

Abstract
Cerebral ischemia triggers secondary ischemia/reperfusion injury and endoplasmic reticulum stress initiates cell apoptosis. However, the regulatory mechanism of the signaling pathway remains unclear. We hypothesize that the regulatory mechanisms are mediated by the protein kinase-like endoplasmic reticulum kinase/eukaryotic initiation factor 2α in the endoplasmic reticulum stress signaling pathway. To verify this hypothesis, we occluded the middle cerebral artery in rats to establish focal cerebral ischemia/reperfusion model. Results showed that the expression levels of protein kinase-like endoplasmic reticulum kinase and caspase-3, as well as the phosphorylation of eukaryotic initiation factor 2α, were increased after ischemia/reperfusion. Administration of atorvastatin decreased the expression of protein kinase-like endoplasmic reticulum kinase, caspase-3 and phosphorylated eukaryotic initiation factor 2α, reduced the infarct volume and improved ultrastructure in the rat brain. After salubrinal, the specific inhibitor of phosphorylated eukaryotic initiation factor 2α was given into the rats intragastrically, the expression levels of caspase-3 and phosphorylated eukaryotic initiation factor 2α in the were decreased, a reduction of the infarct volume and less ultrastructural damage were observed than the untreated, ischemic brain. However, salubrinal had no impact on the expression of protein kinase-like endoplasmic reticulum kinase. Experimental findings indicate that atorvastatin inhibits endoplasmic reticulum stress and exerts neuroprotective effects. The underlying mechanisms of attenuating ischemia/reperfusion injury are associated with the protein kinase-like endoplasmic reticulum kinase/eukaryotic initiation factor 2α/caspase-3 pathway.
Keywords: nerve regeneration, neuroprotection, protein kinase-like endoplasmic reticulum kinase, eukaryotic initiation factor 2α, endoplasmic reticulum stress, focal cerebral ischemia/reperfusion, atorvastatin, apoptosis
Introduction
Cerebral infarction leads to the necrosis of brain cells and loss of neurological function. Recent discoveries suggest that endoplasmic reticulum stress is closely involved in apoptosis. The endoplasmic reticulum is an organelle responsible for protein folding, homeostasis and lipid synthesis. Its functions are influenced by a variety of factors, including oxidative stress, ischemia and electrolyte imbalance (Gargalovic et al., 2006; Zhang and Kaufman, 2006; Hotamisligil, 2010; Chen and Brandizzi, 2013; Mandl et al., 2013). These factors contribute to the accumulation of unfolded and misfolded proteins within the endoplasmic reticulum, trigger endoplasmic reticulum stress (Walter and Ron, 2011; Lu et al., 2014; Pincus et al., 2014), induce pro-apoptotic transcription factor C/EBP homologous protein, and activate C-JNK kinase, cleavage apoptotic protein 12 and caspase-3 (Bernales et al., 2006; Xu et al., 2013; Yang et al., 2014b), ultimately triggering cell apoptosis or death.
There are three signaling pathways involved in endoplasmic reticulum stress, namely protein kinase-like endoplasmic reticulum kinase (PERK), inositol-requiring kinase 1 and activating transcription factor 6 (Oyadomari and Mori, 2003; Senkal et al., 2010; Hetz et al., 2011; Mei et al., 2013; Sone et al., 2013). PERK is a serine/threonine kinase and can be activated by endoplasmic reticulum stress (Schröder and Kaufman, 2005; Malhotra and Kaufman, 2007; Clarke et al., 2012; Gardner et al., 2013; Liu et al., 2013; Sano and Reed, 2013). In addition, PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), thereby blocking the mRNA translation process (Prostko et al., 1992; Kaufman, 1999). Caspase-3 is one of the key enzymes leading to cell apoptosis and one of the crucial downstream factors executing the apoptotic program (Nakagawa et al., 2000; Zhang et al., 2012; Seo et al., 2014). Therefore, we hypothesize that endoplasmic reticulum stress-caused apoptosis after ischemia/reperfusion is associated with caspase-3 and that the PERK/eIF2α/caspase-3 pathway underlies the mechanism of injury.
