(1→2)-α-Manp-(1→5)-β-Araf-(1→2)-α-Araf-(1→5)-α-Araf-(1→5)-α-Araf-octyl-NHCOCF3 | 1094 | 4.4 ± 3.9 |
| 3 | β-Araf-(1→2)-α-Araf-(1→5)[β-Araf-(1→2)-α-Araf-(1→3)]-α-Araf-(1→5)-α-Araf-octyl-NHCOCF3 | 1034 | 3.6 ± 0.4 |
| 4 | β-Araf-(1→2)-α-Araf-(1→5)-α-Araf-(1→5)-α-Araf-octyl-NHCOCF3 | 770 | NB2 |
| 5 | α-Araf-(1→5)-α-Araf-(1→5)-α-Araf-octyl-NHCOCF3 | 638 | 7.6 ± 3.2 |
| 6 | α-Araf-(1→5)-[α-Araf-(1→3)]-α-Araf-(1→5)-α-Araf-octyl-NHCOCF3 | 770 | 2.4 ± 0.7 |
| 7 | β-Araf-(1→2)-α-Araf-(1→3)-α-Araf-(1→5)-α-Araf-octyl-NHCOCF3 | 770 | 1.1 ± 0.2 |
| 8 | α-Araf-(1→5)-α-Araf-(1→5)-α-Araf-(1→5)-α-Araf-octyl-NHCOCF3 | 669 | 6.9 ± 0.8 |
| 9 | α-Manp-(1→2)-α-Manp-(1→2)-α-Manp-(1→5)-β-Araf-(1→2)-α-Araf-(1→5)-α-Araf-(1→5)-α-Araf-octyl-NHCOCF3 | 1256 | 2.6 ± 1.1 |
1Fucose (Fuc) and rhamnose (Rha) residues are in the L-configuration; mannose (Man), arabinose (Ara) and glucose (Glc) residues are in the D-configuration.
2NB – no binding detected.
The affinity of Gp6 toward the oligosaccharide fragments that were positive for binding was several orders of magnitude lower than the affinity of the salmonella phage P22 tailspike protein32 or the receptor binding protein from the lactococcal phage47 toward their cognate receptor oligosaccharides. Further studies are needed to find the native oligosaccharide ligand of Gp6 since the oligosaccharides tested in the current study may represent parts of the actual phage receptor molecules. Notably, it was shown that the oligomannose-capped arabinan is present only in slow growing mycobacteria such as M. avium, M. marinum, M. tuberculosis or M. leprae whereas phosphoinositol-capped arabinan was found in fast-growing M. smegmatis.48-50 The ability of the immobilized recombinant Gp6 to bind M. smegmatis but not MAP or M. marinum cells may possibly be explained by the differences in cap structure of the lipoarabinomannan component of the cell wall assuming that Gp6 is indeed recognizing this molecule in vivo.
Taking into account the ease of the one-step affinity purification, the high yield of soluble recombinant Gp6 and its cell and carbohydrate binding properties, one can also utilize this protein as a scaffold for the development of artificial lectins via directed evolution methods.
The peptidoglycan binding properties of recombinant Gp10 were tested using the commercially available peptidoglycan from B. subtilis that was shown earlier to bind mycobacterial peptidoglycan hydrolases.43 It was possible to selectively pull-down recombinant Gp10 from the solution after incubating with a suspension of peptidoglycan obtained from B. subtilis cell walls (Fig. 4). Notably, the 60 kDa contaminant did not show any peptidoglycan binding properties. More structural and functional studies are obviously needed to determine the binding and catalytic specificity of the Gp10 lysin in more detail. The emerging phage lysin-based technologies look very promising for the detection of staphylococci and listeria.51,52 The current proof-of-principle work adds mycobacteria to the list.
Figure 4.
