

Abstract
The human ribosomal P complex, which consists of the acidic ribosomal P proteins RPLP0, RPLP1, and RPLP2 (RPLP proteins), recruits translational factors, facilitating protein synthesis. Recently, we showed that overexpression of RPLP1 immortalizes primary cells and contributes to transformation. Moreover, RPLP proteins are overexpressed in human cancer, with the highest incidence in breast carcinomas. It is thought that disruption of the P complex would directly affect protein synthesis, causing cell growth arrest and eventually apoptosis. Here, we report a distinct mechanism by which cancer cells undergo cell cycle arrest and induced autophagy when RPLP proteins are downregulated. We found that absence of RPLP0, RPLP1, or RPLP2 resulted in reactive oxygen species (ROS) accumulation and MAPK1/ERK2 signaling pathway activation. Moreover, ROS generation led to endoplasmic reticulum (ER) stress that involved the EIF2AK3/PERK-EIF2S1/eIF2α-EIF2S2-EIF2S3-ATF4/ATF-4- and ATF6/ATF-6-dependent arms of the unfolded protein response (UPR). RPLP protein-deficient cells treated with autophagy inhibitors experienced apoptotic cell death as an alternative to autophagy. Strikingly, antioxidant treatment prevented UPR activation and autophagy while restoring the proliferative capacity of these cells. Our results indicate that ROS are a critical signal generated by disruption of the P complex that causes a cellular response that follows a sequential order: first ROS, then ER stress/UPR activation, and finally autophagy. Importantly, inhibition of the first step alone is able to restore the proliferative capacity of the cells, preventing UPR activation and autophagy. Overall, our results support a role for autophagy as a survival mechanism in response to stress due to RPLP protein deficiency.
Keywords: autophagy, cancer, ER stress, ROS, RPLP0, RPLP1, RPLP2, RPLP proteins, UPR
Abbreviations
- 3-MA
3-methyladenine
- AKT1
v-akt murine thymoma viral oncogene homolog 1
- ATF4/ATF-4
activating transcription factor 4
- ATF6/ATF-6
activating transcription factor 6
- BECN1/Beclin 1
Beclin 1, autophagy related
- CHX
cycloheximide
- CQ
chloroquine
- DOXO
doxorubicin
- DTT
dithiothreitol
- EIF2AK3/PERK
eukaryotic translation initiation factor 2-α kinase 3
- EIF2S1/EIF2
α
- ER
endoplasmic reticulum
- G6PD
glucose-6-phosphate-1-dehydrogenase
- HSPB1
heat shock protein 27kDa protein 1
- HSPA1A
heat shock protein 70kDa protein 1A
- KRAS
Kirsten rat sarcoma viral oncogene homolog
- KRT17
keratin 17, type I
- LCMSMS
liquid chromatography coupled to tandem mass spectrometry
- MAPK1/ERK2
mitogen-activated protein kinase 1
- MTOR
mechanistic target of rapamycin
- NAC
N-acetyl-L-cysteine
- NT
non-target
- OHT
tamoxifen
- RPs
ribosomal proteins
- RPLP proteins
acidic ribosomal proteins RPLP0, RPLP1 and RPLP2
- qRT-PCR
quantitative real-time PCR
- ROS
reactive oxygen species
- Sc
scrambled
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- Sts
staurosporine
- TEM
transmission electron microscopy
- Tg
thapsigargin
- TOF
time of flight
- TP53
tumor protein p53
- TXN
thioredoxin
- UPR
unfolded protein response.
Introduction
The large subunit of the ribosome has a lateral protuberance known as the ribosomal stalk that plays an important role in translation. In eukaryotes, the ribosomal stalk consists of the acidic ribosomal proteins (RPs) RPLP0, RPLP1 and RPLP2, with 2 copies of RPLP1 and RPLP2 heterodimers bound to the C-terminal region of RPLP0 to form a pentameric P complex.1,2 Eukaryotic RPLP1 and RPLP2 proteins also exist in free form in the cytoplasm, and the exchange between the ribosome-bound RPLP1 and RPLP2 proteins and the cytoplasmic pools is thought to regulate the activity of the ribosome.3
Though the precise biological functions of RPLP proteins are still unclear, it is known that all RPLP proteins contain a conserved motif at the C terminus that is involved in the recruitment of both translation factors and ribosome-inactivation proteins to the ribosomal stalk.4-6
Whereas most RPs are transported to the nucleus and shaped into ribosomes, RPLP1 and RPLP2 are located in the Golgi/endoplasmic reticulum (ER) structure and are assembled in the cytoplasm in the very last step in ribosome biogenesis.1 RPLP0, RPLP1, and RPLP2 also have their own specific characteristics on both expression profiling and amino acid composition analysis compared with the major RP proteins. RPLP protein genes (RPLP0, RPLP1, RPLP2) are expressed in a tissue-specific manner and their average frequencies of encoded lysine and arginine are lower than for the main RP genes.7 Additionally, because only RPLP1 and RPLP2 form dimers, they are also believed to have gene expression machinery different from that of the other RP genes. These individual characteristics, together with earlier experimental evidence in yeast and Drosophila, suggest that RPLP proteins have the potential to carry out extraribosomal functions as independent polypeptides.8-10
Strikingly, ribosomal stalk proteins have been linked to several pathological conditions, including autoimmune diseases and human malignancies. For example, autoantibodies against the C-terminal peptide common to RPLP proteins are present in 15% of systemic lupus erythematosus patients.11 RPLP0 mRNA is found overexpressed in human colorectal and hepatocellular carcinomas, and overexpression of RPLP1 mRNA is observed in human lymphoid cell lines containing mutated TP53 (tumor protein p53).12,13 In previous studies, we have reported that RPLP1 overexpression allows primary mouse embryonic fibroblasts to bypass replicative senescence through a TP53/TRP53/p53-independent mechanism and through the increased activity of the E2F1 promoter and the upregulation of CCNE1.14 In addition, we have found that RPLP1 cooperates with KRASG12V in the malignant transformation of murine NIH3T3 cells.14 More recently, we have reported that RPLP protein expression is significantly increased in breast, skin, colon, lung, and ovarian tumors with respect to the corresponding normal tissue. We have also found positive correlations between the expression of RPLP proteins and the presence of metastasis in different subtypes of gynecological cancer.15
Despite mounting evidence of RPLP protein overexpression in cancer cells and a link between their downregulation and specific drug responses,16 it remains unknown how RPLP proteins contribute to these specific cellular changes in human tumors. In the present study, we inhibited the P complex in cancer cells and studied the underlying molecular events that are directly associated with RPLP protein downregulation, including their potential regulatory role in cell cycle arrest and their ability to induce autophagy. Autophagy, while initially considered a cell death mechanism, is being described, in an emerging body of research, as a survival response triggered by certain stress conditions.17-20 Importantly, our data show that RPLP protein knockdown provokes a stress response in which cells ultimately survive by autophagy and that there is no role for autophagy in cell death. The possible implications of these findings in cancer are discussed.
Results
Downregulation of RPLP proteins affects cell proliferation and cell cycle progression
We have previously reported that RPLP proteins are highly overexpressed in most (>80%) breast carcinomas (n = 46), as well as in 61% of colon (n = 35) and ovarian (n = 140) cancers, with respect to their corresponding normal tissues.15 To examine whether the downregulation of RPLP proteins has the converse effect (i.e., prevents cancer cell growth), we used cancer cell lines of breast (MCF-7 and MDA-MB-231), colon (HCT116 TP53−/− and HT-29), and ovarian carcinoma (OV-90). All siRNAs tested targeting RPLP0, RPLP1, or RPLP2 genes were able to inhibit the corresponding protein by >80% (Fig. S1A). Downregulation of each RPLP protein by siRNA- or shRNA-targeting of the corresponding mRNA, inhibited cell growth (by approximately 76 ± 11%) in all cancer cell lines assessed (Figs. 1A and 2A, and Fig. S1B and C). Similarly, RPLP0, RPLP1, and RPLP2 shRNA decreased colony formation in the MCF-7 cell line by up to 75 ± 4%, 82 ± 5%, and 86 ± 4%, respectively (Fig. 1B).
Figure 1.

RPLP protein downregulation induces cell growth arrest. (A) Growth curves of MCF-7 cells stably expressing a control non-target shRNA vector (NT shRNA), or shRNA vectors targeting the RPLP0, RPLP1, or RPLP2 genes (RPLP0 shRNA, RPLP1 shRNA, or RPLP2 shRNA, respectively) with the 3T3 protocol.67 The black arrow represents the recovery point from the drug selection. The data presented are the mean ±SD of 3 independent experiments. *, P ≤ 0.05. (B) Colony formation assay. MCF-7 cells were stably infected with the indicated shRNA vectors (as in A), and were plated at a density of 3,000 cells/well. After 20 d, cells were fixed and stained with a crystal violet solution. Only MCF-7 cells expressing NT shRNA were able to form a high number of colonies. All experiments were performed 3 times (n = 3). (C) Relative mRNA levels of RPLP0, RPLP1, and RPLP2 in MCF-7 cells expressing NT shRNA, RPLP0 shRNA, RPLP1 shRNA, or RPLP2 shRNA vectors, related to parental MCF-7 control cells (mRNA levels set as 1). Data indicate mean values ±SD. *, P ≤ 0.05 versus NT shRNA. The bottom figures represent the corresponding expression of RPLP0, RPLP1, and RPLP2 proteins in MCF-7 cells expressing NT shRNA, RPLP0 shRNA, RPLP1 shRNA, or RPLP2 shRNA. (D) Left panel, western blot analysis of RPLP0, RPLP1, and RPLP2 expression levels in lysates of MCF-7 cells expressing RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or control NT shRNA vector. ACTB was used as a loading control. Right panel represents the protein levels of RPLP0, RPLP1, and RPLP2 after their overexpression in their corresponding knock down cell lines as indicated in the figure. O/E, overexpression. (E) Cell cycle profiles of MCF-7 cells after transfection. Cells transfected with RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA were stained with propidium iodide and the effect on cell cycle progression was monitored with FACS analysis. *, P ≤ 0.05. (F) Growth curves of MCF-7 cells stably expressing a control non-target shRNA vector (NT shRNA), or shRNA vectors targeting the RPLP0, RPLP1, or RPLP2 genes (RPLP0 shRNA, RPLP1 shRNA, or RPLP2 shRNA, respectively) plus the cDNA of RPLP0, RPLP1 and RPLP2 in each case. The black arrows represent the recovery point from the double drug selection.
As shown in Figure 1C and D (left panel), MCF-7 cells that were infected with lentiviral particles for the stable targeting of RPLP0, RPLP1, and RPLP2 genes by shRNA showed decreases of about 77%, 86%, and 90% in the relative mRNA levels, respectively, and a similar reduction in the indicated RPLP protein expression (for RPLP0, RPLP1, and RPLP2: 82%, 84%, and 76%, respectively) compared with cells receiving the NT shRNA. Quantification of protein content from Figure 1D (left panel) is shown in Figure S1D. We also observed a concomitant reduction in RPLP1 expression upon RPLP2 knockdown. This is in line with previous work reporting that RPLP2 protects against the rapid degradation of RPLP1.21
The effect of RPLP protein downregulation on cell cycle distribution was then studied with flow cytometry after propidium iodide DNA staining. As observed in Figure 1E, silencing the RPLP proteins in the MCF-7 cells by shRNA (RPLP0 shRNA, RPLP1 shRNA, and RPLP2 shRNA) resulted in the specific accumulation of cells in the G2 phase. The highest proportion of cells arrested at G2 was observed for RPLP2 shRNA (42.2%). These cells also had the most reduced proportion of cells in progression through the S phase (1.5%). G1 traversal was unaffected by RPLP0 shRNA, RPLP1 shRNA, and RPLP2 shRNA. In addition, neither RPLP0 shRNA, nor RPLP1 shRNA, nor RPLP2 shRNA resulted in increased apoptotic cell death, as indicated by the small sub-G1 population (Fig. 1E). Similar results were obtained with MDA-MB-231 and OV-90 cells (data not shown). These data indicate that efficient downregulation of any of the RPLP proteins leads to a dramatic decrease in cell proliferation and a nonapoptotic cell cycle arrest.
