

Abstract
Background and Aims Viny species are among the most serious invasive plants, and better knowledge of how vines grow to dominate landscapes is needed. Patches may contain a single genotype (i.e. genet), a competitively dominant genet or many independent but interacting genets, yet the clonal structure of vining species is often not apparent. Molecular markers can discriminate among the genetic identities of entwined vines to reveal the number and spatial distribution of genets. This study investigated how genets are spatially distributed within and among discrete patches of the invasive vine kudzu, Pueraria montana var. lobata, in the United States. It was expected that ramets of genets would be spatially clustered within patches, and that an increase in the number of genets within a patch would be associated with a decrease in the average size of each genet.
Methods Six discrete kudzu patches were sampled across 2 years, and 1257 samples were genotyped at 21 polymorphic allozyme loci. Variation in genotypic and genetic diversity among patches was quantified and patterns of genet interdigitation were analysed.
Key Results Substantial genotypic and genetic variation occurred within and among patches. As few as ten overlapping genets spanned up to 68 m2 in one patch, while >90 % of samples were genetically unique in another patch. Genotypic diversity within patches increased as mean clone size decreased, although spatially widespread genets did not preclude interdigitation. Eight genets were shared across ≥2 patches, suggesting that vegetative dispersal can occur among patches.
Conclusions Genetically unique kudzu vines are highly interdigitated. Multiple vegetative propagules have become established in spatially discrete patches, probably through the movement of highway construction or maintenance machinery. The results suggest that common methods for controlling invasive vines (e.g. mowing) may inadvertently increase genotypic diversity. Thus, understanding vine architecture and growth has practical implications.
Keywords: Clonal diversity, clonal structure, colonization, dispersal, founder effects, invasion ecology, kudzu (Pueraria montana var. lobata), vines.
INTRODUCTION
With increasing introductions of exotic species, it is important to understand biological invasion processes and fine-tune control efforts. The USA harbours at least 50 000 introduced species. Of these, approx. 5000 are considered invasive, meaning they have integrated into natural ecosystems, thereby posing economic and ecological threats (Pimentel et al., 2000). Knowledge of how populations are colonized and spread can provide insights into the invasive potential of species, processes by which they dominate landscapes and the potential efficacy of control measures (Dlugosch and Parker, 2008).
The spread of invasive species is often associated with a bottleneck in which one or relatively few propagules are responsible for the colonization and subsequent expansion of populations. Plant populations in particular may expand due to vegetative reproduction. Plants capable of vegetative reproduction or self-fertilization are more likely to invade and become established in suitable habitats (van Kleunen et al., 2008; Burns et al., 2011; Hao et al., 2011). While self-pollination and asexual reproduction increase reproductive assurance for newly established plant populations, they are often associated with less genetic variation. Indeed, comparisons of genotypic diversity of native and introduced clonal plants reveals substantially less genotypic diversity in introduced populations (Barrett et al., 2008). However, many invasive plants have been intentionally introduced multiple times from multiple source populations, potentially increasing genetic diversity and contributing to the adaptive potential of invasive populations (Dlugosch and Parker, 2008). Neutral molecular markers can enable inferences of invasion dynamics by elucidating the introduction history, population growth patterns and reproductive systems of established populations.
Vining species are among the most destructive invasive plants, comprising one-third of the worst invasive species in some ecosystems (Gordon, 1998). Many vining species reproduce sexually and through vegetative propagation of genetically identical clones (i.e. genets). Genetic markers can provide insights into the relative contribution of sexual vs. clonal reproduction within vine patches, as well as the extent of interdigitation (i.e. spatial overlap) among genets, which may be greater in vining species than in species with other vegetative growth forms. Vine populations may support a single founding genet, a dominant genet that competitively excludes other genets or many independently established and interacting genets, yet vining growth makes it difficult to distinguish the number and distribution of genets within populations. Patches that expand through phalanx-type growth (i.e. discrete clusters of genetically distinct ramets) should have high clonal structure and dominance, while guerrilla-type growth (i.e. widespread and interdigitating ramets) should produce less clonal structure and dominance (Araki et al., 2009; Ohsako, 2010). Although the contributions of seed dispersal and clonal growth to invasive spread has been previously evaluated using genetic data for several species, including goldenrod (Solidago canadensis; Dong et al., 2006), an apomictic vine (Bryonia alba; Novak and Mack, 2000) and knotweed (Fallopia spp.; Grimsby et al., 2007), few represent spatially explicit analyses of genet interdigitation (but see, for example, McCormick et al., 2010, Phragmites australis; Ohsako, 2010, Carex kobomugi). Using spatially explicit knowledge of genet distribution within patches, it is possible to distinguish between alternative processes by which vines invade, expand and dominate previously established vegetation.
One of the most noxious invasive plant species in the USA is kudzu, Pueraria montana var. lobata (Fabaceae). From its first introduction at the 1876 Centennial Exposition in Philadelphia, kudzu was widely disseminated as an ornamental and later for erosion control, with >85 million seedlings distributed in the southeastern USA (Forseth and Innis, 2004). Its rapid growth, vegetative propagation, high leaf area index and photosynthetic rate render it an aggressive competitor (Forseth and Innis, 2004). Concerted introductions and colonization events that comprise multiple propagules probably contributed to generally high levels of genetic variation within kudzu patches (Pappert et al., 2000; Jewett et al., 2003; Sun et al., 2005). Nevertheless, heterogeneity of genetic diversity among kudzu patches suggests that their local histories may vary widely, potentially due to variation in the number of founders and propensity for sexual reproduction (Pappert et al., 2000). Spatially explicit analyses of clonal growth and overlap have not been considered, yet vining architecture could play a crucial role in kudzu patch dynamics. For example, establishment and growth of competitively dominant genets could preclude sexual reproduction and subsequent recruitment, enhancing founder effects. Conversely, establishment of many interdigitating genets could enhance outcrossing and subsequent recruitment, and consequentially mitigate founder effects. Indeed, seed set, biomass and leaf area were observed to increase in kudzu patches with more genetic variation and/or individuals with more heterozygous loci (Pappert, 1998; Pappert et al., 2000).
We address how kudzu genotypes are spatially distributed over short spatial scales and whether knowledge of its vining architecture is relevant to its control. We expected that: (1) heterogeneity of clonal and genetic diversity occurs among patches; and (2) ramets of genets are spatially segregated within patches (i.e. genets are not highly interdigitated), such that patches containing spatially widespread genets do not also maintain high levels of clonal variation.
MATERIALS AND METHODS
Study species
Kudzu, Pueraria lobata var. montana (Fabaceae), is a twining and climbing vine that forms dominant patches climbing over and shading established herbaceous and woody vegetation. Growth rates of 20–30 m year–1 have been reported (Forseth and Innis, 2004). Kudzu introductions from Asia to the southeastern USA began in the 1870s and continued through the first half of the 20th century until kudzu was placed on the Federal Obnoxious Weed list in 1970 (Forseth and Innis, 2004). Kudzu covers approx. 3 Mha across at least 27 states and continues to expand at a rate of 50 000ha year–1 (Forseth and Innis, 2004).
