

Ubiquitin (Ub) is a well-known eukaryotic protein that exerts diverse signaling functions inside the cells, including targeting its substrates for proteasomal degradation (Hershko and Ciechanover, 1998). Tagging substrates with Ub or ubiquitination occurs via the action of three sequential enzymes called Ub-activating enzymes (E1), Ub conjugating enzymes (E2), and Ub ligases (E3). This entire process is called the Ub conjugation reaction and the substrate can be either monoubiquitinated or polyubiquitinated, which results in different outcomes for the target substrate. Inside the cells, Ub is in dynamic equilibrium between “free Ub” and “conjugated Ub” (Komander et al., 2009; Park and Ryu, 2014). Free Ub refers to a readily available form of Ub for the conjugation reaction, while conjugated Ub refers to single or multiple Ub attached to target substrates. Thus, it needs to be deconjugated or removed from the substrate (i.e., Ub conjugates) to be used for another conjugation reaction. The free Ub pool is generally maintained by de novo Ub synthesis via expression of ubiquitin genes and by Ub removal or recycling from Ub conjugates via various deubiquitinating enzymes (Komander et al., 2009; Park and Ryu, 2014). In mammals, there are two different classes of ubiquitin genes, Ub-ribosomal fusion genes (Uba52 and Uba80) and polyubiquitin genes (Ubb and Ubc). The resulting Ub-ribosomal subunit or (Ub)n fusion proteins are rapidly cleaved by deubiquitinating enzymes. Thus, all four ubiquitin genes are eventually responsible for the generation of free Ub, which is virtually indistinguishable, regardless of the original encoding gene. The contribution of the four different ubiquitin genes toward the constitution of the free Ub pool varies depending on cell types, the tissue origin, or environmental conditions. Although Ub is an abundant protein inside the cells, because of its dynamic nature and the abundance of its target substrates, the maintenance of free Ub above threshold levels or Ub homeostasis is important for the ubiquitination of target substrates in a timely manner. About a decade ago, it was shown that free Ub depletion by cycloheximide treatment in yeast resulted in reduced viability with increased toxicity, which was ameliorated by Ub overexpression (Hanna et al., 2003). Since then, the cellular maintenance of the free Ub pool has been of great interest because the maintenance of free Ub is expected to be important for cellular function and survival. In 2009, the presence of unanchored free Ub chains, which are converted to monomeric free Ub by a deubiquitinating enzyme to maintain the free Ub pool, was proposed (Kimura et al., 2009). An inhibitor of the deubiquitinating enzyme also plays a role in the maintenance of adequate levels of free Ub. The overexpression of the inhibitor was shown to reduce free Ub levels and disrupt Ub homeostasis, which reduced the viability of yeast under stress conditions (Kimura et al., 2009).
For the past several years, the consequence of Ub homeostasis disruption or Ub deficiency in mammals has been investigated via the deletion of two different polyubiquitin genes. Although homozygous deletion of Ubc resulted in mid-gestation embryonic lethality due to defective fetal liver development, homozygous deletion of Ubb resulted in various phenotypes, including infertility, neuronal dysfunction and death in the hypothalamus, and metabolic disorders, including obesity and sleep abnormalities (Ryu et al., 2008, 2010). Ubb knockout phenotypes are closely associated with defects in cell types with high Ubb expression such as germ cells or hypothalamic neurons. Interestingly, Ubb was also highly expressed in a brain region called the locus coeruleus in which Ubb loss also led to the reduction of total Ub levels, whereas free Ub levels were well maintained due to a significant upregulation of Ubc (Park et al., 2012). The maintenance of free Ub levels in the locus coeruleus was sufficient to protect these neurons, in contrast to the hypothalamus in which free Ub levels were reduced due to Ubb disruption, suggesting a critical role for free Ub in the maintenance of neuronal function and survival. Prior to adult-onset hypothalamic neurodegeneration, neuroinflammation, characterized by reactive gliosis, occurs in the hypothalamus, which seems to be closely related to reduced activity and metabolic rate in Ubb knockout mice (Ryu et al., 2010). However, it is unknown why reactive gliosis preceded neurodegeneration in the hypothalamus and whether cell autonomous defects to maintain free Ub pool, but not their altered cellular microenvironment, are responsible for their neural phenotypes. In particular, whether reactive gliosis is the direct outcome of Ubb disruption is of great interest. To address these issues, it is necessary to isolate cells from the brain of mid-gestation Ubb knockout embryos and to investigate their characteristics during culture in vitro.