RPLP protein re-expression reverts the phenotype provoked by their downregulation
We have individually knocked down the expression of RPLP0, RPLP1, and RPLP2 and showed that disruption of any of the above mentioned proteins decreases cell proliferation and causes cell cycle arrest (Fig. 1A–E). As RPLP0, RPLP1, and RPLP2 are part of a functional complex, disruption of any of the factors might cause the above effect. However, to truly link these phenomena to RPLP proteins, we have re-expressed RPLP0, RPLP1, and RPLP2 proteins in their corresponding knockout cell lines. MCF-7 cells were infected with retroviral particles for the stable expression of RPLP0, RPLP1, and RPLP2 genes. The re-expression of RPLP0, RPLP1 and RPLP2 in such context reverts the above mentioned phenotype (Fig. 1D, right panel, and 1F). Thereby RPLP protein deficiency is responsible for the observed decrease in proliferation and cell cycle arrest.
Inhibition of RPLP proteins induces autophagy in cancer cells
Our next question concerned cell death. MCF-7 cells stably expressing RPLP0 shRNA, RPLP1 shRNA, or RPLP2 shRNA were trypsinized, counted, and stained with trypan blue to mark dead cells. RPLP0 shRNA-, RPLP1 shRNA-, or RPLP2 shRNA-expressing cells had similar numbers of dead cells to the control cells (NT shRNA) (Fig. S2). Therefore, our results suggest that cell death was not a consequence of the downregulation of each RPLP protein. To confirm that the observed inhibition of proliferation did not correspond to apoptosis, an ANXA5/annexin V assay coupled to flow cytometry was performed. As observed in Figure S3A, MCF-7 cells expressing RPLP0 shRNA, RPLP1 shRNA, or RPLP2 shRNA had similar numbers of early and late apoptotic events as the control cells (NT shRNA). Accordingly, increases in condensed and fragmented nuclei were not observed by fluorescence microscopy in these cells after staining with Hoechst 33258 (Fig. S3B). These data, in combination with the absence of PARP1 (poly ADP-ribose polymerase 1) cleavage (Fig. S3C), the absence of significant differences in the cell cycle profile in the sub-G1 cell population among RPLP0 shRNA-, RPLP1 shRNA-, or RPLP2 shRNA-expressing cells compared with NT shRNA-expressing cells (Fig. 1E), and the lack of an increase in the number of dead cells (Fig. S2) indicated that RPLP protein deficiency does not cause cell death.
As we previously found that RPLP1 overexpression bypasses replicative senescence in primary cells,14 we hypothesized that the absence of RPLP1 or its partners RPLP0 and RPLP2 could inhibit proliferation by inducing senescence. However, none of the cells with downregulated RPLP proteins had increased senescence-associated β-galactosidase activity compared with NT shRNA controls (Fig. S4A). The absence of apoptosis and senescence was also observed in MDA-MB-231 and OV-90 cells with downregulated expression of RPLP0, RPLP1, or RPLP2 (data not shown).
Cells can undergo nonapoptotic cell death that is accompanied by large-scale autophagic vacuolization.17 To determine whether downregulation of RPLP proteins induces autophagy, LC3-II levels were analyzed by western blot.22 The results showed that downregulation of RPLP proteins induced a clear increase in LC3-II levels in MCF-7, MDA-MB-231, and OV-90 cells (Fig. 2B). As LC3-I can change independently of LC3-II conversion, LC3-II levels were quantified versus ACTB/β-actin and a clear increase was detected (Fig. S4B).23,24 These results were confirmed with a different shRNA targeting RPLP0, RPLP1 and RPLP2 (Fig. S4C). Additional markers of autophagy such as levels of SQSTM1/p62 degradation were analyzed.25,26 Indeed, SQSTM1 degradation occurred in cells induced to express RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA vs. NT shRNA-expressing cells (Fig. 2B). Autophagy was further confirmed in these cells by the observation of a marked increase in acridine orange staining of cytoplasmic intracellular acid vesicles (Fig. 2C). Moreover, to detect the presence of autophagic features by other methods, MCF-7 cells were transfected with a GFP-LC3 plasmid and further induced to express RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA.27 LC3 immunocytochemistry shows a punctate pattern in the cytoplasm when autophagy is induced.28 We observed that in control cells, GFP-LC3 was equally distributed in cytosolic pools. However, upon RPLP protein inhibition, GFP-LC3 was recruited to phagophore membranes, as indicated by the punctate pattern that was hardly detected in control cells. Results are shown in MCF-7 cells expressing 2 independent shRNAs for each RPLP protein (Fig. S5A). Quantification of the punctate pattern indicative of LC3-II aggregation in autophagosomes is shown (Fig. S5B). In addition, BECN1/Beclin 1 is essential for the initial steps of autophagy.29,30 Therefore, BECN1 immunocytochemistry was performed in MCF-7 cells depleted of RPLP0, RPLP1, and RPLP2, and compared with control cells. Visualization of these cells under a fluorescence microscope revealed the presence of punctate staining that was indicative of BECN1 induction, confirming the presence of autophagy in MCF-7 cells depleted of RPLP0, RPLP1, or RPLP2 proteins (Fig. 2D). Quantification of the positive cells for BECN1 is shown (Fig. 2D, right panel).
Figure 2.

RPLP protein downregulation induces autophagy in breast and ovarian cancer cell lines. (A) Numbers of MCF-7, MDA-MB-231, and OV-90 cells after transfection with RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA vector at the 3rd d of selection with puromycin. *, P ≤ 0.01. The data are the mean ±SD of 3 independent experiments. (B) Representative immunoblot for autophagy, which is characterized by the conversion of LC3-I (cytosolic form) into LC3-II (autophagosome membrane-bound form), and SQSTM1. Note the decrease of SQSTM1 protein and the increase in LC3-II levels compared with ACTB in MCF-7, MDA-MB-231, and OV-90 cells expressing RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA vector at 72 h after transfection. (C) Acridine orange staining of MCF-7, MDA-MB-231, and OV-90 cells stably expressing shRNAs (as in A). Cells treated with 2.5 µM OHT for 4 d were used as a positive control for acidic vesicles, in particular the autolysosomes characteristic of autophagy. (D) MCF-7 cells expressing RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA vector were incubated with BECN1 antibody and then analyzed using a fluorescence microscope. Arrows signal the punctate staining indicating BECN1 expression. Positive staining was scored in 100 cells, with error bars indicating mean values ±SD. *, P ≤ 0.05.
To confirm the presence of autophagy by morphological features, transmission electron microscopy (TEM) was performed.19 The presence of autophagosomes, autolysosomes, and other signs of autophagy after fixation and examination of cells stably expressing RPLP0 shRNA, RPLP1 shRNA, or RPLP2 shRNA was analyzed and compared with that of NT shRNA-expressing cells. Clear signs of autophagy were detected in RPLP protein-deficient cells (Fig. 3A and B).
Figure 3.

Detection of autophagy by TEM. (A) MCF-7 cells were induced to express the indicated shRNAs. After selection with puromycin for 3 d, cells were fixed and examined. High power magnification at the indicated scale revealed autophagosomes (arrow heads) and autolysosomes (black arrows), features characteristic of autophagic cells. The white arrow (RPLP0 shRNA) indicates a double-membrane structure engulfing a putative defective mitochondria that resembles an incipient phagophore. N, nucleus; M, mitochondria; ER, endoplasmic reticulum; G, Golgi apparatus. Scale bar: 0.5 μm. (B) Quantifications are based on counting autophagic vacuoles in the field of view. The values are the mean ± S.D. of 3 independent experiments. *, P ≤ 0.05.
Finally, we tested the ability of autophagy inhibitors in our cells. First, 3-methyladenine (3-MA), an autophagy inhibitor that acts at early steps of the autophagy pathway, was used.31 The increased levels of LC3-II induced by downregulation of RPLP0, RPLP1, or RPLP2 in MCF-7 cells could be reversed by treatment with 3-MA, suggesting that autophagy was indeed induced (Fig. 4A). Quantification of LC3-II vs. ACTB demonstrated that LC3-II abundance does not change in these cells following 3-MA treatment (data not shown). However, cell proliferation did not improve (Fig. S4D). With western blotting, we found an increased cleavage of PARP1 in 3-MA-treated cells and a decrease in proCASP6/procaspase-6 proteins, both manifestations of apoptosis (Fig. 4A). Treatment of MCF-7 cells deficient for RPLP0, RPLP1, or RPLP2 with 3-MA led to cytoplasmic shrinkage and a switch from cytoplasmic to nuclear sequestration of acridine orange staining. Results were analyzed by confocal microscopy with samples counterstained with Hoechst (Fig. 4B). Further indications of apoptosis by changes in cell morphology were observed (Fig. 4B). Additional signs of apoptosis were only observed in RPLP protein-deficient cells treated with 3-MA and not in cells with tamoxifen (OHT)-induced autophagy and treated identically with 3-MA (data not shown).
Figure 4.

Inhibition of autophagy in cells with downregulated RPLP protein expression induces apoptosis. (A) Representative immunoblot of LC3, PARP1 cleavage, and proCASP6 in MCF-7 cells expressing RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA vector, in the absence (DMSO) or presence of 10 mM 3-MA for 48 h. ACTB was used as a loading control. Note the presence of PARP1 cleavage and the decrease in proCASP6 in MCF-7 cells treated with Sts and also in MCF-7 cells expressing RPLP0 shRNA, RPLP1 shRNA, and RPLP2 shRNA compared with control cells (NT shRNA) following treatment with 3-MA (as in A), indicative of apoptosis. (B) Acridine orange staining counterstained with Hoechst in MCF-7 cells expressing RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA vector, in the absence or presence of 10 mM 3-MA (as in A). Representative confocal microscopy images of control (DMSO) RPLP protein-deficient cells with punctate staining of autophagosomes (left part). The acridine orange cytoplasmic pattern is blocked by the autophagy inhibitor 3-MA (on the right), resulting in acridine orange-positive nuclear staining and morphological changes that are characteristic of apoptosis. Scale bar: 20 μm.
Accumulation of autophagosomes could either indicate increased autophagic flux or defective autophagy.22 Therefore, we examined if there was sufficient autophagic flux upon RPLP protein knockdown. Autophagic flux is crucial in determining whether the autophagic cargo and assembly finally reaches the lysosomes and is subsequently degraded. One of the ways to determine autophagic flux is by pretreatment with the lysosomal inhibitor of autophagy chloroquine (CQ),22 prior to the efficient knockdown of each RPLP protein. Lysosomal inhibitors increase LC3-II formation, partly by blocking autophagosomal-lysosomal fusion. Therefore, we investigated the effect of lysosomal inhibition on RPLP protein knockdown-induced LC3-II formation. In the presence of 10 μM of CQ, increased time- and dose-dependent expression of LC3-II conversion was found (Fig. 5A and data not shown). Our results showed that CQ increased LC3 turnover in MCF-7 cells at 24 and 48 h, indicating that effective autophagy was initiated and preceded to lysosomal rupture. This result is in agreement with the fact that CQ inhibits a late stage of the autophagy pathway. Moreover, cells treated with CQ die by apoptosis, as observed by PARP1 cleavage and a decrease in the proCASP6 form (Fig. 5A). Proteolysis of PARP1 correlates with the increased nuclear DNA fragmentation and strongly suggests apoptosis.32,33 Accordingly, PARP1 accumulation in the nucleus was observed (Fig. 5B) and quantified (Fig. S4E). Moreover, since levels of LC3 processing and SQSTM1 degradation are dynamically regulated by autophagic flux,25 autophagy flux assays in the presence of protease inhibitors (e.g., pepstatin A plus E64) were performed. Addition of pepstatin A/E64 in cells downregulated for RPLP0, RPLP1, or RPLP2, resulted in a greater increase of LC3-II and diminishes the degradation of SQSTM1 in comparison with untreated cells (DMSO) indicating that autophagic degradation is prevented. This result shows that the absence of each RPLP protein induced the formation of autophagosomes (Fig. 5C). Furthermore, autophagy-related protein ATG7 is thought to be essential for mammalian autophagy.34 To assess the role of ATG7 in the induction of autophagy by RPLP protein knockdown, RNAi-mediated silencing of ATG7 was performed. Inhibition of ATG7 prevented LC3-II accumulation induced by RPLP protein knockdown. Results from 2 independent siRNAs against ATG7 mRNA are shown (Fig. 5D). Moreover, inhibition of ATG7 rescued MCF-7 cells from the autophagic phenotype by causing apoptotic cell death (Fig. 5B and D, and Fig. S4E).
Figure 5.

