

Abstract
Although RIPK1 (receptor [TNFRSF]-interacting protein kinase 1) is emerging as a critical determinant of cell fate in response to cellular stress resulting from activation of death receptors and DNA damage, its potential role in cell response to endoplasmic reticulum (ER) stress remains undefined. Here we report that RIPK1 functions as an important prosurvival mechanism in melanoma cells undergoing pharmacological ER stress induced by tunicamycin (TM) or thapsigargin (TG) through activation of autophagy. While treatment with TM or TG upregulated RIPK1 and triggered autophagy in melanoma cells, knockdown of RIPK1 inhibited autophagy and rendered the cells sensitive to killing by TM or TG, recapitulating the effect of inhibition of autophagy. Consistently, overexpression of RIPK1 enhanced induction of autophagy and conferred resistance of melanoma cells to TM- or TG-induced cell death. Activation of MAPK8/JNK1 or MAPK9/JNK2, which phosphorylated BCL2L11/BIM leading to its dissociation from BECN1/Beclin 1, was involved in TM- or TG-induced, RIPK1-mediated activation of autophagy; whereas, activation of the transcription factor HSF1 (heat shock factor protein 1) downstream of the ERN1/IRE1-XBP1 axis of the unfolded protein response was responsible for the increase in RIPK1 in melanoma cells undergoing pharmacological ER stress. Collectively, these results identify upregulation of RIPK1 as an important resistance mechanism of melanoma cells to TM- or TG-induced ER stress by protecting against cell death through activation of autophagy, and suggest that targeting the autophagy-activating mechanism of RIPK1 may be a useful strategy to enhance sensitivity of melanoma cells to therapeutic agents that induce ER stress.
Keywords: autophagy, cell death, endoplasmic reticulum stress, melanoma, RIPK1
Abbreviations
- 3-MA
3-methyladenine
- AMPK
AMP-activated protein kinase
- ATF6
activating transcription factor 6
- Baf A1
bafilomycin A1
- CAMKK2
calcium/calmodulin-dependent protein kinase kinase 2: β
- EIF2AK3/PERK
eukaryotic translation initiation factor 2-α kinase 3
- ER
endoplasmic reticulum
- ERN1/IRE1
endoplasmic reticulum to nucleus signaling 1
- HSF1
heat shock transcription factor 1
- HSPA5
heat shock 70kDa protein 5 (glucose-regulated protein: 78kDa)
- MAP2K1/MEK1
mitogen-activated protein kinase kinase 1
- MAPK
mitogen-activated protein kinase
- MAPK1/ERK2
mitogen-activated protein kinase 1
- MAPK3/ERK1
mitogen-activated protein kinase 3
- MAPK8/JNK1
mitogen-activated protein kinase 8
- MAPK9/JNK2
mitogen-activated protein kinase 9
- MAPK11/p38β
mitogen-activated protein kinase 11
- MAPK12/p38γ
mitogen-activated protein kinase 12
- MAPK13/p38δ
mitogen-activated protein kinase 13
- MAPK14/p38α
mitogen-activated protein kinase 14
- NFKB1
nuclear factor of kappa light polypeptide gene enhancer in B-cells 1
- PRKAA1
protein kinase AMP-activated: α 1 catalytic subunit
- RIPK1
receptor (TNFRSF)-interacting protein kinase 1
- SQSTM1/p62
sequestosome 1
- TG
thapsigargin
- TM
tunicamycin
- TNFRSF1A/TNFR1
tumor necrosis factor receptor superfamily: member 1A
- UPR
unfolded protein response
- XBP1
x-box binding protein 1
Introduction
RIPK1 (receptor [TNFRSF]-interacting protein kinase 1) is a protein Ser/Thr kinase that mediates both cell survival and death signaling and is emerging as an important determinant of cell fate in response to cellular stress, in particular, to activation of death receptors such as the TNFRSF1A/TNFR1 (tumor necrosis factor receptor superfamily, member 1A).1-4 Upon TNFRSF1A stimulation, RIPK1 is stabilized through K63 (Lys63)-linked polyubiquitination, which activates NFKB1 (nuclear factor of kappa light polypeptide gene enhancer in B-cells 1) and MAPKs (mitogen activated protein kinases) to promote cell survival.5,6 However, on conditions where K63-linked ubiquitination of RIPK1 is inhibited, RIPK1 switches its function to that of promoting apoptosis.2,3 In addition, when activation of CASP8 (caspase 8, apoptosis-related cysteine peptidase) is limited, deubiquitinated RIPK1 recruits RIPK3 leading to programmed necrosis (necroptosis) in some types of cells.2,7 Although many studies in recent years have primarily focused on the role of RIPK1 in induction of cell death,1,2,4 ripk1−/− mice display extensive apoptosis and die at age 1 to 3 d,1 indicative of the importance of a prosurvival role of RIPK1 in cells.
Endoplasmic reticulum (ER) stress is characterized by accumulation and aggregation of unfolded and/or misfolded proteins in the ER lumen. The ER responds to ER stress by activating a range of signaling pathways,8-11 which couples the ER protein folding load with the ER protein folding capacity and is termed the ER stress response or the unfolded protein response (UPR). The UPR of mammalian cells is initiated by 3 ER transmembrane proteins, ATF6 (activating transcription factor 6), ERN1/IRE1 (endoplasmic reticulum to nucleus signaling 1), and EIF2AK3/PERK (eukaryotic translation initiation factor 2-α kinase 3).8-11 The UPR is essentially a cellular protective response, but excessive or prolonged UPR can kill cells primarily by induction of apoptosis.12-14 Of note, previous studies have shown that many types of cancer cells including melanoma cells are relatively resistant to apoptosis triggered by pharmacological ER stress inducers such as tunicamycin (TM) and thapsigargin (TG).15,16 A number of mechanisms, such as upregulation of the prosurvival BCL2 family protein MCL1 and activation of the mitogen-activated protein kinase kinase 1 (MAP2K1)-MAPK1/3 and phosphoinositide 3-kinase (PI3K)-AKT signaling pathways contribute to reduced susceptibility of melanoma cells to pharmacological ER stress-induced cell death.16-19
Besides induction of apoptosis, ER stress can also activate autophagy, which is essentially a lysosomal degrading process through sequestering proteins and organelles into autophagosomes (autophagic vesicles) that eventually fuse with lysosomes for degradation.20-24 Similar to the UPR, autophagy is a prosurvival mechanism that recycles cellular constituents to maintain nutrient supply in cells under stresses such as nutrition deprivation.24 However, prolonged or excessive autophagy is often associated with cell death.25,26 In the pathogenesis of cancer, autophagy is thought to be a double-edged sword, as it functions to promote or inhibit cancer development and progression in a stage- and context-dependent manner. Nevertheless, accumulating evidence has shown that autophagy in general promotes cancer cell survival under various cellular stress conditions including ER stress.27-31
A number of mechanisms have been shown to contribute to activation of autophagy by ER stress, such as induction of HSPA5/GRP78 (heat shock 70kDa protein 5 [glucose-regulated protein, 78kDa]) and activation of the EIF2AK3-EIF2S1-ATF4 axis of the UPR and the calcium-CAMKK-AMPK signaling pathway.20,30,32 Activation of MAPK8/9 by ER stress also activates autophagy through phosphorylation of prosurvival BCL2 family proteins including BCL2 and BCL2L1/BIM thus dissociating them from BECN1/Beclin 1 that plays a critical part in initiation of autophagy flux.33-36 Similarly, the other prosurvival protein MCL1 can also bind to BECN1 and disruption of the association triggers autophagy.37,38 It seems therefore that ER stress may activate autophagy through varying mechanisms depending on cell types and stimuli in question.
HSF1 (heat shock transcription factor 1) is an evolutionarily conserved transcription factor that not only plays an important role in orchestrating cellular responses to heat shock, but is also involved in many other biological processes, such as cell division, insulin signaling, and embryonic development.39 The expression of HSF1 is upregulated in many types of cancers including melanoma that promotes cancer cell survival, invasion, and migration.39,40 Unlike many transcription factors, HSF1 binds to DNA as a trimer.39,41 Binding of each DNA-binding domain in an HSF1 trimer to a single heat shock element (an nGAAn sequence) is necessary for HSF1-mediated transactivation of its target genes. 39,41 Noticeably, activation of HSF1 is primarily regulated by posttranslational modification such as phosphorylation and interactions with various binding partners.42
In this study, we have studied the role of RIPK1 in response of human melanoma cells to pharmacological ER stress induced by TM or TG. We show here that RIPK1 is upregulated in melanoma cells after treatment with TM or TG, and is important for protection of melanoma cells from TM- or TG-induced apoptosis by induction of autophagy. Moreover, we show that RIPK1-triggered autophagy is executed by MAPK8/9-mediated phosphorylation of BCL2L11 thus leading to its disassociation from BECN1, and that upregulation of RIPK1 by ER stress is caused by a transcriptional increase mediated by HSF1 downstream of the ERN1-XBP1 axis of the UPR.
Results
RIPK1 is upregulated in human melanoma cells under ER stress induced by TM or TG
We examined the expression of RIPK1 in Mel-RM and MM200 cells in response to pharmacological ER stress induced by TM or TG. These cells were relatively resistant to cell death induced by TM or TG even when it was used at a concentration (3 μM and 1 μM for TM and TG, respectively) at which induction of cell death reached a “plateau” (Fig. S1A).43 Activation of the UPR was evidenced by induction of HSPA5, the active (spliced) form of XBP1 mRNA, and DDIT3/CHOP, phosphorylation of EIF2S1, and cleavage of ATF6 (Fig. 1A and Fig. S1B and C). Remarkably, induction of ER stress upregulated RIPK1 in both Mel-RM and MM200 cells (Fig. 1A). This was associated with elevation in its mRNA expression that was caused by a transcriptional increase instead of changes in the stability of the mRNA, as indicated by its turnover rates, which remained similar in cells before and after treatment with TM or TG as shown in actinomycin D-chasing assays (Fig. 1B and C). Upregulation of RIPK1 by TM and TG was confirmed in another 4 melanoma cell lines (ME4405, SK-Mel-28, Mel-CV, and IgR3) that were relatively resistant to ER stress-induced cell death (Fig. 1D and Fig. S1D and E).43 However, RIPK1 was not significantly increased by ER stress in Mel-RMu cells and melanocytes that were comparatively sensitive to cell death induced by ER stress, although the UPR was similarly activated in these cells by TM and TG (Fig. 1D and Fig. S1B–E). Of note, cell death induced by TM or TG in melanoma cells and melanocytes was mainly due to apoptosis, as it was markedly inhibited by the general caspase inhibitor z-VAD-fmk (Fig. S1F).
Figure 1.

