

Abstract
Autophagy plays important roles in metabolism, differentiation, and survival in T cells. TNFAIP3/A20 is a ubiquitin-editing enzyme that is thought to be a negative regulator of autophagy in cell lines. However, the role of TNFAIP3 in autophagy remains unclear. To determine whether TNFAIP3 regulates autophagy in CD4 T cells, we first analyzed Tnfaip3-deficient naïve CD4 T cells in vitro. We demonstrated that Tnfaip3-deficient CD4 T cells exhibited reduced MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) puncta formation, increased mitochondrial content, and exaggerated reactive oxygen species (ROS) production. These results indicate that TNFAIP3 promotes autophagy after T cell receptor (TCR) stimulation in CD4 T cells. We then investigated the mechanism by which TNFAIP3 promotes autophagy signaling. We found that TNFAIP3 bound to the MTOR (mechanistic target of rapamycin) complex and that Tnfaip3-deficient cells displayed enhanced ubiquitination of the MTOR complex and MTOR activity. To confirm the effects of enhanced MTOR activity in Tnfaip3-deficient cells, we analyzed cell survival following treatment with Torin1, an MTOR inhibitor. Tnfaip3-deficient CD4 T cells exhibited fewer cell numbers than the control cells in vitro and in vivo. In addition, the impaired survival of Tnfaip3-deficient cells was ameliorated with Torin1 treatment in vitro and in vivo. The effect of Torin1 was abolished by Atg5 deficiency. Thus, enhanced MTOR activity regulates the survival of Tnfaip3-deficient CD4 T cells. Taken together, our findings illustrate that TNFAIP3 restricts MTOR signaling and promotes autophagy, providing new insight into the manner in which MTOR and autophagy regulate survival in CD4 T cells.
Keywords: ATG5, autophagy, CD4, MTOR, TNFAIP3, ubiquitin
Abbreviations
- 4-OHT
4-hydroxytamoxifen
- ACTB/bACT
actin, β
- AKT
v-akt murine thymoma viral oncogene homolog
- PRKAA/AMPK
protein kinase, AMP-activated
- ATG
autophagy related
- BAK1
BCL2-antagonist/killer 1
- BAX
BCL2-associated X protein
- BCL2
B-cell CLL/lymphoma 2
- BCL10
B-cell CLL/lymphoma 10
- CD3E
CD3 antigen, epsilon polypeptide
- CD28
CD28 antigen
- CD44
CD44 antigen
- CD69
CD69 antigen
- CHX
cycloheximide
- EIF4EBP1
eukaryotic translation inhibition factor 4E binding protein 1
- ESR
estrogen receptor
- IFNG
interferon, gamma
- IL2
interleukin 2
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- LPS
lipopolysaccharide
- MALT1
MALT1 paracaspase
- MCL1
myeloid cell leukemia 1
- MFI
mean fluorescence intensity
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NFKB
nuclear factor of kappa light polypeptide gene enhancer in B-cells
- PBS
phosphate-buffered saline
- PI3K
class I phosphoinositide 3-kinase
- PLA
proximity ligation assay
- RIPK1
receptor (TNFRSF)-interacting serine-threonine kinase 1
- ROS
reactive oxygen species
- RPS6KB1
ribosomal protein S6 kinase, polypeptide 1
- TCR
T cell receptor
- TNFAIP3/A20
tumor necrosis factor, α-induced protein 3
- TRAF6
TNF receptor-associated factor 6, E3 ubiquitin protein ligase
Introduction
Autophagy is a lysosomal degradation pathway that is important for cellular metabolism and renovation. A portion of the cytoplasm is engulfed by a phagophore, resulting in the formation of a double-membrane structure known as the autophagosome. ATG5 and ATG12, which associate with ATG16L1, are essential for autophagosome formation. MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) is present on both the inner and outer membranes of the autophagosome and serves as an adaptor for selective substrates.1,2 Tissue-specific deletions of Atg genes revealed that they play specific roles in autophagy in the immune system. T cell-specific Atg5-deficient mice (Atg5fl/fl Lck-Cre) and Atg7-deficient mice (Atg7fl/fl Lck-Cre) exhibit normal thymocyte development and reduced numbers of peripheral T cells. Autophagy-deficient mature T cells display increased mitochondrial content and increased reactive oxygen species (ROS) production.3,4 Although these studies have begun to define the biological mechanism of autophagy, little is known about how T cell receptor (TCR)-induced autophagy is restricted.
T cell metabolism is dynamic and regulates the activation, differentiation, and function of T cells. One of the key molecules of metabolism is MTOR (mechanistic target of rapamycin [serine/threonine kinase]).5 In T cells, MTOR can be regulated by multiple signals.6 CD28-mediated costimulation is a classic activating signal for the PI3K–AKT pathway, which upregulates MTOR activity.7-9 TCR stimulation also activates PRKAA/AMPK (protein kinase, AMP-activated), which suppresses MTOR activity.10-12 In contrast, MTOR also regulates several downstream pathways. RPS6KB1 (ribosomal protein S6 kinase, 70 kDa, polypeptide 1) and EIF4EBP1 (eukaryotic translation inhibition factor 4E binding protein 1), substrates for the MTOR complex, are associated with the initiation of mRNA translation and the control of protein synthesis. The activity of the MTOR complex is canonically measured by the phosphorylation of its substrates, including RPS6KBP1 and EIF4EBP1.13 In addition, the MTOR complex suppresses autophagy under nutrient-rich conditions. Previous studies have demonstrated that the ubiquitination of MTOR is mediated via binding to FBXW7 (F-box and WD repeat domain containing 7, E3 ubiquitin protein ligase) in HCT116 cells14 and that the K63 ubiquitination of MTOR regulates the activation of MTOR by amino acids.15 However, the regulation of ubiquitination of MTOR in the immune system, particularly in T cells, is unclear.
TNFAIP3 (tumor necrosis factor, α-induced protein 3) encodes the TNFAIP3/A20 protein, which is a ubiquitin-modifying enzyme16,17 that is critical for preventing inflammation in vivo. Tnfaip3-deficient (tnfaip3−/−) mice exhibit severe spontaneous multiorgan inflammation, cachexia, and perinatal death.18 TNFAIP3 binds to ubiquitin chains and ubiquitinated signaling complexes and regulates the activity and stability of signaling proteins such as RIPK1 (receptor (TNFRSF)-interacting serine-threonine kinase 1), RIPK2 (receptor-interacting serine-threonine kinase 2), TRAF6 (TNF receptor-associated factor 6, E3 ubiquitin ligase), and CTNNB1/β-catenin (catenin [cadherin-associated protein], β 1, 88kDa).16,17,19,20 TNFAIP3 is expressed in T cells.21 During T cell activation, TNFAIP3 is recruited to the MALT1–BCL10 complex and cleaved by the paracaspase MALT1.22 TNFAIP3 has also been reported to deubiquitinate MALT1 and restrict TCR signals.23 In mice in which TNFAIP3 was specifically deleted in mature conventional T cells, Tnfaip3-deficient CD8 T cells exhibit increased sensitivity to antigen stimulation, as indicated by the increased production of IL2 and IFNG.24 However, the physiological role of TNFAIP3 in T cells is not fully understood. TNFAIP3 may also play important roles in human autoimmune diseases. Recent genetic studies identified TNFAIP3 as a susceptibility gene for autoimmune diseases including inflammatory bowel disease.25-29 Previous reports suggested that TNFAIP3 restricts LPS-induced autophagy in RAW cells and baseline autophagy in HeLa cells.30,31 Given that autophagy plays a critical role during T cell activation, we investigated whether TNFAIP3 regulates autophagy in T cells and the mechanism by which this protein might regulate autophagy signaling. Surprisingly, however, we found that TNFAIP3 restricts MTOR signaling and promotes autophagy in CD4 T cells.
