

Abstract
Opioids are a group of analgesic agents commonly used in clinical practice. There are three classical opioid receptors (DOP, KOP and MOP), while the novel NOP receptor is considered to be a non-opioid branch of the opioid receptor family. Opioids can act at these receptors as agonists, antagonists or partial agonists. Opioid agonists bind to G-protein coupled receptors to cause cellular hyperpolarisation. Most clinically relevant opioid analgesics bind to MOP receptors in the central and peripheral nervous system in an agonist manner to elicit analgesia. Opioids may also be classified according to their mode of synthesis into alkaloids, semi-synthetic and synthetic compounds.
Keywords: Analgesics, opioid/pharmacology, opioid classification, pharmacokinetics
Introduction
Morphine is commonly considered to be the archetypal opioid analgesic and the agent to which all other painkillers are compared. There is evidence to suggest that as long ago as 3000 bc the opium poppy, Papaver somniferum, was cultivated for its active ingredients. It was, however, not until morphine was isolated from opium in 1806 by Sertürner that modern opioid pharmacology was truly born. In 1847 the chemical formula for morphine was deduced and this, coupled with the invention of the hypodermic needle in 1853, led to the more precise and widespread clinical use of morphine.1,2
Classification
Though morphine is the most widely known extract of P. somniferum, four naturally occurring alkaloids (plant-derived amines) can be isolated from it: morphine, codeine, papaverine and thebaine. Following Sertürner’s isolation of morphine as the active component of the opium poppy, simple chemical manipulations of these basic opiate alkaloids began to yield a range of semi-synthetic opioids useful in clinical medicine (agents such as diamorphine, dihydrocodeine, buprenorphine, nalbuphine, naloxone and oxycodone). During the 20th century a number of synthetic opioids were also produced either by design or serendipitously. These synthetic compounds can be divided into four chemical groupings: the morphinan derivatives (levorphanol, butorphanol), the diphenylheptane derivatives (methadone, propoxyphene), the benzomorphan derivatives (pentazocine, phenazocine) and the phenylpiperidine derivatives (pethidine, alfentanil, fentanyl, sufentanil and remifentanil) (Table 1).1,2
Table 1.
Classification of opioids by synthetic process.
| Naturally occurring compounds | Semi-synthetic compounds | Synthetic compounds |
|---|---|---|
| Morphine | Diamorphine (heroin) | Pethidine |
| Codeine | Dihydromorphone | Fentanyl |
| Thebaine | Buprenorphine | Methadone |
| Papaverine | Oxycodone | Alfentanil |
| Remifentanil | ||
| Tapentadol |
Opioids can also be classified according to their effect at opioid receptors. In this manner opioids can be considered as agonists, partial agonists and antagonists. Agonists interact with a receptor to produce a maximal response from that receptor (analgesia following morphine administration is an example). Conversely, antagonists bind to receptors but produce no functional response, while at the same time preventing an agonist from binding to that receptor (naloxone). Partial agonists bind to receptors but elicit only a partial functional response no matter the amount of drug administered (buprenorphine).
Opioid receptors
In addition, opioids can be categorised according to the type of opioid receptor at which they produce their effects. Classically, there are considered to be three opioid receptors. These receptors are all G-protein-coupled receptors, and were originally named mu (after morphine, its most commonly recognised exogenous ligand), delta (after vas deferens, the tissue within which it was first isolated) and kappa (after the first ligand to act at this receptor, ketocyclazocine). In 1996 the International Union of Pharmacology (IUPHAR) renamed the receptors OP1 (the delta receptor), OP2 (the kappa receptor) and OP3 (the mu receptor). In 2000 this nomenclature was again changed to DOP, KOP and MOP (Table 2).3,4 Now, however, owing to the extensive literature previously using the Greek nomenclature for opioid receptors (δ, κ and µ) IUPHAR recommends using this classification and the DOP, KOP and MOP classification of 2000. Some authorities describe the existence of multiple subtypes of the three classical opioid receptors, but this is not a belief held by all researchers within the field.5 The classical opioid receptors are distributed widely within the central nervous system and, to a lesser extent, throughout the periphery, occupying sites within the vas deferens, knee joint, gastrointestinal tract, heart and immune system, amongst others.6
Table 2.
