

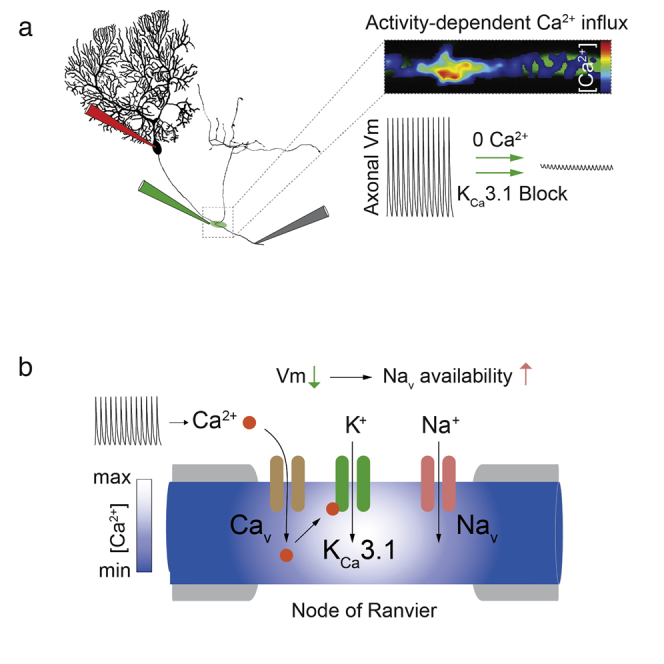
Summary
Functional connectivity between brain regions relies on long-range signaling by myelinated axons. This is secured by saltatory action potential propagation that depends fundamentally on sodium channel availability at nodes of Ranvier. Although various potassium channel types have been anatomically localized to myelinated axons in the brain, direct evidence for their functional recruitment in maintaining node excitability is scarce. Cerebellar Purkinje cells provide continuous input to their targets in the cerebellar nuclei, reliably transmitting axonal spikes over a wide range of rates, requiring a constantly available pool of nodal sodium channels. We show that the recruitment of calcium-activated potassium channels (IK, KCa3.1) by local, activity-dependent calcium (Ca2+) influx at nodes of Ranvier via a T-type voltage-gated Ca2+ current provides a powerful mechanism that likely opposes depolarizing block at the nodes and is thus pivotal to securing continuous axonal spike propagation in spontaneously firing Purkinje cells.
Graphical Abstract
Highlights
-
•
Activity-dependent node of Ranvier Ca2+ influx in Purkinje cell axons
-
•
Cav and KCa3.1 channels required for axonal spike propagation
-
•
Nodal KCa3.1 channels provide repolarizing drive to sustain axonal spike propagation
Functional connectivity between brain regions relies on long-range signaling by myelinated axons. Gründemann and Clark show that local, activity-dependent calcium influx at nodes of Ranvier recruits calcium-activated potassium channels (KCa3.1) that drive repolarization and sustain node excitability, providing a pivotal mechanism to secure spike propagation along Purkinje cell axons.
Introduction
Understanding information transmission within neuronal circuits, and the factors underlying long-range axonal signaling malfunction, relies on identifying the axonal ion channels that are key regulators of node of Ranvier (NoR) excitability. NoRs are highly specialized zones in myelinated axons containing high densities of voltage-gated sodium channels (Navs) and associated cytoskeletal complexes, creating active zones essential for saltatory conduction of action potentials (APs) (Debanne et al., 2011). The repolarizing currents required to sustain Nav availability at NoRs for reliable AP transmission is less clear cut, particularly in the mammalian brain. Both low- and high-voltage-activated potassium (K+) channel subunits (Kv7 and Kv3.1/3) have been anatomically localized to NoRs (Devaux et al., 2003, 2004; Pan et al., 2006), and although their presence can vary between brain regions and axon types (Debanne et al., 2011; Devaux et al., 2003, 2004), recent experiments provide direct evidence that Kv7 channels stabilize NoR membrane potential (Vm) in cortical L5 pyramidal cells (Battefeld et al., 2014) consistent with findings in peripheral nerve (Schwarz et al., 2006). The influence of delayed rectifier (DR) K+ channels (Kv1.1 and Kv1.2), which are widely observed in the juxtaparanodal (JP) zone (Devaux et al., 2003; Ogawa et al., 2010; Rasband, 2010; Zhou et al., 1998), has, however, been difficult to assess without demyelination (Röper and Schwarz, 1989; Wilson and Chiu, 1990), and whether local Vm becomes sufficiently depolarized to recruit JP Kv1 channels during saltatory conduction is disputed (Arancibia-Carcamo and Attwell, 2014). Voltage-gated Ca2+ channels (Cavs), which could provide an additional source of depolarization as well as gating Ca2+-dependent processes including recruitment of Ca2+-dependent K+ channels (KCa), have been described in central myelinated axons and shown to influence excitability at the axon initial segment (AIS) (Bender and Trussell, 2009; Bender et al., 2010; Yu et al., 2010). However, although Cavs have been proposed to influence NoR formation during development (Alix et al., 2008), their presence at mature NoRs in the brain is not established (Zhang et al., 2006).