Results
TNFAIP3 promotes autophagy after TCR stimulation
To determine whether TNFAIP3 regulates autophagy in CD4 T cells, we tested LC3 puncta formation, which is a marker of the autophagosome. We purified naïve CD4 T cells from tnfaip3fl/fl Cd4-Cre, Tnfaip3fl/+ Cd4-Cre, and Tnfaip3+/+ Cd4-Cre mice. Naïve CD4 T cells were stimulated with anti-CD3E plus anti-CD28 in vitro. Viable cells were analyzed for LC3 mobilization by confocal microscopy at 24 h. Tnfaip3fl/+ Cd4-Cre and Tnfaip3+/+ Cd4-Cre T cells displayed similar levels of LC3 puncta formation. Surprisingly, LC3 puncta formation was reduced in tnfaip3fl/fl Cd4-Cre cells after TCR stimulation, whereas no difference was observed at baseline (Fig. 1A). To confirm these results, we analyzed LC3 conformation by immunoblotting. Reduced LC3-II levels were observed in tnfaip3fl/fl Cd4-Cre cells (Fig. 1B). An LC3 flux assay revealed that autophagy occurred in CD4 T cells after stimulation, but its induction was lower in tnfaip3fl/fl Cd4-Cre cells than in Tnfaip3fl/+ Cd4-Cre. Thus, TNFAIP3 promotes autophagy after TCR stimulation in CD4 T cells.
Figure 1.
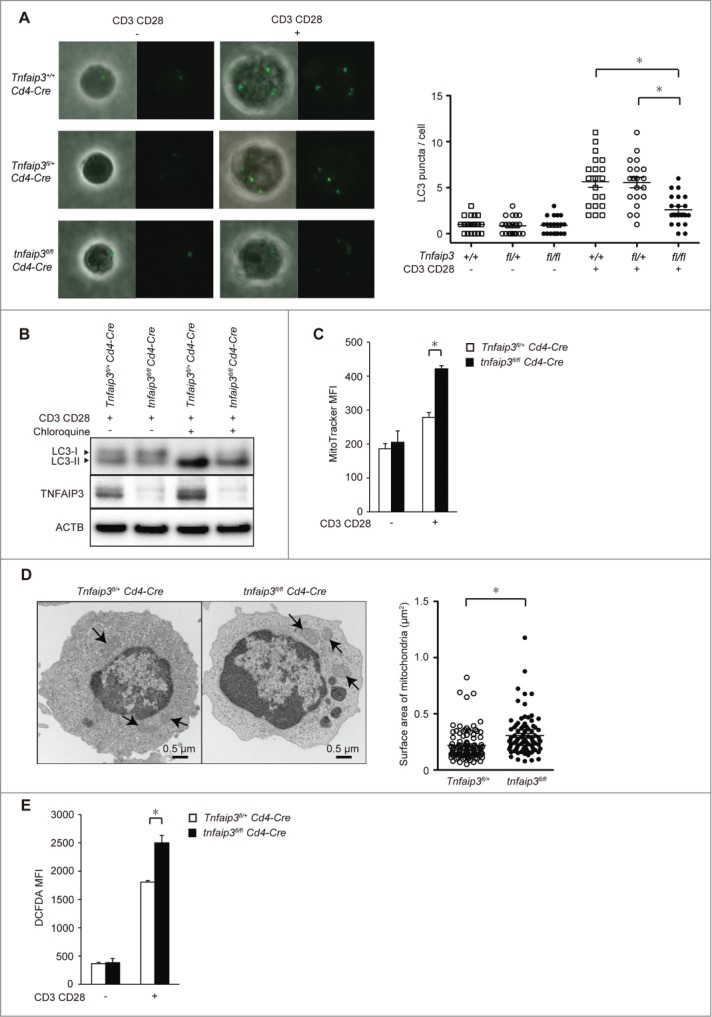
TNFAIP3 promotes autophagy after TCR stimulation. (A) LC3 puncta formation in CD4 T cells. tnfaip3fl/fl Cd4-Cre, Tnfaip3fl/+ Cd4-Cre, and Tnfaip3+/+ Cd4-Cre naïve CD4 T cells were purified from peripheral lymph nodes and spleen and stimulated with anti-CD3E and anti-CD28 antibodies for 24 h. Cells were treated with 40 µM chloroquine for the last 2 h. LC3 puncta were intracellularly stained with anti-LC3 antibody. Images were acquired with a confocal laser microscope (FV10i, Olympus) using a 60× oil-immersion objective lens. Twenty cells from each indicated strain were analyzed. Data are representative of 3 independent experiments. (B) LC3 conversion and turnover assays. tnfaip3fl/fl Cd4-Cre, Tnfaip3fl/+ Cd4-Cre naïve CD4 T cells were stimulated with anti-CD3E and anti-CD28 antibodies for 24 h. The difference in LC3-II levels with and without chloroquine was evaluated. The positions of LC3-I and LC3-II are indicated. Data are representative of 3 independent experiments. (C) MitoTracker mean fluorescence intensity (MFI) in CD4 T cells. tnfaip3fl/fl Cd4-Cre, and Tnfaip3fl/+ Cd4-Cre naïve CD4 T cells were stimulated with anti-CD3E and anti-CD28 antibodies for 24 h. CD4 T cells were stained with MitoTracker Green. Data are representative of 2 independent experiments. (D) Electron micrograph of CD4 T cells. tnfaip3fl/fl Cd4-Cre and Tnfaip3fl/+ Cd4-Cre naïve CD4 T cells were stimulated with anti-CD3E and anti-CD28 antibodies for 24 h. Arrows indicate mitochondria. The surface area was calculated after manually outlining mitochondria using the measure tool in ImageJ software. Twenty cells from each indicated strain were analyzed. Data are representative of 2 independent experiments. (E) ROS accumulation was visualized with the fluorescent dye CM-H2DCFDA. tnfaip3fl/fl Cd4-Cre and Tnfaip3fl/+ Cd4-Cre naïve CD4 T cells were stimulated with anti-CD3E and anti-CD28 antibodies for 24 h. Data are representative of 4 independent experiments. *, p < 0.05 by the Student t test. Error bars indicate standard deviations.
Autophagy is involved in the quality control of mitochondria.1,2 We hypothesized that the reduced autophagy induction in Tnfaip3-deficient cells dysregulates mitochondria and ROS production. We accordingly evaluated mitochondria via staining with a cell-permeable dye and transmission electron microscopy. tnfaip3fl/fl Cd4-Cre cells displayed exaggerated mitochondrial content according to MitoTracker Green staining (Fig. 1C). Additionally, we calculated the mitochondrial surface area by manually outlining mitochondria using a quantification tool in ImageJ. A statistically significant increase in the mitochondrial surface area in tnfaip3fl/fl Cd4-Cre cells was observed when compared with that in Tnfaip3fl/+ Cd4-Cre T cells (Fig. 1D). We next analyzed ROS production. tnfaip3fl/fl Cd4-Cre cells exhibited increased ROS production 24 h after stimulation (Fig. 1E). These findings were similar to those in Atg7-deficient T cells.3 Taken together, these results indicate that the dysregulation of mitochondria and ROS production may arise from the reduction of autophagy in Tnfaip3-deficient cells.