Other evidence shows that fluvastatin inhibits the macrophage apoptosis caused by hypoxia and that the underlying mechanisms are associated with the inhibition of endoplasmic reticulum stress and apoptosis of endothelial cells (Bao et al., 2009). Atorvastatin has a similar molecular structure to fluvastatin, and is a competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase (Chen et al., 2006; Qi et al., 2013). In this study, we hypothesize that atorvastatin can inhibit the hypoxia-caused apoptosis, attenuate cerebral ischemia/reperfusion injury, and exert neuroprotective effects through the endoplasmic reticulum stress pathway. The present study aims to explore the underlying mechanism of atorvastatin in endoplasmic reticulum stress following ischemia/reperfusion injury.
Materials and Methods
Establishing cerebral ischemia/reperfusion model
Sixty clean, healthy, male Sprague-Dawley rats, aged 10 weeks and weighing 189 ± 9 g, were provided by the Animal Laboratory at Xiangya School of Medicine, Central South University (Changsha, Hunan Province, China; license No. SYXK (Xiang) 2013-001). The laboratory conditions were controlled between 40% and 70% humidity and at 20–25°C temperature with a 12-hour day/night cycle. The indoor wind speed was 0.1–0.2 m/s. All experimental protocols were approved by the Experimental Ethics Committee of Mawangdui Hospital of Hunan Province in China. Rats were randomly divided into four groups: sham, ischemia/reperfusion, atorvastatin and atorvastatin + salubrinal. Each group contained 15 rats.
The rat model of focal cerebral ischemia/reperfusion was established in rats of ischemia/reperfusion, atorvastatin and atorvastatin + salubrinal groups. Briefly, Sprague-Dawley rats were anesthetized with 10% chloral hydrate (350 mg/kg). After the right common carotid artery was exposed through a midline neck incision, the common carotid artery, external carotid artery and internal carotid artery were carefully separated from the adjacent tissue and vagus nerve. A gap was made 2 cm lateral to the bifurcation of the external carotid artery adjacent to the common carotid artery and a monofilament was advanced into the internal carotid artery to a depth of 18.5 ± 1.0 mm, until it blocked the blood flow to the middle cerebral artery. At 2 hours after occlusion, the nylon suture was withdrawn to realize reperfusion and the wounds were sutured. Sham-operated animals were prepared in the same way without carotid occlusion.
Drug administration
Rats in the atorvastatin and atorvastatin + salubrinal group were given atorvastatin (5 mg/kg, tablets; Dalian Pfizer, Dalian, Liaoning Province, China) dissolved in 2 mL warm water before intragastrical administration. Rats in the atorvastatin + salubrinal group were given eIF2α specific inhibitor, salubrinal (11.2 mg/kg, intragastrically; ApexBio Technology LLC, Houston, TX, USA), 2 hours after atorvastatin administration, once per day, from preoperative 1 day to postoperative 3 days (Teng et al., 2014). Rats in the sham and ischemia/reperfusion groups were injected with 2 mL of distilled water.
TTC staining of brain infarct volume
At 3 days after cerebral ischemia/reperfusion induction, rats were anesthetized with 10% chloral hydrate and brain tissue was fixed with 4% paraformaldehyde. Then rats were rapidly decapitated and the brain tissue was sliced into 2-mm-thick sections. These sections were incubated in 2% TTC buffer at 37°C in the dark for 30 minutes, then fixed in PBS. Images were captured and analyzed using a HMIAS-2000 high-resolution color image analysis system (Jinma Medical Devices Co., Ltd., Xi’an, Shaanxi Province, China). Brain infarct volume (percentage of total brain volume, %) was measured.