Purkinje cells (PCs) in the cerebellum fire at high rates, both spontaneously and in response to synaptic input, and thus require fast recovery of sodium channels at their NORs. We have obtained direct evidence that, rather than solely relying on voltage-gated potassium channels, activity-dependent, spatially localized Ca2+ influx at NoRs of PC axons recruits an intermediate-type KCa (IK, or KCa3.1) to provide a node-specific repolarizing conductance crucial for axonal spike propagation.
Results
Potassium Channels at Nodes of Ranvier
Using simultaneous somatic and axonal patch-clamp recordings, local pharmacology, and two-photon Ca2+ imaging of cerebellar PC axons, we directly investigated which ion channels are engaged at NoRs during AP propagation. We visualized PC axons in cerebellar slices by dye filling via the somatic recording pipette and recorded axonal APs downstream of NORs (see the Experimental Procedures; Figure 1A) identified by virtue of their presence at axonal branchpoints (Clark et al., 2005). APs are securely transmitted by PC axons at high firing rates, with failures occurring above ≈250 Hz (Khaliq and Raman, 2005; Monsivais et al., 2005). This propagation reliability is retained across axonal branchpoints, with equal limiting frequency in both the main projection axon (257 ± 17 Hz; also Monsivais et al., 2005) and in recurrent axon collaterals (Figures 1B and 1C, 253 ± 14 Hz, 0.05% differentially propagated spikes, n = 6 cells, see also Foust et al., 2010). We used local application (Figure S2A) of various ion channel antagonists to test their impact on AP propagation at NoRs in spontaneously firing PCs (firing rates 20–80 Hz). TTX (10 μM) completely blocked AP propagation, confirming the presence of a NoR at branchpoints (Figure S1D; see also Khaliq and Raman, 2005). In contrast, application of TEA at a concentration (10 mM) that should block a wide variety of K+ channels including Kv1, Kv3, and Kv7 types (Grissmer et al., 1994; Hadley et al., 2000) did not affect AP propagation, having no impact on axonal capacitive current amplitude, firing rate, or conduction velocity (Figures S1B and S1C). This lack of effect was similar at high firing rates, and the limiting frequency for spike propagation was unchanged (Figure S1D). These results are unexpected given that TEA-sensitive K+-channels are thought to contribute to axonal AP repolarization (Devaux et al., 2003; Hille, 1967; Röper and Schwarz, 1989), to stabilize nodal Vm as well as preventing antidromic spike reflection (Goldstein and Rall, 1974), and Kv3.3 subunits have been localized to PC axons (Chang et al., 2007). Potential explanations for these results might be that first, TEA-sensitive K+ channels are absent from PC axons and/or their NoRs and repolarization is mediated by an alternative mechanism. Second, they may be present but at insufficient density to be primary regulators of node excitability. Third, TEA-sensitive K+ channels might be located behind tight myelin junctions in the juxtaparanodes (JPs) (Wang et al., 1993) and contribute to repolarization of the axonal membrane while being inaccessible to TEA, as observed in peripheral nerve (Chiu and Ritchie 1980; Kocsis and Waxman 1980) (however, see Mierzwa et al., 2010). Fourth, these channels may be present in the JPs but weakly activated by the limited Vm changes that are calculated to occur in healthy myelinated axon segments (Arancibia-Carcamo and Attwell, 2014).
Figure 1.

Highly Reliable Spike Propagation along Purkinje Cell Axons Is Secured by Node of Ranvier IK Channels
(A) Schematic of experimental configuration.
(B) Simultaneous whole-cell somatic and cell-attached axonal recordings of a spontaneously firing Purkinje cell in response to a somatic current ramp injection (2 nA). ∗Differentially propagated spike.
(C) Maximal firing frequencies at soma (292 ± 19 Hz, n = 19), main axon (257 ± 17 Hz, n = 9, includes data from Monsivais et al., 2005), and axon collateral (253 ± 14 Hz, n = 19).
(D) Local drug application (green bars) to branchpoint during whole-cell somatic recording (black) and cell-attached axonal recordings (blue) downstream of the targeted branchpoint. Baseline firing rate is indicated above somatic traces.
(E) Averaged whole-cell somatic and cell-attached spikes before (blue), during (green), and after (gray) drug application.
(F) Spike amplitude before (blue, a1) and during (green, a2) drug application.
(G) Data summary of axonal spike suppression.
p < 0.0001 for TRAM 1 μM, 0 Ca2+, Ni2+, p < 0.005 TRAM 500 nM, p < 0.002 TTX. TEA, HBS not significantly different from baseline (Student’s t test). Error bars, ±SEM.