TNFAIP3 restricts MTOR activity in CD4 T cells
We then investigated the mechanism by which TNFAIP3 promotes autophagy signaling. Given that MTOR is a major negative regulator of autophagy, we investigated the change in MTOR activity after TCR stimulation. The activity of the MTOR complex was determined by monitoring the phosphorylation status of RPS6KB1 at Thr389. MTOR activity was enhanced in tnfaip3fl/fl Cd4-Cre cells (Fig. 2A).
Figure 2.
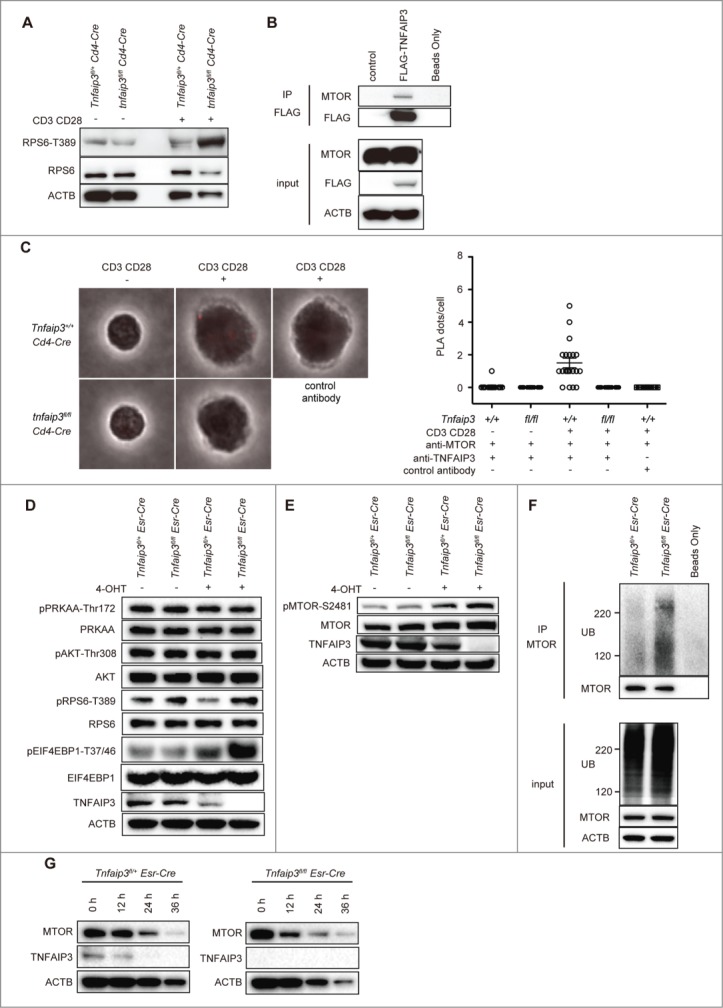
TNFAIP3 restricts MTOR activity in CD4 T cells. (A) phospho-RPS6KB1 expression in Tnfaip3fl/+ and tnfaip3fl/fl Cd4-Cre T cells. Naïve CD4 T cells were stimulated with anti-CD3E and anti-CD28 antibodies for 12 h. RPS6KB1 and ACTB expression are shown as the protein control. (B) Recruitment of TNFAIP3 to the MTOR complex. HEK293T cells were transfected with Flag-TNFAIP3. Protein extracts were immunoprecipitated (IP) with Flag antibody and immunoblotted for the indicated proteins. (C) Duolink PLA demonstrating the close proximity of TNFAIP3 and MTOR in CD4 T cells. Naïve CD4 T cells from tnfaip3fl/fl Cd4-Cre or Tnfaip3+/+ Cd4-Cre mice were stimulated with anti-CD3E antibody plus anti-CD28 for 24 h. Anti-TNFAIP3 and anti-MTOR antibodies were used for the PLA. Twenty cells from each sample were analyzed. Error bars indicate standard deviations. (D and E) MTOR signaling proteins in inducible Tnfaip3-deficient CD4 T cells. Tnfaip3fl/fl Esr-Cre or Tnfaip3fl/+ Esr-Cre naïve CD4 T cells were stimulated with anti-CD3E and anti-CD28 antibodies, and 4-OHT (10 nM) for 48 h. Cell lysates were harvested for immunoblotting. (F) Ubiquitination of the MTOR complex. Tnfaip3fl/fl Esr-Cre or Tnfaip3fl/+ Esr-Cre naïve CD4 T cells were stimulated as described in (D). Each protein extract was immunoprecipitated with MTOR antibody and immunoblotted for the indicated proteins. (G) CHX chase experiment for the MTOR protein. Tnfaip3fl/fl Esr-Cre or Tnfaip3fl/+ Esr-Cre naïve CD4 T cells were stimulated as described in (D). Then, cells were treated with 10 μg/ml CHX, and cell lysates were harvested for immunoblotting at the indicated time points. Data are representative of 2 independent experiments.
To better understand the molecular mechanism by which TNFAIP3 promotes TCR-induced autophagy signaling, we considered TNFAIP3 as a ubiquitin-editing enzyme that regulates complex formation. We thus hypothesized that TNFAIP3 regulates MTOR complex formation. To test this hypothesis, we first investigated whether TNFAIP3 is recruited to the MTOR complex. HEK293T cells were transfected with Flag-TNFAIP3; we found that MTOR was immunoprecipitated with Flag-TNFAIP3 (Fig. 2B). To confirm these interactions in CD4 T cells, we used a proximity ligation assay (PLA). We found that TNFAIP3 and MTOR interact in situ after anti-CD3E plus anti-CD28 stimulation, whereas no PLA foci were detected with control antibody or Tnfaip3-deficient cells (Fig. 2C). These findings indicate that TNFAIP3 interacts with MTOR after TCR stimulation in CD4 T cells.
To control for the potential differences among T cells and avoid the potential problems associated with developmental abnormalities, we sought to eliminate TNFAIP3 expression after T cells developed into mature cells. We accordingly interbred Tnfaip3fl mice with Esr (estrogen receptor)-Cre mice to obtain mice in which TNFAIP3 deletion would occur after treatment with 4-hydroxytamoxifen (4-OHT). Naïve CD4 T cells were purified from Esr-Cre mice and treated with 4-OHT in vitro to effectively ablate TNFAIP3 protein expression (Fig. 2D). We then evaluated the phosphorylation of RPS6KB1 and EIF4EBP1. Consistent with our findings, mature Tnfaip3fl/fl Esr-Cre naïve CD4 T cells that were rendered acutely Tnfaip3-deficient after 4-OHT treatment exhibited exaggerated phosphorylation of RPS6KB1 and EIF4EBP1 (Fig. 2D). These cells also displayed increased phosphorylation of MTOR Ser2481, indicating that Tnfaip3fl/fl Esr-Cre mice exhibited increased MTOR activity. In addition, PI3K-AKT and AMPK can modulate MTOR activity.6 There were no obvious differences in the phosphorylation levels of AKT and AMPK, at least at this time point (Fig. 2D). We infer that TNFAIP3 restricts MTOR activity after TCR stimulation in CD4 T cells.