In 2014, our laboratory provided strong evidence that free Ub level is important for the proper neuronal development as well as for the determination of the fate and self-renewal of neural stem cells. In the October 11th issue of Biochemical and Biophysical Research Communications, we demonstrated that, using cultured cells isolated from the brain of Ubb knockout embryos, neuronal abnormalities characterized by short neurites, poor branching, and impaired synaptic development observed upon Ubb disruption are the simple outcome of the reduced availability of free Ub inside the cells (Ryu et al., 2014a). Restoration of free Ub, but not Ub conjugates, by providing exogenous Ub using lentivirus-mediated delivery was sufficient to abolish apoptosis and improve the neuronal and glial phenotypes observed in Ubb knockout cells. Thus, our in vitro cell culture results support that Ub homeostasis or the maintenance of cellular free Ub above a certain threshold level is essential for the proper neuronal development and survival (Figure 1). Furthermore, in the November 14th issue of Scientific Reports, we also demonstrated that Ubb disruption led to the promotion of premature gliogenesis and the suppression of neurogenesis from neural stem cells with decreased cell numbers during the mid-gestation embryonic stage (Ryu et al., 2014b). Differentiation of neural stem cells into neurons and astrocytes is known to be regulated by Notch signaling (Yoon and Gaiano, 2005). It is known that downregulation of Notch signaling during the embryonic stage promotes neurogenesis and suppresses gliogenesis, while upregulation of Notch signaling during the postnatal stage promotes gliogenesis and neuronal maturation. However, Ubb disruption led to the aberrant activation of Notch signaling even during the embryonic stage, with increased levels of Notch target genes and decreased proneuronal gene expression. This was caused by the increased steady-state levels or accumulation of Notch intracellular domain, probably due to the delayed degradation of Notch intracellular domain that resulted from the reduced function of the proteasome under Ub deficiency. Notch activation is also known to suppress neurogenesis, which, in fact, results in the generation of defective neurons upon Ubb disruption. Defective neurons can activate astrocytes to reactive astrocytes that secrete inflammatory cytokines and proteins, which, in turn, cause the degeneration of defective neurons (Bi et al., 2013). Based on our and other's reports, we determined why reactive gliosis precedes neurodegeneration in Ubb knockout mice. Furthermore, by demonstrating that the phenotypes of Ubb knockout cells can be rescued by suppression of the Notch signaling by pharmacological intervention and by increasing levels of cellular free Ub (unpublished data), we propose that differentiation of neural stem cells can be regulated by the intracellular levels of free Ub. Thus, our data demonstrate that the aberrant activation of Notch signaling due to reduced cellular Ub pools during mid-gestation leads to alterations in the fate of neural stem cells, resulting in the premature onset of gliogenesis and suppression of neurogenesis (Figure 1). Although our Ubb knockout cell culture model is sufficient to investigate the effect of Ub homeostasis disruption, our model may not provide an exact answer regarding the cell types that are responsible for the Ubb knockout phenotypes. It is most likely that Ub deficiency in neural stem cells, i.e., neural stem cell-autonomous defect, is responsible for the dysregulation of their own differentiation. However, we cannot exclude the possibility that Ub-deficient glial cells or neurons, i.e., reactive astrocytes or degenerating neurons, may affect the fate of existing neural stem cells regarding differentiation. Thus, to test this possibility, further investigations such as neural stem cell-specific Ubb knockout or providing exogenous Ub in a neural stem cell-specific manner to Ubb knockout cells are required to fully identify the molecular mechanisms underlying the onset of reactive gliosis caused by Ubb disruption.
Figure 1.
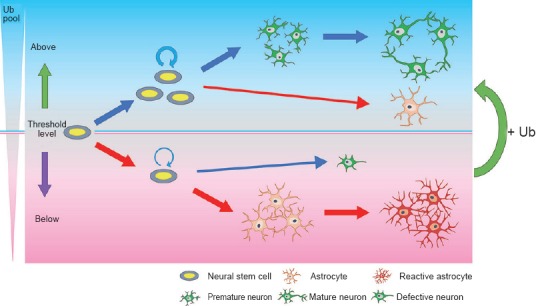
Requirement of free ubiquitin (Ub) above threshold levels for the differentiation of neural stem cells and neuronal development.