Recently, an intermediate-type KCa conductance (IK, KCa3.1), not previously observed in the CNS and with a lower sensitivity to TEA (IC50 = 24 mM) (Wei et al., 2005), was described in cerebellar PCs and shown to contribute to postsynaptic integration (Engbers et al., 2012). We examined whether IK might provide an alternative mechanism for regulating excitability at NoRs and found that local application of the selective IK channel antagonist TRAM-34 (Engbers et al., 2012; Wulff et al., 2000) to axonal branchpoints led to a concentration-dependent block of axonal spike propagation during spontaneous firing (20–80 Hz; Figures 1D–1G), with no observed dependence on firing rate (R = 0.1). Clotrimazole (1 μM), another IK-selective blocker, had similar effects (Figure 1G), while apamin (1 μM) and iberiotoxin (1 μM), which block small (SK) and large (BK) KCa channels, respectively, had no impact (98% ± 0.44% [n = 5] and 97.3% ± 1.2% [n = 4] of control axonal spike amplitude). IK gating depends solely on intracellular [Ca2+] (Kd: 0.1–0.3 μM) (Wei et al., 2005; Joiner et al., 1997), and, since both local removal of extracellular Ca2+ and application of Ni2+ (100 μM) similarly suppressed action potential propagation (Figures 1D–1G), the activation of IK is likely initiated by Ca2+ influx via Cavs. We confirmed that PC axons are immunopositive for the KCa3.1 subunit, the sole molecular entity that constitutes the IK channel, but found, surprisingly, that no hotspots were seen at NoRs and that KCa3.1 labeling was instead relatively uniform along the axon (Figure 2).
Figure 2.

Expression of KCa3.1 in Purkinje Cell Axons
(A) Immunolabeling of KCa3.1 in cerebellar vermis (overview, left), PC soma, and dendrites (middle) and the center of the granule cell layer, which typically corresponds to the anatomical location where Purkinje cell axons have their first axonal branchpoint (70–100 μm) (right, arrowhead: potential axonal branchpoint).
(B) Co-immunolabeling of calbindin and KCa3.1 at PC somata and dendrites. Left, calbindin; Middle, KCa3.1; Right, merge. Maximum intensity projection.
(C) KCa3.1 staining along calbindin-positive axons in the granule cell layer. Maximum intensity projection.
(D) Calbindin-positive axonal branchpoint in the granule cell layer. Single optical plane, image deconvolved.
Activity-Dependent Ca2+ Influx at Nodes of Ranvier
Activity-dependent Ca2+ influx has been observed in optic nerve (Lev-Ram and Grinvald, 1987; Zhang et al., 2006), but the resulting Ca2+ transients are spatially uniform along both NoRs and internodes (Zhang et al., 2006), and the route of Ca2+ entry is uncertain. As yet, there is no direct evidence for Ca2+ influx localized to NoRs in the brain.
Using two-photon Ca2+-imaging, we investigated whether the spatial distribution of calcium signals in PC axons might confer NoR specificity of IK recruitment. We detected prominent activity-dependent increases in intracellular Ca2+ concentration ([Ca2+]i) at NoRs (identified by their location at branchpoints) and at the AIS (see Bender and Trussell, 2009) during trains of evoked APs (Figures 3A–3C; 204 ± 21 Hz). Ca2+ signals were restricted to approximately 5 μm from the center of NoRs (Figures 3C and 3D), and there were no detectable changes in internodal [Ca2+]i (Figure 3B), consistent with the lack of effect on spike propagation of 0 mM Ca2+, 10 mM BAPTA application to the internodes (Figure S2B). [Ca2+]i at NoRs required axonal APs, were suppressed when spontaneous firing was arrested by somatic hyperpolarization (Figure 3G), and could not be driven by somatic depolarization when spikes were blocked with bath-applied TTX (Figures 3E and 3F). Increases in [Ca2+]i were spike rate dependent (Figure S3), slow, and cumulative and lead to sustained [Ca2+]i during continuous activity (Figures 3G and 3H). [Ca2+]i increase was also suppressed by removal of extracellular Ca2+ (16% ± 6% of control n = 4; Figure 3I) and bath application of mibefradil (Figures 3I and 5 μM, 43% ± 14% of control, p = 0.013) but was not sensitive to agatoxin IVa, which blocked Ca2+ transients in PC synaptic boutons as expected (Hillman et al., 1991) (Figures 3I; 9% ± 3% of control, p < 0.0001). This implies that T-type and not P-type CaVs are the primary source of Ca2+ entry at NoRs. Additionally, although prior depolarization of the soma reduced [Ca2+]i at the AIS, AP-triggered nodal Ca2+ signals were unaffected (Figures S3C and S3D), demonstrating that, in PCs, nodal [Ca2+]i is independent of somatic Vm.
Figure 3.

Local, Activity-Dependent Ca2+ Influx at Nodes of Ranvier
(A) Two-photon image of cerebellar Purkinje cell indicating line scan locations.
(B) Ca2+ transients (top, line scans) at locations shown in (A) during current-evoked spike trains (bottom). AIS, axon initial segment. BP1 and BP2, first and second axonal branchpoint.
(C) Frame-scan time series of BP1 during spike train. Green, axon morphology.
(D) Spike train-evoked ΔF/F at ROIs shown in (C) (red boxes). Normalized integrated ΔF/F (ΔF/F∗s) against distance from an axonal branchpoint (bottom, n = 13 neurons).
(E) Soma, AIS, and first BP ΔF/F in response to 500-ms somatic voltage steps (voltage clamp) in bath-applied TTX (0.5 μM).
(F) Pooled data for max ΔF/F versus somatic command potential (soma, black; AIS, red; BP1, blue. n = 6 neurons).
(G) Activity-dependent changes in ΔF/F upon somatic current injection. Baseline holding current: 0 pA.
(H) Summary data for the change in ΔF/F∗s during silence and activity (n = 5 cells).