According to a recent report, MTOR activation is regulated by ubiquitination.15 To determine whether TNFAIP3 regulates MTOR ubiquitination, we stimulated mature Tnfaip3fl/fl Esr-Cre naïve CD4 T cells with anti-CD3E plus anti-CD28 in vitro, immunoprecipitated proteins with MTOR, and immunoblotted for ubiquitin. Tnfaip3fl/fl Esr-Cre cells exhibited significantly increased ubiquitination of the MTOR complex relative to the findings in Tnfaip3fl/+ Esr-Cre cells (Fig. 2F). Next, to confirm MTOR protein stability, we treated Esr-Cre cells with cycloheximide (CHX) (Fig. 2G). Although we found increased ubiquitination of the MTOR complex in Tnfaip3fl/fl Esr-Cre cells, there was no obvious change in MTOR degradation. These data suggested that ubiquitination of MTOR may have other roles than targeting the protein for degradation, which is consistent with the previous paper.15 Taken together, these results indicate that TNFAIP3 restricts ubiquitination of the MTOR complex and MTOR activity.
Inhibition of MTOR restores autophagy and survival in Tnfaip3-deficient T cells in vitro
Torin1 is a highly potent and selective ATP-competitive MTOR inhibitor that can strongly induce autophagy.32 To confirm the effects of enhanced MTOR activity in Tnfaip3-deficient cells, we evaluated LC3 puncta formation and ROS production after Torin1 treatment. Tnfaip3fl/+ Cd4-Cre cells did not display altered LC3 puncta formation. However, LC3 puncta formation was increased (Fig. 3A) and ROS production was reduced after Torin1 treatment (Fig. 3B) in tnfaip3fl/fl Cd4-Cre cells. Thus, inhibition of MTOR enhanced autophagy in Tnfaip3-deficient T cells. As the loss of the essential autophagy gene Atg5 impairs the survival and proliferation of mature T cells in vivo,33 we analyzed cell numbers in vitro. To evaluate cell-intrinsic TNFAIP3 function, we mixed congenically marked naïve CD4 T cells at a 1:1 ratio and stimulated the cells in the same wells. After TCR stimulation, tnfaip3fl/fl Cd4-Cre cell counts were reduced to less than 50%, which meant that the number of tnfaip3fl/fl Cd4-Cre cells was less than that of Tnfaip3+/+ Cd4-Cre cells (Fig. 3C). These reduced cell numbers might be due to the reduction of autophagy in tnfaip3fl/fl Cd4-Cre cells. In addition, tnfaip3fl/fl Cd4-Cre cells displayed increased cell numbers relative to Tnfaip3+/+ Cd4-Cre cells after Torin1 treatment (Fig. 3C). These data indicated that Tnfaip3-deficient T cells exhibited enhanced MTOR activity, resulting in reduced autophagy and impaired cell survival, which is partly suppressed when autophagy is induced by Torin1.
Figure 3.
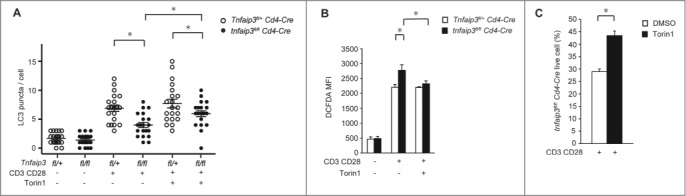
Inhibition of MTOR restored autophagy and survival in Tnfaip3-deficient T cells in vitro. (A) The effect of Torin1 on LC3 puncta formation in CD4 T cells. Naïve CD4 T cells from tnfaip3fl/fl Cd4-Cre or Tnfaip3fl/+ Cd4-Cre mice were stimulated with anti-CD3E and anti-CD28 antibodies, and Torin1 (25 nM) for 24 h. LC3 puncta were intracellularly stained with anti-LC3 antibody. Data are representative of 2 independent experiments. (B) The effect of Torin1 on ROS production in CD4 T cells. Naïve CD4 T cells from tnfaip3fl/fl Cd4-Cre or Tnfaip3fl/+ Cd4-Cre were stimulated as described in (A). CD4 T cells were stained with CM-H2DCFDA. Data are representative of 3 independent experiments. (C) Live cell proportions of Tnfaip3-deficient CD4 T cells in vitro. Naïve CD4 T cells were purified from congenically marked tnfaip3fl/fl Cd4-Cre and Tnfaip3+/+ Cd4-Cre mice and mixed at a 1:1 ratio. The mixed cells were stimulated with anti-CD3E and anti-CD28 antibodies, and Torin1 (25 nM) for 48 h in the same wells. Live cells were quantified by flow cytometry of DAPI-negative cells. Data are representative of 2 independent experiments. * p < 0.05 by the Student t test. Error bars indicate standard deviations.
Tnfaip3-deficient cell survival was ameliorated with Torin1 treatment in vivo
Next, we tested the response of Tnfaip3-deficient and control cells to Torin1 in vivo. Congenically marked tnfaip3fl/fl Cd4-Cre and Tnfaip3+/+ (or Tnfaip3fl/+) Cd4-Cre naïve CD4 T cells were mixed, transferred into Rag2 (recombination activating gene 2)-deficient mice, and then injected intraperitoneally with Torin1. We confirmed the donor tnfaip3fl/fl and Tnfaip3+/+ (or Tnfaip3fl/+) Cd4-Cre naïve CD4 T cells via surface markers and gene expression (Figs. S1A and B). We evaluated T cells in the spleen and lymph nodes on d 20. As the donor naïve T cells displayed a similar phenotype based on traditional markers, we could evaluate cell-intrinsic TNFAIP3 function and control for secondary cytokine effects and individual variability between mice in this system (Fig. 4A). Consistent with our in vitro studies, tnfaip3fl/fl Cd4-Cre cells displayed lower expansion than Tnfaip3+/+Cd4-Cre cells with control treatment (Fig. 4B). These cells express similar levels of CD69, an activation marker (Fig. 4C). However, tnfaip3fl/fl Cd4-Cre T cells exhibited enhanced expansion in vivo with Torin1 treatment (Fig. 4B). Because Tnfaip3fl/+ Cd4-Cre cells may have enhanced responses that are not observed in Tnfaip3+/+ Cd4-Cre cells,34-36 we mixed congenically marked tnfaip3fl/fl Cd4-Cre and Tnfaip3fl/+ Cd4-Cre naïve CD4 T cells. Consistent with the results when we examined Tnfaip3+/+ Cd4-Cre T cells, tnfaip3fl/fl Cd4-Cre T cells displayed lower expansion than Tnfaip3fl/+ Cd4-Cre T cells and enhanced expansion with Torin1 treatment (Fig. 4D). Collectively, reduced LC3 puncta formation in Tnfaip3-deficient cells was ameliorated with Torin1 treatment in vitro, and Tnfaip3-deficient cell survival was ameliorated with Torin1 treatment in vitro and in vivo.
Figure 4.