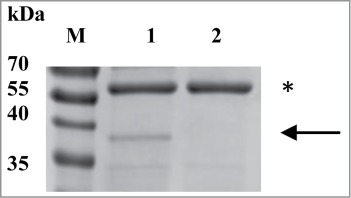
Peptidoglycan binding assay of recombinant Gp10. M, molecular weight markers; lane 1 – the gel image obtained after SDS-PAGE followed by Coomassie R-250 staining of Gp10 purified from the soluble fraction of the expression strain. The arrow indicates the expected position of the recombinant Gp10 and the asterisk labels the ca. 60 kDa contaminant; lane 2, same as lane 1, but the Gp10 sample was pre-incubated with the peptidoglycan.
The addition of a cell concentrating step to the validated PCR-based protocols may increase the sensitivity of MAP detection. Indeed, we recently demonstrated a significant increase in the sensitivity of MAP detection when the Gp10 lysin was coupled to magnetic beads and used to capture and separate MAP cells from complex media before applying the standard PCR protocol.53 Studies are currently underway to determine the ability of both Gp6 and Gp10 to bind clinical isolates of different mycobacteria and to capture the mycobacterial cells from natural matrices such as milk or cattle feces. Our approach can potentially be applied for the development of a rapid culture-independent diagnostic tool for the other M. avium subspecies where Gp10-based biosensors could possibly be used. It is well known that M. avium also causes serious infections in humans that may lead to extensive lung damage.54 Immunocompromised individuals and cystic fibrosis patients are particularly susceptible to such infections that can also be disseminated and damage a number of tissues.55,56 Needless to say that the appearance of novel methods aimed at the early recognition and prevention of these infections is long overdue.
Materials and Methods
Bacteria and phage strains used
Mycobacteria phage L5 (HER-386) was obtained from the Felix D’Herelle Reference Center for Bacterial Viruses (Laval University, Quebec, Canada). Mycobacterium marinum ATCC 927, M. smegmatis mc2 155 and M. avium subsp. paratuberculosis ATCC 19851 were used in the binding studies along with Campylobacter jejuni 11168H, Salmonella enterica subsp. enterica sv. Typhimurium ATCC 19585 and E. coli K-12. E. coli DH5α (Invitrogen) and E. coli BL21(DE3) (Invitrogen) were used for cloning and recombinant protein expression procedures, respectively. All mycobacterial strains were grown in ambient atmosphere. Middlebrook 7H9 (BD Biosciences) broth supplemented with oleic acid-albumin-dextrose catalase (BD Biosciences) and mycobactin J (Allied Monitor Inc.) was used to propagate M. smegmatis at 37°C for 48 hrs as well as MAP cells at 37°C for 10 d. M. marinum was grown for 10 d. at 37°C using Middlebrook 7H9 (BD Biosciences) broth supplemented with oleic acid-albumin-dextrose catalase (BD Biosciences). C. jejuni cells were grown for 18 h under microaerobic conditions (10% CO2, 5% O2, 85% N2) at 37°C on agar plates with Mueller-Hinton medium (BD Biosciences). LB medium (BD Biosciences) was used to propagate S. Typhimurium and E. coli strains overnight at 37°C unless stated otherwise. LB medium containing 25 μg/ml of kanamycin (BioShop Canada Inc.) was used for cloning and protein production procedures.
Bioinformatic analysis
The mycobacterial phage L5 genome22 that is deposited in the National Center for Biotechnology Information (NCBI, USA) database was used as a source of entry data (NCBI reference sequence NC_001335.1). Standard Basic Local Alignment Search Tool (BLAST) analysis25,26 was performed using the web service offered by NCBI. Protein Homology/analogY Recognition Engine (PHYRE), version 2.0 software27 was also used.
