

Abstract
17β-estradiol (E2), a key participant on the initiation of the LH surge, exerts both positive and negative feedback on GnRH neurons. We sought to investigate potential interactions between estrogen receptors alpha (ERα) and beta (ERβ) and gonadotropin releasing hormone receptor (GnRH-R) in GT1-7 cells. Radioligand binding studies demonstrated a significant decrease in saturation E2 binding in cells treated with GnRH agonist. Conversely, there was a significant reduction in GnRH binding in GT1-7 cells treated with E2. In BRET1 experiments, ERα–ERα dimerization was suppressed in GT1-7 cells treated with GnRH agonist (p < 0.05). There was no evidence of direct interaction between ERs and GnRH-R. This study provides the first evidence of reduced ERα homodimerization by GnRH agonist. Collectively, these findings demonstrate significant cross-talk between membrane-initiated GnRH and E2 signaling in GT1-7 cells.
Keywords: GT1-7 cells, Estradiol, Non-classical estrogen signaling, Estrogen receptor alpha, Estrogen receptor beta, GnRH receptor
1. Introduction
Gonadotropin-releasing hormone (GnRH) neurons episodically release gonadotropin-releasing hormone, which drives the pulsatile secretion of pituitary gonadotropins and controls the normal reproductive cycle. Gonadal steroids are principal mediators of GnRH pulsatility and, ultimately, of an appropriately timed LH surge to induce successful ovulation. In particular, 17β-estradiol (E2) is a key determinant and exerts both negative and positive feedback on GnRH neurons. Not only does E2 signal to GnRH neurons indirectly through various neural afferent networks and neuromodulators (Christian and Moenter, 2010; Garcia-Galiano et al., 2012), but E2 also exerts feedback directly on GnRH neurons through both classic nuclear and rapid membrane-associated responses that involve G proteins (Hardy and Valverde, 1994; Hoffman et al., 1990; Kato et al., 1994; Navarro et al., 2003; Rosie et al., 1990; Rothfeld et al., 1989; Russell et al., 2000). However, the mechanism(s) of membrane-initiated E2 signaling in GnRH neurons have not yet been fully elucidated.
Studies of rapid estrogen signaling support the presence of a membrane-associated estrogen receptor, but it remains uncertain whether E2 exerts its rapid action at the plasma membrane through binding with the classical estrogen receptors (ERα and ERβ) or with a distinct membrane receptor. Both ERα and ERβ are expressed in the murine hypothalamus, GnRH neurons, and immortalized GnRH neurons (GT1-7 cells) (Couse et al., 1997; Hu et al., 2008; Navarro et al., 2003). Prior studies in GT1-7 cells and cultured rat hypothalamic GnRH neurons have also demonstrated the presence of ERα and ERβ at the plasma membrane where E2 binding altered pulsatile GnRH secretion via G protein signaling pathways (Hu et al., 2008; Navarro et al., 2003). ERα and ERβ contain a highly conserved motif within the ligand binding domain that facilitates association with caveolin proteins via palmitoylation and allows for translocation to the plasma membrane (Pedram et al., 2007). ERα and ERβ have been shown to translocate to the neuronal membrane where E2 binding induces rapid signaling via interactions between these ERs and metabotropic glutamate receptors (mGluR), which are G protein coupled receptors (GPCR) (Boulware et al., 2005).
The membrane-initiated actions of E2 in GnRH neurons have rapid regulatory effects including changes in electrical properties (Kato et al., 1994), dose-dependent changes in cAMP production, changes in GnRH release, activation of Gi proteins (Navarro et al., 2003), and phosphorylation of cAMP response element binding protein (Kato et al., 1994; Kwakowsky et al., 2014; Navarro et al., 2003). However, while it is clear that E2 exerts both positive and negative feedback on GnRH release, the underpinnings of membrane-initiated estrogen signaling in GnRH neurons remain unclear. Furthermore, a body of literature describes the existence of G-protein coupled receptor (GPCR) heterodimers and oligomers; these associations provide additional mechanisms for the regulation of downstream signaling (Gurevich and Gurevich, 2008; Kaczor and Selent, 2011; Mercier et al., 2002; Wilson et al., 2013). Thus, evidence suggests that E2 could initiate rapid actions on GnRH pulsatility through membrane-initiated binding with ERα and/or ERβ and subsequent direct association of the activated ER with internal residues on the GnRH-R to affect GnRH release, which could further modulate estrogen feedback to GnRH neurons.
Bioluminescence resonance energy transfer (BRET1) is a technique that enables the investigation of protein–protein interactions in live cells and has been used for the study of GPCRs (Ayoub and Pfleger, 2010; Pfleger and Eidne, 2003; Wu and Brand, 1994). BRET1 utilizes the transfer of excited energy from the natural bioluminescence produced during oxidation of the substrate coelenterazine by Renilla luciferase (Rluc), the donor molecule, to enhanced yellow fluorescent protein (YFP), the acceptor molecule (Hart et al., 1978; Pfleger and Eidne, 2003; Xu et al., 1999). The energy transfer can only occur at distances less than 100 Å (10 nm) with an efficiency that is inversely proportional to the distance, thus it is useful for studying molecular interactions in vivo (Gurevich and Gurevich, 2008; Wu and Brand, 1994). Specifically, while ERα and ERβ translocate to the neuronal cell membrane and have been shown to interact with a GPCR to exert some of their rapid signal transduction, no studies have examined their potential association with the GnRH-R. Therefore, we hypothesized that the estrogen receptors, ERα and ERβ, might directly interact with GnRH receptors in GT1-7 cells. To test this hypothesis, we used ER and GnRH-R fusion constructs with BRET1.