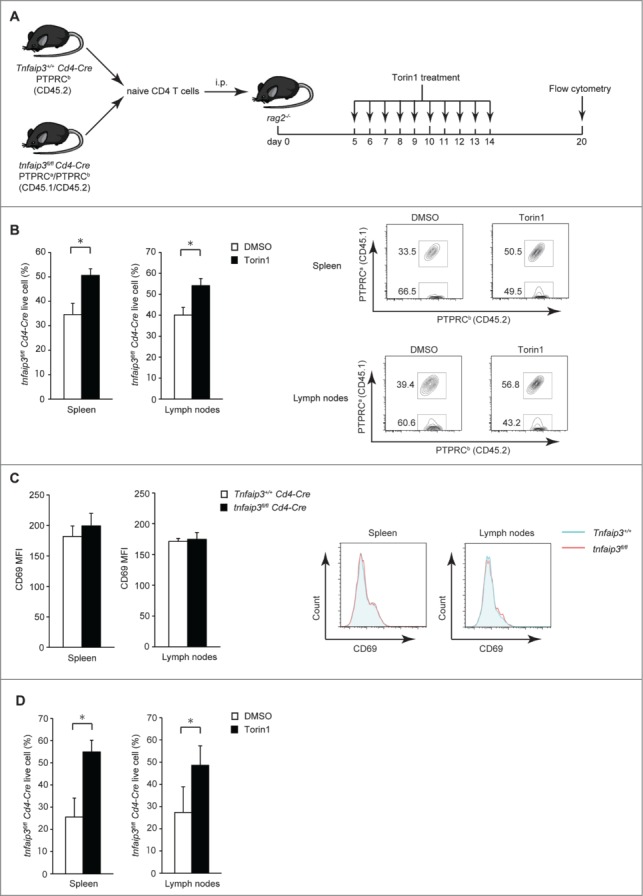
Tnfaip3-deficient cell survival was ameliorated by Torin1 treatment in vivo. (A) Schematic representation of the study. Naïve PTPRCb (CD45.2) Tnfaip3+/+ Cd4-Cre and PTPRCa/PTPRCb (CD45.1/CD45.2) tnfaip3fl/fl Cd4-Cre T cells were mixed at a 1:1 ratio, and 2.5 × 105 cells were adoptively transferred intraperitoneally (i.p.) into rag2−/− mice. Mice were treated for 10 days with 1 mg/kg Torin1 and analyzed on d 20. (B) Live cell proportions of tnfaip3fl/fl Cd4-Cre CD4 T cells in vivo. Live CD4 T cells in the spleen and lymph nodes were quantified by flow cytometry of DAPI-negative cells. Representative flow cytometry plots are shown on the right. Data are representative of 2 independent experiments. (C) Expression of CD69 in live CD4 T cells. CD69 MFI of the indicated cells was analyzed in live CD4 T cells from each tissue. Representative flow cytometry plots are shown on the right. Data are representative of 2 independent experiments. (D) Live cell proportions of tnfaip3fl/fl Cd4-Cre CD4 T cells in vivo. Congenically marked naïve tnfaip3fl/fl Cd4-Cre and Tnfaip3fl/+ Cd4-Cre T cells were mixed at a 1:1 ratio and transferred into rag2-deficient mice. Experiments were performed as described in (A) and (B). Data are representative of 2 independent experiments. * p < 0.05 by the Student t test. Error bars indicate standard deviations.
Enhanced MTOR activity regulates the survival of Tnfaip3-deficient CD4 T cells via autophagy
MTOR regulates many signals other than autophagy. Torin1 inhibits RPS6KB1, the translational inhibitor EIF4EBP1, and other signals, whereas Torin1 can strongly induce autophagy. To confirm whether Torin1 supports survival in Tnfaip3-deficient cell by activation of autophagy, we analyzed Esr-Cre cells in vitro. First, we confirmed cell survival using mixed (1:1) congenically marked Tnfaip3fl/fl Esr-Cre and Tnfaip3fl/+ Esr-Cre naïve CD4 T cells. After 72 h of TCR stimulation, both genotypes of CD4 T cells without 4-OHT increased in size by approximately 50%, indicating that CD4 T cells were similarly expanded. However, Tnfaip3fl/fl Esr-Cre T cells with 4-OHT exhibited lower expansion than Tnfaip3fl/+ Esr-Cre CD4 T cells, which is consistent with the Cd4-Cre cell observation. In addition, Tnfaip3fl/fl Esr-Cre T cells with 4-OHT stimulation displayed enhanced cell numbers relative to Tnfaip3fl/+ Esr-Cre CD4 T cells after Torin1 treatment (Fig. 5A). Taken together, we confirmed that TNFAIP3 deficiency impaired cell survival using an Esr-Cre in vitro deletion system.
Figure 5.

For figure legend, see page 1060.
ATG5 is essential for autophagosome formation. Next, to confirm Torin1's effect on survival in Tnfaip3-deficient cells, we analyzed TNFAIP3 and ATG5 double-deficient CD4 T cells. We generated Tnfaip3fl/fl Atg5fl/fl Esr-Cre mice, in which we could induce deletion of TNFAIP3 and ATG5 in vitro. We confirmed the gene deletion efficiency by real-time PCR (Fig. 5B). We next mixed congenically marked Tnfaip3fl/fl Atg5fl/fl Esr-Cre and Tnfaip3fl/fl Atg5fl/+ Esr-Cre naïve CD4 T cells and then treated them with Torin1. Without 4-OHT treatment no deletion occurred, and Tnfaip3fl/fl Atg5fl/fl Esr-Cre and Tnfaip3fl/fl Atg5fl/+ Esr-Cre naïve CD4 T cells displayed similar expansion. With 4-OHT treatment, however, Tnfaip3fl/fl Atg5fl/fl Esr-Cre cells (Tnfaip3 and Atg5 double-deficient) did not expand compared with Tnfaip3fl/fl Atg5fl/+ Esr-Cre cells (Tnfaip3-deficient). Additionally, these differences were not ameliorated by Torin1 treatment (Fig. 5C). These data indicated that ATG5 enhanced survival in Tnfaip3-deficient cells, and the effects of Torin1 on survival in Tnfaip3-deficient cells were abolished by Atg5 deficiency. To determine whether abnormalities in BCL2 family protein expression are associated with impaired survival in Tnfaip3-deficient cells, we examined the expression of the anti-apoptotic members Bcl2 and Mcl1 and the pro-apoptotic members Bax and Bak1 using inducible Tnfaip3- and/or Atg5-deficient CD4 T cells. There were no differences in the expression of anti-apoptotic and pro-apoptotic molecules, suggesting that the lower expansion of Tnfaip3-deficient cell number might not be due to apoptotic cell death (Fig. 5D). Taken together, the data suggest that the restoration of autophagy by Torin1 contributed to survival in Tnfaip3-deficient cells.
Discussion
Many fine points of autophagy induction in T cells render it different from that in other cell types.37 T cells exhibit enhanced autophagy after TCR stimulation, as measured both by the detection of LC3-II conformation and visualization of LC3 puncta.33,38 Our studies revealed that TNFAIP3 restricts the ubiquitination of the MTOR complex and MTOR activity after TCR stimulation, resulting in enhanced autophagy in CD4 T cells. In addition, reduced LC3 puncta formation and survival in Tnfaip3-deficient cells were ameliorated by the enhancement of autophagy using Torin1. These findings are apparently in contradiction with previously reported results.30,31 TNFAIP3 restricts LPS-induced autophagy in RAW cells and baseline autophagy in HeLa cells. Potential explanations include the possibility that the overexpression of TNFAIP3 indirectly suppresses autophagy induction. Furthermore, cell types and stimulation types differed from those used in our experiments. Collectively, we have clearly demonstrated that TNFAIP3 promotes autophagy after TCR stimulation using primary Tnfaip3-deficient CD4 T cells.