Transmission electron microscopy observation of the ultrastructure of the rat brain
At 3 days after cerebral ischemia/reperfusion induction, rats were decapitated under 10% chloral hydrate anesthesia and brain tissue was fixed with 4% paraformaldehyde and pre-fixed in 2.5% glutaraldehyde-PBS overnight, then rinsed with 0.1 M PBS three times and post-fixed with 1% osmium tetroxide. Subsequently, sections were dehydrated, immersed, embedded, polymerized and trimmed, and observed under transmission electron microscope (HT7700, Hitachi, Tokyo, Japan).
Western blot analysis of PERK, phosphorylated eIF2α and caspase-3 expression in rat brain tissue infarction side
At 3 days after cerebral ischemia/reperfusion induction, rats were polyving sacrificed under 10% chloral hydrate anesthesia, and the brain tissue from the affected side was minced and mixed with modified RIPA lysis buffer. The tissue samples obtained were homogenized and centrifuged at 0°C and the supernatants were divided into small aliquots and stored at –80°C after the precipitates were discarded. 200-μg samples were electrophoresed on a sodium dodecyl sulfate/polyacrylamide gel, in a stacking gel at 80 V and a separating gel at 120 V, and proteins were transferred onto a polyvinylidene difluoride membrane through a semi-drying process. The membrane was blocked with western blocking solution at room temperature for 3 hours, and incubated with rabbit anti-PERK, rabbit anti-phosphorylated eIF2α, rabbit anti-caspase-3 and β-actin polyclonal antibodies (1:4,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight. Then the membrane was rinsed and incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:2,000; Santa Cruz Biotechnology) at room temperature for 2 hours. After 1-hour hybridization, tissues were rinsed and developed. The optical density was measured using LS117 densitometer (Shenzhen Jinpengcheng Software Technology Co., Ltd., Shenzhen, Guangdong Province, China). The expression levels of target proteins were expressed as the ratio of target protein optical density to β-actin optical density.
Statistical analysis
Data were processed using SPSS 16.0 software (SPSS, Chicago, IL, USA) and expressed as the mean ± SD. The difference between groups was compared with one-way analysis of variance followed by the least significant difference test. A P < 0.05 value was considered statistically significant.
Results
Atorvastatin decreased brain infarct volume in rats with cerebral ischemia/reperfusion injury
TTC staining results showed that no infarction was detected in the sham-operated rats; cerebral infarction was visible in the rats from the cerebral ischemia/reperfusion group. Compared with the ischemia/reperfusion group, brain infarct volumes in the atorvastatin and atorvastatin + salubrinal groups were significantly reduced (P < 0.05; Figure 1).
Figure 1.
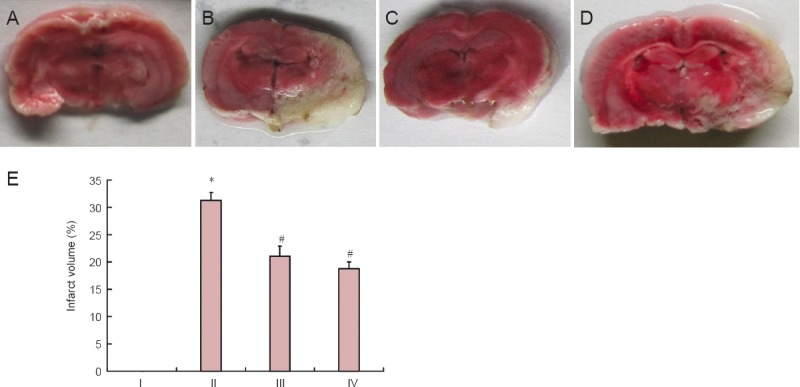
Effect of atorvastatin on brain infarct volume of rats with cerebral ischemia/reperfusion injury.