Gene manipulations
The genes of putative mycobacteriophage L5 cell binding proteins were cloned between the EcoRI and HindIII sites in the pET-30a(+) plasmid (Novagen) as follows. The corresponding genes were amplified by PCR directly from the phage L5 suspension. Taq DNA polymerase (Fermentas) was used for the PCR that was performed using 3 μl of phage lysate per 50 μl of reaction mix in the presence of 0.1 mg/ml of BSA (Fermentas). EcoRI and HindIII restriction sites were introduced during the PCR at the 5′ and 3′ ends. Primers GGCATCGAATTCATGGCCGACCTCGGCAACCCACTCG and GATGCTAAGCTTTTACCTCGGCTGTCGGTAAACGCGGC were used for the amplification of gene 6 as forward and reverse primers, respectively. Primers GGCATCGAATTCATGACCTTCACAGTCACCCGCGAG and GATGCTAAGCTTTCATAGGCCACCTCTTTCTGCGATG were used for the amplification of gene 10 as forward and reverse primers, respectively. All 4 primers were procured from Integrated DNA Technologies. The PCR cycling conditions were: 95°C/2 min, followed by 30 cycles of 95°C/30 sec, 55°C/30 sec, 72°C/1 min and a final elongation step of 72°C/10 min. PCR amplification resulted in a single product for each gene. The PCR products were purified directly from the reaction mixture using the GeneJet plasmid Miniprep spin column kit (Fermentas). The resulting DNA was digested with the appropriate restriction enzymes and was ligated with the linearized pET-30a(+) plasmid using T4 DNA ligase (Fermentas). E. coli DH5α was transformed with the ligation product. The resulting plasmid was then purified and product integrity was confirmed by sequencing in the Molecular Biology Service Unit, Department of Biological Sciences, University of Alberta.
Protein production
His6-tagged Gp6 and Gp10 proteins were expressed in E. coli BL21 cells transformed with the pET-30a(+) plasmid containing either gene 6 or gene 10. Cells were grown at 30°C to an OD600 of 0.5, induced with 0.2 mM IPTG and incubated overnight at room temperature with shaking. Cells were harvested, disrupted by sonication and the soluble fraction was subjected to the standard immobilized metal affinity chromatography (IMAC) procedure. Briefly, cells were resuspended in IMAC buffer A (50 mM sodium phosphate, pH 8.2, 1 M NaCl, 30 mM imidazole) with the Complete Mini, EDTA-free protease inhibitor cocktail (Roche) and then disrupted by sonication. Cell debris was removed by centrifugation at 27000 g for 30 min. The soluble fraction was filtered through a 0.22 μm filter (Millipore) and loaded onto a 1 ml HisTrap HP column (GE Healthcare). The column was washed with 20 column volumes of buffer A and the target protein was eluted with buffer A plus 500 mM imidazole. Both Gp6 and Gp10 proteins were subsequently dialyzed against PBS (phosphate-buffered saline, 1.8 mM KH2PO4, 10 mM Na2HPO4, pH 7.4, 2.7 mM KCl, 137 mM NaCl). The protein concentration was determined by measuring the UV absorbance. The A280/A260 ratio was in the range of 1.7–1.8 for all preparations of Gp6. Extinction coefficients at 280 nm were calculated for the recombinant Gp6 using ProtParam Tool (www.expasy.org) assuming all cysteine residues to be in the reduced state and turned out to be 43430 M−1 cm−1 and A0.1% of 1.093 for the His-tagged Gp6. The Christian-Warburg method was used to determine the total protein concentration of the Gp10 preparation obtained from the soluble cell fraction.
Protein immobilization
Proteins were immobilized onto cysteamine covered gold surfaces that were activated with glutaraldehyde according to the previously described protocol28 where RBP samples were used instead of whole phage particles. Briefly, the gold substrates were fabricated using piranha cleaned silicon substrates by sputtering a 25 nm thick gold layer. The gold substrates were sonicated in acetone, isopropanol, ethanol and MilliQ (Millipore) water for 5 min each prior to their use. The gold substrates were incubated overnight at 40°C in a 50 mM solution of cysteamine hydrochloride (Sigma-Aldrich). The cysteamine self assembled monolayer (SAM) substrates were modified by 2% glutaraldehyde (Sigma-Aldrich) for 1 hr at room temperature and washed twice in PBS. These modified substrates were incubated in a 20 μg/ml solution of Gp10 (or Gp6) in PBS overnight at 60°C. The negative control substrate was incubated in PBS only. To block bacterial nonspecific binding, the substrates were incubated in 1 mg/ml solution of bovine serum albumin (Sigma-Aldrich) and were washed twice in PBS.