Changes in the classification of the classical opioid receptors over time.
| Pre cloning | Post cloning | IUPHAR 1996 | IUPHAR 2000 |
|---|---|---|---|
| δ | DOR | OP1 | DOP |
| κ | KOR | OP2 | KOP |
| µ | MOR | OP3 | MOP |
Soon after the discovery of the opioid receptors, a series of endogenous ligands active at the receptors were discovered in brain extracts. Three pro-hormone precursors provide the parent compounds from which these endogenous ligands are derived. Proenkephalin is cleaved to form met-enkephalin and leu-enkephalin, which bind to the DOP receptor. Dynorphin A and B are derived from prodynorphin and are agonists at the KOP receptor. Pro-opiomelancortin (POMC) is the parent compound for β-endorphin, an agonist at the MOP receptor, though it is capable of displaying agonist activity at all three classical opioid receptors.2,3,7 Two further endogenous peptides act as agonists at the MOP receptor, endomorphin 1 and 2, but no precursor has yet been identified (Table 3). There is significant cross-talk between the endogenous agonists and the three classical receptors.
Table 3.
Opioid receptors and their endogenous ligands and precursors.
| Receptor | Precursor | Peptide |
|---|---|---|
| DOP | Pro-enkephalin | [Met]-enkephalin |
| [Leu]-enkephalin | ||
| KOP | Pro-dynorphin | Dynorphin-A |
| Dynorphin-B | ||
| MOP | POMC | β-Endorphin |
| Unknown | Endomorphin-1 | |
| Endomorphin-2 | ||
| NOP | Pre-pro-nociceptin | N/OFQ |
In 1994 a fourth G-protein-coupled endogenous opioid like receptor was found, and was subsequently named the nociceptin (NOP) receptor. Quickly after this discovery came the isolation from brain extracts of its endogenous ligand, nociceptin/orphanin FQ (N/OFQ). This endogenous ligand is similarly derived from a precursor compound, in this instance from the polypeptide precursor pre-pro-nociceptin. Though the N/OFQ/NOP system does not bind naloxone, nor are its effects reversed by naloxone, it is a G-protein-coupled receptor system that shares a marked similarity to the known amino acid sequences of the classical opioid receptors.3,8 At a cellular level, N/OFQ acts to produce similar actions to those described for the classical opioid receptors above. For these reasons it has been classified as the fourth opioid receptor; however, owing to its lack of response to the classical opioid antagonist (naloxone) some pharmacologists have questioned the wisdom of this classification. IUPHAR considers the NOP receptor to be a non-opioid branch of the opioid receptor family.4
In clinical practice the stimulation of the differing opioid receptors produces a range of effects, which are often dependent upon the location of the receptor, along with analgesia. Agonists binding to MOP receptors may cause analgesia, but also sedation, respiratory depression, bradycardia, nausea and vomiting and a reduction in gastric motility. Activation of DOP receptors can cause spinal and supraspinal analgesia and reduce gastric motility, while KOP receptor stimulation may produce spinal analgesia, diuresis and dysphoria. Spinally, N/OFQ has been shown to produce analgesia and hyperalgesia, dependent upon the administered concentration, and allodynia. Supraspinally, when administered intracerebrovascularly it is thought to produce a pro-nociceptive anti-analgesic effect, owing to an inhibition of endogenous opioid tone.3,9
Intracellular events
Though producing subtly different functional effects, all of these receptors display similar cellular responses following receptor activation. Binding of an opioid agonist to a G-protein-coupled opioid receptor on the transmembrane portion of the receptor causes the α subunit of the G-protein to exchange its bound guanosine diphosphate (GDP) molecule with intracellular guanosine triphosphate (GTP). This then allows the α-GTP complex to dissociate away from the βγ complex. Both of these complexes (α-GTP and βγ) are then free to interact with target proteins. In the case of a classical opioid agonist binding to its G-protein receptor, this results in the inhibition of adenylyl cyclase. This in turn causes a reduction in intracellular cyclic adenosine monophosphate (cAMP) levels. Additionally, these complexes interact with a number of ion channels, producing activation of potassium conductance and an inhibition of calcium conductance. The net effect of these changes is a reduced intracellular cAMP, a hyperpolarisation of the cell in question and, for neuronal cells, reduced neurotransmitter release (Figure 1). In some cell types activation of opioid receptors can also paradoxically lead to an increase in the intracellular calcium concentration.7,10
Figure 1.
