

Abstract
The idea of compartmentalization of genotype and phenotype in cells is key for enabling Darwinian evolution. This contribution describes bioinspired systems that use in vitro compartments—water-in-oil droplets and gel-shell beads—for the directed evolution of functional proteins. Technologies based on these principles promise to provide easier access to protein-based therapeutics, reagents for processes involving enzyme catalysis, parts for synthetic biology and materials with biological components.
Keywords: bioinspiration, cell compartmentalization, synthetic biology, protein engineering, antibodies, microfluidics, emulsion droplets
1. Introduction
The cell membrane separates molecules belonging to a cell from those that are part of the environment. A key role of this compartmentalization is to link the genotype (‘the genetic constitution of an individual’) with its corresponding phenotype (‘a set of observable characteristics of an individual’). This linkage is important for Darwinian evolution, as selection is exerted at the level of the phenotype, but survival and propagation of a selected trait are dependent on the relevant gene being carried forward to subsequent rounds of evolution. Both have to be linked to ensure that a selective advantage conferred by a gene leads to the emergence of improved species.
It can thus be argued that compartmentalization is the organizing principle that enables Darwinian evolution—and that a cell-like compartment is the most basic ‘evolutionary unit’. This contribution describes experimental approaches towards artificial evolution that employ mimics of cellular compartments (figure 1): water-in-oil emulsions, droplets or gel beads that keep together an identifier (i.e. a gene), the functional molecule encoded by the gene (i.e. a protein) and a readout (e.g. an optical signal that distinguishes ‘winners' from ‘losers' in an evolutionary selection round). Such compartments can be made completely in vitro, so that—despite being inspired by cells—they are reducing the complexity of the cell-like compartment to just one purpose: linking genotype and phenotype and allowing an assay for the one function of interest. This type of biological reductionism differs from that originally proposed by Crick (‘to explain all biology in terms of physics and chemistry’) [4]: instead of a molecular understanding of the end products of evolution as the basis for future rational design of equally perfect or even improved constructs, the features of the engine of evolution are to be controlled, understood and ultimately mimicked: constituting a system that provides a route towards functional molecules.
Figure 1.
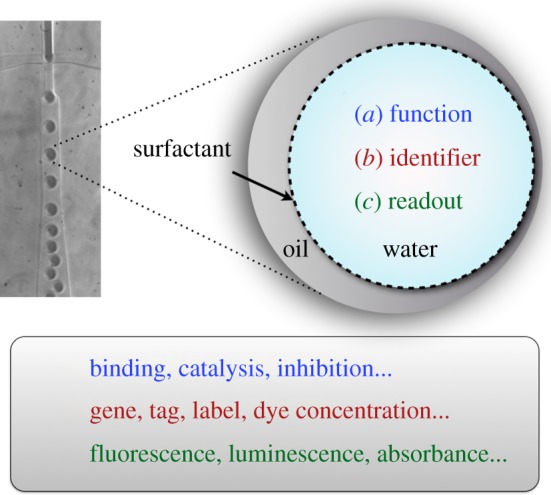
An in vitro compartment (a droplet [1,2] or a bead [3]) combines (a) the function of a molecule (e.g. the catalytic or binding activity of a protein or nucleic acid). (b) The information on its identity (e.g. its sequence encoded by DNA) and (c) a readout to assess the molecule's ability to carry out its function via a miniaturized assay (e.g. based on product fluorescence). Droplet diameters vary between 1 and 200 µm (corresponding to volumes between 0.5 fl and 4 nl).
The ability to evolve functional proteins is assuming an increasingly central role, because rational design of protein binders or catalysts often does not provide efficient solutions, notwithstanding the enormous progress in protein design over the last two decades. For example, antibodies used in therapy are routinely generated by ‘directed evolution’ (i.e. combinatorial selections from large libraries of candidates) and not by design despite a wealth of molecular insight into protein structure. Although we know so much, for example, about the regularity of the antibody structure and its target-binding region from comprehensive databases of primary sequences and structures, antibody binders are made by combinatorial methods (rather than by design in silico). Likewise, computational designs of catalytic proteins [5,6] often require further improvement by directed evolution [7,8] and the contribution of directed evolution can exceed the contribution of design [9]. Of course both approaches—design and selection—are complementary, and each directed evolution experiment will provide rational explanations and contribute in turn to the body of design rules for biological molecules. However, Crick's optimism about the supremacy of molecular design has to be mixed with a dose of realism about its limits and a realistic strategy for the creation of functional biomolecules will have to involve library methods and directed evolution.
Important criteria for a good evolution system are (i) simple to set up, (ii) allowing high throughput (more than 106 experiments within a reasonable experimental timescale, i.e. hours or days) and, as far as possible, (iii) that evolution should be preferably conducted in vitro, because carrying out this process under non-natural conditions can overcome the following constraints:
— the requirement of having to comply with a working biological system (e.g. compatibility with a host organism)—proteins that are toxic to the host cannot be evolved;
— the narrow dynamic range of in vivo selections and the limit that only proteins directly relevant for survival of the host are amenable to in vivo evolution (e.g. in auxotrophic selections); and
— in vivo selections cannot be carried out under non-natural conditions, for example, involving the use of non-natural amino acids, operating at extremes of pH or temperature, or under other desired non-physiological conditions.
To free directed evolution from these constraints and drive it by arbitrarily chosen selection criteria (instead of host cell survival), attention has therefore turned to in vitro compartments for directed evolution that replace the cell compartment with a man-made entity that is equally suited to combine genotype and phenotype.