(I) Ca2+ influx at the axon initial segment, first branchpoint and presynaptic boutons before and after bath application of 0 mM extracellular Ca2+ (n = 4), Agatoxin (AgaTX, n = 4, AIS = 3), and Mibefradil (Mibef, n = 9).
Error bars, ±SEM.
KCa3.1/IK Current Is Sufficient to Sustain Node Excitability
To understand the major impact of IK block on axonal AP propagation, we used a multicompartmental model of a PC (Clark et al., 2005) and tested whether IK, as the sole repolarizing conductance at the NoRs, can support reliable propagation of APs in a continuously firing axon (see the Experimental Procedures for details). At a minimum IK density of 0.05 S/cm2, with an associated T-type CaV with a maximum permeability of 0.002 cm/s (Anwar et al., 2012) and an accompanying, low-density, juxtaparanodal DR K+ conductance (0.002 S/cm2, Clark et al., 2005) to mimic the Kv1.1/Kv1.2 channels commonly observed in immunohistochemical studies (Rasband 2010), AP propagation along the model axon was highly reliable. Complete removal of the IK conductance alone caused NoR depolarization, trapping of sodium channels in inactivated and blocked (Khaliq et al., 2003) states, and AP propagation block (Figure 4A). IK may therefore act to stabilize the NoR Vm during continuous firing and application of the IK channel inhibitor TRAM-34 (Figure 1) could cause depolarization block. Our observed lack of effect of TEA application to NoRs (Figure S1) implies that other K+ channel types previously shown to be present in PC axons (Kv3.1, Chang et al., 2007) and NoRs (Kv7.3, Pan et al., 2006) may not, in the absence of IK, be sufficient to support AP propagation. A direct test of the hypothesis that IK stabilizes nodal Vm requires quantitative measurements of axonal Vm during IK block. Since PC NoRs are too small for patch-clamp recording and voltage-sensitive dye methods cannot be used to detect absolute changes in Vm, we used the AIS as a proxy for NoRs while recording spontaneous firing at the soma, presuming that some IK might be present there, as suggested by our immunohistochemistry data (Figure 2). During TRAM-34 application to the AIS, we observed a reduction in somatic firing rate of 25% ± 5.2% (p < 0.001, n = 15 cells, Figure 4C), accompanied by a small somatic depolarization (1.5 ± 0.2 mV, p < 0.0001) and an increase in AP threshold (1.8 ± 0.16 mV, p < 0.0001). In a minority of cells (4 / 15), TRAM-34 application to the AIS caused sufficient depolarization (ΔVm = 2.56 ± 1.28 mV, mean Vm = −48.4 ± 1.9 mV) to reversibly but completely block firing in some trials. Reduction of firing rate correlated with initial firing rate (R = 0.53, p < 0.05) so that cells with higher baseline rates showed a smaller reduction in spike rate on TRAM-34 application. This may reflect the presence of rate-dependent recruitment of BK and SK currents shown to regulate PC somatic Vm during spontaneous firing (Raman and Bean, 1999). Analysis of the second derivative of the somatic AP revealed a reduction in the component driven by current flow from the AIS (Figure 4D) as well as the local somatic component of the action potential during TRAM-34 application, indicating a reduced recruitment of sodium current at both locations.
Figure 4.

KCa3.1 Sets Node of Ranvier Membrane Potential and Preserves Nodal Excitability
(A) Data obtained from a PC multicompartmental model. NoR (red, sixth node, 1,820 μm from soma) and somatic (gray) APs (Vm, top row) during spontaneous firing in a Purkinje cell model with and without NoR KCa3.1 channels. Left column: AP propagation is sustained by nodal gKCa3.1 (0.05 S/cm2) and juxtaparanodal (JP) delayed rectifier K+ channels (0.002 S/cm2). Right column: lack of axonal KCa3.1 causes depolarization block at the NoR. Second row: Ca2+ current at the NoR. Third row: total K+ current at the JP and NoR, respectively, for each model. Fourth row: fractional resurgent Nav states during spontaneous firing of each respective model. Bottom row: local Ca2+ concentration.
(B) Activity-dependent axonal Ca2+ diffusion in the model axon.
(C) Somatic recording from a spontaneously firing Purkinje cell during local application of 1 μM TRAM-34 to the AIS. TRAM-34 causes a depolarizing shift in Vm and a reduction of firing rate.
(D) Example APs (left) before (black) and during TRAM-34 (blue) application illustrate TRAM-34-induced change in Vm. Average second derivative of somatic APs (middle). Arrowhead indicates first peak originating in the axon. Summary data show a TRAM-induced reduction in the amplitude of the first axonal peak of the second derivative of the somatic action potential (right). Horizontal bars indicate average ±SEM (n = 15), p < 0.0005.