Cell binding assays
The protein covered substrates were exposed to 109 cfu/ml of mycobacterial cells in PBS for 1 hr at room temperature. The immobilized surfaces were washed in 0.05 % Tween 20 (Sigma-Aldrich) before analysis. For fluorescence microscopy, the bacterial cells were stained with 50 μM resazurin (Sigma-Aldrich) for 20 min before exposure to the gold substrates covered with the immobilized proteins. Bacterial cell binding to the gold surface covered with the immobilized protein was assessed as described previously.12 Scanning electron microscopy (SEM) and fluorescent microscopy (FM) were used to estimate the number of bacterial cells bound to the surfaces. The samples were fixed with 2 % glutaraldehyde for 2 hrs at room temperature followed by a gradient of ethanol from 50 % to 100 % before SEM. Finally the samples were dried by nitrogen gas. SEM imaging was performed using a Hitachi S-4800/LEO 1430 microscope. An Olympus IX81 microscope equipped with a FITC filter and a Roper Scientific Cool-Snaps HQ CCD camera was used to record the FM images. ImageJ software (USA NIH) was used to analyze the microscopy images. Average numbers of cells bound to the surface are indicated on the basis of the assessment of the cell number, fields of view, and using 8 gold covered chips per test.
Mass spectrometry
Chemically synthesized fragments of mycobacterial arabinans (Maju Joe and Todd L. Lowary, manuscript in preparation) were used to test the carbohydrate binding ability of recombinant Gp6. Association constants (Ka) for Gp6 binding to carbohydrate ligands were measured using the direct electrospray ionization mass spectrometry (ESI-MS) assay. Complete details of the experimental methodology and data analysis are given elsewhere.29-31 The assay is based on the direct detection and quantification of the abundance of ligand-bound and unbound protein ions in the gas phase. All binding measurements were carried out at 25°C and pH 7.2 using a 9.4T ApexQe FTICR mass spectrometer (Bruker, Billerica, MA).32 ESI was performed in aqueous ammonium acetate (100 mM) solutions prepared from stock solutions of protein and oligosaccharide. The single chain variable fragment (scFv) of the monoclonal antibody Se155–4 was used as a reference protein to distinguish specific from nonspecific ligand binding with the protein during the ESI-MS measurements.29
Peptidoglycan binding assay
Dehydrated peptidoglycan from Bacillus subtilis (Sigma-Aldrich) was suspended in water to obtain a 10 mg/ml stock that was stored at –20°C. This stock was diluted further in water to obtain a 1 mg/ml working stock solution that was used in the experiments and was prepared fresh each time. A 0.1 mg/ml sample of Gp10 in PBS was incubated with the peptidoglycan at a final concentration of 0.1 mg/ml for 30 min at room temperature with occasional stirring. Then, the peptidoglycan was separated from the protein solution by centrifugation (15 min at 18 000 g at 4°C), an aliquot of the supernatant was mixed with the SDS-sample buffer and analyzed by SDS-PAGE.
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interest. A provisional patent describing the novel features and application of Gp6 and Gp10 mycobacteriophage proteins has been filed (US provisional patent application 61861866).
Funding
This work was supported by a grant from the Alberta Livestock and Meat Association (ALMA) to SE and CMS and the National Sciences and Engineering Research Council of Canada (NSERC) to JSK. CMS held an Alberta Innovates Scholar Award and is currently an Alberta Innovates Technology Futures iCORE Strategic Chair in Bacterial Glycomics. Support from the Alberta Glycomics Center and the Bill and Melinda Gates Foundation is also acknowledged. The authors would like to thank Dr. Elena Kitova and Dr. Rambod Daneshfar for helpful discussions.
