

Abstract
Cholesterol confers unique biophysical properties to the plasma membrane bilayer that are essential for maintaining optimal membrane fluidity, which in turn regulate multiple physiological functions required to promote cellular integrity and viability. Conversely, excessive cholesterol causes pathological conditions such as atherosclerosis that can lead to heart attacks. Human atheroma macrophages carry a large burden of free cholesterol (FC) in addition to cholesterol esters. It is recognized that sterols can modulate the levels of other lipids to attain lipid homeostasis; thus, excess FC may play a role in modulating compensatory sphingolipid pathways. Recent studies have shown that excess lipids can cause ER stress and apoptosis. In contrast, autophagy may play a protective role by clearing excess lipids from macrophage foam cell lipid droplets. Interestingly, a macrophage study using a TLR4-specifc agonist showed that de novo sphingolipid biosynthesis is essential for autophagy induction, suggesting links between sphingolipid biosynthesis and autophagy. While the role of autophagy in removing excess lipids has been the focus of many studies, its role in fine-tuning cellular lipid homeostasis remains largely unexplored.
Keywords: atherosclerosis, autophagy, ER stress, free cholesterol, LC3, ORMDL1, p62, serine palmitoyl-CoA transferase, sphingomyelin
Here, we discuss our recent work implicating autophagy in regulating sphingolipid biosynthesis in FC-loaded macrophages. FC-induced cell death in macrophages is mediated in the ER via induction of the unfolded protein response leading to apoptosis. Macrophages adapt to excess FC by increasing the synthesis of phosphatidylcholine (PC) via post-translational dephosphorylation and activation of CTP:phosphocholine cytidylyltransferase, the rate limiting enzyme of PC biosynthesis. It is thought that increased PC might buffer the excess FC and protect from free-cholesterol-mediated cytotoxicity. We confirmed an early report that free-cholesterol loading also induces cellular sphingomyelin (SM) levels. We also observed increased lipid rafts in FC-loaded cells, where excess cholesterol might be sequestered along with SM, also buffering against FC toxicity. Sterols regulate sphingolipid levels via increased recruitment of COL4A3BP/CERT (collagen, type IV, α 3 [Goodpasture antigen] binding protein) from the ER to the Golgi to increase ceramide pools of the Golgi. However, the mechanism by which sterols might activate de novo SM biosynthesis was not known.
The rate-limiting step in sphingolipid biosynthesis is catalyzed by the ER resident enzyme complex serine palmitoyl-CoA transferase (SPT), a heterodimer of SPTLC1 with either SPTLC2 or SPTLC3. In yeast, SPT activity is post-translationally inhibited by and bound to the orosomucoid-like (Orm) proteins. Under conditions of low sphingolipid levels Orm proteins are phosphorylated inactivating their inhibitory activity and allowing increased SPT activity. However, the orthologous mammalian ORMDL proteins have truncated N-termini and lack the phosphorylation sites found in yeast Orm proteins. Thus, the mechanism of the regulation of SPT by ORMDL proteins in mammalian systems may be different from that observed in yeast.
We explored the mechanism of our findings that FC-loaded macrophages have higher SM levels and more lipid rafts as compared to unloaded cells. Increased sphingomyelin could be due to either attenuating its degradation or increasing its de novo biosynthesis. The level of SMPD2, the neutral sphingomyelinase located at the plasma membrane is unchanged by FC loading; however, SPT activity is significantly increased upon FC loading. The levels of SPT enzyme complex proteins are not increased upon FC loading, indicating that increased SPT activity is not due to induced SPT expression. Co-immunoprecipitation experiments showed that SPT is bound by ORMDL1 under normal growth conditions and is thus repressed. However,FC-loaded macrophages have increased ORMDL1 degradation resulting in decreased ORMDL1 protein levels over time. The FC-mediated degradation of ORMDL1 is rescued by treatment with a lysosomal inhibitor but not by proteasome inhibitors, indicating the role of the lysosomal pathway in degrading ORMDL1. Although FC loading causes ER stress, generalized ER stress by tunicamycin treatment fails to increase ORMDL1 degradation, arguing against the notion that ER stress alone is sufficient for degradation of this protein. To further probe the mechanism of FC-mediated degradation of ORMDL1, the localization of ORMDL proteins was determined in FC-loaded cells. Both ORMDL1 and ORMDL3 were translocated out of the ER and found in cytoplasmic puncta upon FC loading. Combined with the lysosomal inhibitor effect, we hypothesized that FC-induced ORMDL protein degradation is mediated by autophagy. Further investigation revealed that the cytoplasmic puncta carrying the ORMDL1 and ORMDL3 proteins, under FC loading conditions, are in fact autophagosomes. Using multiple assays, including SQSTM1/p62 and LC3 western blots and colocalization studies, we found that FC loading induced autophagy and that siRNA-mediated knockdown of ATG7 to inhibit the initiation of autophagy restored ORMDL1 protein levels in FC-loaded cells. We observed that autophagy induced by serum-starvation fails to increase ORMDL1 degradation, indicating some specificity for the FC-induced autophagic degradation of ORMDL1. Autophagy of ORMDL1 in FC-loaded cells occurred without bulk reticulophagy as there was no translocation of the ER marker protein CALR (calreticulin) to cytoplasmic puncta in FC-loaded cells.
FC loading in macrophages appears to evoke 2 independent events, the induction of autophagy and the translocation of ORMDL proteins from the ER to autophagosomes. It is not clear if these events occur sequentially or simultaneously. The consequence of this removal of ORMDL proteins from the ER is activation of SPT leading to increased SM biosynthesis and SM levels, which can buffer excess FC and decrease its cytotoxicity (Fig. 1). However, the consequences of increasing macrophage SPT activity and SM levels may not all be beneficial, as inhibition of SPT with the drug myriocin increases ABCA1-dependent and -independent cholesterol efflux from cultured macrophages and also decreases cholesterol absorption and atherosclerosis in vivo in mouse models. ORMDL proteins are thus novel responders sensing excess FC and relieving negative feedback on the biosynthesis of sphingolipids. We speculate that drugs targeted to the ORMDL protein pathway might be used to prevent excess SM biosynthesis with the ultimate aim of reducing atherosclerosis.
Figure 1.
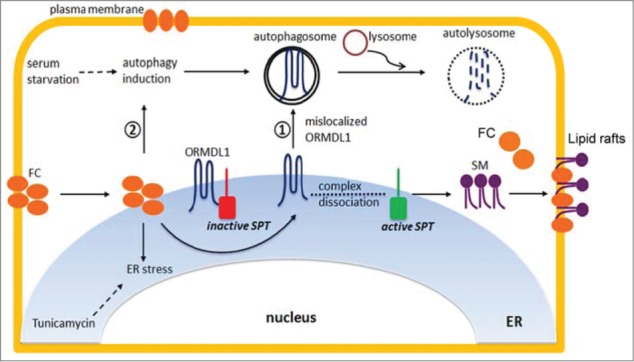
Mechanism for the role of FC-induced autophagy in increasing SM biosynthesis. FC loading in macrophages induces 2 events; induction of autophagy and ejection of ORMDL from ER. These 2 events result in degradation of ORMDL1 and increased SPT activity leading to increased SM biosynthesis.