RIPK1 is upregulated in human melanoma cells under ER stress induced by TM or TG. (A) Whole cell lysates from Mel-RM and MM200 cells with or without treatment with tunicamycin (TM) (3 μM) (left panel) or thapsigargin (TG) (1 μM) (right panel) for the indicated periods were subjected to western blot analysis of RIPK1, HSPA5, and GAPDH (as a loading control). The numbers represent fold changes of HSPA5. The data shown are representative of 3 individual experiments. (B) Total RNA from Mel-RM and MM200 cells with or without treatment with TM (3 μM) (left panel) or TG (1 μM) (right panel) for the indicated periods were subjected to qPCR analysis of RIPK1 mRNA expression. The relative abundance of the RIPK1 mRNA before treatment was arbitrarily designated as 1 (n = 3, mean ±SEM). (C) Total RNA from Mel-RM and MM200 cells treated with TM (3 μM) or TG (1 μM) for 16 h followed by treatment with actinomycin D (100 ng/ml) for the indicated period was subjected to qPCR analysis for the expression of RIPK1 mRNA. The relative abundance of the RIPK1 mRNA without actinomycin D treatment was arbitrarily designated as 1 (n = 3, mean ±SEM, *P < 0.05, Student t test). (D) Whole cell lysates from ME4405, Sk-Mel-28, Mel-CV, IgR3 and Mel-RMu melanoma cells and HEMn-MP melanocytes treated with TM (3 μM) or TG (1 μM) for 16 h were subjected to western blot analysis of RIPK1, HSPA5, and GAPDH (as a loading control). The numbers represent fold changes of HSPA5. The data shown are representative of 3 individual experiments.
RIPK1 protects melanoma cells from killing by TM and TG
We focused on examination of the functional importance of RIPK1 upregulation in response of melanoma cells to pharmacological ER stress by knocking down RIPK1 with 2 individual shRNAs in Mel-RM and MM200 cells (Fig. 2A). Strikingly, RIPK1 knockdown markedly reduced viability of melanoma cells upon treatment with TM or TG (Fig. 2B).44 This was also reflected by reduction in long-term survival in clonogenic experiments (Fig. 2C). Introduction of a construct expressing shRNA-resistant cDNA of RIPK1 reversed the inhibitory effect of RIPK1 knockdown on cell survival (Figs. 2D and E), demonstrating the specificity of the RIPK1 shRNA, and consolidating that RIPK1 plays a role in promoting survival of melanoma cells undergoing TM- or TG-induced ER stress. Consistently, overexpression of RIPK1 enhanced survival of melanocytes upon treatment with TM or TG (Figs. 2F and G). The role of RIPK1 in protection of melanoma cells from cell death induced by pharmacological ER stress was further confirmed by knockdown of RIPK1 in Mel-RMu cells, which were relatively sensitive to ER stress-induced apoptosis. Although RIPK1 knockdown alone did not cause significant cell death in Mel-RMu cells, it further enhanced killing induced by TM or TG (Fig. S2A and B).
Figure 2.

RIPK1 protects melanoma cells from killing by TM or TG. (A) Whole cell lysates from Mel-RM and MM200 cells transduced with the control or RIPK1 shRNA treated with tunicamycin (TM) (3 μM) or thapsigargin (TG) (1 μM) for 16 h were subjected to western blot analysis of RIPK1, HSPA5, and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. The numbers represent fold changes of HSPA5. (B) Mel-RM and MM200 cells transduced with the control or RIPK1 shRNA were treated with TM (3 μM) or TG (1 μM) for 48 h. Cell viability was measured by CellTiter-Glo assays (n = 3, mean ±SEM, *P < 0.05, Student t test). (C) Mel-RM and MM200 cells transduced with the control or RIPK1 shRNA were seeded onto 6-well plates at 2000 cells per well in medium containing TM (1 μM) or TG (0.3 μM). Twelve d later, cells were fixed and stained with crystal violet. The data shown are representative of 3 individual experiments. (D) Mel-RM and MM200 cells stably transduced with the control or RIPK1 shRNA were transiently transfected with a shRNA-resistant mutant form of RIPK1 (pCMV-MYC-mut-RIPK1). Twenty-four h later, whole cell lysates were subjected to western blot analysis of RIPK1 and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (E) Mel-RM and MM200 cells stably transduced with the control or RIPK1 shRNA were transiently transfected with a shRNA-resistant mutant form of RIPK1 (pCMV-MYC-mut-RIPK1). Twenty-four h later, cells were treated with TM (3 μM) or TG (1 μM) for another 48 h. Cell viability was measured by CellTiter-Glo assays (n = 3, mean ±SEM, *P < 0.05; Student t test). (F) Whole cell lysates from HEMn-MP melanocytes transfected with the pCMV-MYC or pCMV-MYC-RIPK1 were subjected to western blot analysis of RIPK1 and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (G) HEMn-MP melanocytes transfected with pCMV-MYC or pCMV-MYC-RIPK1 were treated with TM (3 μM) or TG (1 μM) for 48 h. Cell viability was measured by CellTiter-Glo assays (n = 3, mean ±SEM, *P < 0.05, Student t test).
RIPK1 protects melanoma cells from TM- or TG-induced apoptosis by activation of autophagy
Since autophagy protects against apoptosis induced by ER stress,26,45,46 we examined if RIPK1-mediated protection of melanoma cells upon treatment with TM or TG is associated with activation of autophagy. Indeed, TM or TG triggered autophagy in Mel-RM and MM200 cells as evidenced by conversion of MAP1LC3A (microtubule-associated protein 1 light chain 3 α)-I into MAP1LC3A-II, aggregation of MAP1LC3A-II, formation of double-membrane autophagosomes, and degradation of SQSTM1/p62 (sequestosome 1) (Fig. 3A to C and Fig. S3A).23 However, only moderate activation of autophagy was observed in Mel-RMu cells after treatment with TM or TG (Fig. S3B). Blockade of autophagy at late stages by bafilomycin A1 (Baf A1) or inhibition of autophagy induction by 3-methyladenine (3-MA) rendered Mel-RM and MM200 cells more sensitive to killing by TM or TG (Fig. 3D and E),23 indicating that autophagy plays a role in protection of melanoma cells from TM- or TG-induced cell death. This was further confirmed by knockdown of the autophagy protein ATG5 or ATG7, which similarly sensitized Mel-RM and MM200 cells to cell death induced by TM or TG (Fig. 3F to I). Cell death induced by TM or TG in Mel-RM and MM200 cells with autophagy inhibited was blocked by z-VAD-fmk, indicating that autophagy protects melanoma cells from ER stress by inhibiting apoptosis (Fig. S3C).
Figure 3 (See previous page).

RIPK1 protects melanoma cells from TM- or TG-induced apoptosis by activation of autophagy. (A) Whole cell lysates from Mel-RM and MM200 cells transduced with the control or RIPK1 shRNA were subjected to western blot analysis of RIPK1 and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (B) Mel-RM and MM200 cells transduced with the control or RIPK1 shRNA were treated with tunicamycin (TM) (3 μM) (upper panel) or thapsigargin (TG) (1 μM) (lower panel) for 16 h. Whole cell lysates were subjected to western blot analysis of SQSTM1, MAP1LC3A, and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (C) Electron microscopy images showing ultrastructures of Mel-RM cells transduced with the control or RIPK1 shRNA 1 treated with TM (3 μM) for 24 h. Arrowheads point to double-membraned autophagosomes. The data shown are representative of 3 individual experiments. (D and E) Mel-RM and MM200 cells with or without pretreatment with bafilomycin A1 (Baf A1) (10 nM) (D) or 3-methyladenine (3-MA) (5 mM) (E) for 1 h were treated TM (3 μM) or (TG) (1 μM) for another 48 h. Cell viability was measured by CellTiter-Glo (n = 3, mean ±SEM, *P < 0.05, Student t test). (F) Whole cell lysates from Mel-RM and MM200 cells transfected with the control or ATG5 siRNA were subjected to western blot analysis of ATG5 and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (G) Mel-RM and MM200 cells transfected with the control or ATG5 siRNA were treated with TM (3 μM) or TG (1 μM) for 48 h. Cell viability was measured by CellTiter-Glo (n = 3, mean ±SEM, *P < 0.05, Student t test). (H) Whole cell lysates from Mel-RM and MM200 cells transfected with the control or ATG7 siRNA were subjected to western blot analysis of ATG7 and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (I) Mel-RM and MM200 cells transfected with the control or ATG7 siRNA were treated with TM (3 μM) or TG (1 μM) for 48 h. Cell viability was measured by CellTiter-Glo (n = 3, mean ±SEM, *P < 0.05, Student t test).
To investigate the relationship between RIPK1 upregulation and autophagy triggered by TM or TG, we monitored induction of autophagy by ER stress in Mel-RM and MM200 cells with RIPK1 stably knocked down. Intriguingly, TM- or TG-induced autophagy was mitigated in RIPK1 knockdown Mel-RM and MM200 cells (Fig. 3A to C and Fig. S3A). In contrast, it was enhanced in Mel-RMu cells introduced with exogenous RIPK1 (Figs. 4A and B). These results suggest that RIPK1 is necessary for activation of autophagy in melanoma cells undergoing phamacological ER stress. On the other hand, inhibition of autophagy by 3-MA, Baf A1, or siRNA knockdown of ATG5 or ATG7 abolished protection of Mel-RMu cells from TM- or TG-induced apoptosis by overexpression of RIPK1 (Figs. 4C and D, and Fig. S3D and E), indicating that autophagy is required for RIPK1-mediated protection of melanoma cells. In support, knockdown of RIPK1 did not further sensitize Mel-RM and MM200 cells to killing by TM or TG when autophagy was inhibited (Figs. 4E and F). Collectively, these data reveal that RIPK1 promotes survival of melanoma cells upon ER stress triggered by TM or TG through activation of autophagy. Of note, blockade of autophagy had no effect on upregulation of RIPK1 by TM or TG (Fig. 4G).
Figure 4 (See previous page).

RIPK1 protects melanoma cells from TM- or TG-induced apoptosis by activation of autophagy. (A) Whole cell lysates from Mel-RMu cells stably transfected with pCMV-MYC or pCMV-MYC-RIPK1 were subjected to western blot analysis of RIPK1 and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (B) Mel-RMu cells stably transfected with pCMV-MYC or pCMV-MYC-RIPK1 were treated with tunicamycin (TM) (3 μM) or thapsigargin (TG) (1 μM) for 16 h. Whole cell lysates were subjected to western blot analysis of SQSTM1, MAP1LC3A and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (C) Mel-RMu cells stably transfected with pCMV-MYC or pCMV-MYC-RIPK1 were treated with TM (3 μM) or TG (1 μM) for 48 h with or without pretreatment with bafilomycin A1 (Baf A1) (10 nM) for 1 h. Cell viability was measured by CellTiter-Glo assays (n = 3, mean ±SEM, *P < 0.05, Student t test). (D) Mel-RMu cells stably transfected with pCMV-MYC or pCMV-MYC-RIPK1 were transiently transfected with the control or ATG5 siRNA followed by treatment with TM (3 μM) or TG (1 μM) for 48 h. Cell viability was measured by CellTiter-Glo assays (n = 3, mean ±SEM, *P < 0.05, Student t test). (E) Mel-RM (left panel) and MM200 (right panel) cells transduced with the control or RIPK1 shRNA were treated with TM (3 μM) or TG (1 μM) for 48 h with or without pretreatment with Baf A1 (10 nM) for 1 h. Cell viability was measured by CellTiter-Glo assays (n = 3, mean ±SEM, *P < 0.05, Student t test). (F) Mel-RM and MM200 cells transduced with the control or RIPK1 shRNA were treated with TM (3 μM) (upper panel) or TG (1 μM) (lower panel) with or without pretreatment with Baf A1 (10 nM) for 1 h. Whole cell lysates were subjected to western blot analysis of MAP1LC3A and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (G) Whole cell lysates from Mel-RM and MM200 cells treated with TM (3 μM) or TG (1 μM) for 16 h with or without pretreatment with Baf A1 (10 nM) for 1 h were subjected to western blot analysis of RIPK1 and GAPDH (as a loading control). The data shown are representative of 3 individual experiments.