Joshua Lederberg anticipated the potential of such compartments in classical experiments designed to probe the clonal selection theory [10,11]: by isolating single lymph-node cells in emulsion droplet compartments, the secreted antibody was kept together with the cell producing it, thus providing genotype–phenotype linkage by compartmentalization and permitting assays to test the characteristics of each secreted protein. These groundbreaking studies provided evidence for the ‘one cell-one antibody’ rule [12]. Already at the time, Lederberg suggested that such compartments would ‘find routine applications in any laboratory’, which now, half a century later, is starting to become reality.
The potential of emulsion compartments for molecular evolution was first explored by Tawfik & Griffiths [13]. To obtain ‘monoclonal’ compartments (in which one gene and the corresponding protein encoded by it are unambiguously linked), a gene library is diluted so that each droplet contains no more than one member. Encapsulation of particles and molecules into droplets follows a Poisson distribution. In order to obtain mainly monoclonal compartments, most of the droplets are left empty. For example, a suspension containing on average 0.3 entities (DNA molecules or cells) per droplet results in 74%, 22% and 3% of the droplets containing none, one or two entities, respectively. The compartmentalization makes very large numbers of experiments possible in highly parallelized fashion, and also reduces the cost per assay dramatically (by approx. 106-fold [14]), as the assay volume is reduced to the femto- to picolitre scale through use of microdroplets.
Such water-in-oil emulsion compartments can be made in a number of ways:
— by dispersing an aqueous solution in an oil phase, which produces approximately 109 polydisperse droplets (diameter 1–4 µm) in one experiment—which is simply accomplished with an emulsifier or stirrer—taking only a few minutes [15–18], or
— in a microfluidic droplet generator by break-off from an aqueous stream, in which approximately 107 monodisperse compartments with identical size (typically 10–200 µm, adjustable as a function of the device design and flow rates) are produced per hour [15,19,20].
2. Protein display systems generated in compartments
High-affinity protein binders with defined specificity have become indispensable reagents in basic research, large-scale proteomic studies, and also in therapy, where they represent the fastest growing segment of the pharmaceutical market. The need for protein binders is addressed by display technologies [21,22]. For example, in phage display the protein of interest (POI) is fused to a coat protein, e.g. via the N-terminus of the minor (pIII) or major (pVIII) capsid proteins (figure 2) [24–26]. Protein expression occurs in vivo, but subsequent selections are carried out in vitro. It would be desirable to carry out the entire expression and selection process under in vitro conditions and generate a robust and stable display construct. The benefits of a cell-free format have been demonstrated by comparisons of affinity and diversity of binders generated in display formats that involve a host versus in vitro systems [27,28].
Figure 2.
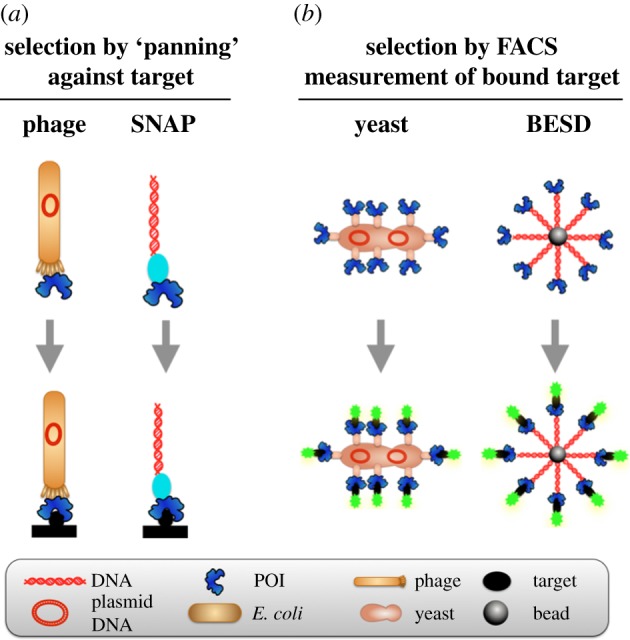
Comparison of natural and artificial display systems distinguished by the method of selection [23]. (a) A functional molecule (i.e. a protein of interest, POI) is connected to a gene encoding it. Selection from a library is based on binding to an immobilized target molecule: if binding molecules can be pulled out from a library based on their affinity by such ‘panning’, the attachment of the coding gene means that the selected clones can be sequenced. In this process, quantitative and direct control of ligand-binding parameters are not possible. Further, labour-intensive measurements are often necessary to assess the strength and specificity of affinity-selected binders. The natural phage display system is contrasted to in vitro SNAP display. (b) Yeast display provides multiple copies of the POI, as does its in vitro equivalent BeSD. Flow cytometry (FACS) measures the number of fluorescently labelled target molecules bound to the display construct and thus screens every mutant in the library, allowing a quantitative threshold to be set as the basis for a considered choice during selection.
In the SNAP display [13,29,30], a display construct is assembled with the help of an in vitro compartment (figure 3a). A link between the POI and DNA is brought about by compartmentalizing a single DNA molecule in each water-in-oil emulsion microdroplet, expressing the POI in vitro and retaining both together by the microdroplet boundary. The corresponding protein is expressed as a fusion with a protein tag that reacts covalently with a label on its coding DNA (a benzylguanine (BG) [34] coupled to DNA) and the droplet compartment keeps gene and cognate protein together (assuring monoclonality). Inspired by the linkage of DNA and POI on a phage, the bare-bones SNAP-display is a reductionist model of the natural phage display system.
Figure 3.