2. Materials and methods
2.1. Cell culture
Immortalized GnRH neurons (GT1-7 cells) were provided by Dr. Richard Weiner (University of California at San Francisco). Cells were cultured in Dulbecco’s modified Eagles’ medium with F12 medium (DMEM/F12) supplemented with 10% heat-inactivated fetal bovine serum. Twenty-four hours before assays, media was replaced by serum- and phenol red-free 1:1 DMEM/F12. HEK293 cells were cultured in a 1:1 mixture of DMEM/F12 containing 10% heat-inactivated fetal bovine serum. All cells were cultured at 37 °C in a humidified incubator containing 5% CO2 and were cultured for at least 7 days prior to use in experiments.
2.2. Plasmid construction
BRET1 fusion constructs were designed as shown. ERα (NM_007956) and ERβ (NM_207707) were extracted from ORFEXPRESS Gateway PLUS Shuttle Clones (GeneCopoeia) by transformation into GCI-5α E. coli cells followed by amplification by PCR, restriction enzyme digestion, and DNA purification using QIAquick Purification Kit. Human ERα-YFP and ERα-BRET constructs contained a linker between the two fusion proteins of 10 amino acids (GGGGSGGGGS). Linker-Rluc and Linker-YFP genes were PCR-amplified using designed primers and cloned into pcDNA3.1-V5-HIS (Invitrogen) at Bam H1 and Age 1 sites for ERα constructs and at Not I and Age I sites for ERβ constructs. ERα-Rluc and ERα-YFP BRET1 constructs were created by ligating the coding sequence of ERα into the Kpn 1 and BamH1 sites of both pcDNA-Rluc and pcDNA-YFP (Kang et al., 2011). ERβ-Rluc, ERβ-YFP, and YFP-ERβ were generated by ligating the coding sequence of ERβ into the Eco R1 and Not 1 sites of both pcDNA-Rluc and pcDNA-YFP. The YFP moiety was attached to both the C and N terminus of ERβ to provide an alternate acceptor configuration, as this can impact BRET1 signal strength. A single nucleotide was then added to the linker of the ERα constructs to maintain amino acid frame using the QuikChange II Site-Directed Mutagenesis Kit. The GnRH-R-Rluc construct had been previously prepared and was used to generate GnRH-R-YFP constructs in a similar fashion as described above. All fusion constructs were verified by direct DNA sequencing, Western Blot analysis, and BRET1 functionality testing (Kang et al., 2011; Neithardt et al., 2006).
2.3. Analysis of construct functionality in HEK293 cells
HEK293 cells were seeded at a density of 3.2 × 105 cells on 12-well tissue culture plates. After 16–18 hours in culture at 60–80% confluence, cells were transfected with 0, 0.2, or 0.8 μg of BRET1 construct and empty pcDNA3.1 to maintain total DNA of 1.0 μg using Lipofectamine 2000 (Invitrogen). Forty-eight hours later, cells were detached with phosphate-buffered saline (PBS) with 0.05% trypsin, washed twice with PBS, suspended in culture media and seeded in 96-well plates at a density of 50,000 cells per well. Twenty-four hours later, cells were washed with PBS, incubated with 5 μM coelenterazine in PBS, and assessed for light emitted between 400 and 600 nm using a Mithras LB940 (Berthold Technologies). Specifically, the total fluorescence and luminescence were evaluated to confirm protein expression and BRET1 functionality of each fusion construct.
2.4. BRET1 analysis in GT1-7 cells
GT1-7 cells were seeded at a density of 8 × 105 cells on 6-well tissue culture plates. After 24 hours in culture, the cells were transfected with various combinations of BRET1 constructs (ERα-YFP, ERα-Rluc, ERβ-YFP, YFP-ERβ, ERβ-Rluc, GnRH-R-Rluc, GnRH-R-YFP), as described above. After 48 hours of transfection, the cells were detached with Versene, suspended in BRET buffer (PBS + 0.1% glucose), and distributed on transparent 96-well plates at a density of 50,000 cells. To assess for interaction between donor and acceptor molecules, cells were incubated with a final concentration of 5 μM coelenterazine in PBS and BRET1 readings were taken immediately.
2.5. Radioligand binding and displacement studies
Saturation binding studies of E2 in GT1-7 cells were performed as previously described (Navarro et al., 2003; Poletti et al., 1994). GT1-7 cells were cultured for 24 hours in 24-well plates at a density of 5 × 105 cells per well. Cells were washed once then treated with serial dilutions of [3H]E2 (70 Ci/mmol, Amersham Pharmacia Biotech, Arlington Heights, IL) for 1 hour at room temperature in serum- and phenol red-free medium containing 0.5% BSA and 1 μM triamcinolone acetonide (Navarro et al., 2003). Cells were then transferred to ice and washed with ice-cold PBS three times, solubilized, and radioligand binding was measured using a scintillation counter. Membrane fractions were treated with serial dilutions of [125I]E2 (2000 Ci/mmol, Amersham Pharmacia Biotech) in a similar fashion and radioligand binding was measured by γ-spectrometry. Cells were either treated with the GnRH antagonist [D-pGlu1, D-Phe2, D-Trp3,6]GnRH (D-pGlu), or GnRH agonist des-Gly10-[D-Ala6]GnRH B-ethylamide (D-Ala6), at 100 nM and compared with untreated cells with saturation binding curves.
GnRH displacement assays were performed as previously described (Nett et al., 1981). Control GT1-7 cells were washed once then treated with [125I]D-Ala6 (2200 Ci/mmol, Amersham Pharmacia Biotech) and non-radioactive D-Ala6 in 100 μl aliquots then incubated for 1 hour at room temperature. Treated cells were washed then treated first with 17 pM of E2 followed by radioligand and non-radioactive agonist. Cells were then rapidly washed with ice-cold PBS three times, solubilized and then analyzed for total radioactive binding by γ-spectrometry.