References
- 1. Sweeney RW. Pathogenesis of paratuberculosis. Vet Clin North Am Food Anim Pract 2011; 27:537-46. [DOI] [PubMed] [Google Scholar]
- 2. Lombard JE. Epidemiology and economics of paratuberculosis. Vet Clin North Am Food Anim Pract 2011; 27:525-35. [DOI] [PubMed] [Google Scholar]
- 3. Chiodini RJ, Chamberlin WM, Sarosiek J, McCallum RW. Crohn's disease and the mycobacterioses: a quarter century later. Causation or simple association? Crit Rev Microbiol 2012; 38:52-93; PMID:22242906; http://dx.doi.org/ 10.3109/1040841X.2011.638273 [DOI] [PubMed] [Google Scholar]
- 4. Brown-Elliott BA, Wallace RJ., Jr. Clinical and taxonomic status of pathogenic nonpigmented or late-pigmenting rapidly growing mycobacteria. Clin Microbiol Rev 2002; 15:716-46; PMID:12364376; http://dx.doi.org/ 10.1128/CMR.15.4.716-746.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh A, Poshtiban S, Evoy S. Recent advances in bacteriophage based biosensors for food-borne pathogen detection. Sensors (Basel) 2013; 13:1763-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tawil N, Sacher E, Mandeville R, Meunier M. Bacteriophages: biosensing tools for multi-drug resistant pathogens. Analyst 2014; 139:1224-36; PMID:24434867; http://dx.doi.org/ 10.1039/c3an01989f [DOI] [PubMed] [Google Scholar]
- 7. Casjens SR, Molineux IJ. Short noncontractile tail machines: adsorption and DNA delivery by podoviruses. Adv Exp Med Biol 2012; 726:143-79; PMID:22297513; http://dx.doi.org/ 10.1007/978-1-4614-0980-9_7 [DOI] [PubMed] [Google Scholar]
- 8. Davidson AR, Cardarelli L, Pell LG, Radford DR, Maxwell KL. Long noncontractile tail machines of bacteriophages. Adv Exp Med Biol 2012; 726:115-42; PMID:22297512; http://dx.doi.org/ 10.1007/978-1-4614-0980-9_6 [DOI] [PubMed] [Google Scholar]
- 9. Leiman PG, Shneider MM. Contractile tail machines of bacteriophages. Adv Exp Med Biol 2012; 726:93-114; PMID:22297511; http://dx.doi.org/ 10.1007/978-1-4614-0980-9_5 [DOI] [PubMed] [Google Scholar]
- 10. Garcia-Doval C, van Raaij MJ. Bacteriophage receptor recognition and nucleic acid transfer. Subcell Biochem 2013; 68:489-518; PMID:23737063; http://dx.doi.org/ 10.1007/978-94-007-6552-8_17 [DOI] [PubMed] [Google Scholar]
- 11. Singh A, Arutyunov D, Szymanski CM, Evoy S. Bacteriophage based probes for pathogen detection. Analyst 2012; 137:3405-21; PMID:22724121; http://dx.doi.org/ 10.1039/c2an35371g [DOI] [PubMed] [Google Scholar]
- 12. Singh A, Arya SK, Glass N, Hanifi-Moghaddam P, Naidoo R, Szymanski CM, Tanha J, Evoy S. Bacteriophage tailspike proteins as molecular probes for sensitive and selective bacterial detection. Biosens Bioelectron 2010; 26:131-8; PMID:20541928; http://dx.doi.org/ 10.1016/j.bios.2010.05.024 [DOI] [PubMed] [Google Scholar]
- 13. Singh A, Arutyunov D, McDermott MT, Szymanski CM, Evoy S. Specific detection of Campylobacter jejuni using the bacteriophage NCTC 12673 receptor binding protein as a probe. Analyst 2011; 136:4780-6; PMID:21955997; http://dx.doi.org/ 10.1039/c1an15547d [DOI] [PubMed] [Google Scholar]
- 14. Javed MA, Poshtiban S, Arutyunov D, Evoy S, Szymanski CM. Bacteriophage receptor binding protein based assays for the simultaneous detection of Campylobacter jejuni and Campylobacter coli. PLoS One 2013; 8:e69770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Poshtiban S, Javed MA, Arutyunov D, Singh A, Banting G, Szymanski CM, Evoy S. Phage receptor binding protein-based magnetic enrichment method as an aid for real time PCR detection of foodborne bacteria. Analyst 2013; 138:5619-26; PMID:23897488; http://dx.doi.org/ 10.1039/c3an01100c [DOI] [PubMed] [Google Scholar]