Autophagy inhibition triggers apoptotic cell death. (A) MCF-7 cells were treated at different time points with CQ, an autophagy inhibitor that prevents the fusion of the autophagosome with the lysosome to form the autolysosome, a late stage in the autophagy cascade. At 24 h and 48 h, CQ was able to prevent autophagy in MCF-7 cells depleted of RPLP0, RPLP1, or RPLP2 while stimulating apoptosis, as observed by the decrease in proCASP6 and the presence of the cleaved PARP1 form characteristic of apoptosis. (B) Immunofluorescence of PARP1 staining in MCF-7 cells treated with ATG7 siRNA with deletion of the indicated RPLP protein. Staining in control cells (NT shRNA) was concentrated in the nucleus (acting as a reservoir). In MCF-7 cells depleted of each RPLP protein (RPLP0, RPLP1, or RPLP2), PARP1 staining was diffuse throughout the nucleus. The morphological appearance of cells treated with CQ is also shown. (C) Addition of E64/pepstatin A in cells depleted of RPLP0, RPLP1 or RPLP2 proteins results in a greater increase of LC3-II and diminishes the degradation of SQSTM1. (D) Effect of 2 ATG7 siRNAs on LC3-II conversion, proCASP6, and the cleaved PARP1 form in MCF-7 cells depleted for the indicated RPLP protein vs. control cells (NT shRNA) and Sc siRNA. These results indicate that ATG7 siRNA is able to avoid the autophagic phenotype by stimulating apoptosis.
Taken together, these data provide strong evidence that autophagy occurs upon RPLP protein inhibition. These observations suggest that RPLP0, RPLP1, or RPLP2 deficiency causes cell cycle arrest accompanied by autophagy that is probably at excessive levels. Moreover, because disruption of autophagosome or lysosome formation with the use of early and late stage autophagy inhibitors (3-MA and CQ, respectively), blocks signs of autophagy, and because cell death occurs by apoptosis, our results suggest that in this scenario autophagy lies as a survival response upstream of apoptotic cell death.
Effect of RPLP protein depletion on translation efficiency
Because RPLP proteins comprise the ribosomal stalk and seem to have an essential role in the recruitment of translational factors,35 it was pertinent to examine how deficiency of the different RPLP proteins affects the ribosomal structure, assembly, or function. The polysome profiles of RPLP protein-deficient cells were not different from those of NT shRNA-expressing cells for the peaks representing the small ribosomal subunit (40 S), large ribosomal subunit (60 S), and monosome (80 S) (Fig. S6A, left panel). The use of DTT as UPR activator and thapsigargin (Tg) as a translation inhibitor verified that in RPLP protein-deficient cells translation is not compromised (Fig. S6A, right panel). Confluent cells were also included as a control of translation inhibition (Fig. S6A, right panel). In NT shRNA-expressing cells, ∼48.7% of RNA was in polysomes, a value that was similar upon RPLP0, RPLP1, or RPLP2 inhibition (Fig. S6B). Moreover, the average number of ribosomes per translated transcript (mRNAs containing 2 or more ribosomes) was not altered and the relative ribosomal content did not decrease, thus indicating a maintained equilibrium between ribosomes and subunits (Fig. S6C and D). To corroborate these results with a different technique, we measured global protein synthesis by [35S]-met radioactivity incorporation. Protein synthesis was not significantly altered upon RPLP0, RPLP1, or RPLP2 inhibition (Fig. S6E; *, P < 0.05). These results suggest that overall translation is not compromised by RPLP protein inhibition.
Inhibition of RPLP proteins inducing autophagy is accompanied by UPR activation
Autophagy can be induced in response to nutrient starvation, protein aggregation, damaged organelles, hypoxia, calcium overload, or ER stress.36 The ER stress response is mediated by 3 receptors located in the ER membrane: EIF2AK3/PERK (eukaryotic translation initiation factor 2-α kinase 3), ATF6/ATF-6 (activating transcription factor 6), and ERN1/IRE1 (endoplasmic reticulum to nucleus signaling 1).37 We found that RPLP0, RPLP1, and RPLP2 protein deficiency leads to increased phosphorylation of EIF2AK3 and its downstream targets, EIF2S1/eIF2 α (eukaryotic translation initiation factor 2 α) and ATF4/ATF-4 (activating transcription factor 4) (Fig. 6A). These results were confirmed with a different shRNA targeting RPLP0, RPLP1 and RPLP2 mRNAs (Fig. 6A). The expression of ATF6 was also upregulated in RPLP protein-deficient cells, whereas alterations in the ERN1 branch of the UPR were not observed (Fig. 6A, and Fig. S7A). These results suggest that RPLP proteins cause ER stress that is associated with the activation of the EIF2AK3, ATF4, and ATF6 branches of the UPR.
Figure 6.

Upregulation of proteins related to the UPR and autophagy. MCF-7 cells expressing RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA vectors, untreated (DMSO) (A) or treated (B) with the autophagy inhibitor 3-MA (10 mM), were immunoblotted for the indicated proteins (phospho- or total). Expressions of p-EIF2AK3, p-EIF2S1, p-MTOR, p-AKT1, and p-MAPK1 were compared with those of total proteins (EIF2AK3, EIF2S1, MTOR, AKT1, and MAPK1, respectively). Expressions of ATF4, ATF6, and TP53 were compared with those of ACTB (used as a loading control). Parental MCF-7 cells treated with Sts (2 µM) were used as a control for apoptosis.
Next, key regulators of autophagy and related proteins were studied, including MTOR (mechanistic target of rapamycin), AKT1 (v-akt murine thymoma viral oncogene homolog 1), MAPK11/12/13/14 (mitogen-activated protein kinase 11, 12, 13, and 14), TP53, and CDKN1A/p21WAF1 (cyclin-dependent kinase inhibitor 1A).36 Downregulation of RPLP0, RPLP1, or RPLP2 in MCF-7 cells led to a reduction in the amount of MTOR and AKT1 phosphorylation, as well as the downregulation of TP53 and its downstream effectors (e.g., CDKN1A) (Fig. 6A, and data not shown). In contrast, MAPK1/ERK2 phosphorylation increased in the RPLP protein-deficient cells (Fig. 6A) without changes in the activation of MAPK11/12/13/14 (data not shown).
To identify specific autophagy mediators, we compared the expression of these modulators in RPLP0 shRNA-, RPLP1 shRNA-, and RPLP2 shRNA-MCF-7s, both with and without treatment with the autophagy inhibitor 3-MA (Fig. 6A and B). 3-MA is a potent class III phosphatidylinositol 3-kinase (PtdIns3k) inhibitor that inhibits the AKT1-MTOR pathway.38 Accordingly, we observed that treatment of RPLP protein-deficient cells with 3-MA resulted in complete inhibition of MTOR phosphorylation and a decrease in AKT1 phosphorylation. We also observed TP53 upregulation and ATF6 downregulation. In addition, the phosphorylation levels of MAPK1 decreased to that of the control (NT shRNA) and parental cells (Fig. 6A and B). Lastly as we observed that inhibition of autophagy with 3-MA blocks EIF2AK3 phosphorylation even though EIF2S1 phosphorylation and ATF4 expression were unchanged, we explore if other kinase such us EIF2AK4 is also involved in the process. We did not observe any change in EIF2AK4 protein in cells where RPLP0, RPLP1 or RPLP2 protein was downregulated (Fig. S7B).
Overall, our data indicate that the autophagic response to RPLP protein inhibition is linked to ER stress/UPR activation. In addition to MTOR, a key modulator of autophagy control,39 our data suggest that MAPK1 phosphorylation may also have a relevant role in the autophagy induced by RPLP protein deficiency.
Inhibition of RPLP proteins induces striking changes in protein expression
To further analyze the specific molecular changes induced by RPLP0, RPLP1, or RPLP2 absence, we performed comparative proteome analyses with 2-dimensional (2-D) gel electrophoresis and mass spectrometry (LCMSMS and TOF/TOF). Statistical analysis revealed common proteins affected by RPLP protein downregulation (Table S1). The most deregulated proteins were identified by sequencing (Fig. 7A). Out of the most significantly deregulated proteins, except for one corresponding to the minichromosome maintenance complex component 7 MCM7, the rest appeared upregulated following RPLP0, RPLP1, and RPLP2 knockdown (Table S1 and Fig. 7A). Among the most upregulated proteins, in decreasing order of significance and as further confirmed by western blot, were: TXN (thioredoxin), KRT17 (keratin 17), HSPB1 (heat shock protein 27kDa protein 1), G6PD (glucose-6-phosphate dehydrogenase), and HSPA1A (heat shock protein 70kDa protein 1A) (Fig. 7A and B).
Figure 7.

Mitochondrial depolarization and redox state perturbed in MCF-7 cells stably expressing RPLP0 shRNA, RPLP1 shRNA, or RPLP2 shRNA vectors. (A) RPLP0 shRNA-, RPLP1 shRNA-, and RPLP2 shRNA-associated alterations in the levels of proteins that are upregulated by a factor of 1.5 or more in MCF-7 cells. Only portions of the 2D gel images are shown in each case (spots relevant to HSPB1, TXN, G6PH, and HSPA1A are highlighted with black circles). The right panel summarizes the proteins that were found to have the most significantly deregulated expression levels out of the 70 differentially expressed proteins (Table S1). (B) Western blotting analysis confirming the upregulation of the proteins identified in (A) as the most significantly deregulated proteins in MCF-7 cells expressing RPLP0 shRNA, RPLP1 shRNA, or RPLP2 shRNA with respect to controls expressing the NT shRNA vector. ACTB was used as a loading control. (C) Enhanced ROS generation after downregulation of RPLP0, RPLP1, or RPLP2 protein in MCF-7 cells. The percentages of DCFH-DA fluorescence represent the levels of intracellular O2−, as described in the Materials and Methods section. Parental MCF-7 cells treated with H2O2 were used as positive controls. Results were confirmed in 3 independent experiments (n = 3). Significant results were found for RPLP0, RPLP1, and RPLP2 deficient cells in comparison to control cells with *, P ≤ 0.05. (D) Flow cytometry-based evaluation of the change in the mitochondrial potential with the 7-AAD red fluorescent dye from the MitoPotential Red Kit. MCF-7 cells expressing RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA vector and parental MCF-7 cells untreated or treated with 2 µM Sts for 3 h (positive control) were stained following the manufacturer's instructions, as described in the Materials and Methods. Quadrant gates were set up on the control cells and applied to the Sts-treated cells and cells expressing the different shRNAs. RPLP0 shRNA, RPLP1 shRNA, and RPLP2 shRNA samples are shown. The positive control treated with Sts underwent a change in mitochondrial potential that is shown in the graph as a downward shift in Red2 fluorescence and was quantified as the percentage of cells in the lower left quadrant. RPLP0 shRNA, RPLP1 shRNA, and RPLP2 shRNA samples do not show cell death, as shown by the few cells with an increase in red fluorescence. Results were confirmed in 3 independent experiments (n = 3). Significant results were found for RPLP0, RPLP1 and RPLP2 deficient cells in comparison to control cells with *, P ≤ 0.05.
The UPR caused by RPLP protein deficiency is activated by ROS induction
TXN, which was the most significantly deregulated protein in the RPLP protein-deficient MCF-7 cells, is a protein that neutralizes the effect of oxidative damage. Therefore, we hypothesized that oxidative damage would be altered in our cells. To determine whether oxidative damage occurred in our cells, the presence of ROS levels, as a read-out of oxidative damage, was analyzed by flow cytometry. ROS levels were detected upon RPLP protein inhibition at similar levels to those provoked by H2O2, a strong oxidizing agent that induces ROS (Fig. 7C). Moreover, RPLP protein inhibition was associated with mitochondrial depolarization (Fig. 7D).