Flowering occurs in August, with flower visitation by multiple native and naturalized pollinators, frequently including hymenoptera (Pappert et al., 2000; Forseth and Innis, 2004). Self-pollination is possible; however, hand pollination increased fruit set by >10 %, indicating possible pollinator limitation (Abramovitz, 1983; Forseth and Innis, 2004). Vines with arboreal distributions exhibit greater flowering and seed set than vines running along the ground (Forseth and Innis, 2004). Seed production varies widely among patches, from 1 to 1800 seeds m−2, and seed dormancy may allow seed bank formation (Forseth and Innis, 2004). Genets with higher levels of heterozygosity tend to be more likely to set seed, accumulate biomass and produce larger leaf areas (Pappert, 1998; Pappert et al., 2000). Seed dispersal distances of ≤6 m have been recorded within patches (Abramovitz, 1983; Forseth and Innis, 2004), but long-distance dispersal may be relatively common along open corridors such as roads (Pappert et al., 2000). Vegetative reproduction occurs by the rooting of stem nodes, followed by the senescence of connections to other nodes and physiological independence after 1–3 years, enabling rapid and extensive clonal spread (Tsugawa and Kayama, 1976; Tsugawa and Ryosei, 1985; Forseth and Innis, 2004). Large tuberous root systems support perennial growth, and individual stems span 0·01–60 m in length, form a multilayered canopy and can produce ramet densities >10 000 ha–2, making eradication difficult (Hipps, 1994; Mitich, 2000; Forseth and Innis, 2004).
Study sites and sampling
To characterize patterns of clonal and genetic variation within and among kudzu patches, we sampled leaf tissue from six patches in both 2010 and 2011. Study sites were located in the vicinity of Athens, GA, USA (N33°55', W83°23'; mean pairwise distance 4·8 km; Fig. 1). For clarity, site names (POR, HDP, TRL, MWS, MOR and BDR) are appended with 10 or 11 when referring to 2010 and 2011 collections, respectively.
Fig. 1.
Map of the kudzu patch locations in Athens, GA, USA. The map shows the proximity of highways (thick lines) and local roads (thin lines) to the patches that were analysed (US Census Bureau, 2005).
Study plots were situated in monodominant (>90 % cover) kudzu patches in disturbed roadside habitats that varied in size (0·18–2·87 ha), habitat characteristics and land use histories (Supplementary Data Table S1). Patch POR has open and level edges that descend into a ravine, with the upper, flatter areas occasionally mowed. Patch HDP grows over thickets across a steep knoll surrounded by pine forest on three sides. Patch TRL covers a ravine and adjacent slopes with a cycle path through its centre. Patch MWS grows over thickets in a large open hillside. Patch MOR grows in a relatively flat, open and grassy highway cloverleaf interchange that is mowed annually during winter. Patch BDR grows over thickets in a post-agricultural field. Aerial photographs spanning the years 1938–1993, obtained from University of Georgia archives, have provided historic documentation of land use and the timing of roadway construction over 73 years preceding the current study (Supplementary Data Table S1). Anthropogenic disturbance was evident at all sites in 1938, at which time HDP, TRL and BDR were used agriculturally, POR and MOR were open roadside sites, and MWS was used residentially, although apparent uses varied through time (Supplementary Data Table S1). Five patches growing along a stretch of highway (POR, HDP, TRL, MWS and MOR) have a maximum age of 22 years, as they did not exist prior to highway construction in 1988. By 1993, all were open roadside habitats (Supplementary Data Table S1).
Sampling in 2010 and 2011 differed to address complementary questions about clonal patterns. Both sets of samples were taken during the growing season (August and July, respectively). In 2010, we employed a regularly spaced sampling scheme, where one leaf was sampled at 5 m grid intervals measured across the patch from a baseline transect near the patch edge. This sampling scheme provided insight into spatial patterns of clonal and genetic variation. In 2011, sampling points were spaced at 10 m intervals across the patch from the same baseline transect (i.e. approximately grid lines 1, 3, 5, etc. of the 2010 sampling scheme). Five leaves were collected at each grid point: one from the focal grid point and one from four additional points at 1 m from each 90° angle surrounding the focal point. This sampling scheme was designed to examine fine-scale genet interdigitation. In 2010 we sampled 80–98 leaves per patch and in 2011 we sampled 96–120 leaves per patch (Table 1).
Table 1.
Clonal variation and size statistics
| Population | n | nC | Psex,max | G | GU | %GU | R | D | E | β | Dc | Dc,min | Dc,max | Size | Sizemax | Rep. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| POR10 | 96 | 71 | 2·16 × 10–3 | 38 | 26 | 68·4 | 0·46 | 0·89 | 0·65 | 0·32 | 0·81 | 0·38 | 1·00 | 22·37 | 46·10 | |
| POR11 | 120 | 106 | 2·14 × 10–3 | 43 | 33 | 76·7 | 0·42 | 0·85 | 0·63 | 0·26 | 0·35 | 0·25 | 0·64 | 34·60 | 59·36 | 4 |
| HDP10 | 80 | 77 | 8·66 × 10–2 | 10 | 7 | 70·0 | 0·12 | 0·26 | 0·06 | 0·03 | 0·86 | 0·86 | 0·86 | 33·31 | 68·01 | |
| HDP11 | 96 | 96 | 1·38 × 10–1 | 18 | 12 | 66·6 | 0·19 | 0·45 | 0·20 | 0·06 | 0·65 | 0·50 | 0·80 | 25·71 | 64·20 | 2 |
| TRL10 | 98 | 89 | 5·33 × 10–1 | 25 | 16 | 64·0 | 0·27 | 0·81 | 0·68 | 0·23 | 0·58 | 0·33 | 0·76 | 30·82 | 60·42 | |
| TRL11 | 120 | 120 | 6·64 × 10–3 | 36 | 21 | 58·3 | 0·35 | 0·75 | 0·51 | 0·16 | 0·62 | 0·13 | 1·00 | 24·39 | 59·17 | 1 |
| MWS10 | 96 | 93 | 8·28 × 10–4 | 51 | 38 | 74·5 | 0·55 | 0·95 | 0·81 | 0·54 | 0·71 | 0·50 | 1·00 | 23·09 | 53·15 | |
| MWS11 | 119 | 119 | 1·50 × 10–3 | 71 | 62 | 87·3 | 0·62 | 0·94 | 0·68 | 0·42 | 0·53 | 0·50 | 0·56 | 22·33 | 54·42 | 0 |
| MOR10 | 96 | 86 | 5·97 × 10–5 | 56 | 46 | 82·1 | 0·60 | 0·90 | 0·47 | 0·29 | 0·93 | 0·78 | 1·00 | 16·23 | 58·52 | |
| MOR11 | 120 | 116 | 2·49 × 10–6 | 104 | 94 | 90·4 | 0·90 | 1·00* | 0·82 | 1·47 | 0·35 | 0·30 | 0·40 | 10·04 | 49·82 | 1 |
| BDR10 | 96 | 92 | 3·39 × 10–3 | 29 | 21 | 72·4 | 0·32 | 0·77 | 0·57 | 0·20 | 0·78 | 0·63 | 0·88 | 18·76 | 49·24 | |
| BDR11 | 120 | 120 | 1·88 × 10–4 | 53 | 45 | 84·9 | 0·46 | 0·80 | 0·37 | 0·16 | 0·51 | 0·20 | 1·00 | 30·57 | 59·17 | 2 |
| Mean10 | 93·7 | 84·7 | 1·04 × 10–1 | 34·8 | 25·7 | 71·9 | 0·39 | 0·76 | 0·54 | 0·27 | 0·78 | 0·58 | 0·92 | 24·10 | 55·91 | NA |
| Mean11 | 115·8 | 112·8 | 2·47 × 10–2 | 54·2 | 44·5 | 77·4 | 0·49 | 0·80 | 0·54 | 0·42 | 0·50 | 0·31 | 0·73 | 24·61 | 57·69 | 1·7 |
| Total10 | 562 | 508 | NA | 205 | 145 | 70·7 | 0·57 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Total11 | 695 | 677 | NA | 321 | 259 | 80·7 | 0·64 | NA | NA | NA | NA | NA | NA | NA | NA | 10 |
n, total number of samples; nC, number of complete multilocus genotypes; Psex,max, maximum probability of sex; G, number of genets; GU, number of unique genets; R, clonal richness; D, Simpson’s diversity; E, Fager’s evenness; β, parameter describing the shape of the Pareto distribution; Dc, Dc,min, Dc,max, mean, minimum and maximum of the clonal dominance index, respectively; size, mean size of clones (m2); sizemax, maximum detected size of a clone (m2); Rep, number of clones detected in both 2010 and 2011 within a patch; NA, not applicable.