- 16. Catalao MJ, Gil F, Moniz-Pereira J, Sao-Jose C, Pimentel M. Diversity in bacterial lysis systems: bacteriophages show the way. FEMS Microbiol Rev 2013; 37:554-71; PMID:23043507; http://dx.doi.org/ 10.1111/1574-6976.12006 [DOI] [PubMed] [Google Scholar]
- 17. Fischetti VA. Exploiting what phage have evolved to control gram-positive pathogens. Bacteriophage 2011; 1:188-94; PMID:23050211; http://dx.doi.org/ 10.4161/bact.1.4.17747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Drulis-Kawa Z, Majkowska-Skrobek G, Maciejewska B, Delattre AS, Lavigne R. Learning from bacteriophages - advantages and limitations of phage and phage-encoded protein applications. Curr Protein Pept Sci 2012; 13:699-722; PMID:23305359; http://dx.doi.org/ 10.2174/138920312804871193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pastagia M, Schuch R, Fischetti VA, Huang DB. Lysins: the arrival of pathogen-directed anti-infectives. J Med Microbiol 2013; 62:1506-16; PMID:23813275 [DOI] [PubMed] [Google Scholar]
- 20. Kretzer JW, Lehmann R, Schmelcher M, Banz M, Kim KP, Korn C, Loessner MJ. Use of high-affinity cell wall-binding domains of bacteriophage endolysins for immobilization and separation of bacterial cells. Appl Environ Microbiol 2007; 73:1992-2000; PMID:17277212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmelcher M, Shabarova T, Eugster MR, Eichenseher F, Tchang VS, Banz M, Loessner MJ. Rapid multiplex detection and differentiation of Listeria cells by use of fluorescent phage endolysin cell wall binding domains. Appl Environ Microbiol 2010; 76:5745-56; PMID:20622130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hatfull GF, Sarkis GJ. DNA sequence, structure and gene expression of mycobacteriophage L5: a phage system for mycobacterial genetics. Mol Microbiol 1993; 7:395-405; PMID:8459766 [DOI] [PubMed] [Google Scholar]
- 23. Sarkis GJ, Jacobs WR, Jr., Hatfull GF. L5 luciferase reporter mycobacteriophages: a sensitive tool for the detection and assay of live mycobacteria. Mol Microbiol 1995; 15:1055-67; PMID:7623662 [DOI] [PubMed] [Google Scholar]
- 24. Fullner KJ, Hatfull GF. Mycobacteriophage L5 infection of Mycobacterium bovis BCG: implications for phage genetics in the slow-growing mycobacteria. Mol Microbiol 1997; 26:755-66; PMID:9427405 [DOI] [PubMed] [Google Scholar]
- 25. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215:403-10; PMID:2231712 [DOI] [PubMed] [Google Scholar]
- 26. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997:3389-402; PMID:9254694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 2009; 4:363-71; PMID:19247286 [DOI] [PubMed] [Google Scholar]
- 28. Singh A, Glass N, Tolba M, Brovko L, Griffiths M, Evoy S. Immobilization of bacteriophages on gold surfaces for the specific capture of pathogens. Biosens Bioelectron 2009; 24:3645-51; PMID:19520565 [DOI] [PubMed] [Google Scholar]
- 29. Sun J, Kitova EN, Wang W, Klassen JS. Method for distinguishing specific from nonspecific protein-ligand complexes in nanoelectrospray ionization mass spectrometry. Anal Chem 2006; 78:3010-8; PMID:16642987 [DOI] [PubMed] [Google Scholar]
- 30. Wang W, Kitova EN, Klassen JS. Influence of solution and gas phase processes on protein-carbohydrate binding affinities determined by nanoelectrospray Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem 2003; 75:4945-55; PMID:14708765 [DOI] [PubMed] [Google Scholar]