RIPK1-mediated activation of MAPK8/9 contributes to induction of autophagy by ER stress in melanoma cells
Since MAPK8/9 is involved in ER stress-induced autophagy in many systems,20,37,47-49 we examined whether it similarly plays a role in activation of autophagy by RIPK1 in melanoma cells undergoing ER stress induced by TM or TG. Indeed, induction of ER stress caused activation of MAPK8/9 (Fig. 5A). Moreover, knockdown of MAPK8/9 attenuated induction of autophagy by TM or TG in Mel-RM and MM200 cells (Figs. 5B and C), suggesting that activation of MAPK8/9 is involved in TM- or TG-induced autophagy. Consistent with this, inhibition of MAPK8/9 by the specific inhibitor SP600125 or knockdown of MAPK8/9 reduced viability of Mel-RM and MM200 cells upon treatment with TM or TG (Figs. 5D and E).
Figure 5.

RIPK1-mediated activation of MAPK8/9 contributes to induction of autophagy by ER stress in melanoma cells. (A) Mel-RM and MM200 cells transduced with the control or RIPK1 shRNA were treated with tunicamycin (TM) (3 μM) or thapsigargin (TG) (1 μM) for 16 h. Whole cell lysates were subjected to western blot analysis of RIPK1, pMAPK8/9, MAPK8/9, and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (B) Whole cell lysates from Mel-RM and MM200 cells transfected with the control or MAPK8/9 siRNA were subjected to western blot analysis of MAPK8, MAPK9 and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (C) Mel-RM and MM200 cells transfected with the control or MAPK8/9 siRNA were treated with TM (3 μM) or TG (1 μM) for 16 h. Whole cell lysates were subjected to western blot analysis of RIPK1, SQSTM1, MAP1LC3A, and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (D) Mel-RM and MM200 cells were treated with TM (3 μM) or TG (1 μM) for 48 h with or without pretreatment with the MAPK8/9 inhibitor SP600125 (10 μM) for 1 h. Cell viability was measured by CellTiter-Glo assays (n = 3, mean ±SEM, *P < 0.05, Student t test). (E) Mel-RM and MM200 cells transfected with the control or MAPK8/9 siRNA 1 were treated with TM (3 μM) or TG (1 μM) for 48 h. Cell viability was measured by CellTiter-Glo assays (n = 3, mean ±SEM, *P < 0.05, Student t test). (F) Mel-RMu cells stably transfected with pCMV-MYC or pCMV-MYC-RIPK1 were treated with TM (3 μM) or TG (1 μM) for 16 h with or without pretreatement with SP600125 (10 μM) for 1 h. Whole cell lysates were subjected to western blot analysis of pMAPK8/9, MAPK8/9, SQSTM1, MAP1LC3A, and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (G) Whole cell lysates from Mel-RM cells treated with TM (3 μM) or TG (1 μM) for 16 h were immunoprecipitated by RIPK1 antibody. The resulting precipitates were subjected to western blot analysis of RIPK1 and MAPK8/9. The data shown are representative of 3 individual experiments.
We further examined the role of RIPK1 in activation of MAPK8/9 in melanoma cells upon treatment with TM or TG. While knockdown of RIPK1 inhibited activation of MAPK8/9 in Mel-RM and MM200 cells (Fig. 5A), overexpression of RIPK1 promoted MAPK8/9 activation in Mel-RMu cells by TM or TG (Fig. 5F). On the other hand, inhibition of MAPK8/9 did not impinge on TM- or TG-induced upregulation of RIPK1 in Mel-RM and MM200 cells (Fig. 5C). These results indicate that activation of MAPK8/9 by ER stress is mediated by RIPK1 in melanoma cells. In support, MAPK8/9 was coimmunoprecipitated with RIPK1 in melanoma cells upon treatment with TM or TG (Fig. 5G). Moreover, inhibition of MAPK8/9 suppressed ER stress-induced autophagy in Mel-RMu cells overexpressing RIPK1 (Fig. 5F), substantiating the role of activation of MAPK8/9 in RIPK1-mediated ER stress-induced autophagy in melanoma cells.
We also tested whether MAPK8/9-mediated activation of autophagy leads to formation of the ripoptosome and triggers necroptosis in melanoma cells undergoing pharmacological ER stress, as does it in other types of cancer cells under different conditions.50 Exposure of Mel-RM and MM200 cells to TM or TG did not affect the expression levels of BIRC2/cIAP1 (baculoviral IAP repeat-containing 2) and BIRC3/cIAP2, nor did it trigger formation of a ripoptosome, as neither TM nor TG triggered recruitment of CASP8 and FADD (Fas [TNFRSF6]-associated via death domain), which are key components of the ripoptosome, to RIPK1 (Fig. S4A and B). In contrast, RIPK1 and CASP8 and FADD were readily coprecipitated in cells treated with the genotoxic stress trigger etoposide that was included as a control (Fig. S4A and B).51 Consistent with these finding, TM- or TG-induced cell death in melanoma cells with or without RIPK1 knocked down could not be inhibited by the RIPK1 kinase inhibitor necrostatin-1, whereas it was blocked by z-VAD-fmk, indicative of induction of apoptosis rather than necroptosis (Fig. S4C).52 Therefore, induction of the formation of the ripoptosome and necroptosis by MAPK8/9-mediated activation of autophagy is highly cell type- and context-dependent.
MAPK8/9 activation phosphorylates BCL2L11 and disassociates it from BECN1 in melanoma cells undergoing ER stress
Disruption of the physical association between prosurvival BCL2 family proteins and BECN1 triggers autophagy.37 We examined whether this contributes to ER stress-induced autophagy in melanoma cells. While BCL2L1 did not appear to be physically associated with BECN1, BCL2 was coimmunoprecipitated with BECN1 (Fig. 6A). However, ER stress did not alter the amount of BCL2 associated with BECN1 (Fig. 6A). These results suggest that displacement of BCL2L1 and BCL2 from BECN1 is not likely to play a major role in ER stress-induced autophagy in melanoma cells. On the other hand, although ER stress caused reduction in the association between MCL1 and BECN1, this change was not affected by knockdown of RIPK1 (Fig. 6A), suggesting that displacement of MCL1 from BECN1 similarly does not play a role in RIPK1-mediated induction of autophagy by ER stress in melanoma cells.
Figure 6 (See previous page).

MAPK8/9 activation phosphorylates BCL2L11 and dissociates it from BECN1 in melanoma cells undergoing ER stress. (A) Whole cell lysates from Mel-RM cells transduced with control or RIPK1 shRNA 1 cells treated with tunicamycin (TM) (3 μM) for 16 h were immunoprecipitated by BECN1 antibody. The resulting precipitates were subjected to western blot analysis of BECN1, BCL2L1, BCL2, and MCL1. The data shown are representative of 3 individual experiments. (B) Whole cell lysates from Mel-RM cells treated with TM (3 μM) for 16 h with or without pretreatment with the MAPK8/9 inhibitor SP600125 (10 μM) for 1 h were subjected to western blot analysis of RIPK1, pMAPK8/9, MAPK8/9, pBCL2L11 (using a phosphorylated BCL2L11-specific antibody), and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (C) Whole cell lysates from Mel-RM cells treated with TM (3 μM) for 16 h with or without pretreatment with the MAPK8/9 inhibitor SP600125 (10 μM) for 1 h were subjected to immunoprecipitation with an antibody against BECN1. The resulting precipitates were subjected to western blot analysis of BECN1 and BCL2L11. The data shown are representative of 3 individual experiments. (D) Whole cell lysates from Mel-RM and MM200 cells transduced with the control or BCL2L11 shRNA were subjected to western blot analysis of BCL2L11 and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (E) Mel-RM and MM200 cells stably transduced with the control or BCL2L11 shRNA were transduced with RIPK1 shRNA 1 and treated with TM (3 μM) or thapsigargin (TG) (1 μM) for 16 h. Whole cell lysates were subjected to western blot analysis of SQSTM1, MAP1LC3A, or GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (F) Mel-RM and MM200 cells stably transduced with the control or BCL2L11 shRNA were treated with TM (3 μM) or TG (1 μM) for 16 h with or without pretreatment with the MAPK8/9 inhibitor SP600125 (10 μM) for 1 h were subjected to western blot analysis of SQSTM1, MAP1LC3A, or GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (G) Mel-RM and MM200 cells with BCL2L11 stably knocked down were transduced with the control or BECN1 shRNA. Twenty-four h later, whole cell lysates were subjected to western blot analysis of BECN1 or GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (H) Mel-RM and MM200 cells with or without BCL2L11 and BECN1 co-knocked down were treated with TM (3 μM) or TG (1 μM) for 16 h. Whole cell lysates were subjected to western blot analysis of SQSTM1, MAP1LC3A, and GAPDH (as a loading control). The data shown are representative of 3 individual experiments.
The BH3-only protein BCL2L11 recruits BECN1 to microtubule and thus suppresses activation of autophagy.35 However, phosphorylation of BCL2L11 disassociates the binding thus freeing BECN1 that initiates autophagy.35 Indeed, associated with the increase in activation of MAPK8/9, BCL2L11 phosphorylation was enhanced in melanoma cells upon induction of ER stress (Fig. 6B), pointing to a role of MAPK8/9 in ER stress-induced BCL2L11 phosphorylation. This was confirmed by inhibition of BCL2L11 phosphorylation with the MAPK8/9 inhibitor SP600125 (Fig. 6B). The increase in BCL2L11 phosphorylation was associated with a decrease in the amount of BCL2L11 coimmunoprecipitated with BECN1 (Fig. 6C), suggesting that displacement of BCL2L11 from BECN1 is involved in induction of autophagy in melanoma cells undergoing ER stress. Indeed, shRNA knockdown of BCL2L11 enhanced ER stress-induced autophagy even when RIPK1 was also knocked down or MAPK8/9 was inhibited (Figs. 6D–F). Nevertheless, BCL2L11 knockdown did not enhance autophagy induction by ER stress when BECN1 was also knocked down (Figs. 6D, G and H), consolidating the role of BECN1 in activation of autophagy mediated by disassociation of BCL2L11 in melanoma cells under ER stress.