Discussion
KCa channels have been implicated in control of excitability in both unmyelinated (Lüscher et al., 1996) and myelinated (Lev-Ram and Grinvald, 1987) mammalian axons, although the evidence for the latter is indirect and the physiological impact unknown. Our results provide the first direct evidence for local, activity-dependent Cav-mediated Ca2+ influx at NoRs and for the recruitment of an intermediate KCa current (IK, KCa3.1) that is an essential component for spike propagation. Our model indicates that IK alone can sustain the Nav availability required for propagation security, although it is conceivable that other conductances might also play a role, for example, in setting the nodal Vm in the absence of firing. The sensitivity of AP conduction to low concentrations of Ni2+ and the suppression of the Ca2+ transients by mibefradil is consistent with the involvement of a T-type Cav, as seen in association with dendritic IK in PCs (Engbers et al., 2012). During sustained firing, this channel type is expected to be largely inactivated but could provide a small window current for Ca2+ entry. This might be of advantage since, due to the higher affinity of IK for Ca2+ (Wei et al., 2005), a small Ca2+ influx can be effective but less expensive for an energetically demanding cellular compartment. IK may provide advantages over other K+ channel types in axons that continuously transmit APs, often at high rates. IK lacks intrinsic voltage dependence and does not inactivate, but, by virtue of the activity dependence of Ca2+ influx, it could be recruited and sustained at levels controlled by mean spike rate. Its localized recruitment by Ca2+ may also be amplified by Ca2+-induced Ca2+ release (Llano et al., 1994). It has recently been shown that PCs also express BK channels under the myelin in the paranode region (Hirono et al., 2015). These channels appear to become functionally relevant at firing frequencies above 100 Hz, suggesting that they may act as a complementary partner to IK under conditions when [Ca2+]i is sufficient for their activation. In the absence of fast voltage-gated K+ currents, APs at NoRs may also be broader, as generated by the model, than the exceptionally narrow somatic APs in PCs, potentially further facilitating Ca2+ entry at these sites. Besides its role in axonal electrogenesis, nodal Ca2+ influx could also be important in regulating NoR structure and functional properties. For example, activity-dependent local Ca2+ levels might be crucial to target and maintain Cavs (Forti et al., 2000) and Navs (Hund et al., 2010) at NoRs (Wang et al., 2007) and to preserve or modify nodal architecture and myelin distribution (Alix et al., 2008; Einheber et al., 1997). Ca2+ signaling is thought to underlie the activity-dependent changes in mitochondrial motility observed in mammalian axons (Chiu, 2011), notably in PCs (Ohno et al., 2011). Importantly, given their spike frequency dependence, NoR Ca2+ signals could provide a readout of neuronal circuit activity, which could result in Ca2+-dependent axonal plasticity beyond the AIS (Grubb and Burrone, 2010; Gründemann and Häusser, 2010; Kole, 2011; Kuba et al., 2010). In providing direct evidence for activity-dependent Ca2+ entry at NoRs and its recruitment of IK, our findings show that Ca2+ may play both short- and long-term roles in regulating axonal excitability, crucial for long-range signaling in neuronal circuits.
Experimental Procedures
Electrophysiological Recordings
Methods used for preparing and recording from PCs and PC axons in cerebellar slices (200–250 μm) from P18–P43 C57Bl/6 mice were as previously described (Monsivais et al., 2005) and carried out under institutional and national approval. PC axons visualized by dye-filling and AP-associated cell-attached axonal capacitive currents were recorded in voltage clamp mode at 34°C ± 1°C. Amplitude of capacitive currents reflects the rate of rise of the axonal action potential and therefore pharmacological reduction in the amplitude reflects reduced sodium channel availability.
Drug Application
Drugs were diluted in ACSF and bath applied or diluted in HEPES-buffered or standard ACSF and pressure-applied locally via patch electrodes using a Picospritzer (0.5–10 psi, Parker). 30 μM Alexa Fluor 488 or 594 was included in the solution to help adjust pressure and pulse duration to target localized drug ejection. Ca2+ was replaced by Mg2+ in 0 mM Ca2+ solutions.
Two-Photon Ca2+ Imaging
Two-photon Ca2+ imaging and simultaneous electrophysiological recordings were performed using a custom-built dual galvanometer-based laser-scanning microscope (Prairie Technologies). A Ti:sapphire pulsed laser (MaiTai, Spectra Physics) tuned to 810 nm was used for two-photon excitation. 200 μM Oregon green 488 BAPTA-1 (OGB-1, Invitrogen) replaced EGTA in the pipette solution, which also contained 50 μM Alexa Fluor 594 hydrazide (Sigma-Aldrich) to visualize morphology. PCs were dialyzed for at least 15 min after establishing whole-cell mode before imaging.
Data Acquisition and Analysis
Data were digitized (ITC-18, InstruTECH, Heka) at 50–100 kHz and acquired using AxoGraph X (http://www.axographx.com/). Analysis was performed using custom-written routines in MATLAB (MathWorks) or IGOR Pro 6 (Wavemetrics) in combination with Neuromatic (http://www.thinkrandom.com/).
Two-photon imaging data were acquired with custom-written MATLAB software either as line scans (2 ms/line, 500 Hz) or as XYT frame scans for a chosen region of interest (5–20 frames/s), digitized with a BNC-2090 board (National Instruments), and analyzed with custom-written scripts in MATLAB, ImageJ, and IGOR Pro 6. Line scan and XYT frame-scan data were filtered using a boxcar running average or 2D-averaging filter, respectively. Relative changes in Ca2+-sensitive fluorescence (OGB-1, ΔF/F) were calculated as raw fluorescence Fraw minus baseline fluorescence F0 normalized to the background (FB) subtracted baseline fluorescence (Yasuda et al., 2004): ΔF / F = Fraw – F0 / F0 – FB. In some cases, data are displayed as raw OGB-1 fluorescence values or relative changes in OGB-1 fluorescence normalized to the Alexa Fluor 594 red fluorescence (ΔF/R, Figure S3). Line scans are presented as averages of three consecutive trials.