Since high ROS levels correlated with the presence of mitochondrial damage, the contribution of ROS and p-MAPK1, as potential key players in UPR induction and autophagy, was further studied in our cells. For this experiment, we inhibited ROS levels with N-acetyl-L-cysteine (NAC) and MAP2K1 (the direct MAPK1 activator) with U0126 inhibitor. Effective inhibition of ROS and p-MAPK1 was verified by western blot (data not shown). Remarkably, NAC treatment almost completely reversed (by ∼90%) the autophagic phenotype in MCF-7 cells with downregulated expression of RPLP0, RPLP1, and RPLP2, as shown by the absence of autophagosomes and autolysosomes by using TEM (Fig. 8A and B). In contrast, MAPK1 inhibition did not rescue the autophagic phenotype (Fig. 8A and B). Concomitant inhibition of ROS and p-MAPK1 reversed the autophagic phenotype, similar to that observed following ROS inhibition alone (Fig. 8A and B). These results were confirmed by LC3-II conversion and acridine orange staining in the indicated cell lines (Fig. 8C and Fig. S8). Moreover, NAC treatment in cells with downregulated expression of RPLP0, RPLP1, or RPLP2 was able to revert cell cycle distribution previously observed (Fig. S9). To understand whether the observed reversal in the phenotype following treatment with NAC or NAC plus U0126 also involved a downregulation of the UPR, the expressions of ATF4, ATF6, p-EIF2S1, and p-EIF2AK3 proteins were analyzed by western blot. The results obtained in cells treated with NAC and NAC plus U0126 were consistent with the decreased activation of EIF2AK3, p-EIF2S1, ATF4, and ATF6 (Fig. 9). In contrast to NAC inhibition, the effect of MAPK1 inhibition was negligible.
Figure 8.

ROS-mediated autophagy in MCF-7 cells with downregulated RPLP proteins. (A) TEM analysis of MCF-7 cells expressing RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA vector, untreated or treated with 20 mM NAC, 10 µM U0126, or NAC plus U0126, as described in the Materials and Methods section. The lack of signs of autophagy such as autophagosomes (arrowheads) and autolysosomes (black arrows) are features of RPLP0 shRNA, RPLP1 shRNA, and RPLP2 shRNA samples treated with NAC, an ROS scavenger. Scale bar: 0.5 μm. (B) Quantifications are based on counting autophagic vacuoles in the field of view. The values are the mean ± S.D. of 3 independent experiments. *, P ≤ 0.05. (C) Western blot analysis of the LC3-II conversion form compared with ACTB in samples treated as in (A).
Figure 9.
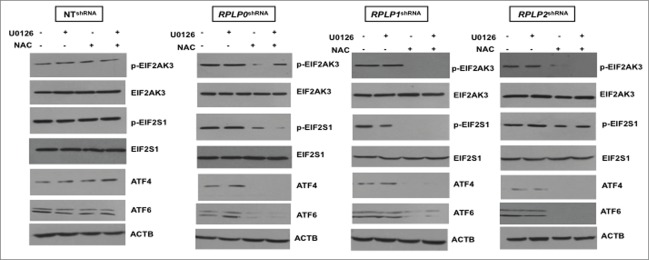
Downregulation of proteins related to the UPR in NAC-treated MCF-7 cells with downregulated RPLP proteins. Western blot analysis of the indicated UPR-related proteins in MCF-7 cells expressing RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA vector, treated as in Figure 8. Expressions of p-EIF2AK3 and p-EIF2S1 were compared with those of EIF2AK3 and EIF2S1 total proteins, respectively. Expressions of ATF4 and ATF6 were compared with those of ACTB.
These results indicate that the contribution of ROS levels upon downregulation of each RPLP protein is crucial for the generation of an autophagic response potentially mediated by UPR.
RPLP protein deficiency involves a sequence of events that results in autophagy
It was unclear if RPLP protein knockdown simply activates ROS, UPR, and autophagy at once as a stress reaction or if these events follow a hierarchical order. To determine how ROS, ER stress, and autophagy are related processes upon RPLP protein inhibition, MCF-7 cells were transduced with scrambled (Sc) siRNA and siRNAs against RPLP0, RPLP1, and RPLP2. Specifically, we used the siRNAs from which the lentiviral vectors were initially constructed (see Materials and Methods). First, we studied ROS levels at 12, 24, and 48 h after transfection. At 12 h, when evidence of RPLP protein inhibition was clear (Fig. S1A), ROS levels started to accumulate at very low concentrations, before further increasing (Fig. 10). At 12 h, there was no evidence of ER stress or UPR activation as there was no increase in ATF4, ATF6, p-EIF2S1, or p-EIF2AK3 protein levels (Fig. 11A). At 12 h, signs of autophagy were also absent in cells with downregulated RPLP proteins in comparison with NT shRNA-expressing cells, with a lack of LC3-II conversion (Fig. 11A and data not shown). The levels of these UPR-related proteins appeared to further increase at 24 and 48 h (Fig. 11A).
Figure 10.

ROS accumulates gradually in MCF-7 cells with downregulated RPLP proteins. (A) MCF-7 cells were transduced with the indicated siRNAs and analyzed for the presence of ROS levels at 12, 24, and 48 h after transfection. (B) The graph represents results from (A). Note that ROS levels accumulate gradually in RPLP protein-depleted cells to reach 18 to 28%.
Figure 11.

UPR- and autophagy-related proteins during ROS accumulation in MCF-7 cells with downregulated RPLP proteins. (A) MCF-7 cells were transduced with the indicated siRNAs (treated as in Fig. 10) and analyzed for the expression of p-EIF2AK3, p-EIF2S1, ATF4, ATF6, and LC3-II conversion. (B) Effect of EIF2AK3 inhibition by treatment with EIF2AK3 inhibitor GSK2606414 (10 μM) on p-EIF2AK3, ATF4, ATF6, LC3, and PARP1 proteins. ACTB was used as a loading control. (C) Proposed model for autophagy activation in stress-related conditions due to RPLP protein deficiency. Black arrows represent the preferred option used by ROS to activate autophagy. In a UPR-defective context, other pathways might operate alternatively to stimulate survival by autophagy (dashed arrows).
To observe the effect of ER stress/UPR inhibition on ROS levels and autophagy, we inhibited p-EIF2AK3 by using 2 different strategies. First, p-EIF2AK3 inhibition was performed with an siRNA approach, and verified by RNA and protein content (data not shown). Concomitant transfection of p-EIF2AK3 siRNA with the siRNA of each RPLP protein gene was performed and compared with Sc siRNA. p-EIF2AK3 inhibition did not influence ROS accumulation at 24 h after transfection (Fig. S10). At other time points, ROS levels were unaltered compared with control cells (data not shown). Moreover, depletion of p-EIF2AK3 decreased the activation of its target genes: EIF2S1, ATF4, and ATF6 in RPLP protein-deficient cells vs. control cells (ATF6 is also influenced by p-EIF2AK3)40 (data not shown).
The second strategy involved the p-EIF2AK3 inhibitor GSK2606414.41 The effect of the drug was tested in MCF-7 cells at different concentrations and time points to verify the optimal conditions for efficient inhibition of p-EIF2AK3 without cytotoxicity (data not shown). Furthermore, MCF-7 cells were induced to express RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, and control shRNA (NT shRNA) and treated for 24 h with 1 μM and 10 μM of GSK2606414. Cell cycle profile and apoptosis were studied by flow cytometry. We observed that GSK2606414 did not change the autophagic phenotype and did not induce cell cycle arrest or apoptosis compared with control (untreated) cells (Fig. S11 and data not shown). Overall, our results suggest that the biological processes that result from inhibition of each RPLP protein follow a sequential order: first ROS, then UPR, and finally autophagy. Moreover, UPR inhibition does not change the autophagy fate or replace it with apoptosis (Fig. 11B, Figs. S10 and S11). We propose a model in which ROS activates autophagy rapidly and efficiently by UPR activation. However, in UPR-defective cells, ROS is still able to stimulate autophagy by alternative pathways that may involve MAPK1 activation or MTOR inhibition (Fig. 11C).
Discussion
Although the structural organization of RPLP0, RPLP1, and RPLP2 proteins when forming the ribosomal stalk has been intensively investigated, little is known of the biological roles of these molecules. The importance of RPLP proteins inhibition yields in the fact that their downregulation might have important therapeutic consequences in cancer, particularly in hormone-dependent tumors.16 Moreover some proteins such as ricin are natural RPLP protein inhibitors.42 The present study demonstrates that the downregulation of RPLP0, RPLP1, or RPLP2 in human cancer cells leads to cell cycle arrest, with the cells ultimately surviving via autophagy. Interestingly, the deficiency of RPLP0, RPLP1, or RPLP2 RPs does not affect the monosome and polysome profiles. No abnormalities in ribosomal function, overall protein synthesis, or mRNA translation were detected. These observations are in line with a previous study showing that concomitant inhibition of RPLP1 and RPLP2 in human cells does not result in significant changes in mRNA translation and protein synthesis.21,43 In particular, these authors do not observe a significant reduction in the number of viable cells. This difference from our results might be due to methodological reasons (the inducible system they employed) and/or the cell types used. Indeed, the study was performed in human embryonic kidney (HEK-293T) cells and derived subclones. The kidney is reported to be one of the human tissues with the lowest gene expression levels for RPLP0, RPLP1, and RPLP2 proteins. This is in contrast to breast and ovarian cancers where RPLP protein levels are consistently upregulated with respect to the normal counterparts.7 Moreover, similar results have been recently described by us in an in vivo knockout model of the RPLP1 protein where overall protein synthesis is not affected.44 Overall, our data support the notion that RPLP proteins might have extraribosomal functions, which appear in stress-related conditions.
The absence of any of the RPLP proteins in the breast cancer cell lines MCF-7 and MDA-MB-231, the colon cancer cell lines HCT116 TP53−/− and HT-29, and the ovarian cancer cell line OV-90 has drastic consequences for cell proliferation, with the cells entering cell cycle arrest and showing autophagic features. Interestingly, inhibition of autophagy provokes apoptosis. In addition to the evidence of autophagy induction in this system from the results obtained with TEM, LC3 expression, and acridine orange staining; the autophagy inhibitor treatment and the fact that ATG7 silencing effectively suppresses autophagy confirms that RPLP protein inhibition provokes an autophagic response. Overall, our results confirm that there is no role for autophagy in cell death, removing any possibility that cell death occurs by excessive autophagy.
The autophagy induced by the deficiency of RPLP proteins was likely to be triggered by the accumulation of misfolded proteins because ER stress and the consequent cascade of reactions related to the UPR were activated. The activation of the ER stress response involved the p-EIF2AK3 and ATF6 branches of the UPR, but not the ERN1, and is independent of the L11-L23-L5-S7-TP53-MDM2 axis. As previously reported,45 the ERN1 signaling pathway is preferentially triggered as a cellular response to low levels of ER stress. In contrast, activation of EIF2AK3 and/or ATF6 is reported to occur when the stress is acute or severe enough to require more than a moderate increase in the level of ER chaperones.46 The cellular stress induced by the absence of RPLP0, RPLP1, or RPLP2 must be so severe that the cell defense mechanism triggers a prosurvival response, flipping the switch to apoptosis if autophagy is inhibited.
Ribosomal stress has been reported to couple the UPR to TP53-dependent cell cycle arrest.47 Certain RPs can be released into the nucleoplasm and bind to MDM2, leading to TP53 stabilization and activation.48-50 Our data show that TP53 is not activated in response to RPLP0, RPLP1, or RPLP2 depletion and it is not responsible for the G2/M cycle arrest that occurs after RPLP protein downregulation because the cells that do not express active TP53 (RPLP protein-deficient MDA-MB-231 and HCT116 TP53−/−) also arrest in G2/M and undergo autophagy. However, the increase in TP53 and CDKN1A is probably responsible for the apoptosis that is specifically induced in RPLP protein-deficient MCF-7 cells treated with 3-MA. These RPLP protein-deficient cells have high levels of intracellular ROS, and it is recognized that deregulation of the cellular redox state can impact the levels and activity of TP53.51,52 Higher levels of ROS appear to be part of a feed-forward loop that stabilizes TP53, which may occur in MCF-7 cells treated with 3-MA, resulting in increased TP53 activity that interferes with mitochondrial function and/or integrity, and eventually contributes to cell death.53 Recent research reveals that autophagy is activated by TP53 and members of the MAPK11/12/13/14 family.54,55 Moreover, the PIK3CA-AKT1-MTOR pathway suppresses autophagy.56 However, the role of all of these proteins in autophagy is complex as they are also involved in apoptotic cell death.56-59 Accordingly, we also found a striking MTOR and AKT1 downregulation with 3-MA treatment that results in cell death.