Formats for artificial covalent genotype–phenotype linkages based on droplet compartmentalization. The key initial step of both display methods is that a DNA library (coding for SNAP-tag-fused variants of the POI) is compartmentalized in water-in-oil emulsion droplets, so that each compartment contains no more than one DNA template (Poisson distribution). (a) In SNAP display [29–33], the POI is in vitro expressed from a single gene in fusion to the SNAP-tag (1). The SNAP-tag of the expressed fusion protein then reacts with its substrate, BG, that has been covalently linked to the DNA template. As a result, the SNAP-tag connects genotype and the displayed protein (responsible for the phenotype). (2) SNAP-tagged proteins are de-emulsified and challenged for binding against an antigen by affinity panning. (3) After non-binders are washed away, binders are eluted together with their encoding genes that can feed the next round of selection. (b) In BeSD display [23], the DNA is amplified by ePCR (using appropriate labelled primers), captured on the beads via a biotin–streptavidin linkage and the POI is in vitro expressed. After the emulsion is broken, beads are incubated with the labelled target and the affinity for the target is measured via fluorescence-activated sorting (FACS). The binding affinity of each recovered variant can be measured by subsequent FACS analysis on the bead display construct. The bead connects genotype and the megavalently displayed protein (responsible for the phenotype).
Selections are performed by ‘panning’ under in vitro conditions: the display construct is passed over immobilized target molecules and the binders stick (with their DNA attached—and can thus be decoded). These selections are based on off-rates (koff) and highly dependent on the conditions employed (e.g. the duration and number of washes in the panning procedure). Variants are recovered if their affinity is above a pre-set threshold, but this threshold is not necessarily precisely defined (i.e. a function of the experimental protocol and the operator's handling).
In a further variation of SNAP display, the ‘panning’ step is replaced with more quantitative, direct readouts of a binding constant (KD). When display constructs contain a larger number of proteins—e.g. approximately 104 copies displayed on bacteria [35–40] or 30 000 copies on yeast [41]—selections can be based on the measurement of the number of bound target molecules (counted by quantification of an optical label for every clone): flow cytometry is employed to rank and sort binders. Variation of the concentration of a fluorescent ligand incubated with the display construct and measurement of the extent to which it sticks, determines selection pressure akin to Kd titrations. This ranking gives access to populations of weaker and stronger binders depending on the chosen fluorescence threshold in flow cytometry (figures 2b and 3b).
Inspired by yeast and bacterial display, a megavalent variation of SNAP display (dubbed BeSD, bead surface display) provides an in vitro equivalent to the multivalent natural display systems (figure 2b) [23]. Again, single genes are compartmentalized in emulsion droplets—but now amplification is performed in the droplet compartment and up to a million copies of DNA and protein are assembled on a bead in a multi-step procedure. The compartment is responsible for keeping the cognate gene and POI together and the resulting construct reminding us of yeast display, but bears more protein copies and is completely generated in vitro. Libraries of such constructs can now be analysed by flow cytometry and binders identified at a throughput of approximately 107 per hour.
Both SNAP methods avoid shortcomings of in vivo display systems, e.g. low transformation efficiency, toxicity of the displayed protein to the host or lack of display construct stability.
3. Selections for enzyme catalysts in compartments
Instead of providing a template for the genotype–phenotype linkage that is later used for selection, the droplet compartment can also be maintained until selection, which makes it eminently suitable for selections of enzyme catalysts. Figure 4 shows how a substrate is co-compartmentalized with the protein catalyst in a droplet, multiple turnovers occur: now selections can be carried out based on product detection. To make product detection as precise as possible, microdroplets are prepared in monodisperse form in microfluidic devices (made, for example, conveniently by soft lithography from polydimethylsiloxane [15,58,59]) and interfaced with analytical systems. Figure 5 shows building blocks of integrated microfluidic devices that have recently been built. Many steps that are normally carried out in manual laboratory routines by pipetting are now automated in ‘lab-on-a-chip’ devices that process the bioinspired cell-like droplets on-chip on an assembly line at ultra-high throughput. In addition to droplet formation, the microfluidic format allows a number of other unit operations that are summarized in figure 4. Droplets are formed at rates well above 1 kHz [52,60] and can then be divided [44], fused [45–50], incubated [48,51], analysed [52–55], sorted [14,56,57] and broken up. An attractive feature of the microfluidic droplet platform is its modularity, where individual elements of a workflow correspond to experimental steps that are represented as jigsaw pieces [43]. Each piece of the jigsaw represents a unit operation and their integration translates a macroscopic workflow to the miniaturized scale within a microfluidic device. Integration of these steps with control over timing can potentially create a versatile system for directed evolution in which complex selection schemes can be realized.
Figure 4.

A workflow for directed evolution of a hydrolase by lysate screening in droplets [42]. (a) Unit operations from the ‘toolbox’ (figure 5) are assembled to miniaturize the steps necessary for single-cell assays of library members for directed evolution. (b) Workflow: (1) the protein of interest (POI), in this case an enzyme, is expressed in E. coli; (2) single cells are compartmentalized, together with substrate and cell lysis agents; (3) droplets are incubated to generate fluorescent product and (4) re-injected into a sorting device, where hits are detected by laser-induced fluorescence and steered into the upper channel by a variable electric field; (5) plasmid DNA from selected droplets is electroporated into E. coli. Repetition of such cycles increases the stringency of selection and enriches hits gradually to identify improved enzyme variants.
Figure 5.