(A–D) Infarct volume was measured by TTC staining: normal brain tissue was red and infarcted tissue was white. (A) Sham group; (B) ischemia/reperfus group; (C) atorvastatin group; (D) atorvastatin + salubrinal group. (E) Quantitative analysis of brain infarct volume in the rat brain. Data are represented as the mean ± SD (n = 15 rats in each group). The differences among groups were compared with one-way analysis of variance followed by the least significant difference test. *P < 0.05, vs. I; #P < 0.05, vs. II. I: Sham group; II: ischemia/reperfusion group; III: atorvastatin group; IV: atorvastatin + salubrinal group.
Atorvastatin improved ultrastructure of brain tissue in rats with cerebral ischemia/reperfusion injury
Under transmission electron microscopy, we found that brain tissue in the sham-operated rats was normal in the ischemia/reperfusion group of rats, the mitochondria, Golgi complex and endoplasmic reticulum were swollen with some vacuoles and debris, mitochondria were unclear or had disappeared, and nuclear membrane depression and nuclear enrichment were observed. In the atorvastatin and atorvastatin + salubrinal groups, the mitochondria, Golgi complex and endoplasmic reticulum were swollen in the brain tissue at the infarction side, but their structure was intact and no nuclear condensation was found (Figure 2).
Figure 2.
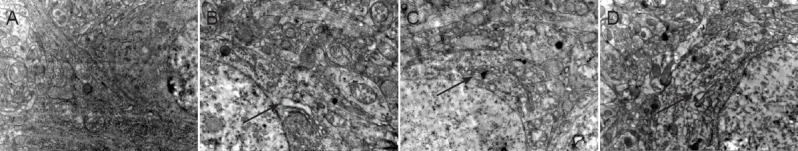
Effect of atorvastatin on the ultrastructure of neurons in infarcted brain tissue of rats with cerebral ischemia/reperfusion injury (transmission electron microscopy, × 20,000).
Neuronal damage was most obvious in the ischemia/reperfusion group, showing the swelling, vacuoles and debris of mitochondria, Golgi complex and endoplasmic reticulum (arrows), unclear or disappeared mitochondria, and nuclear enrichment. (A) Sham group; (B) ischemia/reperfusion group; (C) atorvastatin + salubrinal group; (D) atorvastatin group.
Atorvastatin reduced caspase-3, phosphorylated eIF2α and PERK expression in infarcted brain tissue of rats with cerebral ischemia/reperfusion injury
Western blot assay results showed that ischemia/reperfusion injury obviously increased protein expression of PERK, caspase-3 and phosphorylated eIF2α in rats compared with the sham-operated rats (P < 0.01). Atorvastatin significantly decreased protein expressions of PERK, caspase-3 and phosphorylated eIF2α in rat brain tissue (P < 0.05). Salubrinal, a specific inhibitor of eIF2α acidification, significantly inhibited eIF2α protein phosphorylation and caspase-3 activity in the brain tissue of rats with cerebral ischemia/reperfusion injury (P < 0.05), but had no impact on PERK expression (P > 0.05; Figure 3).
Figure 3.
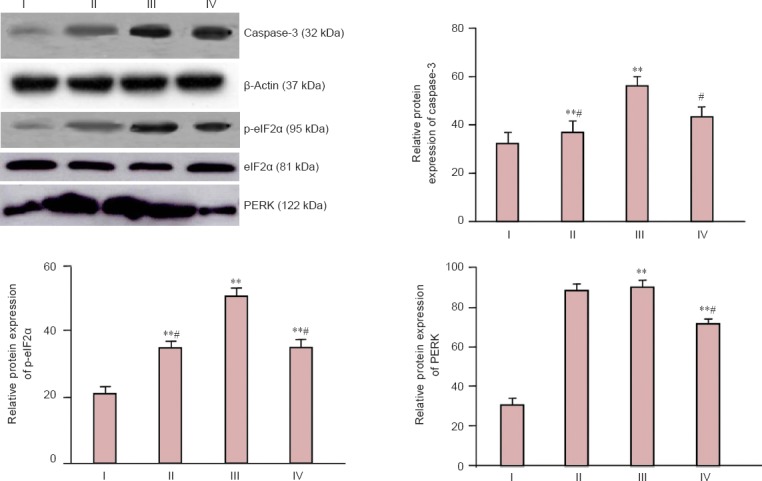
Effect of atorvastatin on caspase-3, p-eIF2α and PERK protein expression in infarcted brain tissue of rats with cerebral ischemia/reperfusion injury (western blot assay).