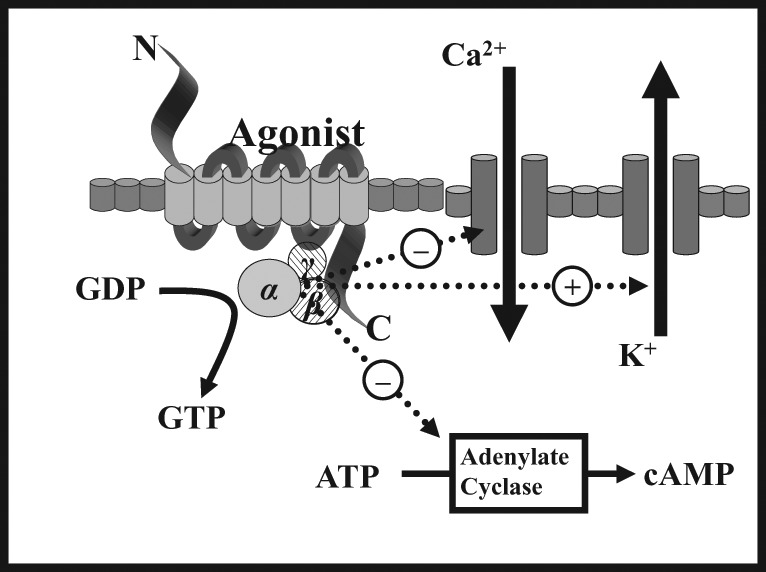
Intracellular changes occurring following the binding of an opioid agonist to a G-protein-coupled opioid receptor.
Opioid-mediated analgesia
Opioid receptors are distributed throughout the central nervous system and within peripheral tissue of neural and non-neural origin. Centrally, the periaqueductal grey (PAG), locus ceruleus and rostral ventral medulla show high concentrations of opioid receptors, and opioid receptors are also present in the substantia gelatinosa of the dorsal horn.
Within the central nervous system, activation of MOP receptors in the midbrain is thought to be a major mechanism of opioid-induced analgesia. Here, MOP agonists act by indirectly stimulating descending inhibitory pathways which act upon the periaqueductal grey (PAG) and nucleus reticularis paragigantocellularis (NRPG) with the net effect of an activation of descending inhibitory neurons. This leads to greater neuronal traffic through the nucleus raphe magnus (NRM), increasing stimulation of 5-hydroxytryptamine and enkephalin-containing neurons which connect directly with the substantia gelatinosa of the dorsal horn. This in turn results in a reduction of nociceptive transmission from the periphery to the thalamus. Exogenous and endogenous opioids can also exert a direct inhibitory effect upon the substantia gelatinosa (in the dorsal horn) and peripheral nociceptive afferent neurones, reducing nociceptive transmission from the periphery (Figure 2). This series of cellular events and mechanisms produces much of the analgesic effect commonly seen following the administration of MOP agonists.
Figure 2.
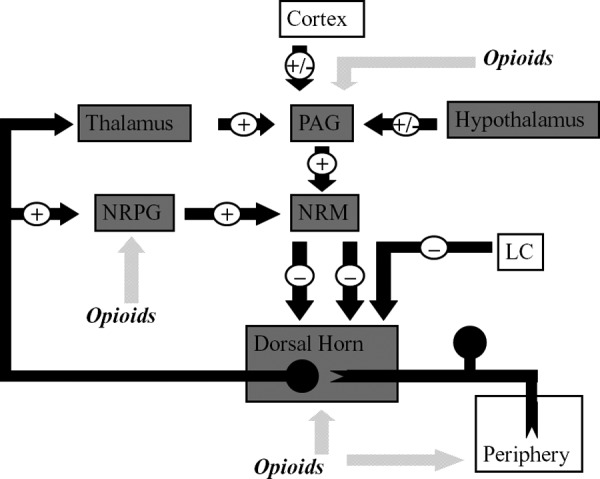
This figure shows schematically the descending inhibitory pathways. Areas shaded grey display a high expression of opioid receptors and their endogenous ligands. MOP agonists produce analgesia either by indirectly increasing neuronal traffic through the descending pathway at the NRPG and PAG, or by directly inhibiting nociceptive afferents in the periphery. MOP agonists act at the NRM to indirectly inhibit spinal pain transmission and, in addition, reduce spinal nociception. PAG = periaqueductal grey; NRPG = nucleus reticularis paragigantocellularis; NRM = nucleus raphe magnus; LC = locus correolus.