Our results suggest that the survival response driven by autophagy generated by RPLP protein downregulation is triggered by ROS. High ROS levels might also contribute to HSP activation (i.e., HSPA1A and HSPB1), similar to that which occurs under other stress conditions.60 Our results are in agreement with those of Wong et al.,61 who report that autophagic induction can occur via ROS production. Moreover, we showed that there is the following sequence of events: first ROS accumulation, which triggers the UPR response, and finally autophagy. ROS inhibition alone is able to reverse the stress in these cells and restore the standard growth conditions without signs of autophagy activation as a survival response. However, UPR inhibition does not change the fate of the cells that still experience autophagy. We hypothesize that in such a case a different pathway than UPR activates the autophagy triggered initially by ROS (Fig. 11C).
Our results suggest that RPLP proteins have potential extraribosomal functions related to the maintenance and/or restoration of ER homeostasis, a process that occurs outside the ribosome but within the ER compartment. This is supported by a recent report where RPLP1 protein deficiency is associated with the activation of proteins related with protein folding and UPR.44 To our knowledge, the present research is the first report that stresses the importance of RPLP proteins for cell survival in human cells, as deficiency of RPLP0, RPLP1, or RPLP2 leads to cell cycle arrest accompanied by autophagy. The UPR activation and the drastic autophagic phenotype caused by the absence of RPLP proteins can be reversed by an ROS scavenger. This indicates that the redox imbalance is the central event leading to autophagy and cell cycle arrest in cancer cells depleted of RPLP0, RPLP1, or RPLP2. Moreover, our results suggest that MAPK1 can act as a sensor molecule of ribosomal stress, and is potentially able to contribute to the autophagic response if UPR is inhibited.
The role of autophagy in cancer is complex. Although some studies indicate that autophagy inhibition by drug treatment or genetic downregulation of autophagy genes sensitizes cancer cells to death, there is increasing evidence that supports the view that autophagy also suppresses tumor growth.62,63 The pro-oncogenic or tumor suppressor function of autophagy is likely to be context-dependent. With respect to cancer cells where downregulation of RPLP proteins is induced, the upregulation of autophagy by ER stress that ultimately leads to cell survival represents a mechanistic view of how autophagy can be exploited in cancer strategies. In support of this concept, we have previously reported that RPLP proteins seem to play an important role in breast cancer, which agrees with previous research from other research groups concerning RPLP proteins in tumorigenesis.15,16,64-66 The fact that the overexpression of RPLP proteins in cancer can be potentially related to a defect in autophagy suggests that translational approaches of autophagy-based therapies should be considered. For example, it would be very interesting to explore if the antiproliferative effect of gonadotropin-releasing hormone analogs that inhibit RPLP1 and RPLP2 is linked to an autophagic response.16 If so, downregulation of RPLP proteins and enhanced autophagy may be useful for improving cancer responses to therapy.
Materials and Methods
Cell lines and antibodies
MCF-7, MDA-MB-231, HeLa, OV-90, HT-29, and HEK-293T cells were obtained from American Type Culture Collection (ATCC, HTB-22, HTB-26, CCL−2, CRL-11732, HTB-38, CRL-11268 respectively). HCT116 TP53−/− cells were a gift from B. Vogelstein (The John Hopkins Oncology Center, Baltimore, MD).
MCF-7, MDA-MB-231, HeLa, OV-90, HT-29, and HCT116 TP53−/− cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Lonza, L01102–500) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. HEK-293T cells were cultured in DMEM/F12 (Lonza, BE 12–615F) with 10% FBS (Lab Clinics, 51810–500) and 2 mM L-glutamine (Lab Clinics, X0550–100). The cells were maintained in a humidified atmosphere of 5% CO2 at 37°C.
Antibodies were obtained or purchased from the following sources: rabbit polyclonal anti-G6PD, rabbit polyclonal anti-MAPK1, rabbit polyclonal anti-PARP1, mouse monoclonal anti-TXN, rabbit polyclonal anti-ATF6, and rabbit polyclonal anti-ATF4 from Santa Cruz Biotechnology (sc-373886, sc-154, sc-7150, sc-365658, sc-22799, sc-22800); rabbit polyclonal anti-phospho-MAPK1, rabbit polyclonal anti-phospho-AKT1, mouse monoclonal anti-AKT1, rabbit polyclonal anti-phospho-MTOR, rabbit polyclonal anti-MTOR, rabbit polyclonal anti-phospho-EIF2S1, rabbit polyclonal anti-EIF2S1, rabbit monoclonal anti-phospho-EIF2AK3, rabbit monoclonal anti-EIF2AK3, mouse monoclonal anti-HSPB1, and rabbit polyclonal anti-HSPA1A from Cell Signaling Technology (9101, 3787, 2920, 2971, 2972, 9721, 9722, 3179, 3192, 2402, 4873); rabbit polyclonal anti-LC3, rabbit polyclonal anti-RPLP0, rabbit polyclonal anti-RPLP1, and mouse polyclonal anti-ACTB from Sigma-Aldrich (L7543, HPA003512, HPA003368, A5616); rabbit polyclonal anti-RPLP2 from Abgent (AP9327a); mouse monoclonal anti-TP53 from Dako (N1581), rabbit monoclonal proCASP6 from Novus Biologicals (EP1325Y), rabbit polyclonal anti-BECN1 from Sigma-Aldrich (B6186), rabbit polyclonal anti-SQSTM1 from Sigma-Aldrich (P0067), rabbit polyclonal anti-EIF2AK4 from Cell Signaling Technology (3302) and goat anti-rabbit Alexa Fluor 667 from Life Technologies (A21244).
siRNA
The following Ambion Silencer predesigned small interfering RNAs (siRNAs) (Ambion, AM16708A) were used: RPLP0 siRNA1 CCCUGAAGUG CUUGAUAUC and RPLP0 siRNA2 CGGGUACAAA CGAGUCCUGtt for the specific targeting of RPLP0 mRNA; RPLP1 siRNA1 GGAGAAGAAA GUGGA-AGCA, RPLP1 siRNA2 GGAGUCUGAU GAUGACAUGtt, and RPLP1 siRNA3 GGAGUCUGAA GAUGACAUGtt for RPLP1; and RPLP2 siRNA1 GGAGGAGUCU GAAGAGUCA, RPLP2 siRNA2 GGUUAUCAGU GAGCUGAAUtt, and RPLP2 siRNA3 GGAGUCUGAA GAGUCAGAUtt for RPLP2 mRNA, ATG7 siRNA1 CGCUUAACAU UGGAGUUCAG and ATG7 siRNA2 GUGUUUAUGA ACUGCCAGGU (Ambion, AM16708), and EIF2AK3 siRNA CAACAAGAAU AUCCGCAAAtt (Ambion, AM4390824). Controls consisted of Silencer negative control siRNA (Sc) (Ambion, AM4611) and Silencer positive control Cy3-labeled GAPDH siRNA (Ambion, AM4623).
MCF-7, MDA-MB-231, HCT116 TP53−/−, HT-29, HeLa, and OV-90 cells were plated in 12-well plates and grown to 50 to 60% confluence, followed by serum starvation for 16 h. Cells were transfected with siRNAs (100 nM) in the presence of Lipofectamine 2000 reagent (Life Technologies, 1248311). Three independent siRNAs for each RPLP protein gene were tested for each transfection versus control siRNAs. MCF-7 cells were plated in 6-well plates and concomitantly transduced with EIF2AK3 siRNA plus the siRNAs corresponding to each RPLP protein gene vs. EIF2AK3 siRNA plus Sc siRNA as control. In all cases, transfection efficiency was ∼80 to 90%, as indicated by the Cy3-labeled siRNA (data not shown). After transfection, cells were harvested at 24 h, 48 h, and 72 h for additional analysis. The efficiency of the siRNA silencing was assessed with total RNA from all samples, which was converted to cDNA, and analyzed with quantitative real-time PCR (qRT-PCR) with TaqMan® Gene Expression Assays (see RNA extraction and quantitative real-time PCR section). All experiments were performed in triplicate, and representative results are reported.
Lentiviral constructs and transduction
For lentiviral transduction, RPLP0 siRNA1 (CCCUGAAGUG CUUGAUAUC) and RPLP0 siRNA2 CGGGUACAAA CGAGUCCUGtt targeting RPLP0, RPLP1 siRNA1 (GGAGAAGAAA GUGGAAGCA) and RPLP1 siRNA2 GGAGUCUGAU GAUGACAUGtt targeting RPLP1, and RPLP2 siRNA1 (GGAGGAGUCU GAAGAGUCA) and RPLP2 siRNA2 GGUUAUCAGU GAGCUGAAUtt targeting RPLP2 -selected for their highest levels of transcript reduction- were cloned into the pLKO.1-puro backbone (Sigma-Aldrich, SHC001) to obtain the following short hairpin RNA (shRNA) vectors: RPLP0 shRNA, gatccGCCCT GAAGTGCTTG ATATCTTCAA GAGAGATATC AAGCACTTCA GGGTTTTTTA CGCGTg; RPLP1 shRNA, gatccGGAGA AGAAAGTGGA AGCATTCAAG AGATGCTTCC ACTTTCTTCT CCTTTTTTAC GCGTg; and RPLP2 shRNA, gatccGGAGG AGTCTGAAGA GTCATTCAA GAGATGACTC TTCAGACTCC TCCTTTTTTA CGCGTg. A nontargeting shRNA vector, NT shRNA (Sigma-Aldrich, SHC002), was used as a control for lentiviral infections.
For the production of lentiviral particles, HEK-293T cells were cotransfected with pCMV-dR8.91 dvpr, pCMV-VSV-G (kindly provided by Prof. D. Trono, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland), and RPLP0, RPLP1 or RPLP2-pLKO.1 shRNA with JetPei (Genycell Biotech, 101–05). The supernatant was collected 48 h after transfection and concentrated by ultracentrifugation in Centricon Plus-100 filters (Millipore, 831826).
Cloning of RPLP0, RPLP1, and RPLP2 genes
Total RNA was isolated from normal human mammary tissue (see RNA extraction section). Further the coding region corresponding to human RPLP0, RPLP1 and RPLP2 genes was obtained by PCR amplification using the Superscript III-One Step kit (Invitrogen, 12574–030). The primers used were: for RPLP0 FORWARD: TGGAAGTGAATTCGTCTTTAA-ACCCTGC, REVERSE: TAAAACTCGAGAAGTTGGTTGC, for RPLP1 FORWARD: TCGGGAATTCCGAGGAAGC, REVERSE: CAGCTTTTTCTCGAGCATG and for RPLP2 FORWARD: ACGCGTGAGAATTCTCCG, REVERSE: TAAAAAGGCTCGAGTTGCAGGGGAGC. The enzymes used for the cloning were EcoRI (inserted in the Forward primer; New England Biolabs, R3101) and XhoI (inserted in the Reverse primer; New England Biolabs, R0146) and the vector used for their expression in human cells was pMaRx as previously reported.14 After cloning, results were confirmed by sequencing (data not shown).
Lentiviral/retroviral infection and cell treatments
For the generation of stable cell lines with downregulated expression of RPLP0, RPLP1, and RPLP2, MCF-7 cells were infected with viral particles at a 1:4 dilution in the presence of 8 µg/ml polybrene (Sigma-Aldrich, 107689). After 24 h, cells harboring the RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, or NT shRNA cassettes were selected in the presence of puromycin (1.5 µg/ml; Sigma-Aldrich) for 3 d. Expression of the constructs was confirmed by fluorescence microscopy and western blotting. For stable expression of the RPLP0, RPLP1 and RPLP2 proteins, MCF-7 cells were infected by retrovirus as described.14
For additional analysis (as indicated in the text and figures), puromycin-selected cells were plated, grown for 36 h, and treated with 2 mM DTT (Sigma-Aldrich, D9779) for 1.5 h; 1 µM taxol (Sigma-Aldrich, T1912) for 3 days; 2.5 µM OHT (Sigma-Aldrich, T5648) for 4 days; 0.75 µM doxorubicin (Sigma-Aldrich, D1515) for 2 h; 10 mM 3-MA (Sigma-Aldrich, M9281) for 48 h; 10 µM CQ (Sigma-Aldrich, C6628) for 12 h, 24 h, and 48 h; 10 µM MAP2K1 inhibitor U0126 (Sigma-Aldrich, U120) for 15 h; 20 mM NAC (Sigma-Aldrich, A7250) for 48 h; 2 µM staurosporine (Sts; Sigma-Aldrich, S5921) for 3 h; 1 μM and 10 μM EIF2AK3 inhibitor GSK2606414 (VWR International, 516535) for 24 h; 100 µg/ml cycloheximide (CHX; Sigma-Aldrich, C1988) for 4 h; and a combination of the lysosomal enzyme inhibitors E64 (Sigma-Aldrich, E3132) at 10 µg/ml and pepstatin A (Sigma-Aldrich, P5318) at 10 µg/ml for 24 h.