*The actual value is 0·996 < 1·00.
Genotyping
Genotype identity and genetic diversity were evaluated using allozymes. We chose cost-efficient allozymes because the number of polymorphic loci (21) adequately differentiated unique multilocus genotypes (MLGs) for a large number of samples (1257). Samples were stored at 4 °C for ≤48 h before leaves were ground with a mortar and pestle. Enzymes were solubilized and stabilized in an extraction buffer (Wendel and Parks, 1992), absorbed onto Whatman filter paper, placed in 96-well plates, and stored at −70 °C until analysis. Enzyme electrophoresis was conducted in 11 % starch gels. Eleven enzyme systems yielded 21 polymorphic loci: aconitase (ACO; two loci), phosphoglucoisomerase (PGI; one locus), UTP-glucose-1-phosphate (UGPP; two loci), diaphorase (DIA; two loci), menadione reductase (MNR; three loci), triose-phosphate isomerase (TPI; two loci), aspartate aminotransferase (AAT; two loci), glutamate dehydrogenase (GDH; one locus), isocitrate dehydrogenase (IDH; two loci), malate dehydrogenase (MDH; two loci) and phosphoglucomutase (PGM; two loci). Enzymes were resolved with buffer systems 6 (TPI, MNR, DIA), 8 (AAT, GDH), 4 (PGI, UGPP, ACO) and 11 (PGM, MDH, IDH), all of which follow Soltis et al. (1983).
Clonal analysis
To characterize clonal variation within and among patches, we identified all putative genets. We grouped all matching MLGs in the 1257 samples using GenAlEx v.6.2 (Peakall and Smouse, 2006). Sixty-eight samples (5·4 %) had missing data at ≥1 locus, requiring additional consideration for clonal assignment. We used a simple and transparent procedure that assigned samples with missing data to either (1) one, and only one, known genotype in the patch or (2) a unique genotype (i.e. the alleles at ≥1 locus differed from all other genotypes in the patch). We conservatively deleted samples from further analysis if they could not be assigned in this way. Prior to analysis of clonal structure, we followed the recommendations of Arnaud-Haond et al. (2007). First, we evaluated the power of our genetic markers to discriminate among MLGs in a sample by evaluating the minimum, maximum and mean number of MLGs identified in each of 1000 randomly sampled combinations of l loci, where l ranges from 1 to the full battery of loci (Alberto et al., 2005). Random sampling was conducted in R (R Core Development Team, 2013). Secondly, we determined whether distinct MLGs (i.e. genets) separated by small allelic distances (i.e. possible somatic mutations or genotyping errors) should be grouped into a single multilocus lineage. To do this, we evaluated the frequency distribution of allelic distances separating MLGs, using GenAlEx to calculate pairwise allelic differences, where a bimodal distribution with a small peak at small allelic distances would indicate that MLGs separated by smaller allelic distances should be grouped into single multilocus lineages (Arnaud-Haond et al., 2007). In the absence of such a secondary peak, it was not possible to merge MLGs separated by a pre-determined allelic distance threshold into a single multilocus lineage. We nevertheless quality checked the data set and manually grouped samples with similar MLGs (differing at approx. 1 locus) and abundances across years into a single multilocus lineage to prevent overestimation of genotypic diversity (Arnaud-Haond et al., 2007). Finally, we calculated the probability that MLG matches are derived independently through sexual reproduction, assuming random mating, by calculating the probability of sex (Psex) using GenAlEx v.6.2 (Peakall and Smouse, 2006), where Psex estimates the probability of obtaining n repeated MLGs in a sample of N by random mating:
| (1) |
where Pgen = (Πpi) 2h, pi = the frequency of each allele in an MLG, and h = the number of heterozygous loci (Parks and Werth, 1993).
Clonal variation was compared within and among patches. We quantified the number of unambiguously assigned genets (G), unique genets (GU) and percentage of unique genets (%GU) within patches. We calculated clonal richness (R), evenness (E) and the complement of the slope of the Pareto distribution (β), which provide the most parsimonious and non-redundant descriptions of clonal variation (Arnaud-Haond et al., 2007). Clonal richness was calculated by R = (G – 1)/(N – 1) (Dorken and Eckert, 2001), and was standardized to the minimum sample size (N = 79) using 1000 bootstrap resamples in R (Arnaud-Haond et al., 2007). Clonal richness ranges from 0 in monotypic patches to 1 if all samples are distinct MLGs, and resampling is required to compare patches with different sample sizes (Arnaud-Haond et al., 2007). Clonal diversity was calculated using Simpson’s diversity index (D), which was used to calculate Fager’s evenness (E = Simpson’s V) (Fager, 1972). Simpson’s D = 1 − Σ[Nj (Nj − 1)/Nr (Nr − 1)], where Nj is the number of samples of the jth MLG and Nr is the total number of samples from that patch. Simpson’s D varies from 0 in monomorphic patches to 1 in patches where all samples are genetically unique. Fager’s E = (D − Dmin)/(Dmax − Dmin), where Dmin = (G − 1) (2Nr − G)/Nr(Nr − 1), Dmax = Nr(G − 1)/G(Nr − 1), and the new parameter G is the number of unique MLGs in that patch. Fager’s E varies from 0 in a monomorphic patch to 1 in a patch with completely uniform MLG frequencies (Eckert and Barrett, 1993). The Pareto distribution is a continuous approximation to describe the distribution of sampled clones into discrete size classes (the number of times that clone was sampled), as described by N≥X = aX–β, where N≥X = the number of sampled ramets belonging to clonal lineages containing X or more sampled ramets in the patch, and a and β are fitted by regression (Arnaud-Haond et al., 2007). The power slope (–β) was derived from the slope of fitted log–log regressions between the cumulative frequency of ramets belonging to each size class or larger (Arnaud-Haond et al., 2007), which we performed in the base package of R. Parameter β is influenced by both richness and evenness. A steep slope (high β) occurs when clonal lineages have low richness and/or high evenness with comparable sizes. A shallow slope (decreasing β) occurs with high richness and/or a skewed distribution with few large and many small MLGs (Arnaud-Haond et al., 2007).
We further characterized the spatial distribution of genets by calculating the index of clonal dominance, Dc = (NS – 1)/(NT – 1), where NS is the number of ramets belonging to a focal genet and NT is the total number of ramets sampled within the range of the focal genet (Ohsako, 2010). This value was calculated for genets represented by ≥3 ramets in a patch, where a convex hull could circumscribe each genet. Clonal dominance of a genet reaches a maximum of 1·0 when its range is exclusively filled with its own ramets (NT = NS). Clonal dominance approaches zero as the frequency of ramets belonging to other genets increases within its range. We used GenAlEx to calculate the minimum and maximum observed area across which clones occurred within patches. We also determined which genets occurred in multiple patches and/or were detected in both 2010 and 2011.
Genetic variation analysis
We compared genetic variation among patches. In this analysis, each MLG was represented only once within a given patch. We calculated genetic diversity as the percentage of polymorphic loci (P), mean number of alleles per locus (A), observed and expected heterozygosity (Ho and He, respectively) and the fixation index (F) in GenAlEx. To compare patches with different sample sizes, mean allelic richness (AR) was calculated using heirfstat in R (Goudet, 2011), with rarefaction to the fewest genotypes/locus/patch (n = 8; two HDP10 MLGs had missing data at a locus). We calculated the proportion of total genetic diversity among patches and between years (FST) using analysis of molecular variation (AMOVA), testing for significance with 1000 permutations in Arlequin v.3.11 (Excoffier et al., 2005).