Protein expression levels were expressed as the optical density ratio of caspase-3, p-eIF2α and PERK to β-actin. Data are represented as the mean ± SD (n = 15 rats per group). The difference among groups was compared using one-way analysis of variance followed by the least significant difference test. **P < 0.01, vs. I; #P < 0.05, vs. II. PERK: Protein kinase-like endoplasmic reticulum kinase; p-eIF2α: phosphorylated eukaryotic initiation factor 2α; I: sham group; II: atorvastatin + salubrinal group; III: ischemia/reperfusion group; IV: atorvastatin group.
Discussion
PERK is a serine-threonine kinase that phosphorylates eIF2α after stress (Lee and Kim, 2013), thereby blocking the mRNA translation, such as activating transcription factor 4, inducing unfolded protein response-related genes, and reducing the unfolded protein levels. Caspase-12 has been shown to be highly involved in the process of endoplasmic reticulum stress (Ma and Hendershot, 2001; Morishima et al., 2002; Li et al., 2010; Walter and Ron, 2011), which triggers caspase-12 precursor cleavage and activation, and further stimulates caspase-12 and other factors to promote the mitochondrial-dependent cell death. Caspase-3 is one of the key enzymes and major executors leading to cell apoptosis, but it cannot be activated by autocatalysis or self-splicing. Little research has focused on the PERK/eIF2α/caspase-3 pathway. The present study found that in the brains of the cerebral ischemia/reperfusion injured rat group, necrotic cells and swelling organelles were observed under the electron microscope; western blot assay results showed that PERK protein expression, eIF2α dephosphorylation and caspase-3 activity were all increased, indicating apoptosis. When the necrosis occurred, endoplasmic reticulum stress was activated, including PERK; at the same time eIF2α phosphorylation was enhanced, which blocked the protein translation process; caspase-3, an apoptosis factor, was also initiated. The correlation of endoplasmic reticulum stress, eIF2α phosphorylation and caspase-3 activity after cerebral ischemia/reperfusion deserves further exploration. In this study, we found that salubrinal administration decreased caspase-3 activity and accordingly inhibited cell apoptosis, but it had no impact on PERK. Therefore we hypothesized that the PERK/eIF2α/caspase-3 pathway is involved in the apoptotic pathway. C/EBP homologous protein is the most studied molecule among the endoplasmic reticulum-mediated apoptosis signaling molecules. The pro-apoptotic mechanism of C/EBP homologous protein largely depends on activating transcription factor 4 and activating transcription factor 6 (Zhu et al., 2012) and is also mediated by the transcriptional inhibition and activation of Bcl-2 family proteins, thereby inhibiting the expression of anti-apoptotic Bcl-2 proteins (Weston and Puthalakath, 2010). Previous studies have highlighted the contribution of PERK/eIF2α/activating transcription factor 4 in C/EBP homologous protein protein expression; the activated PERK signaling pathway can protect cells and prolong cell survival through inhibiting protein synthesis at the early stage of endoplasmic reticulum stress, but as the endoplasmic reticulum stress time is prolonged, PERK induces C/EBP homologous protein expression and promotes apoptosis (Porter and Jänicke, 1999; Salakou et al., 2007; Park et al., 2014; Trivedi et al., 2014; Wang et al., 2014). Increasing evidence from previous research demonstrated that endoplasmic reticulum-associated kinase plays an important role in apoptosis, and a variety of pathways and factors associated with endoplasmic reticulum stress contribute to apoptosis. Our findings indicate that endoplasmic reticulum stress is involved in apoptosis through the PERK/eIF2α/caspase-3 pathway.