Data are shown as mean ± SEM. Significance is tested using Student’s t test unless otherwise stated, and a p value <0.05 is regarded as significantly different (InStat, GraphPad Software).
Immunohistochemistry and Confocal Microscopy
Mice were perfused with ice-cold PBS for 1 min and then with 4% paraformaldehyde (PFA) in PBS (10 min). Brains were dissected and 80 μm slices of cerebellar vermis were cut using a vibratome (Leica). Slices were washed in PBS and blocked in 10% goat serum for 2 hr before 48 hr antibody incubation in 2% goat serum (KCa3.1, mouse monoclonal, Santa Cruz Biotechnology, Calbindin D-28k, rabbit, Swant) followed by second antibody incubation for 24 hr (633 goat anti-rabbit, 488 as well as 633 goat anti-mouse, Invitrogen). Confocal image stacks were acquired using a 63× objective (NA 1.4, z-step: 130 nm, LSM700, Zeiss). High-magnification images of branchpoints were deconvolved with Huygens Software (SVI).
Multicompartmental Modeling
AP propagation along PC axons was simulated in NEURON using a previously published multicompartmental PC model (Clark et al., 2005), which included a resurgent sodium conductance typical of PCs (Khaliq et al., 2003). Additionally, at NoRs the model included a low-threshold Ca2+ conductance (CaVT, 0.002 cm/s) (Anwar et al., 2012) as well as an intermediate type KCa (IK, KCa3.1). Nodal KCa3.1 was based on a previously published BK conductance (Solinas et al., 2007), and its kinetics were adjusted to a four-state model to match previously published experimental recordings of IK (Bailey et al., 2010; Hirschberg et al., 1998). The nodal KCa3.1 current reversal potential was set to −80 mV. The simulations were run for 500 ms, and values shown in Figure 4 were measured at the center of the compartment.
Author Contributions
J.G. and B.A.C. planned and performed experiments and wrote the manuscript.
Acknowledgments
We thank Tiago Branco for discussion, technical advice, and two-photon acquisition software; Christoph Schmidt-Hieber and Arnd Roth for critical discussion; and Michael Häusser for discussion, support, and comments on the manuscript. Work was supported by grants from the Wellcome Trust (WT094077), the European Research Council (AdG 250345), and the Gatsby Charitable Foundation (GAT2919). J.G. was supported by the Wellcome Trust 4 year PhD Programme and an EMBO and Marie Curie Actions postdoctoral fellowship and is currently an Ambizione fellow of the Swiss National Science Foundation.
Published: September 3, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes three figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.08.022.
Supplemental Information
References
- Alix J.J.P., Dolphin A.C., Fern R. Vesicular apparatus, including functional calcium channels, are present in developing rodent optic nerve axons and are required for normal node of Ranvier formation. J. Physiol. 2008;586:4069–4089. doi: 10.1113/jphysiol.2008.155077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar H., Hong S., De Schutter E. Controlling Ca2+-activated K+ channels with models of Ca2+ buffering in Purkinje cells. Cerebellum. 2012;11:681–693. doi: 10.1007/s12311-010-0224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancibia-Carcamo I.L., Attwell D. The node of Ranvier in CNS pathology. Acta Neuropathol. 2014;128:161–175. doi: 10.1007/s00401-014-1305-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey M.A., Grabe M., Devor D.C. Characterization of the PCMBS-dependent modification of KCa3.1 channel gating. J. Gen. Physiol. 2010;136:367–387. doi: 10.1085/jgp.201010430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battefeld A., Tran B.T., Gavrilis J., Cooper E.C., Kole M.H.P. Heteromeric Kv7.2/7.3 channels differentially regulate action potential initiation and conduction in neocortical myelinated axons. J. Neurosci. 2014;34:3719–3732. doi: 10.1523/JNEUROSCI.4206-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender K.J., Trussell L.O. Axon initial segment Ca2+ channels influence action potential generation and timing. Neuron. 2009;61:259–271. doi: 10.1016/j.neuron.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender K.J., Ford C.P., Trussell L.O. Dopaminergic modulation of axon initial segment calcium channels regulates action potential initiation. Neuron. 2010;68:500–511. doi: 10.1016/j.neuron.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S.Y., Zagha E., Kwon E.S., Ozaita A., Bobik M., Martone M.E., Ellisman M.H., Heintz N., Rudy B. Distribution of Kv3.3 potassium channel subunits in distinct neuronal populations of mouse brain. J. Comp. Neurol. 2007;502:953–972. doi: 10.1002/cne.21353. [DOI] [PubMed] [Google Scholar]
- Chiu S.Y. Matching mitochondria to metabolic needs at nodes of Ranvier. Neuroscientist. 2011;17:343–350. doi: 10.1177/1073858410393740. [DOI] [PubMed] [Google Scholar]
- Chiu S.Y., Ritchie J.M. Potassium channels in nodal and internodal axonal membrane of mammalian myelinated fibres. Nature. 1980;284:170–171. doi: 10.1038/284170a0. [DOI] [PubMed] [Google Scholar]
- Clark B.A., Monsivais P., Branco T., London M., Häusser M. The site of action potential initiation in cerebellar Purkinje neurons. Nat. Neurosci. 2005;8:137–139. doi: 10.1038/nn1390. [DOI] [PubMed] [Google Scholar]