Our findings revealed that TNFAIP3 restricts MTOR signaling in CD4 T cells. We found that TNFAIP3 was recruited to the MTOR complex. A recent study illustrated that MTOR polyubiquitination is required for MTORC1 activation and that TRAF6 ubiquitinates MTOR in HEK293T cells.15 SQSTM1/p62 (sequestosome 1) modulates the K63-linked ubiquitination of MTOR to regulate its kinase activity, but not its levels.15 We discovered in this study that TNFAIP3 restricted the ubiquitination of the MTOR complex and MTOR activity under more physiological conditions. Moreover, TNFAIP3 did not regulate MTOR protein turnover based on a CHX chase assay, suggesting that TNFAIP3 may modulate K63-linked ubiquitination of the MTOR complex. Taken together, our results reveal an unexpected and novel function of TNFAIP3.
TNFAIP3 expression was observed with strikingly high levels in lymphoid organs.21 A previous paper demonstrated that Tnfaip3-deficient thymocytes were more sensitive to TNF/TNFα (tumor necrosis factor).8 During T cell activation, TNFAIP3 regulates the ubiquitination of MALT1, resulting in NFKB signaling. Tnfaip3 deletion in CD8 T cells greatly enhances their capacity to produce IL2 and IFNG,24 which is also regulated by NFKB. We uncovered TNFAIP3's novel physiological role in T cells. TNFAIP3 is a regulator of both NFKB and MTOR. TNFAIP3 restricts MTOR activity, resulting in the promotion of autophagy after TCR stimulation. In addition, we found that the enhancement of autophagy by Torin1 improves survival in Tnfaip3-deficient CD4 T cells in vitro and in vivo. Naïve T cells from atg7fl/fl Lck-Cre mice display an imbalance of BCL2 family expression.3 Although we observed a survival defect in Tnfaip3-deficient cells, we could not detect any difference in BCL2 family expression with inducible deletion systems. There might be another mechanism other than the reduced autophagy induction to explain the decrease in Tnfaip3fl/fl Cd4-Cre cell numbers. Further investigation is required to clarify the mechanism by which TNFAIP3 supports survival in CD4 T cells.
MTOR is a crucial regulator of cell growth, proliferation, autophagy, and many physiological events. MTOR regulates the diverse functions of professional antigen-presenting cells, such as dendritic cells and has important roles in the activation of effector T cells.39 MTOR inhibition is a promising therapeutic strategy for the prevention of transplant rejection and the treatment of autoimmune diseases, and in the present study we confirmed the improved survival of Tnfaip3-deficient CD4 T cells after Torin1 treatment. Our results suggest that the inhibition of MTOR supports the survival of Tnfaip3-deficient CD4 T cells via the induction of autophagy. Genome-wide association studies have demonstrated that TNFAIP3 is a susceptibility gene for some autoimmune diseases including inflammatory bowel disease.25-29 In the future, MTOR inhibitors might be used for the treatment of these diseases.39 Conversely, our findings caution against the use of MTOR inhibitors in patients with specific single nucleotide polymorphisms.
In conclusion, we identified a unique mechanism by which TNFAIP3 restricts MTOR complex activation via ubiquitination, and enhanced MTOR activity negatively regulates the survival of Tnfaip3-deficient CD4 T cells via the suppression of autophagy. These findings provide further insight into the manner by which MTOR and autophagy regulate survival in CD4 T cells.
Materials and Methods
Animals
The generation of mice bearing a “floxed” allele of Tnfaip3 (Tnfaip3fl mice) has been described.34 Atg5fl mice40 were provided by Noboru Mizushima (University of Tokyo). Breeding Tnfaip3fl mice with Cd4-Cre transgenic mice generated mice lacking TNFAIP3 specifically in T cells. Mice featuring a tamoxifen-inducible deletion of Tnfaip3 and Atg5 were obtained by breeding Tnfaip3fl and Atg5fl mice with Gt(ROSA)26Sortm1(Cre/Esr1) transgenic mice (Esr-Cre mice). For mixed experiments, we bred the C57BL/6 (PTPRCb/CD45.2) and PTPRCa/CD45.1 congenic strains. For these experiments, naïve CD4 T cells were bead-enriched from congenically marked mice and mixed at a ratio of 1:1, and cells were stimulated in the same well for in vitro experiments. For in vivo experiments, mixed cells were adoptively transferred intraperitoneally (i.p.) into Rag2 (recombination activating gene 2)-deficient mice. For drug administration, we dissolved Torin1 (TOCRIS, 4247) in 4% methyl-β-cyclodextrin (Sigma, C4555) solution in phosphate-buffered saline (PBS; Nacalai Tesque, 14249-24). All mice were generated and/or maintained in a C57BL/6J inbred background. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Tokyo Medical and Dental University.
Flow cytometry
Single-cell suspensions were prepared from the thymus, spleen, and peripheral lymph nodes of mice aged 6–10 wk. Cells were analyzed by flow cytometry using the following antibodies: anti-CD4-PEcy7 (100528), anti-CD4-APC (100516), anti-CD8-PEcy7 (100722), anti-ITGAM/CD11b-PEcy7 (101216), anti-Ly-6G/Ly-6C-APC (108412), anti-PTPRCa/CD45.1-APC (110714), anti-PTPRCb/CD45.2-FITC (109806), anti-SELL/CD62L-FITC (553150), anti-SELL/CD62L-APC (104411), anti-CD69-FITC (104506), and anti-PTPRC/B220-PEcy7 (103222), which were all purchased from BioLegend. Anti-CD44-FITC (553133) and anti-CD44-PE (553134) were purchased from BD PharMingen. DAPI (D3571), MitoTracker Green FM (M7514), and CM-H2DCFDA (C6827) were all purchased from Invitrogen. Data were acquired using a FACS CantoII flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star).
In vitro T cell assay
For in vitro T cell assays, naïve CD4 T cells were purified by bead enrichment from the pooled spleen and lymph node tissues of the indicated strains of mice. In experiments using bead-enriched cells, naïve T cells were enriched by negative selection (EasySep, StemCell Technology, 19765A) (>95% purity). Inducible deletion of Tnfaip3 from T cells was achieved by treating purified naïve T cells from Tnfaip3fl/fl Esr-Cre mice with 10 nM 4-OHT (Sigma Aldrich, H7904) for 48 h. For in vitro stimulation, 5.0 × 104 T cells were cultured in 96-well plates containing plate-bound 2.5 µg/ml anti-CD3E (BD, 553058) and 5 µg/ml anti-CD28 (BD, 553295). Live cells were quantified by flow cytometry of DAPI-negative cells. Chloroquine was purchased from Sigma (C6628). In CHX chase experiments, naïve CD4 T cells purified from Esr-Cre mice were stimulated for 48 h with 4-OHT and then treated with 10 μg/ml CHX. Cell lysates were harvested for immunoblotting at the indicated time points. CHX was purchased from Nacalai Tesque (06741-91).