2.6. Statistical analysis
The total fluorescence and luminescence were used as relative measures of the expression level of the acceptor (YFP) and donor (Rluc) proteins, respectively. For comparison of total fluorescence and luminescence, one-way ANOVA and the linear trend test was used to assess for significant difference in expression level between different construct concentrations. The BRET ratio was calculated using the following formula: (emission at 535 nm) – (emission at 485 nm) × Cf/(emission at 485 nm), where Cf corresponds to (emission at 535 nm)/(emission at 485 nm) for the receptor-Rluc construct expressed alone. To evaluate the specificity of the receptor interactions, saturation assays were performed in which cells were co-transfected with a stable amount of Rluc construct and increasing amounts of YFP construct. Saturation curves were plotted with GraphPad Prism4 software using a nonlinear repression curve assuming one-site binding. Statistical analysis of differences between curves was assessed using a comparison of fits with set alpha of 0.05 and the null hypothesis that one curve was adequate for all data sets and the alternative hypothesis that different curves were best suited for each data set.
3. Results
3.1. Radioligand binding and displacement assays
Radioligand binding and displacement assays were performed to evaluate for changes in E2 or GnRH binding properties in GT1-7 cells treated with labeled GnRH agonist or E2, respectively. Membrane fractions revealed an attenuation in E2 binding sites when cells were co-treated with D-Ala6 (GnRH agonist), consistent with the conclusion that ligand-induced activation of the GnRH-R reduced E2 binding (Fig. 1A). Saturation binding studies in GT1-7 cells demonstrated a single high-affinity estrogen binding site with EC50 of 201 pM, similar to previous findings (Navarro et al., 2003), that was unchanged by treatment with GnRH antagonist (Fig. 1B). Additionally, displacement-binding studies exhibited specific, high-affinity binding of [125I]-D-Ala6 that was inhibited by GnRH in a dose-, time-, and temperature-dependent manner. When treated concomitantly with the GnRH agonist and E2, the number of GnRH-binding sites available at saturation was significantly decreased from 467 ± 21.2 fmol/mg protein to 262.7 ± 42.7 fmol/mg protein with unchanged EC50 (Fig. 1C). These experiments indicate reciprocal modulation of the binding properties of E2 and GnRH; e.g., each agonist led to a reduction in the binding sites available at saturation. We interpret the findings to suggest possible cross-talk between the ER and GnRH-R signaling pathways, which might be important for an appropriately timed LH surge.
Fig. 1.

Radioligand saturation binding curves in cell or membrane lysates from GT1-7 cells. (A) Saturation binding studies of E2 in control and GnRH agonist-treated GT1-7 cell membrane fractions. Concomitant treatment of GT1-7 cells with GnRH agonist des-Gly10-[D-Ala6]GnRH B-ethylamide (D-Ala6), at 100 nM results in a significant blunting of number of E2 binding sites. (B) Saturation binding studies of E2 in control and GnRH-antagonist [D-pGlu1, D-Phe2, D-Trp3,6]GnRH (D-pGlu) treated GT1-7 cell lysates at 100 nM. E2 binding affinity in GT1-7 cells was unchanged in the presence of D-pGlu, a GnRH-antagonist (EC50 201 vs 203 pM). (C) Displacement binding studies of GnRH agonist analog in control and estrogen treated GT1-7 cells. In samples treated with E2 at 17 pM in addition to the unlabeled GnRH agonist, the number of GnRH binding sites was suppressed (467 ± 21.2 fmol/mg protein vs 262.7 ± 42.7 fmol/mg protein).
3.2. Constitutive and ligand-induced interactions between estrogen receptors
To evaluate this possibility, we used BRET1 fusion constructs for ERα, ERβ, and GnRH-R. As a control, we tested for the expected dimerization interaction between ERs in order to confirm BRET1 construct functionality. BRET1 fusion construct expression and functionality (Fig. 2A) was assessed by BRET1 analysis of total luminescence and fluorescence in HEK293 cells transiently transfected with the BRET1 ERα-Rluc fusion construct (Supplementary Fig. S1A–B). A constitutive ERα BRET1 signal was observed in GT1-7 cells after transient transfection with ERα-Rluc and ERα-YFP (Fig. 2B). BRET1 measurements were taken in triplicate and mean total luminescence was significantly increased based on amount of construct added. YFP constructs were similarly tested and mean net YFP fluorescence was also confirmed to be dose-dependent (Kang et al., 2011). Other constructs demonstrated similar expression and dose-dependent functionality (data not shown).
Fig. 2.

Design of BRET1 fusion constructs for ER and GnRH-R and saturation binding studies. (A) Schematic representation of the BRET fusion protein constructs. Acceptor and donor molecules, Renilla luciferase (Rluc) and enhanced yellow fluorescent protein (YFP) were used with linker of 10 amino acids (GGGGSGGGGS) between the two fusion proteins. Linker-Rluc and Linker-YFP genes were PCR-amplified using designed primers and cloned into pcDNA3.1-V5-HIS (Invitrogen) at Bam H1 and Age 1 sites for ERα constructs and at Not I and Age I sites for ERβ constructs. ERα-Rluc and ERα-YFP BRET1 constructs were created by ligating the coding sequence of ERα into the Kpn 1 and BamH1 sites of both pcDNA-Rluc and pcDNA-YFP. ERβ-Rluc, ERβ-YFP and YFP-ERβ were generated by ligating the coding sequence of ERβ into the Eco R1 and Not 1 sites of both pcDNA-Rluc and pcDNA-YFP. The YFP moiety was attached to both the C and N terminus of ERβ to provide an alternate acceptor configuration, as this can impact BRET1 signal strength. (B) ERα-Rluc + ERα-YFP: Specific BRET1 ratio between ERα-Rluc donor and ERα-YFP acceptor in GT1-7 cells transiently transfected with constant amount of ERα-Rluc and increasing amounts ERα-YFP plasmid DNA. The specific BRET1 ratio increased with increasing amounts of plasmid DNA. (C) ERα-Rluc + ERα-YFP: BRET1 saturation binding assay performed in GT1-7 cells transiently transfected with 0.2 μg ERα-Rluc and 0–1.0 μg of ERα-YFP. BRET1 measurements were then obtained in triplicate in both untreated and E2 treated cells (100 nM for 30 minutes) and saturation curves were plotted, demonstrating a hyperbolic curve that is characteristic of a specific protein–protein interaction. E2 treatment of 100 nM for 30 minutes resulted in a significant increase in the maximum BRET1 signal (0.725 ± 0.013 to 0.114 ± 0.006, p < 0.01) with unchanged BRET50. (D) ERα-Rluc + YFP-ERβ: BRET1 saturation binding assay performed in GT1-7 cells transiently transfected with ERα-Rluc and YFP-ERβ. Saturation curves also demonstrate a specific BRET1 signal that increased with E2 treatment (BRETmax 0.228 ± 0.037 to 0.335 ± 0.021, p = 0.0048).
