

Abstract
Oxidative stress (OS) stimulates autophagy in different cellular systems, but it remains controversial if this rule can be generalized. We have analyzed the effect of chronic OS induced by the parkinsonian toxin paraquat (PQ) on autophagy in astrocytoma cells and primary astrocytes, which represent the first cellular target of neurotoxins in the brain. PQ decreased the basal levels of LC3-II and LC3-positive vesicles, and its colocalization with lysosomal markers, both in the absence and presence of chloroquine. This was paralleled by increased number and size of SQSTM1/p62 aggregates. Downregulation of autophagy was also observed in cells chronically exposed to hydrogen peroxide or nonlethal concentrations of PQ, and it was associated with a reduced astrocyte capability to protect dopaminergic cells from OS in co-cultures. Surprisingly, PQ treatment led to inhibition of MTOR, activation of MAPK8/JNK1 and MAPK1/ERK2-MAPK3/ERK1 and upregulation of BECN1/Beclin 1 expression, all signals typically correlating with induction of autophagy. Reduction of OS by NMDPEF, a specific NQO2 inhibitor, but not by N-acetylcysteine, abrogated the inhibitory effect of PQ and restored autophagic flux. Activation of NQO2 by PQ or menadione and genetic manipulation of its expression confirmed the role of this enzyme in the inhibitory action of PQ on autophagy. PQ did not induce NFE2L2/NRF2, but when it was co-administered with NMDPEF NFE2L2 activity was enhanced in a SQSTM1-independent fashion. Thus, a prolonged OS in astrocytes inhibits LC3 lipidation and impairs autophagosome formation and autophagic flux, in spite of concomitant activation of several pro-autophagic signals. These findings outline an unanticipated neuroprotective role of astrocyte autophagy and identify in NQO2 a novel pharmacological target for its positive modulation.
Keywords: astrocytes, macroautophagy, NQO2, paraquat, parkinson disease, ROS
Abbreviations
- Ab
antibody
- AVs
autophagic vacuoles
- BNAH
benzyldihydronicotinamide riboside
- CQ
chloroquine
- CA-DCF-DA
5(6)-carboxy-2′,7′ dichlorofluorescein diacetate
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- FACS
flow cytometry
- GFAP
glial fibrillary acidic protein
- GFP
green fluorescent protein
- K3
menadione
- MAPK
mitogen-activated protein kinase
- MFI
mean fluorescence intensity
- MitoSOX
3,8-phenanthridinediamine, 5-(6′-triphenylphosphoniumhexyl)-5,6 dihydro-6-phenyl
- MPTP
1-methyl 4-phenyl 1,2,3,6-tetraidro-piridine
- NMDPEF
N-[2-(2-methoxy-6H-dipyrido[2,3-a:3,2-e]pyrrolizin-11-yl)ethyl]-2-furamide]
- NFE2L2
nuclear factor, erythroid 2-like 2
- OS
oxidative stress
- PBS
phosphate-buffered saline
- p-
phosphorylated
- PQ
paraquat
- ROS
reactive oxygen species
- RT
room temperature
- shRNA
short harpin ribonucleic acid
- siRNA
small interfering ribonucleic acid
- SN
substantia nigra
- TTBS
Tween-Tris buffered saline
- WB
western blotting
Introduction
Parkinson disease (PD) is a common neurodegenerative disorder that is primarily characterized by progressive loss of dopaminergic neurons in substantia nigra (SN) pars compacta and consequent reduced dopamine production. The glial cells (astrocytes and microglia) play a complex role in dopaminergic neurotoxicity.1-5 Both microglia and astrocytes can degrade dopamine with consequent generation of oxidative stress (OS) and detrimental effects on dopaminergic neurons.6,7 While parkinsonian neurotoxins activate microglial cells5 leading to neuroinflammation in nigrostriatal area, the protective effects of astrocytes in development and regeneration of the nigrostriatal dopamine system largely outweigh the pro-inflammatory role of the glia.3,8,9 Astrocytes contribute to the blood-brain barrier8 and protect neurons from OS induced by hydrogen peroxide (H2O2), toxic metabolites and neurotransmitters, including dopamine, 6-hydroxydopamine, and glutamate.10-12 This property has been associated with a basally high expression of NFE2L2 (nuclear factor, erythroid 2-like 2) that leads to increased synthesis and secretion of glutathione.13