Clinical opioids
All opioids used in clinical practice today exert their action, at least in part, at the MOP receptor, with some having additional activity at one or more further opioid receptor or receptors distinct from the opioid family. Of those drugs used in clinical practice, morphine, though generally considered to be the archetypal MOP agonist to which all other analgesics are compared, also displays some degree of activity at additional receptors, acting as an agonist at MOP receptors, but also having activity at both DOP and KOP receptors. While this MOP receptor agonism is responsible for the majority of the analgesic properties of opioids, activity at opioid receptors also accounts for many of the commonly observed side-effects seen with their use. Opioids may cause a reduction in conscious level and euphoria, making them drugs of abuse. They also exert effects on the respiratory system, reducing respiratory rate and obtunding airway reflexes, an effect which is considered advantageous during anaesthesia. Although opioids are generally considered to preserve cardiac stability, histamine release and the associated reductions in systemic vascular resistance and blood pressure are marked with morphine. Amongst many other side-effects, opioids can also cause constipation, nausea, vomiting, urinary retention, pruritus, muscular rigidity, miosis and dysphoria in certain individuals. This list is by no means comprehensive, but, despite their numerous drawbacks to this day, opioids remain the yardstick against which all other clinically effective analgesics are measured.
In clinical practice, morphine is frequently administered via oral or intravenous routes, although subcutaneous, transdermal, sublingual, intramuscular, epidural, intrathecal and intra-articular routes are also commonly utilised depending upon the setting. However, owing to its low lipid solubility, morphine penetrates the blood–brain barrier slowly, causing it to have a relatively slow onset of effect if administered via a route beyond this anatomical barrier. This means that even following intravenous administration, peak analgesic effect will not be achieved for some time. Oral administration of morphine will further act to slow this onset of action and reduce morphine’s bioavailability. Typically, 40–60% of orally ingested morphine will fail to reach the systemic circulation as a result of significant first-pass metabolism in the liver and gut wall. Here, morphine is metabolised, predominantly via glucoronidation, to active metabolites excreted in urine. These metabolites, particularly in the case of morphine-6-glucuronide, can be longer lived and more potent than the parent compound, morphine. In health, therefore, morphine displays an elimination half-life of around 150 minutes, and, although this value may be altered by age, concomitant use of other medications and derangements of renal and hepatic function, in a clinical setting it is often necessary to provide relatively frequent doses of morphine to allow for a consistent plasma and effect site concentration of morphine to optimise the analgesic effect.
Of the other opioids commonly encountered in an acute hospital setting, alfentanil, fentanyl and remifentanil all effectively act clinically as MOP receptor agonists, differing from morphine primarily in their pharmacokinetic properties. Alfentanil and fentanyl are both highly lipid soluble with a far more rapid onset of action than morphine. In the palliative care and chronic pain settings, such is fentanyl’s lipophilicity that it is often delivered via the sublingual or transdermal routes to avoid oral or intravenous administration. As with all highly lipid soluble drugs, prolonged administration of alfentanil or fentanyl may result in sequestration of the drug to fat stores. This in turn may result in an extended period of recovery during which the drug is cleared from the body as it is returned to the vascular compartment from the fatty tissues prior to excretion renally, on cessation of administration. Remifentanil differs in this respect as, although significantly more lipid soluble than morphine, it is rapidly metabolised extrahepatically by non-specific esterases in blood and tissue. Owing to this novel mechanism of metabolism, remifentanil is often chosen as a rapid short-acting opioid analgesic agent in theatre and intensive care, where patients may be sedated for prolonged periods of time and rapid clearance of drug is beneficial.
Although there is good evidence from clinical trials to suggest that opioids are effective at treating pain in the short and medium term, there is a lack of high-quality evidence supporting their use for chronic pain states, with a series of recent Cochrane reviews questioning the use opioids in this setting.11–14 These reviews report weak evidence to support opioid management of long-term non-cancer pain and show only a small to moderate benefit of opioid administration in osteoarthritis coupled with an increased frequency of adverse events. Similarly, the use of opioids in the management of low back pain is not supported by high-quality evidence, while their use in neuropathic pain conditions is supported only by evidence for use in the intermediate term.