Statistical analyses
To compare the 2010 and 2011 sampling events, we conducted paired t-tests to determine if genotypic or genetic diversity parameters significantly differed between years. Parameters included: number of genets (G), number of unique genets (GU), clonal richness (R), Simpson’s diversity (D), Fager’s evenness (E), shape of the Pareto distribution (β), mean, minimum and maximum of the clonal dominance index (Dc, Dc,min and Dc,max, respectively), mean and maximum clone sizes (m2), percentage polymorphic loci (%P), mean alleles per locus (A), allelic richness (AR), observed heterozygosity (Ho), expected heterozygosity (He) and fixation index (F). While differences between sampling schemes are confounded with temporal changes in patches, neither changes in patch parameter estimates between years nor the difference in parameter estimates under different sampling schemes is a central focus of this study. These comparisons simply aid evaluation of whether complementary biological information may be provided by different sampling strategies.
To characterize clonal patterns further, we evaluated the hypothesis that widespread genets within patches are spatially dominant (i.e. exclude other, potentially competing genotypes), such that patches with widespread genets maintain little genotypic variation. We inferred correlations of average clone size (m2) with clonal variation (R and β) and mean clonal dominance (Dc) across patches. Parameter R provides a rarefied measure of genotypic diversity, parameter β provides an integrative measure of clonal variation, and parameter Dc describes the average degree of interdigitation among genets. We used analysis of covariance (ANCOVA) considering sampling year, mean clone size and the interaction between sampling year and clone size. A log-transformation of β was required to obtain an approximately normal distribution. A positive relationship between mean clone size and mean clonal dominance and a negative relationship between mean clone size and genotypic variation (R and β) would be consistent with our hypothesis that widespread clones are spatially dominant and could inhibit genotypic variation within patches. If slopes were similar between samples (i.e. non-significant interaction), the interaction was removed and the model was refit to test for different intercepts between years (i.e. parallel lines). If no significantly different intercept was determined between years, a model was fit to consider the same slope and intercept.
RESULTS
Clonal analysis
Our 21 polymorphic allozyme loci distinguished MLGs with high probability. The 68 samples that were not genotyped at all 21 loci had missing data at a mean of 3·3 loci. It was possible to assign unique clonal identities to 16 (23·5 %) of these samples and match 19 (27·9 %) more with a single fully resolved genotype in its patch. After conservatively dropping 33 samples (48·5 %) with missing data, the data set contained 540 MLGs and 1224 samples. Overall genotypic resolution initially increased rapidly with the number of loci but approached an asymptote after addition of the 20th locus (Supplementary Data Fig. S1A). However, gains in genotypic resolution may have continued with additional loci in POR and TRL, where increases in genotypic resolution did not asymptote with 21 loci (Supplementary Data Fig. S1B). The overall frequency distribution of allelic distances separating MLGs did not suggest that somatic mutation or scoring errors were common (Supplementary Data Fig. S2A). However, in patches with low genetic and genotypic diversity, a bimodal pattern indicated that most genotypes were separated by a few allelic differences while some genotypes were separated by many more allelic differences (POR, HDP and, TRL; Supplementary Data Fig. S2B). This pattern supported manual data set curation in which common MLGs across years differing by one or two alleles were merged to produce a single MLG. Overall, 50 MLGs were grouped into 11 genets prior to further analysis (POR and TRL = 6; HDP = 5; MOR = 9; MWS = 23; BDR = 7). Of the 515 MLGs ultimately distinguished, 404 (78·4 %) were detected once and 111 (21·6 %) were detected in ≥2 samples (Fig. 2; Supplementary Data Fig. S3). The probability of an MLG being encountered twice with random mating (Psex) was low in all cases, with the maximum (Psex,max; i.e. the most likely MLGs) ranging from 5·33 × 10–1 in TRL10 to 2·49 × 10–6 in MOR11 (Table 1). Thus, erroneous assignment of matching genotypes to the same clone is unlikely.
Fig. 2.

Maps of clones within patches HDP and MOR. Patch HDP (A and B) contained the least and MOR (C and D) the greatest numbers of clones in 2010 and 2011. Genets are shown in boxes coloured according to their frequency (red ≥20 occurrences; blue = 2–9; green = 1). The distribution of genets that were detected infrequently overall, but which occurred in multiple populations, is highlighted with unique colours (Table 2). Corresponding clonal maps of all patches are shown in Supplementary Data Fig. S3.
Patches differed in clonal structure, but clonal variation metrics were similar within most patches between 2010 and 2011. The number of genotypes per patch (G) ranged broadly among sites (2010, 10–56; 2011, 18–104; Table 1) and significantly differed between years (2010 mean = 34·8; 2011 mean = 54·2; t = –2·908, d.f. = 5, P = 0·030). The number of genotypes sampled only once within a patch (GU) ranged from seven in HDP10 to 94 in MOR11 [mean 2010 = 25·7 (71·9 %); mean 2011 = 44·5 (77·4 %); Table 1] and significantly differed between years (t = –2·731, d.f. = 5, P = 0·041). Clonal richness (R) ranged from 0·12 in HDP10 to 0·90 in MOR11 (2010 mean = 0·39; 2011 mean = 0·49; Table 1), but did not significantly differ between years (t = – 2·249, d.f. = 5, P = 0·074). Most patches had high genet diversity (Simpson’s D ≥ 0·750), although Simpson’s D in HDP varied between 0·26 and 0·45 between years, and did not significantly differ between years (2010 mean = 0·76; 2011 mean = 0·80; Table 1; t = –0·901, d.f. = 5, P = 0·409). Evenness was more variable and did not significantly differ between years (2010, 0·06–0·81; mean =0·54; 2011, 0·20–0·82 mean = 0·54; Table 1; t = 0·057, d.f. = 5, P = 0·957). The overall distribution of genet size classes conforms to the Pareto distribution (r2 = 0·884; P = 0·017). The β coefficient describing the Pareto distribution ranged from 0·03 to 1·47, and was similar between years for all patches except MOR, which changed from 0·29 to 1·47 (2010 mean = 0·27; 2011 mean = 0·42; Table 1; Fig. 3). Pareto parameter β did not significantly differ between years (t = –0·743, d.f. = 5, P = 0·491). Mean clonal dominance (Dc) ranged from 0·35 to 0·93 (2010 mean = 0·78; 2011 mean = 0·50), with individual genets yielding Dc,min as low as 0·13 but often reaching Dc,max of 1·00 (Table 1). There was a significant difference in mean Dc (t = 3·490, d.f. = 5, P = 0·017) and Dc,min (t = 3·490, d.f. = 5, P = 0·018), but not Dc,max (t = 1·345, d.f. = 5, P = 0·236). The spatial extent of genets also varied broadly (mean 2010, 16·23–33·31 m2; mean 2011, 10·04–34·6 m2; max 2010, 46·1–68·0 m2; max 2011, 49·8–64·2 m2; Table 1; Fig. 2). Neither mean clone size (t = –0·135, d.f. = 5, P = 0·898) nor maximum size (t = –0·523, d.f. = 5, P = 0·623) differed significantly between years. In sum, more genets were detected with the sampling scheme of 2011 than that of 2010. It is possible that these additional and potentially rare genets were detected simply due to the greater number of samples collected in 2011. Yet the significant decrease in mean and minimum clonal dominance with the finer grained sampling scheme of 2011 suggests that spatially restricted genets are highly interdigitated with widespread genets. Thus, sampling at multiple and smaller spatial scales may provide greater detail to portray patch composition more accurately.
Fig. 3.