In saturation assay studies, transient transfection of constant amounts of ERα-Rluc with increasing concentrations of ERα-YFP (Fig. 2C) or ERα-Rluc with YFP-ERβ (Fig. 2D) showed a specific BRET1 signal, demonstrating homodimer and heterodimer formation, respectively. Addition of 100 nM E2 for 30 minutes resulted in a significant increase in the BRETmax for both homodimer (0.725 ± 0.013 to 0.114 ± 0.006, p < 0.01) and heterodimer (0.228 ± 0.037 to 0.335 ± 0.021, p < 0.05) formation, suggesting an increase in dimerization (Fig. 2C and D).
3.3. Evidence of cross-talk between GnRH and ER signaling
Based on findings that GnRH agonist suppressed E2 saturation binding in GT1-7 cells (Fig. 1A), BRET1 studies were performed to evaluate for an effect of GnRH upon ER dimerization. Saturation assay studies resulted in a specific BRET1 signal for all samples, again demonstrating ERα homodimerization (Fig. 3). Notably, cotreatment with GnRH agonist resulted in suppression of the E2-induced increase in signal (dotted line) as denoted by the change in BRETmax (0.311 ± 0.046 vs. 0.507 ± 0.045 vs. 0.336 ± 0.025, p = 0.018). These findings suggest a rapid mechanism of cross-talk between the GnRH and E2 signaling pathways such that GnRH agonist treatment resulted in a decrease in ligand-induced ERα–ERα interaction. We interpret the results to indicate that GnRH treatment decreased estrogen-dependent ERα homodimerization.
Fig. 3.
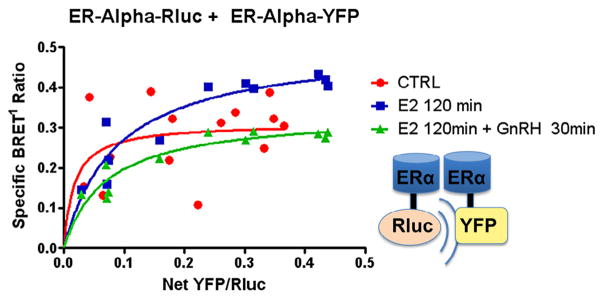
Effect of GnRH on ERα dimerization. ERα-Rluc + ERα-YFP: GT1-7 cells in serum-and phenol red-free 1:1 DMEM/F12 medium were transiently transfected with 0.2 μg of ERα-Rluc and increasing amounts from 0 to 1.0 μg of ERα-YFP (y-axis). BRET1 measurements were then taken in triplicate of untreated cells (x-axis), cells treated with E2 100 nM for 2 hours or cells treated with E2 100 nM for a total of 120 min, with GnRH agonist 100 nM added for the last 30 min of incubation. Saturation curves were then plotted as shown, demonstrating a decrease in ligand-induced ERα–ERα dimerization (dotted line, square boxes) when cells were also treated with GnRH (BRETmax 0.311 ± 0.046 vs. 0.507 ± 0.045 vs 0.336 ± 0.025, p = 0.018).
3.4. Constitutive and ligand-induced interactions between GnRH receptors
Next, BRET1 experiments were performed to test for interaction between GnRH-R fusion constructs in GT1-7 cells based on previous observations of GnRH-R microaggregation in other cell types (Cornea and Conn, 2002; Cornea et al., 2001). As expected due to the inherent ability of GT1-7 cells to secrete GnRH, a constitutive GnRH-R BRET signal was observed in GT1-7 cells after transient transfection with GnRH-R-Rluc and GnRH-R-YFP. In saturation assay studies, transient transfection with GnRH-R-Rluc and increasing concentrations of GnRH-R-YFP resulted in a specific BRET1 signal. Treatment with 100 nM GnRH for 30 minutes resulted in a non-significant decrease in the maximum BRET ratio (0.060 ± 0.013 vs. 0.048 ± 0.011, p = 0.129) (Fig. 4).
Fig. 4.
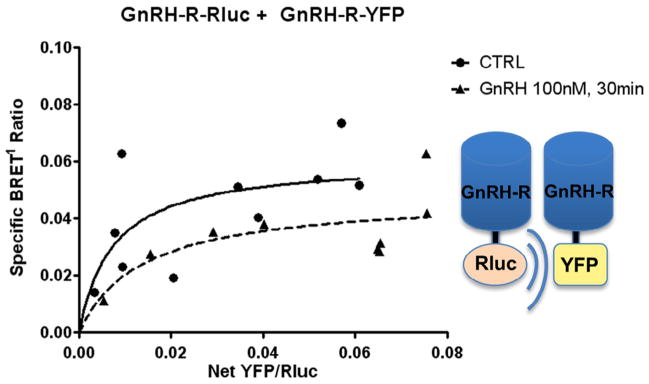
GnRH-R fusion construct expression and functionality. GnRH-R-Rluc + GnRH-R-YFP: Saturation binding assays were performed with GnRH-R-Rluc and GnRH-R-YFP. The hyperbolic curves demonstrate specific protein–protein interaction between BRET constructs, confirming construct functionality and GnRH-R homodimerization in GT1-7 cells. We did not detect a significant change with GnRH agonist treatment at 100 nM for 30 minutes (BRETmax 0.060 ± 0.013 vs 0.048 ± 0.011, p = 0.129).