Unit operations for generating and handling in vitro compartments [43]. In addition to droplet formation (a), the microfluidic format allows a number of other unit operations. Droplets can be divided [44], fused [45–50], incubated [48,51], analysed [52–55], sorted [14,56,57] and broken up. An attractive feature of this approach is its modularity, where individual elements of a workflow correspond to experimental steps that are represented as jigsaw pieces. Each piece of the jigsaw represents a unit operation and their integration translates a macroscopic workflow to the miniaturized scale within a microfluidic device.
Much recent work has been devoted to meeting the challenge of integration of the physical droplet processing steps with standard biological operations that may later be part of an integrated workflow for directed evolution. First, compartmentalization of cells is possible: single bacteria or yeast cells can be cultivated in droplets and recovered alive [61]. Second, in vitro protein expression from a single template (with up to 30 000 protein molecules expressed per DNA molecule in a droplet) has been demonstrated [51]. Kinetic parameters for several enzymes were also determined in microfluidic droplets, providing the facility to evaluate individual mutants kinetically [53,54].
An entire workflow to miniaturize rounds of directed evolution is shown in figure 6: in a single-cell lysate protocol [42], single cells (each cell representing one library member) were compartmentalized with lysis reagents and substrate, so that after cell rupture compartmentalized enzymatic reactions catalysed by the protein produced by a single cell can be monitored, and subsequently sorted. Catalysts can be incubated in a delay line (with several point measurements) [42] or—for slow reactions—after offline storage for several days [62]. This procedure was exemplified by the successful evolution of a promiscuous hydrolase [42] in two rounds of genetic diversification and selection, which led to improved expression and activity by an order of magnitude each. The genotype–phenotype linkage provided by the droplet boundary was maintained until de-emulsification after selection.
Figure 6.

GSBs for directed enzyme evolution [3]. (a) Assembly of GSBs: monodisperse water-in-oil emulsion droplets are produced with a microfluidic emulsion generator. The aqueous solution from which droplets are derived contains agarose and the polyanion alginate (red zig-zag line) that gelates upon cooling (from 37°C to 4°C), so that a solid bead is formed within the droplet template. The droplet boundary is removed by breaking the emulsion in the presence of the polycation poly(allylamine hydrochloride) (PAH; blue zig-zag line). Upon spontaneous encounter of the anionic alginate and the cationic PAH at the surface of the agarose template, a polyelectrolyte complex forms that maintains compartmentalization and substitutes the oil/water interface of the former emulsion droplet with a semipermeable surface layer, functionally resembling a semipermeable cell membrane. (b) Workflow of directed evolution in GSBs: (1) the protein-of-interest (POI), in this case an enzyme, is expressed in E. coli cells prior to droplet formation. (2) Single cells are then encapsulated into monodisperse water-in-oil emulsion droplets produced by a microfluidic emulsion generator (with Poisson distribution governing the occupancy). (3) Within the droplet, cells are lysed to allow encounter with reagents and substrate: now the enzymatic reaction can take place during an incubation period. (4) The gel-droplet emulsion is broken, or de-emulsified, in the presence of PAH to form the semipermeable polyelectrolyte shell layer, (5) GSBs harbouring active enzyme are distinguished from those with inactive enzyme variants by their fluorescence (and can be selected by flow cytometry, FACS). (6) The GSBs are broken up for genotype recovery.
4. Compartments turned to gel-shell beads: materials with evolvable components
Natural cells can be considered as incredible examples of functional materials, because their architecture equips them with functions of everyday survival, such as converting foodstuffs to energy, sensing or movement. In addition, they are vehicles for Darwinian evolution, providing for long-term development of an organism (or its components) by selection. By contrast, materials or devices are typically designed as such, but no mechanism for adaptation is built in. While future generations of cells will invariably evolve (e.g. in response to an environmental challenge), man-made materials will be limited to the original design: improvements are possible, but the designer has to intervene to specifically improve its properties.
In an attempt to turn droplet compartments into composite materials, we devised gel-shell beads (GSBs) that resemble minimalist versions of a natural cell [3]: a shell surrounds its interior, where functional molecules and their code (DNA) are lodged. To this end, microfluidic devices were used to produce large numbers of cell-sized droplets, which contain agarose that forms a stable structure (similar to the cytoskeleton): upon lowering the temperature, additional ingredients—agarose and alginate—solidify creating agarose microspheres (Ø ∼ 25 µm) in droplets and ‘immortalize’ the monoclonal nature of the original droplet. Addition of a functional polyelectrolyte shell with selective permeability (like the semipermeable cell membrane) completes the synthesis of biomimetic compartments. The shell—created by layer-by-layer technology [63–65]—is capable of selective retention (with permeability only for molecules less than 2 kDa) that co-compartmentalizes genotype and phenotype, thereby keeping the coding DNA, the enzyme and its (fluorescent) reaction product together. Most importantly, the beads are robust and can be easily screened/sorted using standard flow cytometry, a feature that sets GSBs apart from any existing high-throughput screening system currently available.
As above, single bacteria were encapsulated with substrate in microdroplets and lysed to liberate the POI and successful selections of a bioremediation catalyst, a phosphotriesterase, were carried out [3].
GSBs can be seen as containers for biocatalysts for cost-effective and sustainable applications that require easy recovery and repeated use of enzymes [66]. The stable catalyst cage of a GSB can contain single proteins, but may also encapsulate multiple components, e.g. sequential enzyme cascades or tandem reactions [67–71], enzymatic pathways [72–74] or synthetic gene circuits [75], that can be evolved directly in this format.