Pareto distribution summarizing clonal variation within and among patches. Graphs show the cumulative frequency of genotypes represented by ≥X number of ramets on a log–log scale. The β coefficient describing the Pareto distribution, regression coefficient (r2) and significance of the regression (P) are shown for each analysis.
Sixteen genets were detected in multiple patches and/or sampling events (Table 2). Ten genets from 2010 were detected a second time within their original patch (range = 0–4; Table 1), and eight genets were detected in multiple patches (Table 2). Of 16 repeatedly detected genets, only four were particularly common (≥10 samples) and four were relatively rare (two samples; Table 2). Thus, regardless of changes to our sampling scheme between years, it is clear that the identity, abundance, distribution and spatial dominance of individual genets differed within patches between years (Fig. 2).
Table 2.
Summary of 16 matching multilocus genotypes obtained from multiple patches and/or years
| ID | n | Patch | Genotype* |
|---|---|---|---|
| H1 | 3 | BDR10 (2) BDR11 (1) | 343544443434333333443444453355343344444444 |
| I1 | 97 | BDR10 (43) BDR11 (54) | 343544443434333333443444453355343444444444 |
| F2 | 4 | POR10 (1) TRL10 (3) | 444544443344333333443534443345444444444444 |
| H2 | 3 | TRL10 (1) POR10 (1) MOR10 (1) | 444544443344333333443534443355443444444444 |
| I2 | 2 | TRL10 (1) POR11 (1) | 444544443344333333443534443355444444444445 |
| O2 | 163 | POR10 (27) POR11 (40) TRL10 (36) TRL11 (60) | 444544443344333433443534443355444444444444 |
| G3 | 2 | TRL11 (1) POR10 (1) | 444544443444333333443534443335443444444444 |
| H3 | 2 | TRL11 (1) POR11 (1) | 444544443444333333443534443345443444444445 |
| J3 | 7 | POR10 (1) POR11 (6) | 444544443444333333443534443355443444444444 |
| K3 | 8 | POR11 (7) TRL11 (1) | 444544443444333333443534443355443444444445 |
| Q3 | 2 | POR10 (1) POR11 (1) | 444544443444333334443534443345443444444444 |
| S3 | 3 | POR10 (2) POR11 (1) | 444544443444333334443534443355444444444444 |
| W3 | 9 | POR11 (5) TRL11 (4) | 444544444444333333443534443355444444444445 |
| N4 | 4 | HDP10 (1) HDP11 (3) | 445544443433333335463444443345443444443444 |
| O4 | 139 | HDP10 (68) HDP11 (71) | 445544443433333335463444443355443444443444 |
| W4 | 37 | MOR10 (30) MOR11 (7) | 454534443333343344663434443355443444443444 |
ID, name given to each multilocus genotype; n, total number of samples in which that genotype appeared; patch, patches in which that clone occurred (the number of samples from that patch); genotype, multilocus genotype of each clone, with each allele at a locus indicated by a single digit.
*Genotypes are designated as a string, with each allele coded as a single number with the 21 loci arranged in the following order: ACO1, ACO2, PGI1, UGPP1, UGPP4, DIA1, DIA2, MNR1, MNR2, MNR3, TPI1, TPI2, AAT1, AAT2, GDH1, IDH1, IDH2 and MDH1.
Genetic variation analysis
Levels of genetic diversity ranged broadly among patches and differed between sampling schemes. Percentage polymorphic loci (P) ranged from 42·9 to 76·2 % in 2010 and from 47·6 to 76·2 % in 2011 (2010 total = 85·7 %; 2011 total = 90·5 %; Table 3), significantly differing between years (t = –2·989, d.f. = 5, P = 0·031). The mean number of alleles per locus (A) ranged from 1·48 to 1·95 in 2010 and from 1·52 to 2·38 in 2011 (means = 1·68 and 1·89; Table 3), significantly differing between years (t = –3·093, d.f. = 5, P = 0·027). Allelic richness (AR) ranged from 1·41 to 1·82 in 2010 and from 1·46 to 1·85 in 2011 (means = 1·58 and 1·62; Table 3), significantly differing between years (t = –3·508, d.f. = 5, P = 0·017). Observed heterozygosity (Ho; range = 0·199–0·370) was greater than expected heterozygosity (He; range = 0·142–0·305) in all patches, and both Ho and He were significantly greater in 2011 (t = –4·320, d.f. = 5, P = 0·008; t = –3·223, d.f. = 5, P = 0·023, respectively). The fixation index (F) was always negative (range = –0·488 to −0·049; Table 3) and differed significantly between years (t = 3·128, d.f. = 5, P = 0·026).
Table 3.
Summary of genetic diversity
| Population | n | P | A | AR | Ho | He | F |
|---|---|---|---|---|---|---|---|
| POR10 | 38 | 42·9 | 1·57 | 1·51 | 0·237 | 0·172 | –0·307 |
| POR11 | 43 | 61·9 | 1·71 | 1·52 | 0·269 | 0·190 | –0·277 |
| HDP10 | 10 | 42·9 | 1·48 | 1·43 | 0·259 | 0·169 | –0·427 |
| HDP11 | 18 | 47·6 | 1·52 | 1·46 | 0·288 | 0·180 | –0·488 |
| TRL10 | 25 | 47·6 | 1·52 | 1·41 | 0·199 | 0·142 | –0·238 |
| TRL11 | 36 | 52·4 | 1·67 | 1·49 | 0·254 | 0·174 | –0·319 |
| MWS10 | 51 | 57·1 | 1·66 | 1·62 | 0·279 | 0·232 | –0·089 |
| MWS11 | 71 | 66·7 | 2·00 | 1·68 | 0·296 | 0·237 | –0·158 |
| MOR10 | 56 | 76·2 | 1·95 | 1·82 | 0·330 | 0·297 | –0·049 |
| MOR11 | 104 | 76·2 | 2·05 | 1·85 | 0·370 | 0·305 | –0·172 |
| BDR10 | 29 | 66·7 | 1·91 | 1·67 | 0·277 | 0·236 | –0·138 |
| BDR11 | 53 | 76·2 | 2·38 | 1·78 | 0·363 | 0·278 | –0·247 |
| Mean10 | 34·83 | 55·6 | 1·68 | 1·58 | 0·264 | 0·208 | –0·208 |
| Mean11 | 54·17 | 635 | 1·89 | 1·63 | 0·307 | 0·227 | –0·277 |
| Total10 | 209 | 85·7 | 2·33 | 1·87 | NA | 0·285 | NA |
| Total11 | 325 | 90. | 2·71 | 1·91 | NA | 0·309 | NA |
n, number of unique genotypes included in the analysis; P, percentage polymorphic loci; A, average number of alleles per locus; AR, allelic richness (mean per locus); Ho, observed heterozygosity; He, expected heterozygosity; F, fixation index; NA, not applicable.
The proportion of total genetic variation partitioned among patches was significant in both 2010 (FST = 0·237; P ≤ 0·001) and 2011 (FST = 0·226; P ≤ 0·001). Pairwise FST values were significant (P ≤ 0·001) between all pairs of patches in both years (mean 2010 = 0·209–0·269; 2011 =0·195–0·293; Table 4). Pairwise FST values were much lower between years within patches (range = 0·007–0·107; mean =0·060; Table 4).
Table 4.