We also examined whether the CAMKK2 (calcium/calmodulin-dependent protein kinase kinase 2, β)-AMP-activated protein kinase (AMPK) signaling pathway that has been implicated in ER stress-induced autophagy in other types of cells is involved in autophagy induced by pharmacological ER stress in melanoma cells.53,54 Although treatment with TM or TG caused phosphorylation of PRKAA1 (protein kinase, AMP-activated, α 1 catalytic subunit) in Mel-RM and MM200 cells, indicative of activation of calcium-CAMKK2-AMPK signaling (Fig. S5), the CAMKK2 inhibitor STO-609 did not affect the induction of autophagy and upregulation of RIPK1 by TM or TG (Fig. S5), suggesting that the calcium-CAMKK2-AMPK pathway does not play a major role in TM- or TG-induced upregulation of RIPK1 and activation of autophagy in melanoma cells.
Activation of HSF1 downstream of XBP1 is responsible for transcriptional upregulation of RIPK1 by TM or TG in melanoma cells
To identify the signaling pathway(s) of the UPR responsible for transcriptional upregulation of RIPK1 in melanoma cells under ER stress, we took advantage of melanoma cell lines deficient in the expression of ERN1, ATF6, or EIF2AK3, which were established by shRNA knockdown with lentiviral transduction (Fig. 7A).14 Knockdown of ERN1 or ATF6, but not knockdown of EIF2AK3, partially inhibited upregulation of RIPK1 by TM and TG (Fig. 7B). Moreover, deficiency in ERN1 or ATF6 recapitulated partially the inhibitory effect of RIPK1 knockdown on ER stress-triggered autophagy (Figs. 4F and 7B). Thus, RIPK1 upregulation in melanoma cells under ER stress is mediated by the ERN1 and ATF6 branches of the UPR.
Figure 7.

XBP1 plays an important role in RIPK1 upregulation in melanoma cells upon ER stress. (A) Whole cell lysates from Mel-RM and MM200 cells transduced with the control, ERN1, ATF6, or EIF2AK3 shRNA were subjected to western blot analysis of ERN1 (left panel), ATF6 (middle panel) or EIF2AK3 (right panel) and GAPDH (as a loading control). The numbers represent knockdown efficiencies. The data shown are representative of 3 individual western blot analyses. (B) Mel-RM and MM200 cells transduced with the control, ERN1, ATF6, or EIF2AK3 shRNA were treated with tunicamcyin (TM) (3 μM) or thapsigargin (TG) (1 μM) for 16 h. Whole cell lysates were subjected to western blot analysis of HSF1, RIPK1, SQSTM1, MAP1LC3A, and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (C) Mel-RM and MM200 cells transfected with the control or XBP1 siRNA were subjected to RT-PCR for the analysis of XBP1 mRNA. GAPDH was used as a loading control. The data shown are representative of 3 individual qPCR analyses. (D) Mel-RM and MM200 cells transfected with the control or XBP1 siRNA were treated with TM (3 μM) or TG (1 μM) for 16 h. Whole cell lysates were subjected to western blot analysis of HSF1, RIPK1, SQSTM1, MAP1LC3A, and GAPDH (as a loading control). The data shown are representative of 3 individual experiments.
The ERN1 and ATF6 pathways of the UPR converge on XBP1.8,55 We thus tested whether XBP1 plays a part in ER stress-induced RIPK1 upregulation in Mel-RM and MM200 cells with XBP1 knockdown (Fig. 7C). Indeed, XBP1 deficiency blunted upregulation of RIPK1, and recapitulated the inhibitory effect of RIPK1 knockdown on activation of autophagy (Figs. 4F and 7D). These results point to an important role of XBP1 in RIPK1 upregulation in melanoma cells undergoing pharmacological ER stress. However, as a transcription factor, XBP1 does not seem to be directly responsible for the transcriptional increase in RIPK1, as no potential XBP1 binding site was identified in the RIPK1 promoter by in silico analysis.
To search for the active region of the RIPK1 promoter in response to ER stress, an array of incremental deletion luciferase reporter constructs (pGL3-RIPK1-) included in the −1643 to +25 fragment of the promoter were transiently transfected into Mel-RM and MM200 cells (Fig. 8A and Fig. S6A and B). Induction of ER stress enhanced transcriptional activity in all the constructs but pGL3-RIPK1-65 to +25 (Fig. 8A and Fig. S6B). The smallest fragment that was responsive to ER stress was pGL3-RIPK1-168 to +25 (Fig. 8A and Fig. S6B). Therefore, the fragment between −168 and −65 is necessary for the ER stress-induced transcriptional increase in RIPK1 in melanoma cells.
Figure 8.

Heat shock factor protein 1 (HSF1) is responsible for transcriptional upregulation of RIPK1 in melanoma cells upon ER stress. (A) Mel-RM (left panel) and MM200 (right panel) cells transfected with the indicated pGL3-basic based reporter constructs were treated with tunicamycin (TM) (3 μM) for 16 h followed by measurement of the luciferase activity (n = 3, mean ±SEM, *P < 0.05, Student t test). (B) Formaldehyde-cross-linked chromatin of Mel-RM and MM200 with or without treatment with TM (3 μM) for 16 h was subjected to immunoprecipitation with antibody against HSF1 (top panel) or XBP1 (bottom panel). The precipitates were subjected to PCR amplification using primers for the −168 to +25 region of the RIPK1 promoter. The data shown are representative of 3 individual experiments. (C) Formaldehyde-cross-linked chromatin of Mel-RM and MM200 with or without treatment with TM (3 μM) for 16 h was subjected to immunoprecipitation with antibody against HSF1. The precipitates were subjected to PCR amplification using primers for the HSP70-1 promoter. The data shown are representative of 3 individual experiments. (D) Mel-RM and MM200 cells transfected with indicated pGL3-basic based reporter constructs were treated with TM (3 μM) for 16 h followed by measurement of the luciferase activity (n = 3, mean ±SEM, *P < 0.05, Student t test). (E) Whole cell lysates of Mel-RM and MM200 cells transfected with the control or HSF1 siRNA were subjected to western blot analysis of HSF1 and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (F) Mel-RM and MM200 cells transfected with the control or HSF1 siRNA were treated with TM (3 μM) or thapsigargin (TG) (1 μM) for 16 h. Whole cell lysates were subjected to western blot analysis of RIPK1, SQSTM1, MAP1LC3A, pMAPK8/9, MAPK8/9, and GAPDH (as a loading control). The data shown are representative of 3 individual experiments. (G) Mel-RM and MM200 cells transfected with the control or HSF1 siRNA were treated with TM (3 μM) or TG (1 μM) for 48 h. Cell viability was measured by CellTiter-Glo (n = 3, mean ±SEM, *P < 0.05, Student t test).
The −168 to −65 region of the RIPK1 promoter has 3 predicted binding sites for the transcription factor HSF1 (Fig. S6C), which is known to promote survival of cells under various stress.56-58 Indeed, HSF1 was upregulated by ER stress in Mel-RM and MM200 cells but there was no noticeable alteration in HSF1 expression in Mel-RMu cells upon ER stress (Fig. 7B and D and Fig. S6D). ChIP assays demonstrated that HSF1 bound to the −168 to −65 fragment of the RIPK1 promoter, similar to its binding to the promoter of its bona fide target, HSPA1A/HSP70-1, which was included as a control (Figs. 8B and C). In contrast, XBP1 was not found to be associated with the promoter (Fig. 8B). Consistent with the notion that binding of each DNA-binding domain in an HSF1 trimer to a single heat shock element is necessary for HSF1-mediated transactivation of its target genes,39,41 mutation of any of the binding sites diminished the transcriptional activity of pGL3-RIPK1-168 to −65 even upon induction of ER stress (Fig. 8D and Fig. S6C and E). These results indicate that binding of HSF1 to the −168 to −65 region is required for ER stress-triggered RIPK1 promoter activation in melanoma cells. In support, knockdown of HSF1 inhibited upregulation of the RIPK1 protein in Mel-RM and MM200 cells by TM or TG (Fig. 8E and F). Similarly, it also inhibited TM- or TG-induced upregulation of the RIPK1 mRNA, which was due to inhibition of transcription of the RIPK1 gene rather than changes in RIPK1 mRNA stability, as the turnover rates of the RIPK1 transcript remained unaltered in cells with or without HSF1 knockdown (Fig. S7A and B). Moreover, knockdown of HSF1 blocked TM- or TG-triggered activation of MAPK8/9 and induction of autophagy, and sensitized Mel-RM and MM200 cells to TM- or TG-induced cell death (Fig. 8E–G).
The findings that RIPK1 was upregulated by TM or TG downstream of XBP1 and that HSF1 similarly played an important role in TM- or TG-triggered upregulation of RIPK1 suggest that HSF1 and XBP1 are functionally related in regulation of RIPK1 by pharmacological ER stress in melanoma cells (Figs. 7D and 8F). To confirm this, we tested the reciprocal effects of knockdown of XBP1 and HSF1 on the expression of each other in Mel-RM and MM200 cells upon treatment with TM or TG. The results showed that knockdown of XBP1 blocked upregulation of HSF1 by TM or TG (Fig. S7C). This was due to inhibition of HSF1 transcription, as XBP1 knockdown reduced the levels of HSF1 mRNA, which was not caused by changes in its stability as shown in actinomycin-D chasing assays (Fig. S7D). In contrast, knockdown of HSF1 did not affect activation of XBP1 in melanoma cells triggered by TM or TG (Fig. S7E). These data suggest that HSF1 is transcriptionally upregulated downstream of XBP1 in melanoma cells under pharmacological ER stress. Importantly, along with the increase in its expression, phosphorylation of HSF1 at Ser230 and Ser326 that is known to enhance its transactivation activity was increased in Mel-RM and MM200 cells upon treatment with TM or TG,39,42 which was however inhibited when XBP1 was knocked down (Fig. S7F). In contrast, phosphorylation of HSF1 was not altered in Mel-RMu cells.
Discussion
In this report, we provide evidence that RIPK1 plays a critical role in survival of human melanoma cells undergoing pharmacological ER stress induced by TM or TG through activation of autophagy. While RIPK1 was upregulated in melanoma cells relatively resistant to TM- or TG-induced apoptosis, knockdown of RIPK1 inhibited activation of autophagy and rendered the cells susceptible to killing by TM or TG. Conversely, introduction of exogenous RIPK1 into melanocytes and melanoma cells that were sensitive to ER stress-induced apoptosis enhanced induction of autophagy and protected the cells from ER stress-induced killing. Moreover, our results showed that induction of autophagy by RIPK1 in melanoma cells upon treatment with TM or TG was due to activation of BECN1 as a result of its disassociation from BCL2L11 that was phosphorylated by MAPK8/9, and that TM- or TG-induced upregulation of RIPK1 was caused by a transcriptional increase mediated by HSF1 downstream of the ERN1-XBP1 axis of the UPR (Fig. 9).
Figure 9.