- 31. El-Hawiet A, Kitova EN, Klassen JS. Quantifying carbohydrate-protein interactions by electrospray ionization mass spectrometry analysis. Biochemistry 2012; 51:4244-53; PMID:22564104 [DOI] [PubMed] [Google Scholar]
- 32. El-Hawiet A, Kitova EN, Arutyunov D, Simpson DJ, Szymanski CM, Klassen JS. Quantifying ligand binding to large protein complexes using electrospray ionization mass spectrometry. Anal Chem 2012; 84:3867-70; PMID:22507285 [DOI] [PubMed] [Google Scholar]
- 33. Barsom EK, Hatfull GF. Characterization of Mycobacterium smegmatis gene that confers resistance to phages L5 and D29 when overexpressed. Mol Microbiol 1996; 21:159-70; PMID:8843442 [DOI] [PubMed] [Google Scholar]
- 34. Mediavilla J, Jain S, Kriakov J, Ford ME, Duda RL, Jacobs WR, Jr., Hendrix RW, Hatfull GF. Genome organization and characterization of mycobacteriophage Bxb1. Mol Microbiol 2000; 38:955-70; PMID:11123671; http://dx.doi.org/ 10.1046/j.1365-2958.2000.02183.x [DOI] [PubMed] [Google Scholar]
- 35. Payne KM, Hatfull GF. Mycobacteriophage endolysins: diverse and modular enzymes with multiple catalytic activities. PLoS One 2012; 7:e34052; PMID:22470512; http://dx.doi.org/ 10.1371/journal.pone.0034052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bracho MA, Moya A, Barrio E. Contribution of Taq polymerase-induced errors to the estimation of RNA virus diversity. J Gen Virol 1998; 79 (Pt 12):2921-8; PMID:9880005 [DOI] [PubMed] [Google Scholar]
- 37. Kropinski AM, Arutyunov D, Foss M, Cunningham A, Ding W, Singh A, Pavlov AR, Henry M, Evoy S, Kelly J, et al. . Genome and proteome of Campylobacter jejuni bacteriophage NCTC 12673. Appl Environ Microbiol 2011; 77:8265-71; PMID:21965409; http://dx.doi.org/ 10.1128/AEM.05562-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barbirz S, Becker M, Freiberg A, Seckler R. Phage tailspike proteins with beta-solenoid fold as thermostable carbohydrate binding materials. Macromol Biosci 2009; 9:169-73; PMID:19148901; http://dx.doi.org/ 10.1002/mabi.200800278 [DOI] [PubMed] [Google Scholar]
- 39. Bartual SG, Garcia-Doval C, Alonso J, Schoehn G, van Raaij MJ. Two-chaperone assisted soluble expression and purification of the bacteriophage T4 long tail fibre protein gp37. Protein Expr Purif 2010; 70:116-21; PMID:19913618; http://dx.doi.org/ 10.1016/j.pep.2009.11.005 [DOI] [PubMed] [Google Scholar]
- 40. Sanchez-Ruiz JM. Protein kinetic stability. Biophys Chem 2010; 148:1-15. [DOI] [PubMed] [Google Scholar]
- 41. Manning M, Colon W. Structural basis of protein kinetic stability: resistance to sodium dodecyl sulfate suggests a central role for rigidity and a bias toward beta-sheet structure. Biochemistry 2004; 43:11248-54; PMID:15366934; http://dx.doi.org/ 10.1021/bi0491898 [DOI] [PubMed] [Google Scholar]
- 42. Rohman M, Harrison-Lavoie KJ. Separation of copurifying GroEL from glutathione-S-transferase fusion proteins. Protein Expr Purif 2000; 20:45-7; PMID:11035949; http://dx.doi.org/ 10.1006/prep.2000.1271 [DOI] [PubMed] [Google Scholar]
- 43. Both D, Schneider G, Schnell R. Peptidoglycan remodeling in Mycobacterium tuberculosis: comparison of structures and catalytic activities of RipA and RipB. J Mol Biol 2011; 413:247-60; PMID:21864539; http://dx.doi.org/ 10.1016/j.jmb.2011.08.014 [DOI] [PubMed] [Google Scholar]