Growth curves
MCF-7, MDA-MB-231, HeLa, OV-90, HCT116 TP53−/−, and HT-29 cells were seeded at 1 × 106 cells/10-cm plate after siRNA transfection or shRNA infection and selection. In cells transfected with siRNA, proliferation was determined by a trypan blue (Sigma-Aldrich, T8154) exclusion test. Cells were harvested after transfection with siRNA on d 3, 6, and 9. Following a 3 to 5 min incubation in trypan blue, the viable cells were counted under a microscope.
In the case of cells expressing shRNA, cells were counted every 3 d and seeded at the same density as that indicated by the 3T3 protocol.67 The relative number of cells was considered a measure of the number of cells per passage related to the initial number of cells seeded per plate. In addition, duplicates were stained with crystal violet (Sigma-Aldrich, HT90132) at each passage, destained with 10% acetic acid (Sigma-Aldrich, A6283), and quantified at 595 nm to count relative cell numbers.
To assess cell proliferation by another method, colony formation assays were performed. Once cells were infected and selected with puromycin, cells were seeded at a density of 0.3 × 104 cells/per well in a 6-well plate. Cells were continuously cultured, and growth media was changed every 3 d. After 20 d, cells were fixed with 0.5% glutaraldehyde (Sigma-Aldrich, G5882), stained with crystal violet, and cell proliferation was assessed by observing the appearance of the growing colonies.
RNA extraction and quantitative real-time PCR
Cells were collected by centrifugation and total RNA was isolated with the RNeasy Mini Kit (Qiagen, 74104) following the manufacturer's instructions. Random primers of the Revert Aid H Minus cDNA Synthesis Kit (Fermentas, K1631) were used to carry out cDNA synthesis from 1.5 µg of total RNA according to the manufacturer's instructions. RPLP0, RPLP1, and RPLP2 expression was detected with the TaqMan Gene Expression Assay (Applied Biosystems, Hs99999902_m1, Hs01653088_g1, and Hs01115130_g1, respectively). An ABI PRISM 7000 instrument (Applied Biosystems, Foster City, CA, USA) was used to perform the relative quantification analysis. Data were analyzed with the 7000 Sequence Detection Software, v.1.2.3 (Applied Biosystems). The PCR cycling program consisted of denaturing at 95°C for 10 min and 40 cycles at 95°C for 15 s, and annealing and elongation at 60°C for 1 min. The PCR was performed in triplicate and 2 different probes for assessing endogenous mRNA levels were used: ACTB/BETA-ACTIN (Hs99999903_m1; Applied Biosystems) and POLR2A (Hs00172187_m1; Applied Biosystems).
Western blot analysis
Cultured cells were washed in phosphate-buffered saline (PBS; Labs Clinics, X0515–500) and lysed on ice for 15 min in lysis buffer containing 50 mM HEPES-NaOH, pH 7.4, 1% NP-40 (Sigma-Aldrich, 9016–45–9), 10% glycerol, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, protease inhibitors cocktail (Sigma-Aldrich, P2340), and phosphatase inhibitors cocktail (Millipore, 524625–1SFT). Cell lysates were centrifuged at 15,000 × g for 30 min at 4°C and the supernatant fraction was collected. The protein concentrations were determined with a Bradford assay (Bio-Rad, 500–0006), and equivalent amounts of protein were denatured, resolved by SDS-PAGE (6–12%), and transferred to PVDF membrane (Millipore, 88518). The membranes were blocked for 1 h in 5% nonfat dry milk in Tris-buffered saline (50 mM Tris-Cl, ph 7.6, 150 mM NaCl), 0.1% (v/v) Tween-20 (TBST) (Sigma-Aldrich, P9416) before an overnight incubation at 4°C with primary antibodies. HRP-conjugated secondary antibodies were incubated at room temperature (RT) for 1 h (Cell Signaling Technology, anti-rabbit 7074 and anti-mouse 7056). Proteins were detected with an enhanced chemiluminescence kit (Millipore, 345818) according to the manufacturer's instructions.
Analysis of senescence
Senescence-associated β-galactosidase (SA-β-gal) activity68 was determined with a Senescence β-galactosidase commercial assay following the manufacturer's instructions (Cell Signaling Technology, 9860).
Detection of apoptosis
Apoptosis was determined by studying morphological changes in the nuclear chromatin of cells stained with Hoechst 33258 (bisbenzimide; Sigma-Aldrich, 861405). Cells were fixed for 10 min in 4% paraformaldehyde and permeabilized for 2 min in 100% ethanol. Following 2 washes with PBS, cells were incubated in TBST buffer containing 0.1 µg/ml Hoechst 33258 for 10 min at RT. The results were expressed as the percentage of apoptotic cells over the total number of cells counted. Similar results were obtained in at least 3 independent experiments. Analysis of apoptotic cells was also performed with the ANXA5/annexin V-FITC Apoptosis Detection Kit (eBioscience, 88–8007–72) according to the manufacturer's instructions. Samples were analyzed with a FACSCalibur flow cytometer (BD FACSCaliburTM, BD Biosciences, San Jose, CA, USA). For each cell line, 20,000 cells were collected and analyzed with the FCS Express Software.
Apoptosis induction was also evaluated according to the cleaved status of PARP1 on western blot.
Detection of autophagy
To detect acidic vesicular organelles, including the autolysosomes that are characteristic of autophagy, cells were stained with acridine orange as described previously.22 Cells were seeded in round coverslips, washed with PBS, and mounted in a microscope slide where a suspension of 1 μg/ml of acridine orange (Sigma-Aldrich, A6014) was applied. Cells were viewed under a fluorescent microscope Olympus IX71 (OLYMPUS IBERIA, S.A.U., Barcelona, Spain). The distinctive red staining that is due to acridine orange staining of autolysosomes in the cytoplasm was considered indicative of late autophagy. At least 200 cells were counted for each treatment. Three independent experiments were performed. Autophagy evaluation was also performed using a confocal microscope Olympus FV1000 (OLYMPUS IBERIA, S.A.U., Barcelona, Spain) in cells stained with both acridine orange and Hoechst. Autophagy induction was also evaluated according to LC3-I conversion to LC3-II by immunoblotting as previously described.36 Autophagy was also detected by confocal microscopy by using 2 different approaches: (a) BECN1 immunocytochemistry in cells expressing RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, and NT shRNA; and (b) transfection of MCF-7 cells with a GFP-LC3 plasmid. Transfected cells were sorted by flow cytometry and then induced to express RPLP0 shRNA, RPLP1 shRNA, RPLP2 shRNA, and NT shRNA. Finally, autophagy was evaluated by using TEM as previously described.19
Cell cycle analysis
For DNA content analysis, cells were harvested after treatment. One million cells were fixed in 70% ethanol for 15 min at −20°C. Afterward, cells were treated with 100 μg/ml RNase A (Sigma-Aldrich, R4875) and stained with 50 μg/ml of propidium iodide (Sigma-Aldrich, 81845). DNA content was analyzed on a FACSCalibur flow cytometer (BD Biosciences). For each cell line, 20,000 cells were collected and the percentage of cells in each phase of the cell cycle was determined with the FCS Express Software.
Protein synthesis measurement
Cells were washed twice with PBS and incubated for 2 h with DMEM without methionine (Invitrogen, 21013–024) and supplemented with 10% dialyzed FBS (Invitrogen, 26400–044). Afterward, cells were incubated for 30 min in the same medium supplemented with 50 µCi/ml [35S]-methionine (PerkinElmer Life Sciences, NE6709A500UC). The cells were then washed 3 times with ice-cold PBS, lysed for protein extraction, and the protein was precipitated with trichloroacetic acid (Sigma-Aldrich, 91228) and acetone (Sigma-Aldrich, 650501). The total radioactive isotope incorporation was measured with liquid scintillation counting and normalized to cpm/µg total protein. A positive control involved cells treated with 100 µg/ml of CHX (Sigma-Aldrich, C4859) for 4 h.
Reverse transcription PCR analysis of XBP1 mRNA splicing
Total RNA was extracted for cDNA synthesis. To detect human unspliced and spliced XBP1 mRNA, PCR was performed with the primers 5′-CTG GAA CAG CAA GTG GTA GA-3′ and 5′-CTG GGT CCT TCT GGG TAG AC-3′ as described previously.69 Unspliced (XBP1, 398 bp) and spliced (XBP1-s, 424 bp) XBP1 fragments were separated on 2% agarose gels, stained with ethidium bromide (Sigma-Aldrich, E7637), and photographed (Bio-Rad Fluor-S Multimager).
Analysis of polysomes
The number of ribosomes within the polysomal mRNA fraction (mRNA containing 2 or more ribosomes) is a reflection of de novo protein synthesis. Ribosome profiles were prepared from a total of 2 × 107 MCF-7 cells at 80% confluence, which were washed with ice-cold PBS containing 100 μg/ml CHX to block ribosomes in the elongation step. Cells were lysed with buffer A (15 mM Tris-HCl, pH 7.4, 80 mM KCl, 5 mM MgCl2, and 100 μg/ml CHX) containing 1% (v/v) Triton X-100 (Sigma-Aldrich, T8787), 40 U/ml RNasin (Promega, N2114), and protease inhibitors (Sigma-Aldrich, P2340). Cytoplasmic extracts were obtained after centrifugation at 15,000 × g for 30 min at 4°C. They were loaded on to a linear 10 to 50% (w/v) sucrose gradient in buffer A and then centrifuged at 192,072 x g in an SW40 Ti rotor (Beckman Coulter, Brea, CA, USA) for 2 h at 4°C. Gradients were fractionated by upward displacement with 87% (v/v) glycerol in an ISCO density-gradient fractionator. Absorbance at 260 nm was monitored continuously with an ISCO UA-5 UV monitor (Teledyne ISCO, Lincoln, NE, USA). The overall translation efficiency, the average number of ribosomes per mRNA, and the relative ribosome content were calculated as described previously.21
Intracellular ROS detection
ROS was detected by DCFH-DA (5–6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester; Molecular Probes, C10491) as described previously.70 Briefly, cells were loaded with 10 µmol/L DCFH-DA in PBS for 30 min at 37°C in 5% CO2. Cells were washed in PBS and measured immediately for the formation of the fluorescent-oxidized derivative of DCFH-DA at an excitation wavelength of 488 nm and an emission wavelength of 525 nm on a FACSCalibur flow cytometer and FCS Express Software. For each sample, at least 10,000 events were analyzed in each of 3 independent experiments.
Mitochondrial assay
The Millipore FlowCellect™ MitoPotential Red Kit (Millipore, FCCH100105) was used to determine the percentage of cells with a depolarized membrane potential in culture. Cells with an intact mitochondrial membrane potential show high Red2 fluorescence (emission, 650 nm), whereas cells with an impaired mitochondrial membrane potential show a lower Red2 fluorescence. The assay was performed following the manufacturer's instructions. Briefly, cells were collected and resuspended at 1 × 106 cells/ml in 1X HSC Assay Buffer (Millipore, 4700–1325). For every 100 μl of cells in suspension, 100 µl of MitoPotential Red Working Solution was added. Cells were incubated for 15 min at 37°C in a CO2 incubator. After incubation, cells were centrifuged and washed with 1X HSC Assay Buffer. Seven-AAD reagent (5 µl; Sigma-Aldrich, A9400) was added to the samples before immediate acquisition on a FACSCalibur flow cytometer equipped with 488 nm and 633 nm lasers.71
2-D electrophoresis and mass spectrometry
Sample preparation
Control MCF-7 cells (NT shRNA) and MCF-7 cells expressing shRNAs for RPLP0, RPLP1, and RPLP2 vectors were lysed by the addition of 400 µl of lysis buffer (7 M urea [Sigma-Aldrich, U6504], 2 M thiourea [Sigma-Aldrich, T8656], 4% CHAPS [Life Technologies, 28300], 30 mM Tris, pH 8.5). The mixture was sonicated 8 times for 10 s on ice and then centrifuged at 12,000 × g at 4°C for 3 min. Each protein extract (100 μl) was further purified by a modified TCA-acetone precipitation kit (2-D-CleanUp kit; GE Healthcare, 80–6484–51) and finally resuspended in lysis buffer. Protein concentration was determined by using the Bio-Rad RCDC Protein Assay (Bio-Rad, 500–0112-MSDS).