Opioids are, however, frequently prescribed within the community, where codeine, oxycodone and buprenorphine are commonly used for chronic pain states. All act primarily on MOP receptors, but codeine first needs to be metabolised to morphine by the body for it to display any activity. Between 5% and 10% of the population are estimated to lack the ability to perform this conversion, so derive limited, if any, pain relief from it. Oxycodone, a potent semi-synthetic derivative of thebaine that mediates its analgesic properties through both MOP and KOP receptors, has a high oral bioavailability, which can be manufactured in a time-release preparation, allowing it to be administered twice a day.15 In contrast, buprenorphine, another agent commonly prescribed for chronic pain patients, is one of the few opioid partial agonists available for medical administration.3,16 In practice, this means that it produces analgesic effects at lower plasma concentrations via its interaction with the MOP receptor, but anti-analgesic effects at high doses through interactions with the KOP and NOP receptors. Along with the ability to deliver buprenorphine via a transdermal route, these properties make it a useful drug within the pain clinic, where its lower potential for respiratory depression and overdose than pure MOP agonists is highly advantageous.
Tramadol and methadone are two further commonly prescribed opioid receptor agonists that, in addition to MOP effects, also have activity at other non-opioid sites. Tramadol, a phenylpiperidine analogue of codeine with comparable analgesic effect, is thought to work through modulation of serotonin and norepinephrine reuptake, in addition to its action as an MOP receptor agonist. Although tramadol displays many of the side-effects associated with MOP receptor agonists, it is purported to produce less respiratory depression and fewer gastrointestinal side-effects than pure MOP agonists of comparable analgesic potency. It may, however, also interact with drugs that inhibit serotonin and noradrenaline reuptake centrally, leading to seizures. The synthetic opioid methadone, meanwhile, with its long duration of action, limited first-pass metabolism, high bioavailability and more limited potential to induce euphoria, is often used as an oral opioid substitute in individuals addicted to intravenous opioids. In addition to its role in addiction medicine, methadone is sometimes used in the treatment of chronic pain where its postulated antagonistic activity at the N-methyl-d-aspartate (NMDA) receptor may account for some of its effectiveness in neuropathic pain states. A third dual-action centrally-acting analgesic agent, Tapentadol, has also recently been released in the UK for use in moderate to severe acute pain and as a prolonged-release preparation for chronic pain. Tapentadol displays MOP receptor agonist and noradrenaline reuptake inhibitor properties and is purported to have comparable analgesic efficacy to controlled-release oxycodone.
In various medical settings, reversal of the effects of opioid analgesia may be required, particularly for patients with markedly depressed respiratory function or conscious level. Naloxone and naltrexone can be used to achieve this reversal through their action as antagonists at all three of the classical opioid receptors. They do not, however, bind to the NOP receptor, and therefore would not modulate the effects of any future NOP agonists or partial agonists.
Conclusion
Opioid-based medications have been used by man for over 5000 years. However, with the greater understanding of opioid pharmacology gained over the past two centuries, modern opioids now provide the clinician with a range of different agents, with pharmacokinetic and dynamic properties tailored to a variety of medical settings. This ensures that, despite their longevity, opioids remain the gold standard to which all other analgesics are compared.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
The authors declare that they do not have any conflict of interest.
Further reading
Bailey PL, Egan TD and Stanley TH. In: Miller RD (ed) Anesthesia (5th ed). Philadelphia: Churchill Livingstone, 2000.
Collett BJ. Chronic opioid therapy for non-cancer pain. Br J Anaesth 2001; 87: 133–143. Eguchi M. Recent advances in selective opioid receptor agonists and antagonists. Med Res Rev 2004; 24: 182–212.
The British Pain Society. Opioids for persistent pain: good practice. A consensus statement prepared on behalf of the British Pain Society, the Faculty of Pain Medicine of the Royal College of Anaesthetists, the Royal College of General Practitioners and the Faculty of Addictions of the Royal College of Psychiatrists. January 2010.
Multiple-choice questions
Which of the following statements are correct? More than one answer may be correct for each question.