Microscopy analysis
Cells were washed with PBS and fixed in 4% paraformaldehyde (Nacalai Tesque, 09154-85) for 10 min at 4°C. Fixed cells were permeabilized with 50 µg/ml digitonin (Invitrogen, BN2006) in PBS for 5 min, blocked with 3% bovine serum albumin (Nacalai Tesque, 01863-48) in PBS for 30 min, and incubated with LC3 antibody (MBL, M152-3) for 1 h. After washing, cells were incubated with Alexa Fluor 488-conjugated anti-mouse antibody (Invitrogen, A11001) for 30 min. Images were acquired with a confocal laser microscope (FV10i, Olympus) using a 60× oil-immersion objective lens. For transmission electron microscopy, cells were prefixed in 1% paraformaldehyde overnight and fixed in 1% osmium buffer for 2 h. Thin sections (80–90 nm) were cut, mounted on copper grids, and post-stained with uranyl acetate and lead citrate. Micrographs were taken using JEM-1200 EX (JEOL, Ltd). We calculated the mitochondrial surface area in manually outlined mitochondria using a quantification tool in ImageJ.
Detection of protein interactions by in situ PLA
To detect protein interactions in CD4 T cells, a Duolink PLA in situ kit (Sigma-Aldrich, 92101) was used according to the manufacturer's instructions. The primary antibodies were rabbit anti-MTOR antibody (Cell Signaling Technology, 2983) and mouse anti-TNFAIP3 monoclonal antibody (Abcam, ab13597). As a control, the primary antibody was mouse control IgG (Vector Laboratories, I-2000). Images were acquired with a confocal laser microscope (FV10i, Olympus), using a 60× oil-immersion objective lens.
Cell culture and reagents
HEK293T cells were cultured in Dulbecco's modified Eagle's medium (Sigma, D5796), with fetal bovine serum (BioWest, S1820) and penicillin/streptomycin (nacalai tesque, 26253-84) at 37°C. pCMV-Flag-TNFAIP3 has been described previously.12 HEK293T cells were transiently transfected using TransIT (Mirus, MIR2300) as indicated by the supplier.
Quantitative RT-PCR
Total RNA from naïve or stimulated CD4 T cells was isolated using the RNeasy Mini Kit (QIAGEN, 74106). cDNA was synthesized using a QuantiTect Reverse Transcription Kit (QIAGEN, 205313). qPCR was performed with a QuantiTect SYBR Green Master Mix (QIAGEN, 204145) and the StepOnePlus Real-Time PCR System (Applied Biosystems, StepOnePlus-01). Gene expression was normalized to expression of the housekeeping gene Actb. The expression of genes was determined using the following primers: Bcl2, 5′-AGTACCTGAACCGGCATCTG-3′ and 5′-CATGCTGGGGCCATATAGTT-3′; Bax, 5′-TGGAGATGAACTGGACAGCA-3′ and 5′-TGAAGTTGCCATCAGCAAAC-3′; Bak1, 5′-CCACATCTGGAGCAGAGTCA-3′ and 5′-TGTCCAGATGCCATTTTTCA-3′; Ifng, 5′-CAATGAACGCTACACACTGC-3′ and 5′-CCACATCTATGCCACTTGAG-3′; Actb, 5′-GACAGGATGCAGAAGGAGA-3′ and 5′-GTACT- TGCGCTCAGGAGGAG-3′; Mcl1, 5′-GTGGAGTTCTTCCACGTACAGGA-3′ and 5′-AGCAACACCCGCAAAAGC-3′; Il17, 5′-CTCCAGAAGGCCCTCAGACTAC-3′ and 5′-AGCTTTCCCTCCGCATTGACACAG-3′; Atg5, 5′-AACTGAAAGAGAAGCAGAACCA-3′ and 5′-TGTCTCATAACCTTCTGAAAGTGC-3′; and Tnfaip3, 5′-AAACCAATGGTGAT- GGAAACTG-3′ and 5′-GTTGTCCCATTCGTCATTCC-3′
Immunoblotting
CD4 T cells or thymocytes were incubated in lysis buffer (either 20 mM HEPES, pH 7.5, 150 mM NaCl, 0.5% CHAPS [Nacalai Tesque, 07957-64], 10% glycerol, 2 mM N-ethylmaleimide, and Halt protease and phosphatase inhibitor cocktail [Pierce, 1861280] or 20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.2% NP-40 [Nacalai Tesuque, 25223-04], 10% glycerol, 2 mM N-ethylmaleimide, and protease inhibitors) on ice for 20 min and centrifuged at 14,000 ×g for 20 min. For Flag immunoprecipitation, cell lysates were incubated with anti-Flag M2 beads (Sigma, A2220). For MTOR immunoprecipitation, anti-MTOR antibody (Cell Signaling Technology, 2983) was coupled with Dynabeads Protein G (Novex, 10004D) in PBS. Cell lysates were incubated with the pre-coupled beads for 10 h at 4°C. Samples were resolved on NuPage precast 4%-12% Bis-Tris gels (Invitrogen, NP0323) and transferred to PVDF membranes. The following antibodies and reagents were used for immunoprecipitation and immunoblotting studies: anti-ACTB (Sigma, A5441), anti-Flag (Sigma, F7425), anti-ATG5 (MBL, PM050), anti-LC3 (MBL, PD014), anti-TNFAIP3 (Cell Signaling Technology, 5630), anti-phospho-PRKAA/AMPKα (Thr172) (Cell Signaling Technology, 2535), anti-PRKAA/AMPKα (Cell Signaling Technology, 2532), anti-phospho-AKT (Thr308) (Cell Signaling Technology, 2965), anti-AKT (Cell Signaling Technology, 9272), anti-RPS6KB1 (Cell Signaling Technology, 2708), anti-phospho-RPS6KB1 (Thr389) (Cell Signaling Technology, 9206), anti-phospho-MTOR (Ser2481) (Cell Signaling Technology, 2974), anti-MTOR (Cell Signaling Technology, 2983), anti-phospho-EIF4EBP1 (Thr37/46) (Cell Signaling Technology, 2855), anti-EIF4EBP1 (Cell Signaling Technology, 9452), and anti-Ub (P4D1) (Santa Cruz Biotechnology, sc-8017). Secondary antibodies (mouse anti-rabbit and goat anti-mouse, 211-032-171 and 115-035-174, respectively) were purchased from Jackson Laboratories.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Barbara Malynn for critically reading the manuscript. We also thank Takeshi Kaizuka, Eisuke Itakura, and Noboru Mizushima for technical assistance.
Funding
This work was supported by JSPS KAKENHI Grant Number 25460946, Takeda Science Foundation, the Research Center Network Program for Realization of Regenerative Medicine, from the Japan Science and Technology Agency (JST)/Japan Agency for Medical Research and Development (AMED), the Japan Foundation for Applied Enzymology, Foundation for Advancement of International Science, The Mochida Memorial Foundation for Medical and Pharmaceutical Research, and the Health and Labour Sciences Research Grants for research on intractable diseases from Ministry of Health, Labour and Welfare of Japan.