- Debanne D., Campanac E., Bialowas A., Carlier E., Alcaraz G. Axon physiology. Physiol. Rev. 2011;91:555–602. doi: 10.1152/physrev.00048.2009. [DOI] [PubMed] [Google Scholar]
- Devaux J., Alcaraz G., Grinspan J., Bennett V., Joho R., Crest M., Scherer S.S. Kv3.1b is a novel component of CNS nodes. J. Neurosci. 2003;23:4509–4518. doi: 10.1523/JNEUROSCI.23-11-04509.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux J.J., Kleopa K.A., Cooper E.C., Scherer S.S. KCNQ2 is a nodal K+ channel. J. Neurosci. 2004;24:1236–1244. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einheber S., Zanazzi G., Ching W., Scherer S., Milner T.A., Peles E., Salzer J.L. The axonal membrane protein Caspr, a homologue of neurexin IV, is a component of the septate-like paranodal junctions that assemble during myelination. J. Cell Biol. 1997;139:1495–1506. doi: 10.1083/jcb.139.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engbers J.D.T., Anderson D., Asmara H., Rehak R., Mehaffey W.H., Hameed S., McKay B.E., Kruskic M., Zamponi G.W., Turner R.W. Intermediate conductance calcium-activated potassium channels modulate summation of parallel fiber input in cerebellar Purkinje cells. Proc. Natl. Acad. Sci. USA. 2012;109:2601–2606. doi: 10.1073/pnas.1115024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forti L., Pouzat C., Llano I. Action potential-evoked Ca2+ signals and calcium channels in axons of developing rat cerebellar interneurones. J. Physiol. 2000;527:33–48. doi: 10.1111/j.1469-7793.2000.00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust A., Popovic M., Zecevic D., McCormick D.A. Action potentials initiate in the axon initial segment and propagate through axon collaterals reliably in cerebellar Purkinje neurons. J. Neurosci. 2010;30:6891–6902. doi: 10.1523/JNEUROSCI.0552-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein S.S., Rall W. Changes of action potential shape and velocity for changing core conductor geometry. Biophys. J. 1974;14:731–757. doi: 10.1016/S0006-3495(74)85947-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissmer S., Nguyen A.N., Aiyar J., Hanson D.C., Mather R.J., Gutman G.A., Karmilowicz M.J., Auperin D.D., Chandy K.G. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- Grubb M.S., Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465:1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründemann J., Häusser M. Neuroscience: A plastic axonal hotspot. Nature. 2010;465:1022–1023. doi: 10.1038/4651022a. [DOI] [PubMed] [Google Scholar]
- Hadley J.K., Noda M., Selyanko A.A., Wood I.C., Abogadie F.C., Brown D.A. Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. Br. J. Pharmacol. 2000;129:413–415. doi: 10.1038/sj.bjp.0703086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. The selective inhibition of delayed potassium currents in nerve by tetraethylammonium ion. J. Gen. Physiol. 1967;50:1287–1302. doi: 10.1085/jgp.50.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman D., Chen S., Aung T.T., Cherksey B., Sugimori M., Llinás R.R. Localization of P-type calcium channels in the central nervous system. Proc. Natl. Acad. Sci. USA. 1991;88:7076–7080. doi: 10.1073/pnas.88.16.7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirono M., Ogawa Y., Misono K., Zollinger D.R., Trimmer J.S., Rasband M.N., Misonou H. BK channels localize to the paranodal junction and regulate action potentials in myelinated axons of cerebellar Purkinje cells. J. Neurosci. 2015;35:7082–7094. doi: 10.1523/JNEUROSCI.3778-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg B., Maylie J., Adelman J.P., Marrion N.V. Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J. Gen. Physiol. 1998;111:565–581. doi: 10.1085/jgp.111.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hund T.J., Koval O.M., Li J., Wright P.J., Qian L., Snyder J.S., Gudmundsson H., Kline C.F., Davidson N.P., Cardona N. A β(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 2010;120:3508–3519. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner W.J., Wang L.Y., Tang M.D., Kaczmarek L.K. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc. Natl. Acad. Sci. USA. 1997;94:11013–11018. doi: 10.1073/pnas.94.20.11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq Z.M., Raman I.M. Axonal propagation of simple and complex spikes in cerebellar Purkinje neurons. J. Neurosci. 2005;25:454–463. doi: 10.1523/JNEUROSCI.3045-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq Z.M., Gouwens N.W., Raman I.M. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J. Neurosci. 2003;23:4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocsis J.D., Waxman S.G. Absence of potassium conductance in central myelinated axons. Nature. 1980;287:348–349. doi: 10.1038/287348a0. [DOI] [PubMed] [Google Scholar]
- Kole M.H. First node of Ranvier facilitates high-frequency burst encoding. Neuron. 2011;71:671–682. doi: 10.1016/j.neuron.2011.06.024. [DOI] [PubMed] [Google Scholar]
- Kuba H., Oichi Y., Ohmori H. Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature. 2010;465:1075–1078. doi: 10.1038/nature09087. [DOI] [PubMed] [Google Scholar]