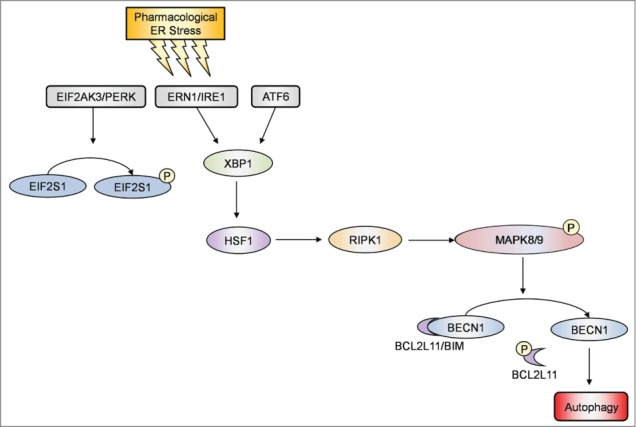
Induction of autophagy by RIPK1 in melanoma cells upon pharmacological ER stress was due to activation of BECN1 as a result of its disassociation from BCL2L11. A schematic illustration of the proposed pharmacological ER stress-XBP1-HSF1-RIPK1-MAPK8/9-autophagy pathway.
Although RIPK1 can mediate both cell survival and death signaling, many studies in recent years focused on its role in induction of apoptosis and programmed necrosis (necroptosis) by various cellular stresses.2,4,59 In particular, the role of RIPK1 in apoptosis and necrosis of cancer cells in response to therapeutic agents is being increasingly reported.2,4 Indeed, overexpression of RIPK1 causes apoptosis in many types of cells,51,60 suggesting that RIPK1 may function primarily as an antisurvival protein in cells. Nevertheless, ripk1−/− mice display extensive apoptosis in selected tissues and die at age 1 to 3 d, suggesting that RIPK1 plays a crucial role in cell survival in a context- and cell type-dependent fashion.1 Indeed, we found that RIPK1 was important for survival of melanoma cells undergoing pharmacological ER stress. This not only reveals the biological role of RIPK1 in melanoma cells in response to ER stress, but also bears important practical implications, as it is known that melanoma cells have in general adapted to ER stress, and that this adaptation contributes to melanoma development, progression, and resistance to treatment.61 Therefore, these results identify RIPK1, similar to upregulation of MCL1 and activation of PI3K-AKT and MAP2K1-MAPK1/3 signaling,16-18 as another adaptive mechanism that confers resistance of melanoma cells to apoptosis triggered by pharmacological ER stress, and suggests that targeting the prosurvival mechanism of RIPK1 may be a useful approach to overcome adaptation of melanoma cells to ER stress.
How does RIPK1 promote melanoma cell survival upon pharmacological ER stress? Our results showed that this was due to activation of autophagy that protects cells against ER stress-induced apoptosis.20,46 Evidence in support of this came from the findings that knockdown of RIPK1 inhibited induction of autophagy in melanoma cells resistant to cell death induced by TM or TG, whereas inhibition of autophagy abolished the RIPK1-mediated protection. Moreover, overexpression of RIPK1 enhanced activation of autophagy and inhibited killing by ER stress in sensitive melanoma cells. In the case of activation of TNFRSF1A or DNA damage, RIPK1 promotes cell survival primarily through activation of NFKB1.1,62 In addition, activation of MAPK1/3 may also be involved.1 However, although inhibition of NFKB1 or MAPK1/3 activation enhanced ER stress-induced apoptosis,16,63 neither of them appeared to play a part in RIPK1-mediated protection of melanoma cells, as knockdown of RIPK1 did not alter the activation levels of NFKB1 or MAPK1/3 in melanoma cells when ER stress was induced (Fig. S8 and data not shown). This suggests that RIPK1 does not have a role in activation of NFKB1 or MAPK1/3 by ER stress triggered by TM or TG in melanoma cells. The mechanism(s) involved in the differential effects of RIPK1 on activation of NFKB1 or MAPK1/3 in cells under different cellular stresses remains to be investigated. Regardless, our results clearly demonstrated an important role of RIPK1 in activation of autophagy in melanoma cells by TM or TG.
BECN1 that plays a crucial role in autophagosome formation is a BH3-only protein and can be in physical association with prosurvival BCL2 family proteins such as BCL2L1, BCL2, and MCL1.37,38,64,65 This represents an important negative regulatory mechanism of BECN1-dependent autophagy. Indeed, displacement of BCL2 and BCL2L1 from BECN1 has been shown to contribute to ER stress-induced autophagy.31 Given that BCL2 and MCL1 are both upregulated in melanoma cells by ER stress,17 it seems likely that disassociation of one or more of these BCL2 family proteins from BECN1 is similarly involved in RIPK1-mediated, TM- or TG-induced autophagy in melanoma cells. However, BCL2L1 did not appear to bind to BECN1, whereas the association of BCL2 with BECN1 was not altered by induction of ER stress in melanoma cells. Although the amount of MCL1 associated with BECN1 was reduced by TM or TG, this was not related to RIPK1. Therefore, none of these prosurvival BCL2 family proteins plays a part in regulation of autophagy by RIPK1 in melanoma cells upon pharmacological ER stress.
Contrary to the observations with prosurvival BCL2 family proteins, we found that displacement of the BH3-only protein BCL2L11 is involved in activation of autophagy by ER stress in melanoma cells. Indeed, BCL2L11 was coimmunoprecipitated with BECN1 in melanoma cells and the amount of BCL2L11 associated with BECN1 was reduced upon ER stress. Moreover, shRNA knockdown of BCL2L11 enhanced ER stress-induced autophagy in melanoma cells even when RIPK1 was also knocked down. BCL2L11 is known to bridge the interaction between BECN1 and the dynein light chain LC8 and thus inhibits autophagy through targeting BECN1 to the cytoskeleton.35 Nevertheless, phosphorylation of BCL2L11 abrogates binding of LC8 to BCL2L11, causing their disassociation and activation of autophagy.35 We found that ER stress increased phosphorylation of BCL2L11, which conceivably contributed activation of autophagy by freeing BECN1. In support, knockdown of BECN1 diminished ER stress-triggered activation of autophagy in melanoma cells even when BCL2L11 was also knocked down. As a BH3-only protein, BCL2L11 plays a role in apoptosis induction by directly neutralizing prosurvival BCL2 family proteins.66,67 Our results suggest that BCL2L11 also plays an indirect role to facilitate ER stress-induced killing of melanoma cells by inhibition of autophagy.
BCL2L11 can be phosphorylated by the MAPKs MAPK1/3, MAPK8/9, and MAPK14/p38α,68-70 which have all been reported to contribute to autophagy.20,47,71-75 However, similar to MAPK1/3 activation, activation of MAPK11/12/13/14 by ER stress was not affected by knockdown of RIPK1 in melanoma cells under ER stress as detected using an antibody against phosphorylated MAPK11/12/13/14 in western blot analysis, suggesting that neither MAPK1/3 nor MAPK11/12/13/14 plays a part in RIPK1-mediated induction of autophagy by ER stress (Fig. S8). On the other hand, activation of MAPK8/9 by ER stress was responsible for activation of autophagy downstream of RIPK1 through phosphorylation of BCL2L11. This was demonstrated by 1) ER stress enhanced MAPK8/9 activation in melanoma cells; 2) knockdown of RIPK1 inhibited activation of MAPK8/9 by ER stress; 3) inhibition of MAPK8/9 abolished ER stress-induced autophagy even when RIPK1 was overexpressed; 4) MAPK8/9 was coimmunoprecipitated with RIPK1 in melanoma cells upon treatment with TM or TG; and 5) inhibition of MAPK8/9 reduced BCL2L11 phosphorylation by ER stress. Consistent with the role of MAPK8/9 in induction of autophagy, inhibition of MAPK8/9 reduced viability of melanoma cells under ER stress. Although this is in contrast to the notion that MAPK8/9 contributes to induction of apoptosis by ER stress,76 it is conceivable that the involvement of MAPK8/9 in regulating cell survival and death is cell type- and context-dependent. MAPK8/9 has been reported to contribute to cell survival under various experimental conditions.77,78 Our results demonstrated that, although the mechanism(s) involved remains undefined, RIPK1 preferentially utilizes MAPK8/9 to phosphorylate BCL2L11 and activate autophagy thus protecting against apoptosis in melanoma cells under pharmacological ER stress. However, whether other factors such as HSPA5 and DDIT3 are involved in RIPK1-mediated, ER stress-induced autophagy in melanoma cells needs to be clarified, but the expression of these proteins was not affected by RIPK1 knockdown in melanoma cells upon treatment with TM or TG (data not shown).
We identified HSF1 as the transcription factor responsible for transcriptional upregulation of RIPK1 during pharmacological ER stress in melanoma cells resistant to ER stress-induced apoptosis. Evidence in support of this includes, 1) the smallest fragment of the RIPK1 promoter that was activated in response to ER stress had 3 consensus HSF1 binding region; 2) HSF1 directly bound to this fragment; 3) mutation of the any of the HSF1 binding sites abolished activation of the RIPK1 promoter by ER stress; 4) HSF1 knockdown inhibited RIPK1 upregulation by ER stress; and 5) phosphorylation of HSF1 at Ser230 and Ser326 that is known to enhance its transactivation activity was also increased by TM or TG in resistant melanoma cells. HSF1 expression is increased in many forms of cancers and plays a role in promoting cell survival.56,79,80 Our finding that HSF1 was not activated by pharmacological ER stress in melanoma cells sensitive to apoptosis induced by TM or TG suggests that additional mechanisms contribute to regulation of HSF1 in melanoma cells, which are nevertheless differentially regulated in the cells undergoing pharmacological ER stress. Further studies to clarify the mechanisms involved are clearly warranted.
TM- or TG-induced upregulation of RIPK1 was partly inhibited in melanoma cells with ERN1 or ATF6 knocked down, suggesting that these pathways of the UPR contributes to the increase in RIPK1 caused by pharmacological ER stress in melanoma cells. Upon activation, ERN1 cleaves XBP1 mRNA, producing a splicing form that encodes a transcription factor.81 Activated ATF6, as a transcription factor, activates downstream target genes of the UPR such as XBP1.82 The conjunction of the ERN1 and ATF6 arms of the UPR on XBP1 suggests that XBP1 may be responsible for the increase in HSF1 triggered by the UPR. This was confirmed in melanoma cells by knockdown of XBP1, which inhibited upregulation of HSF1 by TM or TG. These results are consistent with the important role of XBP1 in transcriptional upregulation of some other prosurvival factors including ETS1 and MCL1 by ER stress in melanoma cells.17,19
In summary, we have demonstrated that RIPK1 promotes melanoma cell survival upon pharmacological ER stress induced by TM or TG through activation of autophagy that is mediated by disassociation of BCL2L11 from BECN1 as a consequence of phosphorylation of BCL2L11 by MAPK8/9. Moreover, we have shown that RIPK1 upregulation by ER stress in melanoma cells is mediated by HSF1 downstream of the ERN1-XBP1 axis of the UPR. Therefore, RIPK1 appears to be a novel adaptive mechanism to ER stress in melanoma cells by acting as a key player of a signaling pathway of ERN1-XBP1-HSF1-RIPK1-MAPK8/9-BECN1 that mediates ER stress-triggered activation of autophagy through displacement of BCL2L11 from BECN1 (Fig. 9). However, as TM and TG are not applicable in the clinic and do not represent the main mechanisms of therapeutic induction ER stress, the potential impact of our observations on anticancer therapy needs to be further validated using clinically relevant ER stress inducers.