- 44. Besra GS, Khoo KH, Belisle JT, McNeil MR, Morris HR, Dell A, Brennan PJ. New pyruvylated, glycosylated acyltrehaloses from Mycobacterium smegmatis strains, and their implications for phage resistance in mycobacteria. Carbohydr Res 1994; 251:99-114; PMID:8149383; http://dx.doi.org/ 10.1016/0008-6215(94)84279-5 [DOI] [PubMed] [Google Scholar]
- 45. Khoo KH, Suzuki R, Dell A, Morris HR, McNeil MR, Brennan PJ, Besra GS. Chemistry of the lyxose-containing mycobacteriophage receptors of Mycobacterium phlei/ Mycobacterium smegmatis. Biochemistry 1996; 35:11812-9; PMID:8794763; http://dx.doi.org/ 10.1021/bi961055+ [DOI] [PubMed] [Google Scholar]
- 46. Rademacher C, Shoemaker GK, Kim HS, Zheng RB, Taha H, Liu C, Nacario RC, Schriemer DC, Klassen JS, Peters T, et al. . Ligand specificity of CS-35, a monoclonal antibody that recognizes mycobacterial lipoarabinomannan: a model system for oligofuranoside-protein recognition. J Am Chem Soc 2007; 129:10489-502; PMID:17672460; http://dx.doi.org/ 10.1021/ja0723380 [DOI] [PubMed] [Google Scholar]
- 47. Bebeacua C, Tremblay D, Farenc C, Chapot-Chartier MP, Sadovskaya I, van Heel M, Veesler D, Moineau S, Cambillau C. Structure, adsorption to host, and infection mechanism of virulent lactococcal phage p2. J Virol 2013; 87:12302-12; PMID:24027307; http://dx.doi.org/ 10.1128/JVI.02033-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nigou J, Gilleron M, Puzo G. Lipoarabinomannans: from structure to biosynthesis. Biochimie 2003; 85:153-66; PMID:12765785; http://dx.doi.org/ 10.1016/S0300-9084(03)00048-8 [DOI] [PubMed] [Google Scholar]
- 49. Pitarque S, Herrmann JL, Duteyrat JL, Jackson M, Stewart GR, Lecointe F, Payre B, Schwartz O, Young DB, Marchal G, et al. . Deciphering the molecular bases of Mycobacterium tuberculosis binding to the lectin DC-SIGN reveals an underestimated complexity. Biochem J 2005; 392:615-24; PMID:16092920; http://dx.doi.org/ 10.1042/BJ20050709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mishra AK, Driessen NN, Appelmelk BJ, Besra GS. Lipoarabinomannan and related glycoconjugates: structure, biogenesis and role in Mycobacterium tuberculosis physiology and host-pathogen interaction. FEMS Microbiol Rev 2011; 35:1126-57; PMID:21521247; http://dx.doi.org/ 10.1111/j.1574-6976.2011.00276.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chibli H, Ghali H, Park S, Peter YA, Nadeau JL. Immobilized phage proteins for specific detection of staphylococci. Analyst 2014; 139:179-86; PMID:24255915; http://dx.doi.org/ 10.1039/c3an01608k [DOI] [PubMed] [Google Scholar]
- 52. Schmelcher M, Loessner MJ. Application of bacteriophages for detection of foodborne pathogens. Bacteriophage 2014; 4:e28137; PMID:24533229; http://dx.doi.org/ 10.4161/bact.28137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Singh U, Arutyunov D, Basu U, dos Santos Seckler H, Szymanski CM, Evoy S. Mycobacteriophage lysin-mediated capture of cells for the PCR detection of Mycobacterium avium subspecies paratuberculosis Anal Meth 2014; 6:5682-9 [Google Scholar]
- 54. Weiss CH, Glassroth J. Pulmonary disease caused by nontuberculous mycobacteria. Expert Rev Respir Med 2012; 6:597-612; quiz 3; PMID:23234447; http://dx.doi.org/ 10.1586/ers.12.58 [DOI] [PubMed] [Google Scholar]
- 55. Griffith DE. Therapy of nontuberculous mycobacterial disease. Curr Opin Infect Dis 2007; 20:198-203; http://dx.doi.org/ 10.1097/QCO.0b013e328055d9a2 [DOI] [PubMed] [Google Scholar]
- 56. Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, et al. . An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007; 175:367-416. [DOI] [PubMed] [Google Scholar]