N-terminal glucose-regulated protein is responsible for the mechanism of fluvastatin reducing the hypoxia-caused apoptosis in macrophages and inhibits PERK phosphorylation that follows endoplasmic reticulum stress (Hayashi et al., 2003; Jia et al., 2012; Yang et al., 2014a). Atorvastatin can regulate blood lipid levels, exert anti-inflammatory and anti-oxidative effects, significantly reduce the incidence and mortality of cerebrovascular diseases, and block HMG-CoA reduction of mevalonic acid. Mevalonic acid is a precursor for the synthesis of geranylgeranyl pyrophosphate and farnesyl pyrophosphate, which are involved in immune regulation and apoptosis (Chen et al., 2008; Qian et al., 2011). Therefore, we speculate that atorvastatin may interfere with endoplasmic reticulum stress-mediated apoptosis. In the present study, results from the atorvastatin-only intervention found that it attenuated the organelle swelling and necrosis, and decreased PERK protein expression, eIF2α dephosphorylation and caspase-3 activity. Previous studies mainly focus on the effects of atorvastatin on regulating blood lipid, reducing inflammation and preventing atherosclerosis progression (Gleissner et al., 2007; Vilahur et al., 2009) rather than investigating anti-apoptosis or protection from ischemia-caused neuronal cell death. Our experimental findings indicate that atorvastatin could prevent apoptosis and attenuate nerve cell injury by acting through the PERK/eIF2α/caspase-3 pathway.
Footnotes
Conflicts of interest: None declared.
Copyedited by Dawes EA, Norman C, Yu J, Yang Y, Li CH, Song LP, Zhao M
References
- Bao XM, Wu CF, Lu GP. Atorvastatin attenuates homocysteine-induced apoptosis in human umbilical vein endothelial cells via inhibiting NADPH oxidase-related oxidative stress-triggered p38MAPK signaling. Acta Pharmacol Sin. 2009;30:1392–1398. doi: 10.1038/aps.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Huang KC, Lin WW. HMG-CoA reductase inhibitors upregulate heme oxygenase-1 expression in murine RAW264. 7 macrophages via ERK, p38 MAPK and protein kinase G pathways. Cell Signal. 2006;18:32–39. doi: 10.1016/j.cellsig.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Chen JC, Wu ML, Huang KC, Lin WW. HMG-CoA reductase inhibitors activate the unfolded protein response and induce cytoprotective GRP78 expression. Cardiovasc Res. 2008;80:138–150. doi: 10.1093/cvr/cvn160. [DOI] [PubMed] [Google Scholar]
- Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013;23:547–555. doi: 10.1016/j.tcb.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R, Cook KL, Hu R, Facey COB, Tavassoly I, Schwartz JL, Baumann WT, Tyson JJ, Xuan J, Wang Y, Wärri A, Shajahan AN. Endoplasmic reticulum stress, the unfolded protein response, autophagy and the integrated regulation of breast cancer cell fate. Cancer Res. 2012;72:1321–1331. doi: 10.1158/0008-5472.CAN-11-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol. 2013;5:a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Baruch-Oren T, Berliner JA, Kirchgessner TG, Lusis AJ. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- Gleissner CA, Leitinger N, Ley K. Effects of native and modified low-density lipoproteins on monocyte recruitment in atherosclerosis. Hypertension. 2007;50:276–283. doi: 10.1161/HYPERTENSIONAHA.107.089854. [DOI] [PubMed] [Google Scholar]