- Lev-Ram V., Grinvald A. Activity-dependent calcium transients in central nervous system myelinated axons revealed by the calcium indicator Fura-2. Biophys. J. 1987;52:571–576. doi: 10.1016/S0006-3495(87)83246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I., DiPolo R., Marty A. Calcium-induced calcium release in cerebellar Purkinje cells. Neuron. 1994;12:663–673. doi: 10.1016/0896-6273(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Lüscher C., Lipp P., Lüscher H.R., Niggli E. Control of action potential propagation by intracellular Ca2+ in cultured rat dorsal root ganglion cells. J. Physiol. 1996;490:319–324. doi: 10.1113/jphysiol.1996.sp021146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierzwa A., Shroff S., Rosenbluth J. Permeability of the paranodal junction of myelinated nerve fibers. J. Neurosci. 2010;30:15962–15968. doi: 10.1523/JNEUROSCI.4047-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsivais P., Clark B.A., Roth A., Häusser M. Determinants of action potential propagation in cerebellar Purkinje cell axons. J. Neurosci. 2005;25:464–472. doi: 10.1523/JNEUROSCI.3871-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y., Oses-Prieto J., Kim M.Y., Horresh I., Peles E., Burlingame A.L., Trimmer J.S., Meijer D., Rasband M.N. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J. Neurosci. 2010;30:1038–1048. doi: 10.1523/JNEUROSCI.4661-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno N., Kidd G.J., Mahad D., Kiryu-Seo S., Avishai A., Komuro H., Trapp B.D. Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. J. Neurosci. 2011;31:7249–7258. doi: 10.1523/JNEUROSCI.0095-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z., Kao T., Horvath Z., Lemos J., Sul J.-Y., Cranstoun S.D., Bennett V., Scherer S.S., Cooper E.C. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman I.M., Bean B.P. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J. Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband M.N. Clustered K+ channel complexes in axons. Neurosci. Lett. 2010;486:101–106. doi: 10.1016/j.neulet.2010.08.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röper J., Schwarz J.R. Heterogeneous distribution of fast and slow potassium channels in myelinated rat nerve fibres. J. Physiol. 1989;416:93–110. doi: 10.1113/jphysiol.1989.sp017751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz J.R., Glassmeier G., Cooper E.C., Kao T.-C., Nodera H., Tabuena D., Kaji R., Bostock H. KCNQ channels mediate IKs, a slow K+ current regulating excitability in the rat node of Ranvier. J. Physiol. 2006;573:17–34. doi: 10.1113/jphysiol.2006.106815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas S., Forti L., Cesana E., Mapelli J., De Schutter E., D’Angelo E. Computational reconstruction of pacemaking and intrinsic electroresponsiveness in cerebellar Golgi cells. Front. Cell. Neurosci. 2007;1:2. doi: 10.3389/neuro.03.002.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Kunkel D.D., Martin T.M., Schwartzkroin P.A., Tempel B.L. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature. 1993;365:75–79. doi: 10.1038/365075a0. [DOI] [PubMed] [Google Scholar]
- Wang H.-G., George M.S., Kim J., Wang C., Pitt G.S. Ca2+/calmodulin regulates trafficking of Ca(V)1.2 Ca2+ channels in cultured hippocampal neurons. J. Neurosci. 2007;27:9086–9093. doi: 10.1523/JNEUROSCI.1720-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei A.D., Gutman G.A., Aldrich R., Chandy K.G., Grissmer S., Wulff H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol. Rev. 2005;57:463–472. doi: 10.1124/pr.57.4.9. [DOI] [PubMed] [Google Scholar]
- Wilson G.F., Chiu S.Y. Ion channels in axon and Schwann cell membranes at paranodes of mammalian myelinated fibers studied with patch clamp. J. Neurosci. 1990;10:3263–3274. doi: 10.1523/JNEUROSCI.10-10-03263.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H., Miller M.J., Hansel W., Grissmer S., Cahalan M.D., Chandy K.G. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc. Natl. Acad. Sci. USA. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R., Nimchinsky E.A., Scheuss V., Pologruto T.A., Oertner T.G., Sabatini B.L., Svoboda K. Imaging calcium concentration dynamics in small neuronal compartments. Sci. STKE. 2004;2004:pl5. doi: 10.1126/stke.2192004pl5. [DOI] [PubMed] [Google Scholar]
- Yu Y., Maureira C., Liu X., McCormick D. P/Q and N channels control baseline and spike-triggered calcium levels in neocortical axons and synaptic boutons. J. Neurosci. 2010;30:11858–11869. doi: 10.1523/JNEUROSCI.2651-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.-L., Wilson J.A., Williams J., Chiu S.Y. Action potentials induce uniform calcium influx in mammalian myelinated optic nerves. J. Neurophysiol. 2006;96:695–709. doi: 10.1152/jn.00083.2006. [DOI] [PubMed] [Google Scholar]
- Zhou L., Zhang C.L., Messing A., Chiu S.Y. Temperature-sensitive neuromuscular transmission in Kv1.1 null mice: role of potassium channels under the myelin sheath in young nerves. J. Neurosci. 1998;18:7200–7215. doi: 10.1523/JNEUROSCI.18-18-07200.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.