Materials and Methods
Cell lines
The human melanoma cell lines Mel-RM, MM200, ME4405, Sk-Mel-28, Mel-CV, IgR3, and Mel-RMu were obtained as described previously.83 They were cultured in Dulbecco's Modified Eagle's Medium (Sigma-Aldrich, D7777) supplemented with 5% fetal bovine serum (Ausgenex, FBS500-S). The human melanocyte line (HEMn-MP) was purchased from Life Technologies (C-102-5C) and cultured as previously described.83 This study was approved by the human ethics committee of the University of Newcastle.
Antibodies, recombinant proteins, and other reagents
The antibodies (Ab) against RIPK1 (sc-7881) used for immunoprecipitation and antibodies against SQSTM1/p62 (sc-28359), BECN1 (sc-48341), HSPA5 (sc-13968), ATF6 (sc-166659), EIF2AK3 (sc-13073), phospho-MAPK1/3 (sc-7383), XBP1 (sc-8015), BIRC2/cIAP1 (sc-271419), BIRC3/cIAP2 (sc-7944) and phospho-HSF1 (Ser230) (sc-30443-R) were purchased from Santa Cruz Biotechnology. The Ab used against RIPK1 for western blotting was from BD Biosciences (551042). The Abs against HSF1 (4356), ATG5 (8540), MAPK8/9 (9252), phospho-MAPK8/9 (9251), phospho-BCL2L11 (4581), MAPK1/3 (9102), phospho-MAPK11/12/13/14 (9216), MAPK11/12/14 (9212), phospho-PRKAA1 (2531), PRKAA1 (2532), phospho-ACACA (3661), ACACA (3662), phospho-EIF2S1 (9721) and EIF2S1 (9722) were purchased from Cell Signaling Technology. The Abs for ERN1 (ab37073), ATG7 (ab52472), MAPK8 (ab10664), MAPK9 (ab76125) and phospho-HSF1 (Ser326) (ab76076) were purchased from Abcam, and the mouse mAb GAPDH antibody was purchased from Ambion (AM4300). The rabbit polyclonal Ab against MAP1LC3A/LC3A (AP1802a) and BCL2L11 (IMG-171) were purchased from Abgent and Imgenex, respectively. Bafilomycin A1 (B1793), 3-MA (M9281), actinomycin D (A9415), tunicamycin (TM) (T7765), thapsigargin (TG) (T9033) and necrostatin-1 (N9037) were purchased from Sigma-Aldrich and the MAPK8/9 inhibitor, SP600125 (420119), the pancaspase inhibitor, z-VAD-fmk (627610), and the CAMKK2 inhibitor, STO-609 (570250), were purchased from Calbiochem.
Cell viability
Cell viability was quantitated using the CellTiter-Glo® Luminescent Cell Viability Assay kit (Promega, G7571) as described previously.84 Briefly, cells were seeded at 5,000 cells per well onto opaque flat-bottomed 96-well culture plates and allowed to grow for 24 h followed by the desired treatment. Cells were then incubated with CellTiter-Glo® for 10 min at room temperature. Luminescence was recorded with a Synergy™ 2 multidetection microplate reader (BioTek, Winooski, VT).
Clonogenic assays
Clonogenic assays were performed as described previously.85 Briefly, cells were seeded at 1,000 cells/well onto 6-well culture plates and allowed to grow for 24 h followed by the desired treatment. Cells were then allowed to grow for a further 12 d before fixation with methanol and staining with 0.5% crystal violet. The images were captured with a Bio-Rad VersaDoc™ image system (Bio-Rad, Gladesville, NSW).
Immunoprecipitation (IP)
The method used was as described previously with minor modification.86 Briefly, 500 μl of lysates were precleared by incubation with 30 μl of Protein A/G PLUS Agarose beads (Santa Cruz, sc-2003) packed beads on a rotator at 4°C for 1 h. Five μg of the specific antibody or corresponding IgG was then added to the precleared lysate and rotated at 4°C for 2 h. Following that, 30 μl of freshly packed beads were added into the mixture and incubated overnight on a rotator at 4°C. The beads were then pelleted by centrifugation and washed 3 times with ice-cold lysis buffer. The resulting immunoprecipitates was then subjected to western blot analysis.
Western blotting
Western blot analysis was carried out as described previously.83 Labeled bands were detected by Luminata™ Crescendo Western HRP substrate (Millipore, WBLUR0100) and images were captured with ImageReader LAS-4000 (Fujifilm, Minato, Tokyo).
Quantitative reverse transcription and real-time PCR (qPCR)
Total RNA was isolated using RNeasy mini kit (Qiagen, 74106) following the manufacturer's instructions. RNA was reverse transcribed into cDNA using qScript (Quanta Biosciences, 95048-500) following the manufacturer's instructions. qPCR was performed using the ABI Prism 7900HT sequence detection system (Life technologies, Carlsbad, CA) with specific-gene primers: Active XBP1 forward 5′- TGC TGA GTC CGC AGC AGG TGC-3′; Active XBP1 reverse 5′- GCT TGG CTG ATG ACG TCC CCA C-3′; RIPK1 forward 5′- AGG CTT TGG GAA GGT GTC TC −3′; RIPK1 reverse 5′- CGG AGT ACT CAT CTC GGC TTT-3′; ACTB (β-actin) forward, 5′-GGC ACC CAG CAC AAT GAA G-3′; ACTB reverse, 5′-GCC GAT CCA CAC GGA GTA CT-3′). The following PCR cycle was used: Standard Fast Cycle 95°C for 20 sec, 40 cycles of 95°C for 1 sec and 60°C for 20 sec using PerfeCTa® SYBR® Green FastMix®, ROX™ (Quanta Biosciences, 95073-012). Cycle threshold (CT) values for specific genes were normalized to the CT value for the housekeeping gene, ACTB. The fold changes of mRNA expressed were determined by comparison with ACTB, where the control sample was arbitrarily designated as 1, and the values of the relative abundance of mRNA of other samples were calculated accordingly. The specificity of the qPCR was controlled using a nontemplate control.
Detection of XBP1 mRNA splicing
The method used for detection of unspliced and spliced XBP1 mRNAs was as described previously.16 Briefly, reverse transcription-PCR (RT-PCR) products of XBP1 mRNA were obtained from total RNA extracted using primers 5′-CGG TGC GCG GTG CGT AGT CTG GA-3′ (forward) and 5′-TGA GGG GCT GAG AGG TGC TTC CT-3′ (reverse). The RT-PCR products were digested with ApaLI to distinguish the active spliced form from the inactive unspliced form. Subsequent electrophoresis revealed the inactive form as 2 cleaved fragments and the active form as a noncleaved fragment.
Small interference RNA
The siRNA constructs for ATG5, ATG7, HSF1, PRKAA1, MAPK8/9 and their nontargeting control were purchased from GenePharma (A10001). The siRNA construct for XBP1 (siGENOME SMARTpool M-009552-02-0010) and nontargeting control (siGENOME SMARTpool D-001206-13-20) was from Dharmacon. Transfection of siRNA was carried out as described previously.83
Short hairpin RNA (shRNA) knockdown
Sigma-Aldrich MISSION® Lentiviral Transduction Particles for shRNA-mediated knockdown of ERN1 (SHCLNV-NM_001433), ATF6 (SHCLNV-NM_007348) and EIF2AK3 (SHCLNV-NM_032025) were purchased from Sigma-Aldrich and used as described previously.14 The human RIPK1 shRNA (TG320591), BCL2L11 shRNA (TG306422), BECN1 shRNA (TG314484) and scrambled shRNA (TG300130), were purchased from Origene, and were used to infect cells according to the manufacturer's protocol.
Plasmid vectors and transfection
The pCMV-MYC-RIPK1 (HG11268-M-M) construct was purchased from Sino Biological Inc.. A mutant RIPK1 construct containing 4 silent mutations within the RIPK1 shRNA1 target sequence was also cloned into pCMV-MYC vector. The mCherry-hLC3B-pcDNA3.1 (40827) was purchased from Addgene. Cells were transfected with 2 μg plasmid in Opti-MEM medium (Life Technologies, 31985070) with Lipofectamine 2000 reagent (Life Technologies, 11668019) according to the manufacturer's protocol.
Detection of RFP-MAP1LC3A puncta
Autofluorescence of cells transfected with the mCherry-hLC3B-pcDNA3.1 vector (Addgene, 40827) with or without treatment were observed under an Olympus FV1000 confocal laser scanning microscope (Olympus, Shinjuku, Tokyo).
Luciferase-reporter constructs
The RIPK1 promoter sequence from 1643 bp upstream to 25 bp downstream of the human RIPK1 gene transcription start site was cloned by genomic PCR using human genomic DNA as a template. Deletions of the promoter were generated by PCR with 5′ primers and a fixed 3′ primer. The sequences of these forward primers were: 5′-TCC CCC GGG TTG CTG AGT AGT ATC CCA TCG-3′ (−1643 to +25), 5′-CCG CTC GAG GCC AGT GTT TGG TGT ATT AGT C-3′ (−1101 to +25), 5′- CGC TCG AGA CAA TGG CTC ACA CCT GTA AT-3′ (−430 to +25), 5′- CGC TCG AGA CTG ACA AAG TGA GAC CCT G-3′ (−168 to +25), 5′-CGC TCG AGT ACA GGG TAC AGC TCT GCC G-3′ (−65 to +25). The reverse primer was: 5′-CCA AGC TTA AGG ACA TGT CTG GTT GCA TTC-3′. Mutagenesis of the RIPK1 binding site was performed by PCR using oligonucleotides carrying mutations at the presumed HSF1 core recognition sites, in combination with the antisense primer. These RIPK1 promoter fragments were cloned into promoterless luciferase reporter plasmid pGL3-Basic Luciferase Vector (Promega, E1751). A 1.6 kb fragment of the RIPK1 promoter was subcloned into the pGL3-Basic Luciferase Vector as described.19 Cells were transiently transfected with luciferase constructs together with the pRL-TK vector (Promega, E2241) as a control for transfection efficiency. The luciferase activity was measured using the Dual Luciferase Reporter Assay System (Promega, E1910) with a Synergy 2 multi-detection microplate reader.