Pairwise and mean pairwise FST values
| POR | HDP | TRL | MWS | MOR | BDR | Mean 2011 | |
|---|---|---|---|---|---|---|---|
| POR | 0·058 | 0·389 | 0·035 | 0·268 | 0·285 | 0·199 | 0·235 |
| HDP | 0·375 | 0·007 | 0·412 | 0·211 | 0·244 | 0·208 | 0·293 |
| TRL | 0·028 | 0·423 | 0·107 | 0·301 | 0·312 | 0·224 | 0·257 |
| MWS | 0·260 | 0·153 | 0·297 | 0·092 | 0·122 | 0·133 | 0·207 |
| MOR | 0·265 | 0·181 | 0·297 | 0·162 | 0·035 | 0·209 | 0·234 |
| BDR | 0·260 | 0·200 | 0·299 | 0·173 | 0·225 | 0·060 | 0·195 |
| Mean 2010 | 0·238 | 0·266 | 0·269 | 0·209 | 0·226 | 0·231 | 0·060 |
Interpatch comparisons for 2010 and 2011 are below and above the (bold) diagonal, respectively. Interpatch comparisons between 2010 and 2011 are on the diagonal. All comparisons are significant (P ≤ 0·001). Mean pairwise values for each patch are listed for each year (2010, bottom; 2011, right) and for each interannual comparison (bottom right).
Statistical analysis
Correlations between clone size and levels of clonal variation and dominance do not support the hypothesis that widespread genets are spatially dominant. Instead, widespread genets were interdigitated with other genets (Figs 2 and 4). ANCOVA did not reveal significant interactions between years and mean clone size for any independent variable tested. Mean clone size was significantly negatively correlated with R (r2 = 0·510; P = 0·009) and log-transformed β (r2 = 0·421; P = 0·022), without significantly different intercepts between years (Fig. 4). The relationship between average clone size and Dc exhibited significantly different intercepts between years (r2 = 0·597; P = 0·006; Fig. 4C). Finer sampling in 2011 often led to detection of multiple MLGs in closer proximity than in 2010, so lower Dc was apparent (Fig. 4C).
Fig. 4.
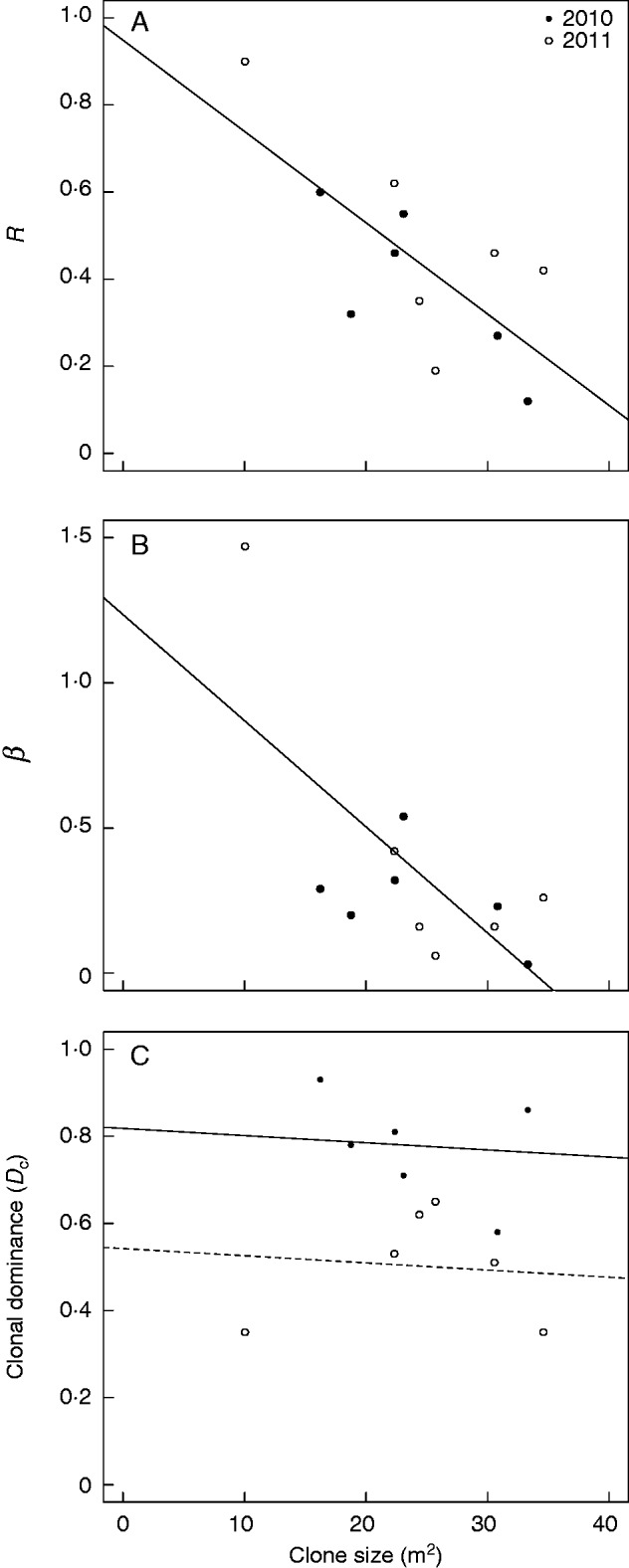
Correlations between mean clone size (m2) and patch characteristics. Patch characteristics include (A) clonal richness (R), (B) the Pareto distribution parameter β (note that β was log-transformed prior to performing ANCOVA) and (C) mean clonal dominance (Dc).
DISCUSSION
Clonal and genetic variation differed among kudzu patches. Patterns of clonal diversity and structure range as broadly among kudzu patches as they do among taxonomically and ecologically disparate species. A meta-analysis of 77 clonal plant species produced mean Simpson’s D = 0·85 and Fager’s E = 0·74 (Honnay and Jacquemyn, 2008), which was similar to the six kudzu patches except HDP (Table 1). Yet the range of mean clonal dominance among kudzu patches (0·35–0·93) exceeded the interspecific range (0·42–0·89) among eight comparable studies (Ohsako, 2010). These kudzu patches also had more variable β values than 15 terrestrial and marine plant species (kudzu = 0·03–1·47; other species = 0·60–1·49; Ohsako, 2010) as well as four sea grass populations with the broadest intraspecific range previously reported (Posidonia oceanica =0·06–1·23; Arnaud-Haond et al., 2007). Across species, clonal diversity and evenness are significantly higher in self-compatible than in self-incompatible species (Honnay and Jacquemyn, 2008), suggesting that new genotypes are consistently produced in populations with greater pollen availability. However, mechanisms that give rise to this pattern may vary within and among species. For example, the rates with which new genotypes arise from self-fertilization probably vary depending upon the MLG composition of self-compatible kudzu patches (Pappert et al., 2000). Similarly, genet overlap should increase outbreeding and seed set, yet a significant association between mating system and clonal structure has not been demonstrated (Charpentier, 2001; Honnay and Jacquemyn, 2008; Ohsako, 2010). Processes shaping kudzu clonal patterns vary at the patch level, providing further evidence that patterns are only loosely determined by intrinsic species characteristics.
Contrary to expectations, both common and less common MLGs were interdigitated. A key process in shaping clonal patterns in a patch is probably initial site invasion, which may be associated with founder effects if few propagules (i.e. seeds or clippings) originally colonize a site. Depending on the number and genetic similarity of founders, colonization can reduce genetic diversity and increase genetic structure among populations. Subsequent changes in clonal and genetic variation occur as a function of the degree of sexual reproduction within patches and gene flow into patches (Hamrick and Nason, 1996). For example, HDP is a relatively small, isolated and recently founded patch that originated following construction of an adjacent highway in 1988. This patch had among the lowest levels of genetic diversity and the lowest clonal diversity, with one widespread genet (O4) interdigitating with other genets across the patch. Conversely, MOR is a larger, less isolated patch that originated with construction of the same highway. This patch exhibited the highest levels of clonal and genetic diversity, with many interdigitating genets. Across the broad range of kudzu clonal patterns, mean clonal dominance was not significantly greater in patches with widespread clones and genets were highly interdigitated in each patch.