Two-dimensional differential in-gel electrophoresis (2D-DIGE)
A pool consisting of equal amounts of each of the samples analyzed in the DIGE experiment was prepared as an internal standard for quantitative comparisons. Tissue samples were labeled with Cy3 (GE Healthcare, 25–8010–83) or Cy5 (GE Healthcare, 25–8010–85 cyanine dyes, whereas the internal standard pooled sample was labeled with Cy2 dye (GE Healthcare, 25–8010–82). The 2D-DIGE was performed using GE Healthcare reagents and equipment (GE Healthcare). First-dimension IEF was performed on IPG strips (GE Healthcare, 17–6002–44) (24 cm; linear gradient pH 3–10) by using an Ettan IPGphor system (Amersham Biosciences, 80–6414–02). Second-dimension SDS-PAGE was run by overlaying the strips on 12.5% isocratic Laemmli home made gels (24 cm × 20 cm) (Bio-Rad, 161–0158), cast in low fluorescence glass plates, on an Ettan DALTsix system (Amersham Biosciences, 80–6485–27). Gels were run at 20°C at a constant power of 2.5 W/gel for 30 min followed by 17 W/gel until the bromophenol blue tracking front reached the end of the gel. Fluorescence images of the gels were acquired on a Typhoon 9400 scanner (GE Healthcare, Freiburg, Germany, 63–0055–78). Cy2, Cy3, and Cy5 images were scanned at 488 nm/520 nm, 532 nm/580 nm and 633 nm/670 nm excitation/emission wavelengths, respectively, at a 100-μm resolution. Image analysis and statistical quantification of relative protein abundances were performed using Progenesis SameSpots v2.0 software (NonLinear Dynamics, Newcastle, UK).
Protein identification
Protein spots of interest were excised from the gel using an automated Spot Picker (GE Healthcare, 18–1145–28). In-gel trypsin digestion was performed using autolysis-stabilized trypsin (Promega, V5280). Tryptic digests were purified using ZipTip microtiter plates (Millipore, ZPC180010). MALDI-MS (matrix-assisted laser desorption/ionization mass spectrometry) analysis of tryptic peptides was performed on an Autoflex Speed TOF-TOF Instrument (Bruker Daltonics, Bremen, Germany). Samples were prepared using α-cyano-4-hydroxy-cinnamic acid as a matrix on anchor-chip targets (Bruker Daltonics, 209512). Calibration was performed in the external mode using a peptide calibration standard kit (Bruker Daltonics, 222570). The spectra were processed using Flex Analysis 3.0 software (Bruker Daltonics). Peak lists were generated using the signals in the m/z 800 to 4000 region, with a signal-to-noise threshold of greater than 3. The SNAP algorithm included in the software was used to select the monoisotopic peaks from the isotopic distributions observed. After removing m/z values corresponding to usually observed matrix cluster ions, an internal statistical calibration was applied. Peaks corresponding to frequently seen keratin and trypsin autolysis peptides were then removed. The resulting final peak list was used for the identification of the proteins by peptide mass fingerprinting. Mascot 2.2 program (Matrix Science Ltd., London, UK) was used to search the SWISS-PROT 55.4 database, limiting the search to human proteins (19,630 sequences). The search parameters were as follows: trypsin cleavages excluding N-terminal to proline, one or 2 missed cleavages allowed, carbamidomethylation set as fixed modification, methionine oxidation as variable modification, mass tolerance less than 50 ppm, and monoisotopic mass values. The criterion for positive identification was a significant Mascot probability score (score >55, P < 0.05). Alternatively, proteins were identified by ion trap mass spectrometry.72
Table 1.Experimental design
| Gel | Cy2 | Cy3 | Cy5 |
|---|---|---|---|
| 1 | pool | MCF-7_NT_EXP 1 | MCF-7_RPLP1_EXP 1 |
| 2 | pool | MCF-7_RPLP0_EXP 1 | MCF-7_RPLP2_EXP 1 |
| 3 | pool | MCF-7_RPLP2_EXP 2 | MCF-7_NT_EXP 2 |
| 4 | pool | MCF-7_RPLP1_EXP 2 | MCF-7_RPLP0_EXP 2 |
| 5 | pool | MCF-7_NT_EXP 4 | MCF-7_RPLP0_EXP 4 |
| 6 | pool | MCF-7_RPLP1_EXP 4 | MCF-7_RPLP2_EXP 4 |
Deregulated proteins common to RPLP0 shRNA, RPLP1 shRNA, and RPLP2 shRNA were further confirmed by western blot.
Statistical procedures
Pairwise differences between groups were analyzed using the Student t test (proliferation curves, mRNA study by qRT-PCR, quantification of BECN1 protein staining by immunocitochemistry and LC3II, RPLP0, RPLP1, and RPLP2 proteins by western blot, quantification of vacuoles by TEM, detection of ROS, changes in the mitochondrial potential, cell cycle profiles, apoptosis, and senescence by flow cytometry). A P value of less than 0.05 or 0.01 (when specified) was considered statistically significant. Pairwise differences between groups were analyzed using the ANOVA test (proteomic analysis which involved 2-D eletrophoresis and mass spectrometry). A P value of less than 0.05 was considered statistically significant. Additionally, the differential spots were filtered for a minimum fold-change of 1.5.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are very grateful to Dr. J.P. García-Ballesta (CSIC, Spain) for his help with the RPLP protein antibody. We are very grateful to Dr. George Thomas and Dr. Antonio Gentilella for their help with the ribosomal profile analysis. We are grateful to the Cryo-Electron Microscopy Unit at the Scientific & Technological Centers of the University of Barcelona. We are grateful to Marta Valeri for her support with confocal microscopy.
Funding
This work was supported by the FIS project PI12/01104. M.E.LL. is a FIS investigator (CP03/00101). AF is funded by a FIS fellowship.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Ballesta JP, Remacha M. The large ribosomal subunit stalk as a regulatory element of the eukaryotic translational machinery. Prog Nucleic Acid Res Mol Biol 1996; 55:157-93; PMID:8787610 [DOI] [PubMed] [Google Scholar]
- 2.Naganuma T, Shiogama K, Uchiumi T. The N-terminal regions of eukaryotic acidic phosphoproteins P1 and P2 are crucial for heterodimerization and assembly into the ribosomal GTPase-associated center. Genes Cells 2007; 12:501-10; PMID:17397397; http://dx.doi.org/ 10.1111/j.1365-2443.2007.01067.x [DOI] [PubMed] [Google Scholar]
- 3.Lee KM, Yu CW, Chan DS, Chiu TY, Zhu G, Sze KH, Shaw PC, Wong KB. Solution structure of the dimerization domain of ribosomal protein P2 provides insights for the structural organization of eukaryotic stalk. Nucleic Acids Res 2010; 38:5206-16; PMID:20385603; http://dx.doi.org/ 10.1093/nar/gkq231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan DS, Chu LO, Lee KM, Too PH, Ma KW, Sze KH, Zhu G, Shaw PC, Wong KB. Interaction between trichosanthin, a ribosome-inactivating protein, and the ribosomal stalk protein P2 by chemical shift perturbation and mutagenesis analyses. Nucleic Acids Res 2007; 35:1660-72; PMID:17308345; http://dx.doi.org/ 10.1093/nar/gkm065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCluskey AJ, Poon GM, Bolewska-Pedyczak E, Srikumar T, Jeram SM, Raught B, Gariepy J. The catalytic subunit of shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain. J Mol Biol 2008; 378:375-86; PMID:18358491; http://dx.doi.org/ 10.1016/j.jmb.2008.02.014 [DOI] [PubMed] [Google Scholar]
- 6.Chiou JC, Li XP, Remacha M, Ballesta JP, Tumer NE. The ribosomal stalk is required for ribosome binding, depurination of the rRNA and cytotoxicity of ricin A chain in Saccharomyces cerevisiae. Mol Microbiol 2008; 70:1441-52; PMID:19019145; http://dx.doi.org/ 10.1111/j.1365-2958.2008.06492.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishii K, Washio T, Uechi T, Yoshihama M, Kenmochi N, Tomita M. Characteristics and clustering of human ribosomal protein genes. BMC Genomics 2006; 7:37; PMID:16504170; http://dx.doi.org/ 10.1186/1471-2164-7-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci U S A 2001; 98:4569-74; PMID:11283351; http://dx.doi.org/ 10.1073/pnas.061034498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yacoub A, Kelley MR, Deutsch WA. Drosophila ribosomal protein PO contains apurinic/apyrimidinic endonuclease activity. Nucleic Acids Res 1996; 24:4298-303; PMID:8932386; http://dx.doi.org/ 10.1093/nar/24.21.4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furukawa T, Uchiumi T, Tokunaga R, Taketani S. Ribosomal protein P2, a novel iron-binding protein. Arch Biochem Biophys 1992; 298:182-6; PMID:1524426; http://dx.doi.org/ 10.1016/0003-9861(92)90110-I [DOI] [PubMed] [Google Scholar]
- 11.Elkon KB, Parnassa AP, Foster CL. Lupus autoantibodies target ribosomal P proteins. J Exp Med 1985; 162:459-71; PMID:2410526; http://dx.doi.org/ 10.1084/jem.162.2.459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnard GF, Staniunas RJ, Mori M, Puder M, Jessup MJ, Steele GD Jr., Chen LB. Gastric and hepatocellular carcinomas do not overexpress the same ribosomal protein messenger RNAs as colonic carcinoma. Cancer Res 1993; 53:4048-52; PMID:8395335 [PubMed] [Google Scholar]
- 13.Loging WT, Reisman D. Elevated expression of ribosomal protein genes L37, RPP-1, and S2 in the presence of mutant p53. Cancer Epidemiol Biomarkers Prev 1999; 8:1011-6; PMID:10566557 [PubMed] [Google Scholar]
- 14.Artero-Castro A, Kondoh H, Fernandez-Marcos PJ, Serrano M, Ramon y Cajal S, Lleonart ME. Rplp1 bypasses replicative senescence and contributes to transformation. Exp Cell Res 2009; 315:1372-83; PMID:19233166; http://dx.doi.org/ 10.1016/j.yexcr.2009.02.007 [DOI] [PubMed] [Google Scholar]
- 15.Artero-Castro A, Castellvi J, Garcia A, Hernandez J, Ramon y Cajal S, Lleonart ME. Expression of the ribosomal proteins Rplp0, Rplp1, and Rplp2 in gynecologic tumors. Hum Pathol 2011; 42:194-203; PMID:21040949; http://dx.doi.org/ 10.1016/j.humpath.2010.04.020 [DOI] [PubMed] [Google Scholar]
- 16.Chen A, Kaganovsky E, Rahimipour S, Ben-Aroya N, Okon E, Koch Y. Two forms of gonadotropin-releasing hormone (GnRH) are expressed in human breast tissue and overexpressed in breast cancer: a putative mechanism for the antiproliferative effect of GnRH by down-regulation of acidic ribosomal phosphoproteins P1 and P2. Cancer Res 2002; 62:1036-44; PMID:11861379 [PubMed] [Google Scholar]
- 17.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004-10; PMID:18971948; http://dx.doi.org/ 10.1038/nrm2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navarro-Yepes J, Burns M, Anandhan A, Khalimonchuk O, del Razo LM, Quintanilla-Vega B, Pappa A, Panayiotidis MI, Franco R. Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid Redox Signal 2014; 21:66-85; PMID:24483238; http://dx.doi.org/ 10.1089/ars.2014.5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim Y, Kim YS, Kim DE, Lee JS, Song JH, Kim HG, Cho DH, Jeong SY, Jin DH, Jang SJ, et al.. BIX-01294 induces autophagy-associated cell death via EHMT2/G9a dysfunction and intracellular reactive oxygen species production. Autophagy 2013; 9:2126-39; PMID:24322755; http://dx.doi.org/ 10.4161/auto.26308 [DOI] [PubMed] [Google Scholar]
- 20.Wilkinson S, O'Prey J, Fricker M, Ryan KM. Hypoxia-selective macroautophagy and cell survival signaled by autocrine PDGFR activity. Genes Dev 2009; 23:1283-8; PMID:19487569; http://dx.doi.org/ 10.1101/gad.521709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Azorin F, Remacha M, Ballesta JP. Functional characterization of ribosomal P1/P2 proteins in human cells. Biochem J 2008; 413:527-34; PMID:18422483; http://dx.doi.org/ 10.1042/BJ20080049 [DOI] [PubMed] [Google Scholar]