Transmission electron microscopy
Transmission electron microscopy (TEM) was used to analyze cell morphology and intracellular structure to determine the type of cell death in melanoma cell lines as described previously.84 Briefly, cells were harvested, chemically fixed in 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.2), washed and then embedded in molten 4% agarose gel. Trimmed agar blocks containing fixed cells were subsequently fixed in 1% osmium tetroxide. En bloc staining of samples was carried out by submerging agar blocks in 2% uranyl acetate. Agar blocks were then rinsed in water and dehydrated. Next, resin infiltration was performed by submerging blocks in increasing gradients of ethanol and Procure Resin (SPI Supplies, 02659-AB), followed by embedding in pure Procure Resin. Samples in resin were then polymerized by incubating them at 60°C for 24 h. Polymerized resin blocks were then cut to 70-nm-thick sections. Sections were mounted onto Formvar non-carbon-coated grids and positively stained with 2% uranyl acetate and lead citrate solution. Stained samples on grids were visualized using a Tecnai™ G2 Spirit BioTwin (FEI, Hillsboro, OR, 9921219/D1275 ).
ChIP analysis
Analysis was performed using the ChIP Assay Kit (Upstate, 17-295). Briefly, cells were cross-linked using 1% formaldehyde at 37°C for 10 min. After washing, cells were resuspended in 200 μl SDS lysis buffer. DNA was sheared to small fragments by sonication. The recovered supernatant fraction was incubated with antibodies overnight on a rotor at 4°C. After washing, the precipitated protein-DNA complexes were dissolved in 1× TE (10mM Tris-HCl, 1mM EDTA, pH 8.0) buffer and incubated at 65°C for 4 h. DNA was then purified with phenol/chloroform, and a fraction was used as PCR template to detect the presence of the promoter sequences between −168 and +25 of RIPK1. The promoter sequences of HSPA1A/HSP70-1 was used as a positive control.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Health and Medical Research Council (NHMRC) (APP1026458), the Cancer Council NSW (RG13-04 and RG 13-15), Cancer Institute NSW, and Hunter Medical Research Institute, Australia. XDZ is a recipient of the National Health and Medical Research Council (NHMRC) senior research fellowship, CCJ is supported by NHMRC training fellowship, HYT is a recipient of the Hunter Medical Research Institute (HMRI) early career support, LJ is supported by the Cancer Institute NSW (CINSW) early career fellowship.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell's decision to live or die. Cell Death Differ 2007; 14:400-10; PMID:17301840; http://dx.doi.org/ 10.1038/sj.cdd.4402085 [DOI] [PubMed] [Google Scholar]
- 2.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133:693-703; PMID:18485876; http://dx.doi.org/ 10.1016/j.cell.2008.03.036 [DOI] [PubMed] [Google Scholar]
- 3.Christofferson DE, Li Y, Hitomi J, Zhou W, Upperman C, Zhu H, Gerber SA, Gygi S, Yuan J. A novel role for RIP1 kinase in mediating TNFalpha production. Cell Death Dis 2012; 3:e320; PMID:22695613; http://dx.doi.org/ 10.1038/cddis.2012.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 2010; 22:263-8; PMID:20045303; http://dx.doi.org/ 10.1016/j.ceb.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y, Xia F, Hermance N, Mabb A, Simonson S, Morrissey S, Gandhi P, Munson M, Miyamoto S, Kelliher MA. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-kappaB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol Cell Biol 2011; 31:2774-86; PMID:21606198; http://dx.doi.org/ 10.1128/MCB.01139-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niu J, Shi Y, Iwai K, Wu ZH. LUBAC regulates NF-kappaB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J 2011; 30:3741-53; PMID:21811235; http://dx.doi.org/ 10.1038/emboj.2011.264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30:689-700; PMID:18570872; http://dx.doi.org/ 10.1016/j.molcel.2008.05.014 [DOI] [PubMed] [Google Scholar]
- 8.Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the Mammalian unfolded protein response. Annu Rev Cell Dev Biol 2002; 18:575-99; PMID:12142265; http://dx.doi.org/ 10.1146/annurev.cellbio.18.011402.160624 [DOI] [PubMed] [Google Scholar]
- 9.Croft A, Tay KH, Boyd SC, Guo ST, Jiang CC, Lai F, Tseng HY, Jin L, Rizos H, Hersey P, et al.. Oncogenic activation of MEK/ERK primes melanoma cells for adaptation to endoplasmic reticulum stress. J Invest Dermatol 2014; 134:488-97; PMID:23921951; http://dx.doi.org/ 10.1038/jid.2013.325 [DOI] [PubMed] [Google Scholar]
- 10.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334:1081-6; PMID:22116877; http://dx.doi.org/ 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol 2012; 197:857-67; PMID:22733998; http://dx.doi.org/ 10.1083/jcb.201110131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ 2006; 13:363-73; PMID:16397583; http://dx.doi.org/ 10.1038/sj.cdd.4401817 [DOI] [PubMed] [Google Scholar]
- 13.Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, et al.. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol 2004; 165:347-56; PMID:15123740; http://dx.doi.org/ 10.1083/jcb.200310015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tay KH, Luan Q, Croft A, Jiang CC, Jin L, Zhang XD, Tseng HY. Sustained IRE1 and ATF6 signaling is important for survival of melanoma cells undergoing ER stress. Cell Signal 2014; 26:287-94; PMID:24240056; http://dx.doi.org/ 10.1016/j.cellsig.2013.11.008 [DOI] [PubMed] [Google Scholar]
- 15.Li B, Pi Z, Liu L, Zhang B, Huang X, Hu P, Chevet E, Yi P, Liu J. FGF-2 prevents cancer cells from ER stress-mediated apoptosis via enhancing proteasome-mediated Nck degradation. Biochem J 2013; 452:139-45; PMID:23448571; http://dx.doi.org/ 10.1042/BJ20120680 [DOI] [PubMed] [Google Scholar]
- 16.Jiang CC, Chen LH, Gillespie S, Wang YF, Kiejda KA, Zhang XD, Hersey P. Inhibition of MEK sensitizes human melanoma cells to endoplasmic reticulum stress-induced apoptosis. Cancer Res 2007; 67:9750-61; PMID:17942905; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-2047 [DOI] [PubMed] [Google Scholar]
- 17.Jiang CC, Lucas K, Avery-Kiejda KA, Wade M, deBock CE, Thorne RF, Allen J, Hersey P, Zhang XD. Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress. Cancer Res 2008; 68:6708-17; PMID:18701495; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0349 [DOI] [PubMed] [Google Scholar]
- 18.Jiang CC, Yang F, Thorne RF, Zhu BK, Hersey P, Zhang XD. Human melanoma cells under endoplasmic reticulum stress acquire resistance to microtubule-targeting drugs through XBP-1-mediated activation of Akt. Neoplasia 2009; 11:436-47; PMID:19412428; http://dx.doi.org/ 10.1593/neo.09208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong L, Jiang CC, Thorne RF, Croft A, Yang F, Liu H, de Bock CE, Hersey P, Zhang XD. Ets-1 mediates upregulation of Mcl-1 downstream of XBP-1 in human melanoma cells upon ER stress. Oncogene 2011; 30:3716-26; PMID:21423203; http://dx.doi.org/ 10.1038/onc.2011.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al.. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26:9220-31; PMID:17030611; http://dx.doi.org/ 10.1128/MCB.01453-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14:230-9; PMID:16794605; http://dx.doi.org/ 10.1038/sj.cdd.4401984 [DOI] [PubMed] [Google Scholar]
- 22.Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344-8; PMID:21127245; http://dx.doi.org/ 10.1126/science.1193497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer 2005; 5:886-97; PMID:16239905; http://dx.doi.org/ 10.1038/nrc1738 [DOI] [PubMed] [Google Scholar]
- 25.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/ 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rikiishi H. Novel Insights into the Interplay between Apoptosis and Autophagy. Int J Cell Biol 2012; 2012:317645; PMID:22496691; http://dx.doi.org/ 10.1155/2012/317645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin S, White E. Tumor suppression by autophagy through the management of metabolic stress. Autophagy 2008; 4:563-6; PMID:18326941; http://dx.doi.org/ 10.4161/auto.5830 [DOI] [PubMed] [Google Scholar]
- 28.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726-34; PMID:16148885; http://dx.doi.org/ 10.1038/nrc1692 [DOI] [PubMed] [Google Scholar]
- 29.Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res 2010; 70:3431-4; PMID:20424122; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-4027 [DOI] [PubMed] [Google Scholar]
- 30.Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, et al.. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 2012; 122:4621-34; PMID:23143306; http://dx.doi.org/ 10.1172/JCI62973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int J Cell Biol 2010; 2010:930509; PMID:20145727; http://dx.doi.org/ 10.1155/2010/930509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ 2008; 15:1460-71; PMID:18551133; http://dx.doi.org/ 10.1038/cdd.2008.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muscarella DE, Bloom SE. The contribution of c-Jun N-terminal kinase activation and subsequent Bcl-2 phosphorylation to apoptosis induction in human B-cells is dependent on the mode of action of specific stresses. Toxicol Appl Pharmacol 2008; 228:93-104; PMID:18201741; http://dx.doi.org/ 10.1016/j.taap.2007.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol 1999; 19:8469-78; PMID:10567572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PP, Sadiq O, Rubinsztein DC. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell 2012; 47:359-70; PMID:22742832; http://dx.doi.org/ 10.1016/j.molcel.2012.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 2009; 10:285-92; PMID:19180116; http://dx.doi.org/ 10.1038/embor.2008.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/ 10.1016/j.cell.2005.07.002 [DOI] [PubMed] [Google Scholar]
- 38.Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS, Cheng AL, Chen PJ, Chen KF. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis 2013; 4:e485; PMID:23392173; http://dx.doi.org/ 10.1038/cddis.2013.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vihervaara A, Sistonen L. HSF1 at a glance. J Cell Sci 2014; 127:261-6; PMID:24421309; http://dx.doi.org/ 10.1242/jcs.132605 [DOI] [PubMed] [Google Scholar]
- 40.Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012; 150:549-62; PMID:22863008; http://dx.doi.org/ 10.1016/j.cell.2012.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pelham HR. A regulatory upstream promoter element in the Drosophila hsp 70 heat-shock gene. Cell 1982; 30:517-28; PMID:6814763; http://dx.doi.org/ 10.1016/0092-8674(82)90249-5 [DOI] [PubMed] [Google Scholar]
- 42.Guettouche T, Boellmann F, Lane WS, Voellmy R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem 2005; 6:4; PMID:15760475; http://dx.doi.org/ 10.1186/1471-2091-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang CC, Mao ZG, Avery-Kiejda KA, Wade M, Hersey P, Zhang XD. Glucose-regulated protein 78 antagonizes cisplatin and adriamycin in human melanoma cells. Carcinogenesis 2009; 30:197-204; PMID:18842681; http://dx.doi.org/ 10.1093/carcin/bgn220 [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012; 148:228-43; PMID:22265414; http://dx.doi.org/ 10.1016/j.cell.2011.11.030 [DOI] [PubMed] [Google Scholar]