3.5. Analysis for direct interaction between GnRH-Rs and ERs
Given the effect of the GnRH ligand on ER dimerization, experiments were next performed to evaluate for direct interaction between GnRH-R and ERα or ERβ. An extremely small BRET1 signal was observed in GT1-7 cells after transient transfection with GnRH-R-Rluc and ERα-YFP. The signal was unchanged by treatment with E2 100 nM, (Fig. 5A). Similarly small BRET1 signals were observed at baseline in saturation assays performed on cells transfected with GnRH-R and ERβ-YFP or YFP-ERβ, detecting no evidence of a specific constitutive interaction (Fig. 5B–C). In studies evaluating for protein–protein interactions between GnRH-R-Rluc and ERβ-YFP, treatment with either E2 or GnRH resulted in no change in the BRETmax (0.009 ± 0.002 vs. 0.038 ± 0.067 vs. 0.023 ± 0.012, p = 0.614) demonstrating no ligand-induced interaction between these two receptors (Fig. 5B). Therefore, these experiments did not provide evidence for a direct interaction between either ERα or ERβ with GnRH-R in transiently transfected GT1-7 cells either in the presence or absence of E2 or GnRH agonist.
Fig. 5.

No association between ERs and GnRH-R by BRET1. (A) GnRH-R-Rluc + ERα-YFP: BRET1 measurements in cells transiently transfected with GnRH-R-Rluc and ERα-YFP demonstrated only a very small constitutive signal that was unchanged by treatment with E2 at 100 nM. (B–C) GnRH-R-Rluc + ERβ-YFP (ERβ CT-YFP) and GnRH-R-Rluc + YFP-ERβ (ERβ NT-YFP): Saturation binding assays performed in GT1-7 cells transiently transfected with GnRH-R-Rluc and increasing amounts (0–1.0 μg) of either ERβ-YFP (CT-YFP) or YFP-ERβ (NT-FFP) demonstrate almost undetectable BRET1 signals and absence of a hyperbolic curve. Treatment with either E2 or GnRH agonist (100 nM for 30 minutes) demonstrated similarly low signals (BRETmax 0.009 ± 0.002 vs 0.038 ± 0.067 vs 0.023 ± 0.012, p = 0.614). Thus, studies did not suggest a constitutive or ligand-induced specific interaction between GnRH-R and ERβ. Findings were similar in cells transfected with GnRH-R and ERα (not shown).
4. Discussion
Considerable evidence indicates that E2 affects GnRH release both by positive and negative feedback upon GnRH-secreting neurons, leading to the mid-cycle LH surge. The mechanism(s) by which E2 exerts direct feedback on GnRH neurons are not completely understood. Here, we tested the mechanism of membrane-initiated E2 signaling and sought to determine whether ERα or ERβ might interact directly with the GnRH-R in GT1-7 cells. With radioligand studies, we found that available E2 binding sites were reduced in the presence of GnRH. In addition, GnRH binding saturation kinetics were suppressed by E2 in GT1-7 cells. Furthermore, BRET1 experiments demonstrated a reduction of ERα–ERα dimerization in GT1-7 cells treated with GnRH agonist. However, we did not detect direct interaction between either of the ERs and the GnRH-R. We interpret these findings to indicate that there is significant cross-talk between membrane-initiated GnRH and E2 signaling, but the mechanism does not appear to occur via a direct interaction between these two receptor types. Rather, downstream signaling events initiated by GnRH binding appear to reduce ER dimerization.
Currently, there are only three known sequenced estrogen receptors – ERα, ERβ, and G protein-coupled estrogen receptor (GPER1 – formerly known as GPR30) (Barton, 2012; Vrtačnik et al., 2014). ERα is known to have at least three and ERβ at least five different isoforms (Irsik et al., 2013; Vrtačnik et al., 2014) encoded by alternatively spliced transcripts of the ESR1 and ESR2 genes such as ESR36 (Wang et al., 2005). Furthermore, membrane subpopulations of ERα and ERβ (mERα, mERβ) have been proposed (Barton, 2012). Each ER isoform has different ligand binding, dimerization, or transcriptional abilities, leading to various E2 mediated effects. GPER1, specifically, is located on the cell surface (Kenealy and Terasawa, 2012) and studies using GPER1 agonist G1 (Bologa et al., 2006), antagonist G15 (Dennis et al., 2009), and siRNA knockdown of GPER1 reveal that GPER1 is at least partly responsible for rapid excitatory E2 action (Kenealy and Terasawa, 2012). Estrogen receptor X (ER-X), and novel membrane ER STX (STX-R) sensitive to the diphenylacrylamide compound (Chakraborty and Roy, 2013) have also been found to mediate rapid excitatory actions of estradiol and directly modify GnRH neuronal activity in primates independently of ERα or ERβ (Cheong et al., 2014; Terasawa and Kenealy, 2012; Terasawa et al., 2009). In our study, classic ERα or ERβ and associated constructs were transfected exclusively (without intentional isoforms) in the GT1-7 cells. However our results reflect rapid-action of E2 signaling on GnRH in line with the finding that classical ERs may also mediate rapid signals induced by E2 (Ding et al., 2014; Pedram et al., 2006; Prossnitz and Barton, 2011).