As the GSBs maintain the elements necessary for the directed evolution of an encapsulated protein, evolvability is programmed into these constructs (GSBs). The evolved catalyst is caged in GSBs, where it remains catalytically active and able to turn over multiple substrate molecules that enter the cage from the outside. After it has done its job, the caged catalyst can be removed, stored (e.g. by freezing) and used again.
In the composite GSBs, the functional components are DNA encoded, so by evolving a caged enzyme, evolvability of a ‘composite material’ (= a functional enzyme in a scaffold) is demonstrated: each composite carries the functional component (the enzyme) together with information that defines the identity of its functional component (DNA). This type of evolvability is key to diving further into ‘functional diversity space’. Where combinatorial approaches exist in materials science, libraries are usually smaller than screened here (almost a million members). The combination of the ability to decode a single species, extreme miniaturization (to pl droplets) and extremely straightforward screening/sorting in a commercial flow cytometer, provides the basis for easy access to molecular diversity, increasing the chances of success and setting the scene for more ambitious searches for novel functional materials.
5. Conclusion
The idea of cell compartmentalization is inspiring a range of practical approaches aimed at making new, functional molecules by Darwinian evolution. The extension of evolutionary principles that are enabled by the compartmentalization in its various guises has the potential to shape our material world as much as evolution has shaped Nature, with the only difference that it is up to us to decide for which purpose bioinspired parts and devices should be used.
Funding statement
This research described in this perspective was funded by the Biological and Biotechnological Research Council (BBSRC) and the Engineering and Physical Sciences Research Council (EPSRC) and the Human Frontier Science Programme. P.Y.C. holds a fellowship of the EU ITN PhosChemRec. F.H. was an ERC Starting Investigator.
References
- 1.Schaerli Y, Hollfelder F. 2009. The potential of microfluidic water-in-oil droplets in experimental biology. Mol. Biosyst. 5, 1392–1404. ( 10.1039/b907578j) [DOI] [PubMed] [Google Scholar]
- 2.Theberge AB, Courtois F, Schaerli Y, Fischlechner M, Abell C, Hollfelder F, Huck WT. 2010. Microdroplets in microfluidics: an evolving platform for discoveries in chemistry and biology. Angew. Chem. Int. Ed. Engl. 49, 5846–5868. ( 10.1002/anie.200906653) [DOI] [PubMed] [Google Scholar]
- 3.Fischlechner M, Schaerli Y, Mohamed MF, Patil S, Abell C, Hollfelder F. 2014. Evolution of enzyme catalysts caged in biomimetic gel-shed beads. Nat. Chem. 6, 791–796. ( 10.1038/nchem.1996) [DOI] [PubMed] [Google Scholar]
- 4.Crick FHC. 1966. Of molecules and men. Seattle, WA: University of Washington Press Cited after Van Regenmortel MHV. 2004 Reductionism and complexity in molecular biology. EMBO Reports 5, 1016–1020 ( 10.1038/sj.embor.7400284) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker D. 2014. Protein folding, structure prediction and design. Biochem. Soc. Trans. 42, 225–229. ( 10.1042/BST20130055) [DOI] [PubMed] [Google Scholar]
- 6.Hilvert D. 2013. Design of protein catalysts. Annu. Rev. Biochem. 82, 447–470. ( 10.1146/annurev-biochem-072611-101825) [DOI] [PubMed] [Google Scholar]
- 7.Khersonsky O, Kiss G, Rothlisberger D, Dym O, Albeck S, Houk KN, Baker D, Tawfik DS. 2012. Bridging the gaps in design methodologies by evolutionary optimization of the stability and proficiency of designed Kemp eliminase KE59. Proc. Natl Acad. Sci. USA 109, 10 358–10 363. ( 10.1073/pnas.1121063109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khersonsky O, Rothlisberger D, Dym O, Albeck S, Jackson CJ, Baker D, Tawfik DS. 2010. Evolutionary optimization of computationally designed enzymes: Kemp eliminases of the KE07 series. J. Mol. Biol. 396, 1025–1042. ( 10.1016/j.jmb.2009.12.031) [DOI] [PubMed] [Google Scholar]
- 9.Blomberg R, Kries H, Pinkas DM, Mittl PR, Grutter MG, Privett HK, Mayo SL, Hilvert D. 2013. Precision is essential for efficient catalysis in an evolved Kemp eliminase. Nature 503, 418–421. ( 10.1038/nature12623) [DOI] [PubMed] [Google Scholar]
- 10.Lederberg J. 1954. A simple method for isolating individual microbes. J. Bacteriol. 68, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nossal GJ, Lederberg J. 1958. Antibody production by single cells. Nature 181, 1419–1420. ( 10.1038/1811419a0) [DOI] [PubMed] [Google Scholar]