References
- 1.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823-30; PMID:20811354; http://dx.doi.org/ 10.1038/ncb0910-823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/ 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol 2009; 182:4046-55; PMID:19299702; http://dx.doi.org/ 10.4049/jimmunol.0801143 [DOI] [PubMed] [Google Scholar]
- 4.Stephenson LM, Miller BC, Ng A, Eisenberg J, Zhao Z, Cadwell K, Graham DB, Mizushima NN, Xavier R, Virgin HW, et al.. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy 2009; 5:625-35; PMID:19276668; http://dx.doi.org/ 10.4161/auto.5.5.8133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maciolek JA, Alex Pasternak J, Wilson HL. Metabolism of activated T lymphocytes. Curr Opin Immunol 2014; 27C: 60-74; PMID:24556090; http://dx.doi.org/ 10.1016/j.coi.2014.01.006 [DOI] [PubMed] [Google Scholar]
- 6.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol 2012; 12:325-38; PMID:22517423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colombetti S, Basso V, Mueller DL, Mondino A. Prolonged TCR/CD28 engagement drives IL-2-independent T cell clonal expansion through signaling mediated by the mammalian target of rapamycin. J Immunol 2006; 176:2730-8; PMID:16493028; http://dx.doi.org/ 10.4049/jimmunol.176.5.2730 [DOI] [PubMed] [Google Scholar]
- 8.Zheng Y, Collins SL, Lutz MA, Allen AN, Kole TP, Zarek PE, et al.. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J Immunol 2007; 178:2163-70; PMID:17277121; http://dx.doi.org/ 10.4049/jimmunol.178.4.2163 [DOI] [PubMed] [Google Scholar]
- 9.Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol 2009; 183:6095-101; PMID:19841171; http://dx.doi.org/ 10.4049/jimmunol.0803510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tamás P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, Cantrell DA. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med 2006; 203:1665-70;PMID:16818670; http://dx.doi.org/ 10.1084/jem.20052469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214-26; PMID:18439900; http://dx.doi.org/ 10.1016/j.molcel.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011; 13:1016-23; PMID:21892142; http://dx.doi.org/ 10.1038/ncb2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12:21-35; PMID:21157483; http://dx.doi.org/ 10.1038/nrm3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008; 321:1499-502; PMID: 18787170; http://dx.doi.org/ 10.1126/science.1162981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell 2013; 51:283-96; PMID:23911927; http://dx.doi.org/ 10.1016/j.molcel.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al.. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004; 430:694-9; PMID:15258597; http://dx.doi.org/ 10.1038/nature02794 [DOI] [PubMed] [Google Scholar]
- 17.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, et al.. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 2004; 5:1052-60; PMID:15334086; http://dx.doi.org/ 10.1038/ni1110 [DOI] [PubMed] [Google Scholar]
- 18.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000; 289:2350-4; PMID:11009421; http://dx.doi.org/ 10.1126/science.289.5488.2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hitotsumatsu O, Ahmad RC, Tavares R, Wang M, Philpott D, Turer EE, Lee BL, Shiffin N, Advincula R, Malynn BA, et al.. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity 2008; 28:381-90; PMID:18342009; http://dx.doi.org/ 10.1016/j.immuni.2008.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao L, Oshima S, Duong B, Advincula R, Barrera J, Malynn BA, Ma A. A20 restricts wnt signaling in intestinal epithelial cells and suppresses colon carcinogenesis. PLoS One 2013; 8: e62223; PMID:23671587; http://dx.doi.org/ 10.1371/journal.pone.0062223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tewari M, Wolf FW, Seldin MF, O'Shea KS, Dixit VM, Turka LA. Lymphoid expression and regulation of A20, an inhibitor of programmed cell death. J Immunol 1995; 154:1699-706; PMID:7836754 [PubMed] [Google Scholar]
- 22.Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, Sun L, Chen ZJ, Marynen P, Beyaert R. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol 2008; 9:263-71; PMID:18223652; http://dx.doi.org/ 10.1038/ni1561 [DOI] [PubMed] [Google Scholar]
- 23.Düwel M, Welteke V, Oeckinghaus A, Baens M, Kloo B, Ferch U, Darnay BG, Ruland J, Marynen P, Krappmann D. A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol 2009; 182:7718-28; PMID:19494296; http://dx.doi.org/ 10.4049/jimmunol.0803313 [DOI] [PubMed] [Google Scholar]
- 24.Giordano M, Roncagalli R, Bourdely P, Chasson L, Buferne M, Yamasaki S, Beyaert R, van Loo G, Auphan-Anezin N, Schmitt-Verhulst AM. The tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20) imposes a brake on antitumor activity of CD8 T cells. Proc Natl Acad Sci U S A 2014. 111:11115-20; PMID:25024217; http://dx.doi.org/ 10.1073/pnas.1406259111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol 2014; 35:22-31; PMID:24246475; http://dx.doi.org/ 10.1016/j.it.2013.10.005 [DOI] [PubMed] [Google Scholar]
- 26.Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol 2012; 12:774-85; PMID:23059429; http://dx.doi.org/ 10.1038/nri3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harhaj EW, Dixit VM. Regulation of NF-kappaB by deubiquitinases. Immunol Rev 2012; 246:107-24; PMID:22435550; http://dx.doi.org/ 10.1111/j.1600-065X.2012.01100.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vereecke L, Beyaert R, van Loo G. Genetic relationships between A20/TNFAIP3, chronic inflammation and autoimmune disease. Biochem Soc Trans 2011; 39:1086-91; PMID:21787353; http://dx.doi.org/ 10.1042/BST0391086 [DOI] [PubMed] [Google Scholar]
- 29.Martin F, Dixit VM. A20 edits ubiquitin and autoimmune paradigms. Nat Genet 2011; 43:822-3; PMID:21874034; http://dx.doi.org/ 10.1038/ng.916 [DOI] [PubMed] [Google Scholar]
- 30.Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal 2010; 3: ra42; PMID:20501938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inomata M, Niida S, Shibata K, Into T. Regulation of Toll-like receptor signaling by NDP52-mediated selective autophagy is normally inactivated by A20. Cell Mol Life Sci 2012; 69:963-79; PMID:21964925; http://dx.doi.org/ 10.1007/s00018-011-0819-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 2009; 284:8023-32; PMID:19150980; http://dx.doi.org/ 10.1074/jbc.M900301200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med 2007; 204:25-31; PMID:17190837; http://dx.doi.org/ 10.1084/jem.20061303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L, Barrera J, Lowell CA, Utz PJ, Malynn BA. et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity 2010; 33:181-91; PMID:20705491; http://dx.doi.org/ 10.1016/j.immuni.2010.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S, et al.. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol 2011; 12:1184-93; PMID:22019834; http://dx.doi.org/ 10.1038/ni.2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bi Y, Zeng N, Chanudet E, Huang Y, Hamoudi RA, Liu H, et al.. A20 inactivation in ocular adnexal MALT lymphoma. Haematologica 2012; 97:926-30; PMID:22207688; http://dx.doi.org/ 10.3324/haematol.2010.036798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLeod IX, Jia W, He YW. The contribution of autophagy to lymphocyte survival and homeostasis. Immunol Rev 2012; 249:195-204; PMID:22889223; http://dx.doi.org/ 10.1111/j.1600-065X.2012.01143.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC, Jin S, Lu B. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol 2006; 177:5163-8; PMID:17015701; http://dx.doi.org/ 10.4049/jimmunol.177.8.5163 [DOI] [PubMed] [Google Scholar]
- 39.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol 2009; 9:324-37; PMID:19390566; http://dx.doi.org/ 10.1038/nri2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al.. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/ 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.