- 45.Gozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H, Mizushima N, Yoshimori T, Kimchi A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ 2008; 15:1875-86; PMID:18806755; http://dx.doi.org/ 10.1038/cdd.2008.121 [DOI] [PubMed] [Google Scholar]
- 46.Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem 2007; 282:4702-10; PMID:17135238; http://dx.doi.org/ 10.1074/jbc.M609267200 [DOI] [PubMed] [Google Scholar]
- 47.Bicknell AA, Tourtellotte J, Niwa M. Late phase of the endoplasmic reticulum stress response pathway is regulated by Hog1 MAP kinase. J Biol Chem 2010; 285:17545-55; PMID:20382742; http://dx.doi.org/ 10.1074/jbc.M109.084681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008; 4:949-51; PMID:18769111; http://dx.doi.org/ 10.4161/auto.6788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Q, Kim YS, Lin Y, Lewis J, Neckers L, Liu ZG. Tumour necrosis factor receptor 1 mediates endoplasmic reticulum stress-induced activation of the MAP kinase JNK. EMBO Rep 2006; 7:622-7; PMID:16680093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He W, Wang Q, Srinivasan B, Xu J, Padilla MT, Li Z, Wang X, Liu Y, Gou X, Shen HM, et al.. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene 2014; 33:3004-13; PMID:23831571; http://dx.doi.org/ 10.1038/onc.2013.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, et al.. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 2011; 43:432-48; PMID:21737329; http://dx.doi.org/ 10.1016/j.molcel.2011.06.006 [DOI] [PubMed] [Google Scholar]
- 52.Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al.. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008; 4:313-21; PMID:18408713; http://dx.doi.org/ 10.1038/nchembio.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang H, Kang R, Wang J, Luo G, Yang W, Zhao Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy 2013; 9:175-95; PMID:23169238; http://dx.doi.org/ 10.4161/auto.22791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al.. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 2007; 25:193-205; PMID:17244528; http://dx.doi.org/ 10.1016/j.molcel.2006.12.009 [DOI] [PubMed] [Google Scholar]
- 55.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem 2005; 74:739-89; PMID:15952902; http://dx.doi.org/ 10.1146/annurev.biochem.73.011303.074134 [DOI] [PubMed] [Google Scholar]
- 56.Dai B, Gong A, Jing Z, Aldape KD, Kang SH, Sawaya R, Huang S. Forkhead box M1 is regulated by heat shock factor 1 and promotes glioma cells survival under heat shock stress. J Biol Chem 2013; 288:1634-42; PMID:23192351; http://dx.doi.org/ 10.1074/jbc.M112.379362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiao X, Zuo X, Davis AA, McMillan DR, Curry BB, Richardson JA, Benjamin IJ. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J 1999; 18:5943-52; PMID:10545106; http://dx.doi.org/ 10.1093/emboj/18.21.5943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dai C, Santagata S, Tang Z, Shi J, Cao J, Kwon H, Bronson RT, Whitesell L, Lindquist S. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest 2012; 122:3742-54; PMID:22945628; http://dx.doi.org/ 10.1172/JCI62727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell 2011; 145:92-103; PMID:21458669; http://dx.doi.org/ 10.1016/j.cell.2011.02.023 [DOI] [PubMed] [Google Scholar]
- 60.Park S, Hatanpaa KJ, Xie Y, Mickey BE, Madden CJ, Raisanen JM, Ramnarain DB, Xiao G, Saha D, Boothman DA, et al.. The receptor interacting protein 1 inhibits p53 induction through NF-kappaB activation and confers a worse prognosis in glioblastoma. Cancer Res 2009; 69:2809-16; PMID:19339267; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-4079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hersey P, Zhang XD. Adaptation to ER stress as a driver of malignancy and resistance to therapy in human melanoma. Pigment Cell Melanoma Res 2008; 21:358-67; PMID:18476909; http://dx.doi.org/ 10.1111/j.1755-148X.2008.00467.x [DOI] [PubMed] [Google Scholar]
- 62.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 1998; 8:297-303; PMID:9529147; http://dx.doi.org/ 10.1016/S1074-7613(00)80535-X [DOI] [PubMed] [Google Scholar]
- 63.Nozaki S, Sledge GW Jr, Nakshatri H. Repression of GADD153/CHOP by NF-kappaB: a possible cellular defense against endoplasmic reticulum stress-induced cell death. Oncogene 2001; 20:2178-85; PMID:11360202; http://dx.doi.org/ 10.1038/sj.onc.1204292 [DOI] [PubMed] [Google Scholar]
- 64.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al.. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007; 26:2527-39; PMID:17446862; http://dx.doi.org/ 10.1038/sj.emboj.7601689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem 2007; 282:13123-32; PMID:17337444; http://dx.doi.org/ 10.1074/jbc.M700492200 [DOI] [PubMed] [Google Scholar]
- 66.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev 2001; 15:1481-6; PMID:11410528; http://dx.doi.org/ 10.1101/gad.897601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Merino D, Giam M, Hughes PD, Siggs OM, Heger K, O'Reilly LA, Adams JM, Strasser A, Lee EF, Fairlie WD, et al.. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol 2009; 186:355-62; PMID:19651893; http://dx.doi.org/ 10.1083/jcb.200905153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hubner A, Barrett T, Flavell RA, Davis RJ. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell 2008; 30:415-25; PMID:18498746; http://dx.doi.org/ 10.1016/j.molcel.2008.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem 2003; 278:18811-6; PMID:12646560; http://dx.doi.org/ 10.1074/jbc.M301010200 [DOI] [PubMed] [Google Scholar]
- 70.Tay KH, Jin L, Tseng HY, Jiang CC, Ye Y, Thorne RF, Liu T, Guo ST, Verrills NM, Hersey P, et al.. Suppression of PP2A is critical for protection of melanoma cells upon endoplasmic reticulum stress. Cell Death Dis 2012; 3:e337; PMID:22739989; http://dx.doi.org/ 10.1038/cddis.2012.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J 2010; 277:2-21; PMID:19843174; http://dx.doi.org/ 10.1111/j.1742-4658.2009.07366.x [DOI] [PubMed] [Google Scholar]
- 72.Ellington AA, Berhow MA, Singletary KW. Inhibition of Akt signaling and enhanced ERK1/2 activity are involved in induction of macroautophagy by triterpenoid B-group soyasaponins in colon cancer cells. Carcinogenesis 2006; 27:298-306; PMID:16113053; http://dx.doi.org/ 10.1093/carcin/bgi214 [DOI] [PubMed] [Google Scholar]
- 73.Sivaprasad U, Basu A. Inhibition of ERK attenuates autophagy and potentiates tumour necrosis factor-alpha-induced cell death in MCF-7 cells. J Cell Mol Med 2008; 12:1265-71; PMID:18266953; http://dx.doi.org/ 10.1111/j.1582-4934.2008.00282.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim DS, Kim JH, Lee GH, Kim HT, Lim JM, Chae SW, Chae HJ, Kim HR. p38 Mitogen-activated protein kinase is involved in endoplasmic reticulum stress-induced cell death and autophagy in human gingival fibroblasts. Biol Pharm Bull 2010; 33:545-9; PMID:20410583; http://dx.doi.org/ 10.1248/bpb.33.545 [DOI] [PubMed] [Google Scholar]
- 75.Zhong W, Zhu H, Sheng F, Tian Y, Zhou J, Chen Y, Li S, Lin J. Activation of the MAPK11/12/13/14 (p38 MAPK) pathway regulates the transcription of autophagy genes in response to oxidative stress induced by a novel copper complex in HeLa cells. Autophagy 2014; 10:1285-300; PMID:24905917; http://dx.doi.org/ 10.4161/auto.28789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011; 13:184-90; PMID:21364565; http://dx.doi.org/ 10.1038/ncb0311-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lamb JA, Ventura JJ, Hess P, Flavell RA, Davis RJ. JunD mediates survival signaling by the JNK signal transduction pathway. Mol Cell 2003; 11:1479-89; PMID:12820962; http://dx.doi.org/ 10.1016/S1097-2765(03)00203-X [DOI] [PubMed] [Google Scholar]
- 78.Yu C, Minemoto Y, Zhang J, Liu J, Tang F, Bui TN, Xiang J, Lin A. JNK suppresses apoptosis via phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol Cell 2004; 13:329-40; PMID:14967141; http://dx.doi.org/ 10.1016/S1097-2765(04)00028-0 [DOI] [PubMed] [Google Scholar]
- 79.Engerud H, Tangen IL, Berg A, Kusonmano K, Halle MK, Oyan AM, Kalland KH, Stefansson I, Trovik J, Salvesen HB, et al.. High level of HSF1 associates with aggressive endometrial carcinoma and suggests potential for HSP90 inhibitors. Br J Cancer 2014; 111:78-84; PMID:24853175; http://dx.doi.org/ 10.1038/bjc.2014.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Santagata S, Hu R, Lin NU, Mendillo ML, Collins LC, Hankinson SE, Schnitt SJ, Whitesell L, Tamimi RM, Lindquist S, et al.. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc Natl Acad Sci U S A 2011; 108:18378-83; PMID:22042860; http://dx.doi.org/ 10.1073/pnas.1115031108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang CC, Chen LH, Gillespie S, Kiejda KA, Mhaidat N, Wang YF, Thorne R, Zhang XD, Hersey P. Tunicamycin sensitizes human melanoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by up-regulation of TRAIL-R2 via the unfolded protein response. Cancer Res 2007; 67:5880-8; PMID:17575157; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-0213 [DOI] [PubMed] [Google Scholar]
- 82.Liu CY, Kaufman RJ. The unfolded protein response. J Cell Sci 2003; 116:1861-2; PMID:12692187; http://dx.doi.org/ 10.1242/jcs.00408 [DOI] [PubMed] [Google Scholar]
- 83.Tseng HY, Jiang CC, Croft A, Tay KH, Thorne RF, Yang F, Liu H, Hersey P, Zhang XD. Contrasting effects of nutlin-3 on TRAIL- and docetaxel-induced apoptosis due to upregulation of TRAIL-R2 and Mcl-1 in human melanoma cells. Mol Cancer Ther 2010; 9:3363-74; PMID:21159614; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-0646 [DOI] [PubMed] [Google Scholar]
- 84.Lai F, Guo ST, Jin L, Jiang CC, Wang CY, Croft A, Chi MN, Tseng HY, Farrelly M, Atmadibrata B, et al.. Cotargeting histone deacetylases and oncogenic BRAF synergistically kills human melanoma cells by necrosis independently of RIPK1 and RIPK3. Cell Death Dis 2013; 4:e655; PMID:23744355; http://dx.doi.org/ 10.1038/cddis.2013.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ye Y, Jin L, Wilmott JS, Hu WL, Yosufi B, Thorne RF, Liu T, Rizos H, Yan XG, Dong L, et al.. PI(4,5)P2 5-phosphatase A regulates PI3K/Akt signalling and has a tumour suppressive role in human melanoma. Nat Commun 2013; 4:1508; PMID:23443536; http://dx.doi.org/ 10.1038/ncomms2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang F, Tay KH, Dong L, Thorne RF, Jiang CC, Yang E, Tseng HY, Liu H, Christopherson R, Hersey P, et al.. Cystatin B inhibition of TRAIL-induced apoptosis is associated with the protection of FLIP(L) from degradation by the E3 ligase itch in human melanoma cells. Cell Death Differ 2010; 17:1354-67; PMID:20300110; http://dx.doi.org/ 10.1038/cdd.2010.29 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.