- 12.Viret C, Gurr W. 2009. The origin of the ‘one cell-one antibody’ rule. J. Immunol. 182, 1229–1230. ( 10.4049/jimmunol.182.3.1229) [DOI] [PubMed] [Google Scholar]
- 13.Tawfik DS, Griffiths AD. 1998. Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 16, 652–656. ( 10.1038/nbt0798-652) [DOI] [PubMed] [Google Scholar]
- 14.Agresti JJ, et al. 2010. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc. Natl Acad. Sci. USA 107, 4004–4009. ( 10.1073/pnas.0910781107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Devenish SRA, Kaltenbach M, Fischlechner M, Hollfelder F. 2013. Droplets as reaction compartments for protein nanotechnology in protein nanotechnology: protocols, instrumentation and applications. J. Gerrard. Ed. 996, 269–286. [DOI] [PubMed] [Google Scholar]
- 16.Kaltenbach M, Hollfelder F. 2011. SNAP display: in vitro protein evolution in microdroplets. Methods Mol. Biol. 805, 101–111. ( 10.1007/978-1-61779-379-0_7) [DOI] [PubMed] [Google Scholar]
- 17.Ghadessy FJ, Holliger P. 2007. Compartmentalized self-replication: a novel method for the directed evolution of polymerases and other enzymes. Methods Mol. Biol. 352, 237–248. [DOI] [PubMed] [Google Scholar]
- 18.Miller OJ, et al. 2006. Directed evolution by in vitro compartmentalization. Nat. Methods 3, 561–570. ( 10.1038/nmeth897) [DOI] [PubMed] [Google Scholar]
- 19.Umbanhowar PB, Prasad V, Weitz DA. 2000. Monodisperse Emulsion Generation via Drop Break Off in a Coflowing Stream. Langmuir 16, 347–351. ( 10.1021/la990101e) [DOI] [Google Scholar]
- 20.Huebner A, Sharma S, Srisa-Art M, Hollfelder F, Edel JB, Demello AJ. 2008. Microdroplets: a sea of applications? Lab Chip 8, 1244–1254. ( 10.1039/b806405a) [DOI] [PubMed] [Google Scholar]
- 21.Douthwaite JA, Jackson RH. 2012. Ribosome display and related technologies: methods and protocols (Methods Mol. Biol.), p. 424 New York, NY: Springer. [Google Scholar]
- 22.Leemhuis H, Stein V, Griffiths AD, Hollfelder F. 2005. New genotype-phenotype linkages for directed evolution of functional proteins. Curr. Opin. Struct. Biol. 15, 472–478. ( 10.1016/j.sbi.2005.07.006) [DOI] [PubMed] [Google Scholar]
- 23.Diamante L, Gatti-Lafranconi P, Schaerli Y, Hollfelder F. 2013. In vitro affinity screening of protein and peptide binders by megavalent bead surface display. Protein Eng. Des. Sel. 26, 713–724. ( 10.1093/protein/gzt039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willats WG. 2002. Phage display: practicalities and prospects. Plant Mol. Biol. 50, 837–854. ( 10.1023/A:1021215516430) [DOI] [PubMed] [Google Scholar]
- 25.Sidhu SS. 2005. Phage display in biotechnology and drug discovery. Boca Raton, FL: CRC Press. [Google Scholar]
- 26.Paschke M. 2006. Phage display systems and their applications. Appl. Microbiol. Biotechnol. 70, 2–11. ( 10.1007/S00253-005-0270-9) [DOI] [PubMed] [Google Scholar]
- 27.Groves M, Lane S, Douthwaite J, Lowne D, Rees DG, Edwards B, Jackson RH. 2006. Affinity maturation of phage display antibody populations using ribosome display. J. Immunol. Methods 313, 129–139. ( 10.1016/j.jim.2006.04.002) [DOI] [PubMed] [Google Scholar]
- 28.Thom G, et al. 2006. Probing a protein–protein interaction by in vitro evolution. Proc. Natl Acad. Sci. USA 103, 7619–7624. ( 10.1073/pnas.0602341103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stein V, Sielaff I, Johnsson K, Hollfelder F. 2007. A covalent chemical genotype–phenotype linkage for in vitro protein evolution. Chembiochem 8, 2191–2194. ( 10.1002/cbic.200700459) [DOI] [PubMed] [Google Scholar]
- 30.Kaltenbach M, Stein V, Hollfelder F. 2011. SNAP dendrimers: multivalent protein display on dendrimer-like DNA for directed evolution. Chembiochem 12, 2208–2216. ( 10.1002/cbic.201100240) [DOI] [PubMed] [Google Scholar]
- 31.Houlihan G, Gatti-Lafranconi P, Kaltenbach M, Lowe D, Hollfelder F. 2014. An experimental framework for improved selection of binding proteins using SNAP display. J. Immunol. Methods 405, 47–56. ( 10.1016/j.jim.2014.01.006) [DOI] [PubMed] [Google Scholar]
- 32.Houlihan G, Lowe D, Hollfelder F. 2013. SNAP display—an in vitro method for the selection of protein binders. Curr. Pharm. Des. 19, 5421–5428. ( 10.2174/1381612811319300012) [DOI] [PubMed] [Google Scholar]
- 33.Kaltenbach M, Hollfelder F. 2011. SNAP display: in vitro protein evolution in microdroplets. In Ribosome display and related technologies: methods and protocols (methods in molecular biology) (eds Jackson RH, Douthwaite JA.), pp. 101–111. London, UK: Humana Press (Springer Protocols). [DOI] [PubMed] [Google Scholar]
- 34.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. 2003. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21, 86–89. ( 10.1038/nbt765) [DOI] [PubMed] [Google Scholar]