The majority of studies on E2 and GnRH regulation describe effects by inhibition or stimulation on the other receptor via both signaling pathways and G-protein-coupled receptors (Barton, 2012; Hu et al., 2008; Kelly and Levin, 2001; Kenealy et al., 2011; Leclercq et al., 2006; Navarro et al., 2003; Perrett and McArdle, 2013; Terasawa and Kenealy, 2012) with outcome measures including changes in subsequent GnRH secretion, pulsatility/frequency, or ER signaling (Boulware et al., 2005; Chu et al., 2009; Hu et al., 2008; Kenealy and Terasawa, 2012; Kwakowsky et al., 2014). Other studies report effects of reciprocal rate of expression of receptor mRNA when treated with either E2 or GnRH (Ng et al., 2009; Otani et al., 2009). These outcome measures have important implications in the scheme of the E2 and GnRH regulatory system; however, our results also highlight the possibility that an additional component of the feedback between E2 and GnRH could be by rapid alteration of receptor number and/or ligand or protein induced changes in ER ligand binding capability. While estrogen receptors are subjected to conformational changes depending on the nature of the molecules with which they transiently interact, some degree of difference in available binding sites is expected at all times (Leclercq et al., 2006). However, our findings imply a rapid GnRH-induced change in ER availability beyond what would be expected by these natural and transient changes in structure.
The mechanism responsible for these findings is unclear, but may be due to consequent changes in estrogen receptor degradation and/or post-transcriptional modifications induced by GnRH via second messengers with consequent conformational changes. Studies in ER degradation reveal biphasic kinetics, first a slow, then more rapid decline – indicating a large population of stable receptors at baseline that are progressively converted into a more labile form subject to degradation (Leclercq et al., 2006). Studies with estrogen agonists and antagonists suggest that this conversion from stable to labile is regulated via cross-talk with other signal transduction pathways (Leclercq et al., 2006). Furthermore, studies in breast cancer cell lines (MCF-7 breast cancer cells) have shown that ligands can induce ER degradation without direct ligand binding (Borras et al., 1996; Leclercq et al., 2006). The mechanism of ligand/second messenger-induced ER turnover is a possible regulatory mechanism that is worthy of more exploration in regard to estrogen and GnRH feedback.
Given this possible mode of interaction and our dimerization findings, we tested the mechanism of membrane-initiated E2 signaling and sought to determine whether ERα or ERβ directly interact with the GnRH-R in GT1-7 cells. We did this by measuring BRET1 signals in GT1-7 cells transfected with GnRH-R-Rluc and ERα-YFP and GnRH-R-Rluc with ERβ-YFP or YFP-ERβ. BRET1 signaling is non-discriminatory of receptor location: cell membrane, nuclear, or other compartments. ERs are highly mobile proteins continuously shuttling between cellular compartments (Leclercq et al., 2007; Levin, 2002). In addition, estrogen receptor dimerization is increased by estradiol, but also by post-translational modification, as has been shown by phosphorylation induced by signal transduction other than the cognate hormone (Kavarthapu et al., 2014). Post-translational estrogen receptor modification could be inhibitory by activation of phosphatases, or via other changes. In efforts to minimize the error in our study we created two ERβ constructs with YFP at the N and C terminuses, but did not detect any difference in BRET1 signals.
Our findings of decreased ERα–ERα dimerization in GT1-7 cells treated with GnRH agonist has not been reported and may comprise one of the negative or inhibitory feedback mechanisms of GnRH on the estradiol-related cellular pathways. The fact that the most stable dimer configuration (ERα homodimer) is inhibited by addition of GnRH agonist implies that a powerful and key step in the dimerization process is at least partially inhibited or suppressed by presumably increased degradation or conformational changes. It is also uncertain as to whether our findings reflect de-novo dimerization rate of strictly the population of unliganded ERs or if stability was altered in existing dimers.
Overall, estrogen and GnRH signaling in GnRH neurons is complex and involves dose, time, and receptor dependent changes in signaling to alter GnRH secretion patterns and receptor availability. Our data indicate that there is significant cross-talk between membrane-initiated GnRH and E2 signaling, but it does not appear to occur via a direct interaction between these two receptors. Rather, downstream signaling events initiated by GnRH agonist influence ER dimerization and/or availability.
Supplementary Material
Highlights.
GnRH binding saturation kinetics were suppressed by E2 in GT1-7 cell extracts.
E2 binding sites and ERα homodimerization decreased following GnRH agonist treatment.
Fusion constructs and BRET1 suggested no direct interaction between ERs and GnRH-R.
Acknowledgments
The authors thank Dr. Alan DeCherney for his support of the project, and Dr. Carlos Medina for approving and facilitating Dr. Gerkowicz’s time at the NIH. This work was supported by the National Institutes of Health (Intramural Research Program of the NICHD), Grants Z01-HD 008737 (Dr. James Segars), Z01-HD 00184 (Dr. Kevin Catt) and ZIA-HD 000150-39 (Dr. Maria Dufau).
Appendix: Supplementary material
Supplementary data to this article can be found online at doi:10.1016/j.mce.2015.01.023.