- Hetz C, Martinon F, Rodriguez D, Glimcher LH. The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiol Rev. 2011;91:1219–1243. doi: 10.1152/physrev.00001.2011. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16:396–399. doi: 10.1038/nm0410-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia F, Wu C, Chen Z, Lu G. Atorvastatin inhibits homocysteine-induced endoplasmic reticulum stress through activation of AMP-activated protein kinase. Cardiovasc Ther. 2012;30:317–325. doi: 10.1111/j.1755-5922.2011.00287.x. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Lee SK, Kim YS. Phosphorylation of eIF2α attenuates statin-induced apoptosis by inhibiting the stabilization and translocation of p53 to the mitochondria. Int J Oncol. 2013;42:810–816. doi: 10.3892/ijo.2013.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZW, Zhu HT, Chen KL, Dong X, Wei J, Qiu C, Xue JH. Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol. 2013;12:158. doi: 10.1186/1475-2840-12-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS, Paton AW, Paton JC, Walter P, Ashkenazi A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. 2014;345:98–101. doi: 10.1126/science.1254312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. The unfolding tale of the unfolded protein response. Cell. 2001;107:827–830. doi: 10.1016/s0092-8674(01)00623-7. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandl J, Mészáros T, Bánhegyi G, Csala M. Minireview: endoplasmic reticulum stress: control in protein lipid and signal homeostasis. Mol Endocrinol. 2013;27:384–393. doi: 10.1210/me.2012-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y, Thompson MD, Cohen RA, Tong X. Endoplasmic reticuium stress and related pathological processes. J Pharmacol Biomed Anal. 2013;1:1000107. [PMC free article] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2003;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Park HJ, Park SJ, Koo DB, Lee SR, Kong IK, Ryoo JW, Park YI, Chang KT, Lee DS. Progesterone production is affected by unfolded protein response (UPR) signaling during the luteal phase in mice. Life Sci. 2014;113:60–67. doi: 10.1016/j.lfs.2014.07.033. [DOI] [PubMed] [Google Scholar]
- Pincus D, Aranda-Díaz A, Zuleta IA, Walter P, El-Samad H. Delayed Ras/PKA signaling augments the unfolded protein response. Proc Natl Acad Sci U S A. 2014;111:14800–14805. doi: 10.1073/pnas.1409588111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Prostko CR, Brostrom MA, Malara EM, Brostrom CO. Phosphorylation of eukaryotic initiation factor (eIF) 2 alpha and inhibition of eIF-2B in GH3 pituitary cells by perturbants of early protein processing that induce GRP78. J Biol Chem. 1992;267:16751–16754. [PubMed] [Google Scholar]
- Qi XF, Zheng L, Lee KJ, Kim DH, Kim CS, Cai DQ, Wu Z, Qin JW, Yu YH, Kim SK. HMG-CoA reductase inhibitors induce apoptosis of lymphoma cells by promoting ROS generation and regulating Akt, Erk and p38 signals via suppression of mevalonate pathway. Cell Death Dis. 2013;4:e518. doi: 10.1038/cddis.2013.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Keyes KT, Long B, Chen G, Ye Y. Impact of HMG-CoA reductase inhibition on oxidant-induced injury in human retinal pigment epithelium cells. J Cell Biochem. 2011;112:2480–2489. doi: 10.1002/jcb.23173. [DOI] [PubMed] [Google Scholar]
- Salakou S, Kardamakis D, Tsamandas AC, Zolota V, Apostolakis E, Tzelepi V, Papathanasopoulos P, Bonikos DS, Papapetropoulos T, Petsas T, Dougenis D. Increased Bax/Bcl-2 ratio up-regulates caspase-3 and increases apoptosis in the thymus of patients with myasthenia gravis. In Vivo. 2007;21:123–132. [PubMed] [Google Scholar]
- Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Senkal CE, Ponnusamy S, Bielawski J, Hannun YA, Ogretmen B. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J. 2010;24:296–308. doi: 10.1096/fj.09-135087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo TB, Kim TW, Shin MS, Ji ES, Cho HS, Lee JM, Kim TW, Kim CJ. Aerobic exercise alleviates ischemia-induced memory impairment by enhancing cell proliferation and suppressing neuronal apoptosis in hippocampus. Int Neurourol J. 2014;18:187–197. doi: 10.5213/inj.2014.18.4.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone M, Zeng X, Larese J, Ryoo H. A modified UPR stress sensing system reveals a novel tissue distribution of IRE1/XBP1 activity during normal Drosophila development. Cell Stress Chaperones. 2013;18:307–319. doi: 10.1007/s12192-012-0383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Chan PH. Induction of GRP78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronal cell death. J Cereb Blood Flow Metab. 2003;23:949–961. doi: 10.1097/01.WCB.0000077641.41248.EA. [DOI] [PubMed] [Google Scholar]
- Teng Y, Gao M, Wang J, Kong Q, Hua H, Luo T, Jiang Y. Inhibition of eIF2α dephosphorylation enhances TRAIL-induced apoptosis in hepatoma cells. Cell Death Dis. 2014;5:e1060. doi: 10.1038/cddis.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi R, Maurya R, Mishra DP. Medicarpin, a legume phytoalexin sensitizes myeloid leukemia cells to TRAIL-induced apoptosis through the induction of DR5 and activation of the ROS-JNK-CHOP pathway. Cell Death Dis. 2014;5:e1465. doi: 10.1038/cddis.2014.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilahur G, Casaní L, Peña E, Duran X, Juan-Babot O, Badimon L. Induction of RISK by HMG-CoA reductase inhibition affords cardioprotection after myocardial infarction. Atherosclerosis. 2009;206:95–101. doi: 10.1016/j.atherosclerosis.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang M, Meng XB, Yu YL, Sun GB, Xu XD, Zhang XP, Dong X, Ye JX, Xu HB, Sun YF, Sun XB. Elatoside C protects against hypoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes through the reduction of endoplasmic reticulum stress partially depending on STAT3 activation. Apoptosis. 2014;19:1727–1735. doi: 10.1007/s10495-014-1039-3. [DOI] [PubMed] [Google Scholar]
- Weston RT, Puthalakath H. Endoplasmic reticulum stress and BCL-2 family members. Adv Exp Med Biol. 2010;687:65–77. doi: 10.1007/978-1-4419-6706-0_4. [DOI] [PubMed] [Google Scholar]
- Xu B, Shan M, Wang F, Deng Y, Liu W, Feng S, Yang TY, Xu ZF. Endoplasmic reticulum stress signaling involvement in manganese-induced nerve cell damage in organotypic brain slice cultures. Toxicol Lett. 2013;222:239–246. doi: 10.1016/j.toxlet.2013.08.001. [DOI] [PubMed] [Google Scholar]
- Yang F, Ma Y, Ang WP, Chen H, Du WD, Wu SB, Lü L, Zhang DQ. Effects of acupuncture intervention on expression of glucose-regulated protein 78 and C/EBP homologous protein in hippocampal CA 1 region in rats with hyperspasmia. Zhen Ci Yan Jiu. 2014a;39:267–271. [PubMed] [Google Scholar]
- Yang X, Xu H, Hao Y, Zhao L, Cai X, Tian J, Zhang M, Han X, Ma S, Cao J, Jiang Y. Endoplasmic reticulum oxidoreductin 1α mediates hepatic endoplasmic reticulum stress in homocysteine-induced atherosclerosis. Acta Biochim Biophys Sin (Shanghai) 2014b;46:902–910. doi: 10.1093/abbs/gmu081. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- Zhang R, Piao MJ, Kim KC, Kim AD, Choi JY, Choi J, Hyun JW. Endoplasmic reticulum stress signaling is involved in silver nanoparticles-induced apoptosis. Int J Biochem Cell B. 2012;44:224–232. doi: 10.1016/j.biocel.2011.10.019. [DOI] [PubMed] [Google Scholar]
- Zhu H, Zhu H, Xiao S, Sun H, Xie C, Ma Y. Activation and crosstalk between the endoplasmic reticulum road and JNK pathway in ischemia-reperfusion brain injury. Acta Neurochir (Wien) 2012;154:1197–1203. doi: 10.1007/s00701-012-1396-z. [DOI] [PubMed] [Google Scholar]