Occurrence of eight genets in multiple patches and 16 genets in multiple years suggests that clones span broad areas and potentially persist for long periods within patches. Patterns and mechanisms of vegetative dispersal by invasive plants is of broad interest and potentially relevant to their control. For example, one geographically widespread clone was reported to occur in multiple populations of knotweed (Fallopia spp.) and it was suggested that this clone exhibited selective advantages that facilitated spread in its invasive range (Grimsby et al., 2007). It is unlikely that kudzu clones have spread to multiple patches due to selective advantage because they comprise only a small proportion of all samples and exhibited changes in frequency between seasons. Similarly, genetic investigation of a vining invasive plant (Bryonia alba) found that clones often occur in >2 populations. (Novak and Mack, 2000), suggesting that vining invasive plant genets may often occur in multiple patches. However, B. alba reproduces both sexually and by apomixis, so it is difficult to isolate the contribution of vegetative dispersal to its clonal distribution. For kudzu, legacies of vegetative dispersal during colonization were evident in the genetic data. All patches sharing MLGs were founded following highway construction and were within 900 m of each other (POR, TRL and MOR; Table 2). Two of these sites (POR and TRL) exhibited quite low pairwise FST (0·028–0·035), within the range of intrapatch FST between years (≤0·107; Table 4). These three sites occurred in the same creek drainage prior to highway construction and are adjacent to roadbeds that were actively filled to construct the roads. Consequently, identical clones may have established from clippings within the earth used to build the roadbeds. Taken together, these data suggest highway construction and maintenance machinery may have inadvertently dispersed kudzu propagules.
We observed substantial variation in genotypic diversity and composition between years in some patches but not others. These changes appeared to be related to disturbance, but could also reflect differences in our sampling schemes. Disturbances, including control measures (e.g. mowing), probably influence patterns of clonal and genetic variation. Kudzu is robust to harsh treatment; in a controlled experiment removal of 50 and 75 % of leaves and shoots did not affect above-ground biomass, and seedlings survived 1–2 months under 0 % direct sunlight (Frye et al., 2012). Since clonal variation was negatively correlated with mean clone size (Fig. 4), actions that reduce kudzu ramet sizes without increasing mortality could enhance genotypic diversity. As a direct consequence of disturbance, subordinate genotypes could gain access to space, light and other resources and could assume a more dominant role in subsequent years. In addition, Pappert (1998) and Pappert et al. (2000) observed that populations with more MLGs had greater seed set and subsequent germination. Recruitment may also be enhanced if activities such as mowing scarifies seeds, which can increase germination rates from 7–17 % to 95–100 % (Susko et al., 2001), or decreases seed damage by insects and fungi, which together limit seedling recruitment to ≤10 % (Forseth and Innis, 2004). Mowing highway perimeters could further facilitate gene flow by dispersing vegetative propagules that subsequently root. Site MOR is located in a highway cloverleaf interchange that has been mowed several times in the last 10 years, including between the 2010 and 2011 surveys (T.R.K. and J.L.H., pers. obs.). Perhaps as a consequence, MOR had the greatest clonal and genetic diversity and exhibited a marked increase in clonal diversity between years. This raises the possibility that some control measures may inadvertently enhance seedling establishment and/or genetic variation, increasing resistance to control. Experimental research into interactions between control measures, genetic variation and the ecological impacts of invasive species over time is necessary to address these observations.
This detailed analysis of the clonal structure of six kudzu patches in Athens, GA provided insights into the establishment and subsequent growth of populations. First, levels of genetic diversity (Table 3) within each patch varied >2-fold among the six patches. Also, the overall genetic differentiation among patches (FST = 0·237) was high for a long-lived outcrossing perennial plant (Hamrick and Godt, 1996) especially at this spatial scale (<15 km). Considerable genetic differentiation occurred even among patches separated by scales of <1 km (mean pairwise FST = 0·219). These FST values are similar to those of kudzu populations ranging from Mississippi to North Carolina, even though the two studies differed in objectives, population sampling design and the number of loci analysed (14 vs. 21) (Pappert et al., 2000). These results would be expected if patches were established by a small number of founding MLGs. Furthermore, these patches are relatively recent in origin and have not experienced enough gene flow to counteract founder effects. The extensive vegetative reproduction within these sites has probably also delayed progress toward gene flow–drift equilibrium. We expect that these results should also be expected for populations of many other invasive viny plant species.
Contrary to expectations, the results did not show the dominance of one or a few genets within a patch. Dominance was generally low (except perhaps HDP; Table 1) and every patch had considerable interdigitation of MLGs. The changes seen between the two years may partially explain the lack of spatial dominance. Although kudzu does not die back during the winter, the loss of its leaves may give each genet a more equal chance to be competitive with the onset of bud break in the spring. The observation that patch MOR, which had been mowed most winters, had the highest number of genets is consistent with the idea that above-ground intergenet competition may start anew after patch-wide disturbance events (i.e. winter or mowing). The stochasticity of such dynamics may make it difficult for a single genet to acquire and maintain dominance within its patch. This hypothesis should be possible to test under experimental conditions. Finally, the observation of a significant excess of heterozygosity in all six patches suggests that successful genets are those that have a higher proportion of heterozygous loci. In particular, the three patches with the greatest excess of heterozygotes (HDP, POR and TRL) have the lowest number of genotypes. These results are consistent with previous work that also demonstrated that populations with fewer genets had higher excesses of heterozygosity (Pappert et al., 1998). Pappert (1998), in a controlled greenhouse experiment, also demonstrated that individuals heterozygous for a high proportion of their allozyme loci produced more biomass and leaf area. Because many viny plant species have three-dimensional architecture in which individual genets grow over each other, these results may be applicable to many plant species.
We found that spatial clonal structure varied broadly among kudzu patches and that disturbance may be associated with the recruitment of additional genotypes. Thus, we suggest that it may be necessary to evaluate more judiciously the efficacy of measures to control invasive vines (e.g. mowing) that could inadvertently increase clonal richness and genetic variation.
SUPPLEMENTARY DATA
Supplementary data are available online at www.aob.oxfordjournals.org and consist of the following. Table S1: history of land use in the area of the sampled patches. Fig. S1: power of the 21 allozyme loci to detect distinct multilocus genotypes. Fig. S2: distribution of allelic distances separating all unique multilocus genotypes. Fig. S3: maps of clones in all patches.
ACKNOWLEDGEMENTS
We thank students of UGA PBIO 8720 for participating in the 2010 portion of this study, and the members of the Hamrick lab, particularly C. Deen, for assistance throughout. C.W. is grateful to J.L.H. for hosting. This work was supported by a University of Georgia Presidential Fellowship to T.R.K., research funds from the UGA Department of Plant Biology; and Yunnan Government Scholarship Funds and the National Science Foundation of China [grant no. 31160080] to C.W.