References
- Ayoub MA, Pfleger KD. Recent advances in bioluminescence resonance energy transfer technologies to study GPCR heteromerization. Curr Opin Pharmacol. 2010;10:44–52. doi: 10.1016/j.coph.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Barton M. Position paper: the membrane estrogen receptor GPER–Clues and questions. Steroids. 2012;77:935–942. doi: 10.1016/j.steroids.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;4:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- Boulware MI, Weick JP, Becklund BR, Kuo SP, Groth RD, Mermelstein PG. Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J Neurosci. 2005;25:5066–5078. doi: 10.1523/JNEUROSCI.1427-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty P, Roy SK. Expression of estrogen receptor alpha 36 (ESR36) in the hamster ovary throughout the estrous cycle: effects of gonadotropins. PLoS ONE. 2013;8:e58291. doi: 10.1371/journal.pone.0058291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong RY, Porteous R, Chambon P, Abrahám I, Herbison AE. Effects of neuron-specific estrogen receptor (ER) α and ERβ deletion on the acute estrogen negative feedback mechanism in adult female mice. Endocrinology. 2014;155:1418–1427. doi: 10.1210/en.2013-1943. [DOI] [PubMed] [Google Scholar]
- Christian CA, Moenter SM. The neurobiology of preovulatory and estradiol-induced gonadotropin-releasing hormone surges. Endocr Rev. 2010;31:544–577. doi: 10.1210/er.2009-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Andrade J, Shupnik MA, Moenter SM. Differential regulation of gonadotropin-releasing hormone neuron activity and membrane properties by acutely applied estradiol: dependence on dose and estrogen receptor subtype. J Neurosci. 2009;29 (17):5616–5627. doi: 10.1523/JNEUROSCI.0352-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornea A, Conn PM. Measurement of changes in fluorescence resonance energy transfer between gonadotropin-releasing hormone receptors in response to agonists. Methods. 2002;27:333–339. doi: 10.1016/s1046-2023(02)00091-9. [DOI] [PubMed] [Google Scholar]
- Cornea A, Janovick JA, Maya-Nunez G, Conn PM. Gonadotropin-releasing hormone receptor microaggregation. Rate monitored by fluorescence resonance energy transfer. J Biol Chem. 2001;276:2153–2158. doi: 10.1074/jbc.M007850200. [DOI] [PubMed] [Google Scholar]
- Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERa) and estrogen receptor-beta (ERβ) messenger ribonucleic acid in the wild-type and ERα-knockout mouse. Endocrinology. 1997;138:4613–4621. doi: 10.1210/endo.138.11.5496. [DOI] [PubMed] [Google Scholar]
- Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, et al. In vivo effects of a GPR30 antagonist. Nat Chem Biol. 2009;6:421–427. doi: 10.1038/nchembio.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Hussain Y, Chorazyczewski J, Gros R, Feldman RD. GPER-independent effects of estrogen in rat aortic vascular endothelial cells. Mol Cell Endocrinol. 2014 doi: 10.1016/j.mce.2014.07.023. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Garcia-Galiano D, Pinilla L, Tena-Sempere M. Sex steroids and the control of the Kiss1 system: developmental roles and major regulatory actions. J Neuroendocrinol. 2012;24:22–33. doi: 10.1111/j.1365-2826.2011.02230.x. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. GPCR monomers and oligomers: it takes all kinds. Trends Neurosci. 2008;31:74–81. doi: 10.1016/j.tins.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy SP, Valverde MA. Novel plasma membrane action of estrogen and antiestrogens revealed by their regulation of a large conductance chloride channel. FASEB J. 1994;8:760–765. doi: 10.1096/fasebj.8.10.8050676. [DOI] [PubMed] [Google Scholar]
- Hart RC, Stempel KE, Boyer PD, Cormier MJ. Mechanism of the enzyme-catalyzed bioluminescent oxidation of coelenterate-type luciferin. Biochem Biophys Res Commun. 1978;81:980–986. doi: 10.1016/0006-291x(78)91447-x. [DOI] [PubMed] [Google Scholar]
- Hoffman GE, Lee WS, Attardi B, Yann V, Fitzsimmons MD. Luteinizing hormone-releasing hormone neurons express c-fos antigen after steroid activation. Endocrinology. 1990;126:1736–1741. doi: 10.1210/endo-126-3-1736. [DOI] [PubMed] [Google Scholar]
- Hu L, Gustofson RL, Feng H, Leung PK, Mores N, Krsmanovic LZ, et al. Converse regulatory functions of estrogen receptor-alpha and -beta subtypes expressed in hypothalamic gonadotropin-releasing hormone neurons. Mol Endocrinol. 2008;22:2250–2259. doi: 10.1210/me.2008-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irsik DL, Carmines PK, Lane PH. Classical estrogen receptors and ERα splice variants in the mouse. PLoS ONE. 2013;8 (8):e70926. doi: 10.1371/journal.pone.0070926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczor AA, Selent J. Oligomerization of G protein-coupled receptors: biochemical and biophysical methods. Curr Med Chem. 2011;18:4606–4634. doi: 10.2174/092986711797379285. [DOI] [PubMed] [Google Scholar]
- Kang JH, Tsia-Morris CH, Dufau M. Complex formation and interactions between transcription factors essential for human prolactin receptor gene transcription. Mol Cell Biol. 2011;31:3208–3222. doi: 10.1128/MCB.05337-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Hiruma H, Kimura F. Acute estradiol modulation of electrical activity of the LHRH pulse generator in the ovariectomized rat: restoration by naloxone. Neuroendocrinology. 1994;59:426–431. doi: 10.1159/000126688. [DOI] [PubMed] [Google Scholar]
- Kavarthapu R, Morris CHT, Dufau ML. Prolactin induces up-regulation of its cognate receptor in breast cancer cells via transcriptional activation of its generic promoter by cross-talk between ERα and STAT5. Oncotarget. 2014;5:9079–9091. doi: 10.18632/oncotarget.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MJ, Levin ER. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol Metab. 2001;12 (4):152–156. doi: 10.1016/s1043-2760(01)00377-0. [DOI] [PubMed] [Google Scholar]