- 35.Chen G, Cloud J, Georgiou G, Iverson BL. 1996. A quantitative immunoassay utilizing escherichia coli cells possessing surface expressed single chain Fv molecules. Biotechnol. Prog. 12, 572–574. ( 10.1021/bp960041s) [DOI] [PubMed] [Google Scholar]
- 36.Löfblom J, Sandberg J, Wernérus H, Ståhl S. 2007. Evaluation of staphylococcal cell surface display and flow cytometry for postselectional characterization of affinity proteins in combinatorial protein engineering applications. Appl. Environ. Microbiol. 73, 6714–6721. ( 10.1128/AEM.01432-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Löfblom J, Wernérus H, Ståhl S. 2005. Fine affinity discrimination by normalized fluorescence activated cell sorting in staphylococcal surface display. FEMS Microbiol. Lett. 248, 189–198. ( 10.1016/j.femsle.2005.05.040) [DOI] [PubMed] [Google Scholar]
- 38.Andreoni C, Goetsch L, Libon C, Samuelson P, Nguyen TN, Robert A, Uhlén M, Binz H, Ståhl S. 1997. Flow cytometric quantification of surface-displayed recombinant receptors on staphylococci. BioTechniques 23, 696. [PubMed] [Google Scholar]
- 39.Rockberg J, Lofblom J, Hjelm B, Uhlen M, Ståhl S. 2008. Epitope mapping of antibodies using bacterial surface display. Nat. Methods 5, 1039–1045. ( 10.1038/nmeth.1272) [DOI] [PubMed] [Google Scholar]
- 40.Christmann A, Wentzel A, Meyer C, Meyers G, Kolmar H. 2001. Epitope mapping and affinity purification of monospecific antibodies by Escherichia coli cell surface display of gene-derived random peptide libraries. J. Immunol. Methods 257, 163–173. ( 10.1016/S0022-1759(01)00461-6) [DOI] [PubMed] [Google Scholar]
- 41.Boder ET, Wittrup KD. 1997. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557. ( 10.1038/nbt0697-553) [DOI] [PubMed] [Google Scholar]
- 42.Kintses B, Hein C, Mohamed MF, Fischlechner M, Courtois F, Laine C, Hollfelder F. 2012. Picoliter cell lysate assays in microfluidic droplet compartments for directed enzyme evolution. Chem. Biol. 19, 1001–1009. ( 10.1016/j.chembiol.2012.06.009) [DOI] [PubMed] [Google Scholar]
- 43.Kintses B, van Vliet LD, Devenish SR, Hollfelder F. 2010. Microfluidic droplets: new integrated workflows for biological experiments. Curr. Opin. Chem. Biol. 14, 548–555. ( 10.1016/j.cbpa.2010.08.013) [DOI] [PubMed] [Google Scholar]
- 44.Link DR, Anna SL, Weitz DA, Stone HA. 2004. Geometrically mediated breakup of drops in microfluidic devices. Phys. Rev. Lett. 92, 054503 ( 10.1103/PhysRevLett.92.054503) [DOI] [PubMed] [Google Scholar]
- 45.Niu X, Gulati S, Edel JB, deMello AJ. 2008. Pillar-induced droplet merging in microfluidic circuits. Lab Chip 8, 1837–1841. ( 10.1039/b813325e) [DOI] [PubMed] [Google Scholar]
- 46.Niu X, Gielen F, deMello AJ, Edel JB. 2009. Electro-coalescence of digitally controlled droplets. Anal. Chem. 81, 7321–7325. ( 10.1021/ac901188n) [DOI] [PubMed] [Google Scholar]
- 47.Mazutis L, et al. 2009. Droplet-based microfluidic systems for high-throughput single DNA molecule isothermal amplification and analysis. Anal. Chem. 81, 4813–4821. ( 10.1021/ac900403z) [DOI] [PubMed] [Google Scholar]
- 48.Mazutis L, Baret JC, Treacy P, Skhiri Y, Araghi AF, Ryckelynck M, Taly V, Griffiths AD. 2009. Multi-step microfluidic droplet processing: kinetic analysis of an in vitro translated enzyme. Lab Chip 9, 2902–2908. ( 10.1039/b907753g) [DOI] [PubMed] [Google Scholar]
- 49.Shim JU, Patil SN, Hodgkinson JT, Bowden SD, Spring DR, Welch M, Huck WT, Hollfelder F, Abell C. 2011. Controlling the contents of microdroplets by exploiting the permeability of PDMS. Lab Chip 11, 1132–1137. ( 10.1039/c0lc00615g) [DOI] [PubMed] [Google Scholar]
- 50.Brouzes E, et al. 2009. Droplet microfluidic technology for single-cell high-throughput screening. Proc. Natl Acad. Sci. USA 106, 14 195–14 200. ( 10.1073/pnas.0903542106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Courtois F, Olguin LF, Whyte G, Bratton D, Huck WT, Abell C, Hollfelder F. 2008. An integrated device for monitoring time-dependent in vitro expression from single genes in picolitre droplets. Chembiochem 9, 439–446. ( 10.1002/cbic.200700536) [DOI] [PubMed] [Google Scholar]
- 52.Huebner A, Srisa-Art M, Holt D, Abell C, Hollfelder F, deMello AJ, Edel JB. 2007. Quantitative detection of protein expression in single cells using droplet microfluidics. Chem. Commun. 2007, 1218–1220. ( 10.1039/b618570c) [DOI] [PubMed] [Google Scholar]
- 53.Huebner A, Olguin LF, Bratton D, Whyte G, Huck WT, de Mello AJ, Edel JB, Abell C, Hollfelder F. 2008. Development of quantitative cell-based enzyme assays in microdroplets. Anal. Chem. 80, 3890–3896. ( 10.1021/ac800338z) [DOI] [PubMed] [Google Scholar]