LITERATURE CITED
- Abramovitz JN. 1983. Pueraria lobata Willd. (Ohwi) Kudzu: limitations to sexual reproduction. Master’s Thesis, University of Maryland, College Park, MD. [Google Scholar]
- Alberto F, Gouveia L, Arnaud-Haond S, Pérens-Lloréns JL, Duarte CM, Serrão EA. 2005. Spatial genetic structure, neighbourhood size and clonal subrange in seagrass (Cymodocea nodosa) populations. Molecular Ecology 14: 2669–2681. [DOI] [PubMed] [Google Scholar]
- Araki K, Shimatani K, Ohara A. 2009. Dynamics of distribution and performance of ramets constructing genets: a demographic–genetic study in a clonal plant, Convallaria keiskei. Annals of Botany 104: 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud-Haond S, Duarte CM, Alberto F, Serrão EA. 2007. Standardizing methods to address clonality in population studies. Molecular Ecology 16: 5115–5139. [DOI] [PubMed] [Google Scholar]
- Barrett SC, Colautti RI, Eckert CG. 2008. Plant reproductive systems and evolution during biological invasion. Molecular Ecology 17: 373–383. [DOI] [PubMed] [Google Scholar]
- Burns JH, Ashman T-L, Steets JA, Harmon-Threatt A, Knight TM. 2011. A phylogenetically controlled analysis of the roles of reproductive traits in plant invasions. Oecologia 166: 1009–1017. [DOI] [PubMed] [Google Scholar]
- Charpentier A. 2001. Consequences of clonal growth for plant mating. Evolutionary Ecology 15: 521–530. [Google Scholar]
- Dlugosch KM, Parker IM. 2008. Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Molecular Ecology 17: 431–449. [DOI] [PubMed] [Google Scholar]
- Dong M, Lu B-R, Zhang H-B, Chen J-K, Li B. 2006. Role of sexual reproduction in the spread of an invasive clonal plant Solidago canadensis revealed using intersimple sequence repeat markers. Plant Species Biology 21: 13–18. [Google Scholar]
- Dorken ME, Eckert CG. 2001. Severely reduced sexual reproduction in northern populations of a clonal plant, Decodon verticillatus (Lythraceae). Journal of Ecology 89: 339–350. [Google Scholar]
- Eckert CG, Barrett SCH. 1993. Clonal reproduction and patterns of genotypic diversity in Decodon verticillatus (Lythraceae). American Journal of Botany 80: 1175–1182. [Google Scholar]
- Excoffier L, Laval G, Schneider S. 2005. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary Bioinformatics 1: 47–50. [PMC free article] [PubMed] [Google Scholar]
- Fager EW. 1972. Diversity: a sampling study. American Naturalist 106: 293–310. [Google Scholar]
- Forseth INJ, Innis AF. 2004. Kudzu (Pueraria montana): history, physiology, and ecology combine to make a major ecosystem threat. Critical Reviews in Plant Sciences 23: 401–413. [Google Scholar]
- Frye MJ, Hough-Goldstein J, Kidd KA. 2012. Response of kudzu (Pueraria montana var. lobata) seedlings and naturalized plants to simulated herbivory. Invasive Plant Science and Management 5: 417–426. [Google Scholar]
- Gordon DR. 1998. Effects of invasive, non-indigenous plant species on ecosystem processes: lessons from Florida. Ecological Applications 8: 975–989. [Google Scholar]
- Goudet J. 2011. hierfstat: estimation and tests of hierarchical F-statistics. R package version 0.04-6. http://CRAN.R-project.org/package=hierfstat. [Google Scholar]
- Grimsby JL, Tsirelson D, Gammon MA, Kesseli R. 2007. Genetic diversity and clonal vs. sexual reproduction in Fallopia spp. (Polygonaceae). American Journal of Botany 94: 957–964. [DOI] [PubMed] [Google Scholar]
- Hamrick JL, Godt MJW. 1996. Effects of life history traits on genetic diversity in plant species. Philosophical Transactions of the Royal Society B: Biological Sciences 351: 1291–1298. [Google Scholar]
- Hamrick JL, Nason JD. 1996. Consequences of dispersal in plants. In: Rhodes OE, Chesser RK, Smith MH, eds. Population dynamics in ecological space and time. Chicago, IL: University of Chicago Press, 203–236. [Google Scholar]
- Hao JH, Qiang S, Chrobock T, van Kleunen M, Liu QQ. 2011. A test of Baker’s law: breeding systems of invasive species of Asteraceae in China. Biological Invasions 13: 571–580. [Google Scholar]
- Hipps CB. 1994. Kudzu: a vegetable menace that started out as a good idea. Horticulture, 72: 36–39. [Google Scholar]
- Honnay O, Jacquemyn H. 2008. A meta-analysis of the relation between mating system, growth form, and genotypic diversity in clonal plant species. Evolutionary Ecology 22: 299–312. [Google Scholar]
- Jewett DK, Jiang CJ, Britton KO, Sun JH, Tang J. 2003. Characterizing specimens of kudzu and related taxa with RAPD’s. Castanea 68: 254–260. [Google Scholar]
- van Kleunen M, Manning JC, Pasqualetto V, Johnson SD. 2008. Phylogenetically independent associations between autonomous self-fertilization and plant invasions. American Naturalist 171: 195–201. [DOI] [PubMed] [Google Scholar]
- McCormick MK, Kettenring KM, Baron HM, Whigham DF. 2010. Spread of invasive Phragmites australis in estuaries with differing degrees of development: genetic patterns, allee effects and interpretation. Journal of Ecology 98: 1369–1378. [Google Scholar]
- Mitich LW. 2000. Intriguing world of weeds. Kudzu [Pueraria lobata (Willd.) Ohwi]. Weed Technology 14: 231–235. [Google Scholar]
- Novak SJ, Mack RN. 2000. Clonal diversity within and among introduced populations of the apomictic vine Bryonia alba (Cucurbitaceae). Canadian Journal of Botany 78: 1469–1481. [Google Scholar]
- Ohsako T. 2010. Clonal and spatial genetic structure within populations of a coastal plant, Carex kobomugi (Cyperaceae). American Journal of Botany 97: 458–470. [DOI] [PubMed] [Google Scholar]
- Pappert R. A. 1998. Population genetic variation and heterotic patterns of Pueraria lobata Ohwi (kudzu). MS thesis, University of Georgia, Athens, Georgia, USA. [Google Scholar]
- Pappert RA, Hamrick JL, Donovan L. 2000. Genetic variation in Pueraria lobata (Fabaceae), an introduced, clonal, invasive plant of the southeastern United States. American Journal of Botany 87: 1240–1245. [PubMed] [Google Scholar]
- Parks JC, Werth CR. 1993. A study of spatial features of clones in a population of bracken fern, Pteridium aquilinum (Dennstaedtiaceae). American Journal of Botany 80: 537–544. [DOI] [PubMed] [Google Scholar]
- Peakall R, Smouse PE. 2006. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes 6: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel D, Lach L, Zuniga R, Morrison D. 2000. Environmental and economic costs of nonindigenous species in the United States. Bioscience 50: 53–65. [Google Scholar]
- R Core Development Team. 2013. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Soltis DE, Haufler CH, Darrow DC, Gastony GJ. 1983. Starch gel electrophoresis of ferns: a compilation of grinding buffers, gel and electrode buffers, and staining schedules. American Fern Journal 73: 9–27. [Google Scholar]
- Sun JH, Li Z-C, Jewett DK, Britton KO, Ye WH, Ge X-J. 2005. Genetic diversity of Pueraria lobata (kudzu) and closely related taxa as revealed by inter-simple sequence repeat analysis. Weed Research 45: 255–260. [Google Scholar]
- Susko DJ, Mueller JP, Spears JF. 2001. An evaluation of methods for breaking seed dormancy in kudzu (Pueraria lobata). Canadian Journal of Botany 78: 197–203. [Google Scholar]
- Tsugawa H, Kayama R. 1976. Studies on population structure of kudzu vine (Pueraria lobata Ohwi). 3. Outline on detachment of rooted nodes. Journal of the Japanese Grassland Society 22: 273–279. [Google Scholar]
- Tsugawa H, Ryosei K. 1985. Studies on population structure of kudzu vine (Pueraria lobata Ohwi). VI. The structure of overwintering aboveground parts of individual plants which constitute a natural kudzu population. Journal of the Japanese Grassland Society 31: 167–172. [Google Scholar]
- US Census Bureau. 2005. TIGER/Line Files, 2005. First Edition http://www.census.gov/geo/www/tiger. [Google Scholar]
- Wendel JF, Parks CR. 1992. Genetic control of isozyme variation in Camillia japonica L. Journal of Heredity 73: 197–204. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.