- Kenealy BP, Terasawa E. Rapid direct action of estradiol in GnRH neurons: findings and implications. Front Endocrinol (Lausanne) 2012;2:106. doi: 10.3389/fendo.2011.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenealy BP, Keen KL, Terasawa E. Rapid action of estradiol in primate GnRH neurons: the role of estrogen receptor alpha and estrogen receptor beta. Steroids. 2011;76 (9):861–866. doi: 10.1016/j.steroids.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwakowsky A, Cheong RY, Herbison AE, Ábrahám IM. Non-classical effects of estradiol on cAMP responsive element binding protein phosphorylation in gonadotropin-releasing hormone neurons: mechanisms and role. Front Neuroendocrinol. 2014;35 (1):31–41. doi: 10.1016/j.yfrne.2013.08.002. [DOI] [PubMed] [Google Scholar]
- Leclercq G, Lacroix M, Laïos I, Laurent G. Estrogen receptor alpha: impact of ligands on intracellular shuttling and turnover rate in breast cancer cells. Curr Cancer Drug Targets. 2006;6 (1):39–64. doi: 10.2174/156800906775471716. [DOI] [PubMed] [Google Scholar]
- Levin ER. Cellular functions of plasma membrane estrogen receptors. Steroids. 2002;67 (6):471–475. doi: 10.1016/s0039-128x(01)00179-9. [DOI] [PubMed] [Google Scholar]
- Mercier JF, Salahpour A, Angers S, Breit A, Bouvier M. Quantitative assessment of beta 1- and beta 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J Biol Chem. 2002;277:44925–44931. doi: 10.1074/jbc.M205767200. [DOI] [PubMed] [Google Scholar]
- Navarro CE, Saeed SA, Murdock C, Martinez-Fuentes AJ, Arora KK, Krsmanovic LZ, et al. Regulation of cyclic adenosine 3′,5′-monophosphate signaling and pulsatile neurosecretion by Gi-coupled plasma membrane estrogen receptors in immortalized gonadotrophin-releasing hormone neurons. Mol Endocrinol. 2003;17:1792–1804. [PubMed] [Google Scholar]
- Neithardt A, Farshori MP, Shah FB, Catt KJ, Shah BH. Dependence of GnRH-induced phosphorylation of CREB and BAD on EGF receptor transactivation in GT1-7 neuronal cells. J Cell Physiol. 2006;208 (3):586–593. doi: 10.1002/jcp.20697. [DOI] [PubMed] [Google Scholar]
- Nett TM, Crowder ME, Moss GE, Duello TM. GnRH-receptor interaction. V Down-regulation of pituitary receptors for GnRH in ovariectomized ewes by infusion of homologous hormone. Biol Reprod. 1981;24:1145–1155. [PubMed] [Google Scholar]
- Ng Y, Wolfe A, Novaira HJ, Radovick S. Estrogen regulation of gene expression in GnRH neurons. Mol Cell Endocrinol. 2009;303 (1–2):25–33. doi: 10.1016/j.mce.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani H, Otsuka F, Takeda M, et al. Regulation of GNRH production by estrogen and bone morphogenetic proteins in GT1-7 hypothalamic cells. J Endocrinol. 2009;203 (1):87–97. doi: 10.1677/JOE-09-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. 2006;9:1996–2009. doi: 10.1210/me.2005-0525. [DOI] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Sainson RC, Kim JK, Hughes CC, Levin ER. A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem. 2007;282:22278–22288. doi: 10.1074/jbc.M611877200. [DOI] [PubMed] [Google Scholar]
- Perrett RM, McArdle CA. Molecular mechanisms of gonadotropin-releasing hormone signaling: integrating cyclic nucleotides into the network. Front Endocrinol (Lausanne) 2013;20:180. doi: 10.3389/fendo.2013.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger KD, Eidne KA. New technologies: bioluminescence resonance energy transfer (BRET) for the detection of real time interactions involving G-protein coupled receptors. Pituitary. 2003;6:141–151. doi: 10.1023/b:pitu.0000011175.41760.5d. [DOI] [PubMed] [Google Scholar]
- Poletti A, Melcangi RC, Negri-Cesi P, Maggi R, Martini L. Steroid binding and metabolism in the luteinizing hormone-releasing hormone-producing neuronal cell line GT1-1. Endocrinology. 1994;135:2623–2628. doi: 10.1210/endo.135.6.7988451. [DOI] [PubMed] [Google Scholar]
- Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;12:715–726. doi: 10.1038/nrendo.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosie R, Thomson E, Fink G. Oestrogen positive feedback stimulates the synthesis of LHRH mRNA in neurones of the rostral diencephalon of the rat. J Endocrinol. 1990;124:285–289. doi: 10.1677/joe.0.1240285. [DOI] [PubMed] [Google Scholar]
- Rothfeld J, Hejtmancik JF, Conn PM, Pfaff DW. In situ hybridization for LHRH mRNA following estrogen treatment. Brain Res Mol Brain Res. 1989;6:121–125. doi: 10.1016/0169-328x(89)90045-4. [DOI] [PubMed] [Google Scholar]
- Russell KS, Haynes MP, Sinha D, Clerisme E, Bender JR. Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc Natl Acad Sci USA. 2000;97:5930–5935. doi: 10.1073/pnas.97.11.5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasawa E, Kenealy BP. Neuroestrogen, rapid action of estradiol, and GnRH neurons. Front Neuroendocrinol. 2012;33 (4):364–375. doi: 10.1016/j.yfrne.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasawa E, Noel SD, Keen KL. Rapid action of oestrogen in luteinising hormone-releasing hormone neurones: the role of GPR30. J Neuroendocrinol. 2009;21:316–321. doi: 10.1111/j.1365-2826.2009.01839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrtačnik P, Ostanek B, Mencej-Bedrač S, Marc J. The many faces of estrogen signaling. Biochem Med. 2014;24 (3):329–342. doi: 10.11613/BM.2014.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem Biophys Res Commun. 2005;336 (4):1023–1027. doi: 10.1016/j.bbrc.2005.08.226. [DOI] [PubMed] [Google Scholar]
- Wilson PC, Lee MH, Appleton KM, El-Shewy HM, Morinelli TA, Peterson YK, et al. The arrestin-selective angiotensin AT1 receptor agonist [Sar1,Ile4,Ile8]-AngII negatively regulates bradykinin B2 receptor signaling via AT1-B2 receptor heterodimers. J Biol Chem. 2013;288:18872–18884. doi: 10.1074/jbc.M113.472381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Brand L. Resonance energy transfer: methods and applications. Anal Biochem. 1994;218:1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- Xu Y, Piston DW, Johnson CH. A bioluminescence resonance energy transfer (BRET) system: application to interacting circadian clock proteins. Proc Natl Acad Sci USA. 1999;96:151–156. doi: 10.1073/pnas.96.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.