- 22.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/ 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- 24.Chen M, Du Y, Qui M, Wang M, Chen K, Huang Z, Jiang M, Xiong F, Chen J, Zhou J, et al.. Ophiopogonin B-induced autophagy in non-small cell lung cancer cells via inhibition of the PI3K/Akt signaling pathway. Oncol Rep 2013; 29:430-6; PMID:23151908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ginet V, Spiehlmann A, Rummel C, Rudinskiy N, Grishchuk Y, Luthi-Carter R, Clarke PG, Truttmann AC, Puyal J. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy 2014; 10:846-60; PMID:24674959; http://dx.doi.org/ 10.4161/auto.28264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nihira K, Miki Y, Ono K, Suzuki T, Sasano H. An inhibition of p62/SQSTM1 caused autophagic cell death of several human carcinoma cells. Cancer Sci 2014; 105:568-75; PMID:24618016; http://dx.doi.org/ 10.1111/cas.12396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141:656-67; PMID:20478256; http://dx.doi.org/ 10.1016/j.cell.2010.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cesen MH, Repnik U, Turk V, Turk B. Siramesine triggers cell death through destabilisation of mitochondria, but not lysosomes. Cell Death Dis 2013; 4:e818; PMID:24091661; http://dx.doi.org/ 10.1038/cddis.2013.361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molejon MI, Ropolo A, Re AL, Boggio V, Vaccaro MI. The VMP1-Beclin 1 interaction regulates autophagy induction. Sci Rep 2013; 3:1055; PMID:23316280; http://dx.doi.org/ 10.1038/srep01055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem 2006; 281:14474-85; PMID:16522639; http://dx.doi.org/ 10.1074/jbc.M600364200 [DOI] [PubMed] [Google Scholar]
- 31.Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann Surg Oncol 2009; 16:761-71; PMID:19116755; http://dx.doi.org/ 10.1245/s10434-008-0260-0 [DOI] [PubMed] [Google Scholar]
- 32.Benavente CA, Schnell SA, Jacobson EL. Effects of niacin restriction on sirtuin and PARP responses to photodamage in human skin. PloS One 2012; 7:e42276; PMID:22860104; http://dx.doi.org/ 10.1371/journal.pone.0042276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sahu U, Sidhar H, Ghate PS, Advirao GM, Raghavan SC, Giri RK. A Novel Anticancer Agent, 8-Methoxypyrimido[4′,5′:4,5]thieno(2,3-) Quinoline-4(3H)-One Induces Neuro 2a Neuroblastoma Cell Death through p53-Dependent, Caspase-Dependent and -Independent Apoptotic Pathways. PloS One 2013; 8:e66430; PMID:23824039; http://dx.doi.org/ 10.1371/journal.pone.0066430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654-8; PMID:19794493; http://dx.doi.org/ 10.1038/nature08455 [DOI] [PubMed] [Google Scholar]
- 35.Bargis-Surgey P, Lavergne JP, Gonzalo P, Vard C, Filhol-Cochet O, Reboud JP. Interaction of elongation factor eEF-2 with ribosomal P proteins. Eur J Biochem 1999; 262:606-11; PMID:10336649; http://dx.doi.org/ 10.1046/j.1432-1327.1999.00434.x [DOI] [PubMed] [Google Scholar]
- 36.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008; 454:455-62; PMID:18650916; http://dx.doi.org/ 10.1038/nature07203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010; 285:10850-61; PMID:20123989; http://dx.doi.org/ 10.1074/jbc.M109.080796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2-11; PMID:22025673; http://dx.doi.org/ 10.1128/MCB.06159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, Wek RC. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell 2011; 22:4390-405; PMID:21917591; http://dx.doi.org/ 10.1091/mbc.E11-06-0510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, et al.. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 2012; 55:7193-207; PMID:22827572; http://dx.doi.org/ 10.1021/jm300713s [DOI] [PubMed] [Google Scholar]
- 42.May KL, Li XP, Martinez-Azorin F, Ballesta JP, Grela P, Tchorzewski M, Tumer NE. The P1/P2 proteins of the human ribosomal stalk are required for ribosome binding and depurination by ricin in human cells. FEBS J 2012; 279:3925-36; PMID:22909382; http://dx.doi.org/ 10.1111/j.1742-4658.2012.08752.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer 2003; 3:179-92; PMID:12612653; http://dx.doi.org/ 10.1038/nrc1015 [DOI] [PubMed] [Google Scholar]
- 44.Perucho L, Artero-Castro A, Guerrero S, Ramon YCS, ME LL, Wang ZQ. RPLP1, a Crucial Ribosomal Protein for Embryonic Development of the Nervous System. PloS one 2014; 9:e99956; PMID:24959908; http://dx.doi.org/ 10.1371/journal.pone.0099956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marcu MG, Doyle M, Bertolotti A, Ron D, Hendershot L, Neckers L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol Cell Biol 2002; 22:8506-13; PMID:12446770; http://dx.doi.org/ 10.1128/MCB.22.24.8506-8513.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 2002; 16:452-66; PMID:11850408; http://dx.doi.org/ 10.1101/gad.964702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang F, Hamanaka RB, Bobrovnikova-Marjon E, Gordan JD, Dai MS, Lu H, Simon MC, Diehl JA. Ribosomal stress couples the unfolded protein response to p53-dependent cell cycle arrest. J Biol Chem 2006; 281:30036-45; PMID:16893887; http://dx.doi.org/ 10.1074/jbc.M604674200 [DOI] [PubMed] [Google Scholar]
- 48.Pan W, Issaq S, Zhang Y. The in vivo role of the RP-Mdm2-p53 pathway in signaling oncogenic stress induced by pRb inactivation and Ras overexpression. PloS One 2011; 6:e21625; PMID:21747916; http://dx.doi.org/ 10.1371/journal.pone.0021625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Llanos S, Serrano M. Depletion of ribosomal protein L37 occurs in response to DNA damage and activates p53 through the L11/MDM2 pathway. Cell Cycle 2010; 9:4005-12; PMID:20935493; http://dx.doi.org/ 10.4161/cc.9.19.13299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell 2009; 16:369-77; PMID:19878869; http://dx.doi.org/ 10.1016/j.ccr.2009.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bee A, Brewer D, Beesley C, Dodson A, Forootan S, Dickinson T, Gerard P, Lane B, Yao S, Cooper CS, et al.. siRNA knockdown of ribosomal protein gene RPL19 abrogates the aggressive phenotype of human prostate cancer. PloS One 2011; 6:e22672; PMID:21799931; http://dx.doi.org/ 10.1371/journal.pone.0022672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhat KP, Itahana K, Jin A, Zhang Y. Essential role of ribosomal protein L11 in mediating growth inhibition-induced p53 activation. EMBO J 2004; 23:2402-12; PMID:15152193; http://dx.doi.org/ 10.1038/sj.emboj.7600247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maillet A, Pervaiz S. Redox regulation of p53, redox effectors regulated by p53: a subtle balance. Antioxid Redox Signal 2012; 16:1285-94; PMID:22117613; http://dx.doi.org/ 10.1089/ars.2011.4434 [DOI] [PubMed] [Google Scholar]
- 54.Jin S. p53, Autophagy and tumor suppression. Autophagy 2005; 1:171-3; PMID:16874039; http://dx.doi.org/ 10.4161/auto.1.3.2051 [DOI] [PubMed] [Google Scholar]
- 55.McClung JM, Judge AR, Powers SK, Yan Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am J Physiol Cell Physiol 2010; 298:C542-9; PMID:19955483; http://dx.doi.org/ 10.1152/ajpcell.00192.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu M, Li CM, Chen ZF, Ji R, Guo QH, Li Q, Zhang HL, Zhou YN. Celecoxib regulates apoptosis and autophagy via the PI3K/Akt signaling pathway in SGC-7901 gastric cancer cells. Int J Mol Med 2014; 33:1451-8; PMID:24676394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng F, Tang Q, Wu J, Zhao S, Liang Z, Li L, Wu W, Hann S. p38alpha MAPK-mediated induction and interaction of FOXO3a and p53 contribute to the inhibited-growth and induced-apoptosis of human lung adenocarcinoma cells by berberine. J Exp Clin Cancer Res 2014; 33:36; http://dx.doi.org/ 10.1186/1756-9966-33-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shrivastava S, Kulkarni P, Thummuri D, Jeengar MK, Naidu VG, Alvala M, Redddy GB, Ramakrishna S. Piperlongumine, an alkaloid causes inhibition of PI3 K/Akt/mTOR signaling axis to induce caspase-dependent apoptosis in human triple-negative breast cancer cells. Apoptosis 2014; 19:1148-64; PMID:24729100; http://dx.doi.org/ 10.1007/s10495-014-0991-2 [DOI] [PubMed] [Google Scholar]
- 59.Bitomsky N, Hofmann TG. Apoptosis and autophagy: Regulation of apoptosis by DNA damage signalling - roles of p53, p73 and HIPK2. FEBS J 2009; 276:6074-83; PMID:19788416; http://dx.doi.org/ 10.1111/j.1742-4658.2009.07331.x [DOI] [PubMed] [Google Scholar]
- 60.Swanlund JM, Kregel KC, Oberley TD. Autophagy following heat stress: the role of aging and protein nitration. Autophagy 2008; 4:936-9; PMID:18758235; http://dx.doi.org/ 10.4161/auto.6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wong CH, Iskandar KB, Yadav SK, Hirpara JL, Loh T, Pervaiz S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PloS One 2010; 5:e9996; PMID:20368806; http://dx.doi.org/ 10.1371/journal.pone.0009996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004; 23:2891-906; PMID:15077152; http://dx.doi.org/ 10.1038/sj.onc.1207521 [DOI] [PubMed] [Google Scholar]
- 63.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10:1533-41; PMID:21878654; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang TW, Chen CC, Chen KY, Su JH, Chang JH, Chang MC. Ribosomal phosphoprotein P0 interacts with GCIP and overexpression of P0 is associated with cellular proliferation in breast and liver carcinoma cells. Oncogene 2008; 27:332-8; PMID:17621266; http://dx.doi.org/ 10.1038/sj.onc.1210651 [DOI] [PubMed] [Google Scholar]
- 65.Sharp MG, Adams SM, Elvin P, Walker RA, Brammar WJ, Varley JM. A sequence previously identified as metastasis-related encodes an acidic ribosomal phosphoprotein, P2. Br J Cancer 1990; 61:83-8; PMID:2153399; http://dx.doi.org/ 10.1038/bjc.1990.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leong S, McKay MJ, Christopherson RI, Baxter RC. Biomarkers of breast cancer apoptosis induced by chemotherapy and TRAIL. J Proteome Res 2012; 11:1240-50; PMID:22133146; http://dx.doi.org/ 10.1021/pr200935y [DOI] [PubMed] [Google Scholar]
- 67.Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol 1963; 17:299-313; PMID:13985244; http://dx.doi.org/ 10.1083/jcb.17.2.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al.. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92:9363-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shang J, Lehrman MA. Discordance of UPR signaling by ATF6 and Ire1p-XBP1 with levels of target transcripts. Biochem Biophys Res Commun 2004; 317:390-6; PMID:15063770; http://dx.doi.org/ 10.1016/j.bbrc.2004.03.058 [DOI] [PubMed] [Google Scholar]
- 70.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension 2007; 49:717-27; PMID:17296874; http://dx.doi.org/ 10.1161/01.HYP.0000258594.87211.6b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmid I, Krall WJ, Uittenbogaart CH, Braun J, Giorgi JV. Dead cell discrimination with 7-amino-actinomycin D in combination with dual color immunofluorescence in single laser flow cytometry. Cytometry 1992; 13:204-8; PMID:1547670; http://dx.doi.org/ 10.1002/cyto.990130216 [DOI] [PubMed] [Google Scholar]
- 72.Colome N, Collado J, Bech-Serra JJ, Liiv I, Anton LC, Peterson P, Canals F, Jaraquemada D, Alvarez I. Increased apoptosis after autoimmune regulator expression in epithelial cells revealed by a combined quantitative proteomics approach. J Proteome Res 2010; 9:2600-9; PMID:20218732; http://dx.doi.org/ 10.1021/pr100044d [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.