If the free Ub amount is above threshold levels, neural stem cell numbers are well maintained, neurogenesis precedes gliogenesis, and mature neurons are generated. However, if the free Ub amount falls below threshold levels, as observed upon Ubb disruption, neural stem cell numbers are reduced due to the impaired self-renewal, gliogenesis occurs prematurely, and defective neurons are generated. Increase in free Ub amount is sufficient to recover neuronal and glial abnormalities to some extent.
Based on our recent reports, we speculate that measuring cellular levels of free Ub will be essential for the optimal differentiation of neural stem cells into neurons and for neuronal development. Researchers who work with patient samples or cultured cells should be aware that the outcome of neural stem cell differentiation can be varied. This will be particularly useful for those who focus on neurons, but not on glial cells, or vice versa. Ub may be one of the key proteins that determine the fate of neural stem cells. Thus, Ub homeostasis should be considered to maximize the efficacy of neural stem cell transplantation. Therefore, our research represents a milestone in the development of a novel and universal therapeutic strategy to overcome various neurodegenerative diseases.
This work was supported by a grant from the Korean Health Technology R & D Projects, Ministry of Health & Welfare, Republic of Korea (HI14C2300) to KYR.
References
- Bi F, Huang C, Tong J, Qiu G, Huang B, Wu Q, Li F, Xu Z, Bowser R, Xia XG, Zhou H. Reactive astrocytes secrete lcn2 to promote neuron death. Proc Natl Acad Sci U S A. 2013;110:4069–4074. doi: 10.1073/pnas.1218497110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Leggett DS, Finley D. Ubiquitin depletion as a key mediator of toxicity by translational inhibitors. Mol Cell Biol. 2003;23:9251–9261. doi: 10.1128/MCB.23.24.9251-9261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Yashiroda H, Kudo T, Koitabashi S, Murata S, Kakizuka A, Tanaka K. An inhibitor of a deubiquitinating enzyme regulates ubiquitin homeostasis. Cell. 2009;137:549–559. doi: 10.1016/j.cell.2009.02.028. [DOI] [PubMed] [Google Scholar]
- Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- Park CW, Ryu KY. Cellular ubiquitin pool dynamics and homeostasis. BMB Rep. 2014;47:475–482. doi: 10.5483/BMBRep.2014.47.9.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CW, Ryu HW, Ryu KY. Locus coeruleus neurons are resistant to dysfunction and degeneration by maintaining free ubiquitin levels although total ubiquitin levels decrease upon disruption of polyubiquitin gene Ubb. Biochem Biophys Res Commun. 2012;418:541–546. doi: 10.1016/j.bbrc.2012.01.063. [DOI] [PubMed] [Google Scholar]
- Ryu HW, Park CW, Ryu KY. Restoration of cellular ubiquitin reverses impairments in neuronal development caused by disruption of the polyubiquitin gene Ubb. Biochem Biophys Res Commun. 2014a;453:443–448. doi: 10.1016/j.bbrc.2014.09.103. [DOI] [PubMed] [Google Scholar]
- Ryu HW, Park CW, Ryu KY. Disruption of polyubiquitin gene Ubb causes dysregulation of neural stem cell differentiation with premature gliogenesis. Sci Rep. 2014b;4:7026. doi: 10.1038/srep07026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KY, Garza JC, Lu XY, Barsh GS, Kopito RR. Hypothalamic neurodegeneration and adult-onset obesity in mice lacking the Ubb polyubiquitin gene. Proc Natl Acad Sci U S A. 2008;105:4016–4021. doi: 10.1073/pnas.0800096105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KY, Fujiki N, Kazantzis M, Garza JC, Bouley DM, Stahl A, Lu XY, Nishino S, Kopito RR. Loss of polyubiquitin gene Ubb leads to metabolic and sleep abnormalities in mice. Neuropathol Appl Neurobiol. 2010;36:285–299. doi: 10.1111/j.1365-2990.2009.01057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K, Gaiano N. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat Neurosci. 2005;8:709–715. doi: 10.1038/nn1475. [DOI] [PubMed] [Google Scholar]