- Regarding opioid use in the community:
- codeine is an active compound
- 100% of the population can derive pain relief from codeine
- buprenorphine is a pure MOP agonist
- oxycodone has a higher oral bioavailability than morphine
- buprenorphine always causes respiratory depression
- Tramadol:
- acts via MOP agonism only
- can cause seizures
- is a natural alkaloid
- is a phenylpiperidine derivative
- inhibits serotonin reuptake
- Morphine has:
- an immediate peak analgesic effect
- potent metabolites
- a high first-pass metabolism
- an analgesic effect that can be reversed with naloxone
- no activity at MOP receptors
- Side-effects commonly encountered with opioids include:
- increased respiratory rate
- nausea and vomiting
- diarrhoea
- dilated pupils
- depression of airway reflexes
- Opioid receptors:
- are G-protein coupled
- are exclusively found in the central nervous system
- the nociceptin receptor is considered a non-opioid branch of the opioid receptor family
- stimulation may produce analgesia
- all have endogenous ligands
Answers
a) False; b) False; c) False; d) True; e) False.
a) False; b) True; c) False; d) False; e) True.
a) False; b) True; c) True; d) True; e) False.
a) False; b) True; c) False; d) False; e) True.
a) True; b) False; c) True; d) True; e) True.
References
- 1. Blakemore PR, White JD. Morphine, the Proteus of organic molecules. Chem Commun 2002; 1159–1168. [DOI] [PubMed] [Google Scholar]
- 2. Charlton JE. (ed.) Opioids: core curriculum for professional education in pain. Seattle: IASP Press, 2005. [Google Scholar]
- 3. McDonald J, Lambert DG. Opioid receptors. Contin Educ Anaesth Crit Care Pain 2005; 5: 22–25. [Google Scholar]
- 4. Dhawan BN, Cesselin F, Raghubir R, et al. International Union of Pharmacology. XII. Classification of opioid receptors. Pharmacol Rev 1996; 48: 567–592. [PubMed] [Google Scholar]
- 5. Dietis N, Rowbotham D, Lambert DG. Opioid receptor subtypes: fact or fiction? British J Anaesth 2011; 107: 8–18. [DOI] [PubMed] [Google Scholar]
- 6. Stein C, Schafer M, Machelska H. Attacking pain at its source: new perspectives on opioids. Nat Med 2003; 9: 1003–1008. [DOI] [PubMed] [Google Scholar]
- 7. Corbett AD, Henderson G, McKnight AT, Paterson SJ. 75 years of opioid research: the exciting but vain quest for the Holy Grail. British J Pharmacology 2006; 147: S153–S162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Calo’ G, Guerrini R, Rizzi A, et al. Pharmacology of nociceptin and its receptor: a novel therapeutic target. Br J Pharmacol 2000; 129: 1261–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pan Z, Hirakawa N, Fields HL. A cellular mechanism for the bidirectional pain-modulating actions of orphanin FQ/nociceptin. Neuron May 26: 515–522. [DOI] [PubMed] [Google Scholar]
- 10. Harrison C, Smart D, Lambert DG. Stimulatory effects of opioids. Br J Anaesth 1998; 81: 20–28. [DOI] [PubMed] [Google Scholar]
- 11. Deshpande A, Furlan AD, Mailis-Gagnon A, Atlas S, Turk D. Opioids for chronic low-back pain. Cochrane Database Syst Rev 2007; 3: CD004959. [DOI] [PubMed] [Google Scholar]
- 12. Nüesch E, Rutjes AWS, Husni E, Welch V, Jüni P. Oral or transdermal opioids for osteoarthritis of the knee or hip. Cochrane Database Syst Rev 2009; 4: CD003115. [DOI] [PubMed] [Google Scholar]
- 13. Eisenberg E, McNicol ED, Carr DB. Opioids for neuropathic pain. Cochrane Database Syst Rev 2006; 3: CD006146. [DOI] [PubMed] [Google Scholar]
- 14. Noble M, Treadwell JR, Tregear SJ, et al. Long-term opioid management for chronic noncancer pain. Cochrane Database Syst Rev 2010; 1: CD006605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nielsen CK, Ross FB, Lotfipour S, Saini KS, Edwards SR, Maree T. Oxycodone and morphine have distinctly different pharmacological profiles: radioligand binding and behavioural studies in two rat models of neuropathic pain. Pain 2007; 132(3): 289–300. [DOI] [PubMed] [Google Scholar]
- 16. Ide S, Minami M, Satoh M, et al. Buprenorphine antinociception is abolished, but naloxone-sensitive reward is retained, in mu-opioid receptor knockout mice. Neuropsychopharmacology 2004; 29: 1656–1663. [DOI] [PubMed] [Google Scholar]