- 54.Huebner A, Bratton D, Whyte G, Yang M, deMello AJ, Abell C, Hollfelder F. 2009. Static microdroplet arrays: a microfluidic device for droplet trapping, incubation and release for enzymatic and cell-based assays. Lab Chip 9, 692–698. ( 10.1039/B813709a) [DOI] [PubMed] [Google Scholar]
- 55.Shim JU, Olguin LF, Whyte G, Scott D, Babtie A, Abell C, Huck WT, Hollfelder F. 2009. Simultaneous determination of gene expression and enzymatic activity in individual bacterial cells in microdroplet compartments. J. Am. Chem. Soc. 131, 15 251–15 256. ( 10.1021/ja904823z) [DOI] [PubMed] [Google Scholar]
- 56.Baret JC, et al. 2009. Fluorescence-activated droplet sorting (FADS): efficient microfluidic cell sorting based on enzymatic activity. Lab Chip 9, 1850–1858. ( 10.1039/b902504a) [DOI] [PubMed] [Google Scholar]
- 57.Fallah-Araghi A, Baret JC, Ryckelynck M, Griffiths AD. 2012. A completely in vitro ultrahigh-throughput droplet-based microfluidic screening system for protein engineering and directed evolution. Lab Chip 12, 882–891. ( 10.1039/c2lc21035e) [DOI] [PubMed] [Google Scholar]
- 58.Mazutis L, Gilbert J, Ung WL, Weitz DA, Griffiths AD, Heyman JA. 2013. Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 8, 870–891. ( 10.1038/nprot.2013.046) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xia Y, Whitesides GM. 1998. Soft lithography. Annu. Rev. Mater. Sci. 28, 153–184. ( 10.1146/annurev.matsci.28.1.153) [DOI] [Google Scholar]
- 60.Shim J, Ranasinghe RT, Smith CA, Ibrahim SM, Hollfelder F, Huck WT, Klenerman D, Abell C. 2013. Utrarapid generation of femtoliter microfluidic droplets for single-molecule-counting immunoassays. ACS Nano 7, 5955–5964. ( 10.1021/nn401661d) [DOI] [PubMed] [Google Scholar]
- 61.Clausell-Tormos J, et al. 2008. Droplet-based microfluidic platforms for the encapsulation and screening of Mammalian cells and multicellular organisms. Chem. Biol. 15, 427–437. ( 10.1016/j.chembiol.2008.04.004) [DOI] [PubMed] [Google Scholar]
- 62.Colin PY, Kintses B, Gielen F, Miton C, Fischer G, Mohamed M, Hyvonen M, Morgavi DP, Janssen DB, Hollfelder F. Submitted. Functional metagenomics in picoliter droplets for ultrahigh-throughput enzyme discovery. [DOI] [PMC free article] [PubMed]
- 63.Peyratout CS, Dahne L. 2004. Tailor-made polyelectrolyte microcapsules: From multilayers to smart containers. Angew. Chem. Int Ed. 43, 3762–3783. ( 10.1002/Anie.200300568) [DOI] [PubMed] [Google Scholar]
- 64.Tong WJ, Song XX, Gao CY. 2012. Layer-by-layer assembly of microcapsules and their biomedical applications. Chem. Soc. Rev. 41, 6103–6124. ( 10.1039/C2cs35088b) [DOI] [PubMed] [Google Scholar]
- 65.Skirtach AG, Yashchenok AM, Mohwald H. 2011. Encapsulation, release and applications of LbL polyelectrolyte multilayer capsules. Chem. Commun. 47, 12 736–12 746. ( 10.1039/c1cc13453a) [DOI] [PubMed] [Google Scholar]
- 66.Dicosimo R, McAuliffe J, Poulose AJ, Bohlmann G. 2013. Industrial use of immobilized enzymes. Chem. Soc. Rev. 42, 6437–6474. ( 10.1039/c3cs35506c) [DOI] [PubMed] [Google Scholar]
- 67.Schoffelen S, van Hest JC. 2013. Chemical approaches for the construction of multi-enzyme reaction systems. Curr. Opin. Struct. Biol. 23, 613–621. ( 10.1016/j.sbi.2013.06.010) [DOI] [PubMed] [Google Scholar]
- 68.Liu Y, et al. 2013. Biomimetic enzyme nanocomplexes and their use as antidotes and preventive measures for alcohol intoxication. Nat. Nanotechnol. 8, 187–192. ( 10.1038/nnano.2012.264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oroz-Guinea I, Garcia-Junceda E. 2013. Enzyme catalysed tandem reactions. Curr. Opin. Chem. Biol. 17, 236–249. ( 10.1016/j.cbpa.2013.02.015) [DOI] [PubMed] [Google Scholar]
- 70.Garcia-Junceda E. 2008. Multi-step enzyme catalysis: biotransformations and chemoenzymatic synthesis, p. 256 Weinheim, Germany: VCH. [Google Scholar]
- 71.Ricca E, Brucher B, Schrittwieser JH. 2011. Multi-enzymatic cascade reactions: overview and perspectives. Adv. Synth. Catal. 353, 2239–2262. ( 10.1002/Adsc.201100256) [DOI] [Google Scholar]
- 72.Zhang Y-HP. 2011. Simpler is better: high-yield and potential low-cost biofuels production through cell-free synthetic pathway biotransformation (SyPaB). ACS Catal. 1, 998–1009. ( 10.1021/cs200218f) [DOI] [Google Scholar]
- 73.Ro DK, et al. 2006. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440, 940–943. ( 10.1038/nature04640) [DOI] [PubMed] [Google Scholar]
- 74.Pirie CM, De Mey M, Jones Prather KL, Ajikumar PK. 2013. Integrating the protein and metabolic engineering toolkits for next-generation chemical biosynthesis. ACS Chem. Biol. 8, 662–672. ( 10.1021/cb300634b) [DOI] [PubMed] [Google Scholar]
- 75.Zhao H. 2013. Synthetic biology. New York, NY: Academic Press. [Google Scholar]