Autophagy, a lysosome-dependent degradation pathway of self-constituents, normally runs at low basal level to ensure cell homeostasis and is activated in response to starvation and metabolic stress to prevent accumulation of damaged organelles and protein aggregates.14-16 Defects in the autophagy machinery have been implicated in several neurodegenerative disorders, including PD.17,18 The progressive accumulation of protein inclusions containing SQSTM1/p62, ubiquitin and/or SNCA/α-synuclein in PD,19-21 Lewy bodies disease, and related disorders21-23 is observed not only in neurons, but also in astrocytes, suggesting that defective autophagy is common to both cell subsets present in affected areas of the brain. The vast majority of studies that address dysregulation of autophagy by toxins mimicking PD are limited to dopaminergic cell lines and primary neurons as cellular models of PD.24,25 However, under physiological conditions, when the blood-brain barrier functions correctly, dopaminergic neurons of the SN are not directly exposed to parkinsonian toxins, which are instead absorbed and metabolized by astrocytes.6,8 In fact, astrocytes are the first target of neurotoxins and thanks to their powerful antioxidant machinery they represent the first line of brain defense against such toxins.13,26-28
Parkinsonian toxins applied to rodents or primates reproduce only certain features of PD.25,29 Among them, SNCA accumulation and Lewy bodies-like protein inclusions are effectively induced only by PQ30 and rotenone.31 All parkinsonian toxins generate reactive oxygen species (ROS) and/or reactive nitrogen species by disruption of the mitochondrial electron transport chain and/or production of toxic metabolites leading to altered energy metabolism.32 In addition, PQ-induced cellular toxicity depends on the presence of oxidoreductases able to reduce PQ by one electron transfer,33 such as plasma membrane NOX 1 (NADPH oxidase 1)34 or mitochondrial COX/ubiquinol: cytochrome c oxidase.35 We have recently identified a cytoplasmic flavoenzyme, NQO2/quinone oxidoreductase 2 (NAD[P]H dehydrogenase, quinone 2), as another critical mediator of PQ-induced OS and toxicity.36 Silencing of NQO2 conferred a partial or full protection in astroglial cells exposed to intermediate concentrations of PQ, and inhibition of NQO2 activity by its specific inhibitor prevented PQ-induced OS, cell death in vitro and animal mortality in models of acute PQ-induced systemic- and SN-toxicity.36 These findings provided further evidence that NQO2 may play a toxifying role in biological systems in the presence of certain xenobiotics.37
ROS generated by H2O2 or MPP+ (the active MPTP metabolite) induce a strong autophagic response in dopaminergic cell lines,38,39 and primary cortical and nigrostriatal neurons,40,41 leading to the activation of a cell death program. Nevertheless, it is unclear if the rule “OS causes autophagy” can be applied to all cell systems and different types of OS. Indeed, several lines of evidence suggest that OS may also have a negative impact on the autophagy machinery, as recently reviewed by us.17,25
Here, we demonstrate that chronic exposure to pro-oxidants, such as PQ and H2O2, reduces basal autophagy in astrocytes. This effect is dose-dependent and occurs also at nonlethal concentrations of PQ. Coculture experiments showed that the impairment of autophagy limited the capacity of astrocytes to protect dopaminergic neuroblastoma cells from a lethal OS. Intriguingly, in spite of a marked decrease in LC3 lipidation, PQ induced a series of proautophagic signals including BECN1 upregulation, and MAPK8 and MAPK1/3 activation. Furthermore, we identify NQO2 as a mediator of the negative effects of PQ on autophagy. Thus, PQ-induced OS generates both positive and negative signals affecting the autophagy machinery and antioxidant capacity of astrocytes, and the balance between these signals can be modulated pharmacologically by NQO2 inhibition. The present findings have important pathophysiological implications: first, they uncover a neuroprotective role of autophagy in astrocytes that outlines the active involvement of these cells in the pathogenesis of OS-related neurodegenerative diseases; second, the fact that inhibition of NQO2 activity can restore protective autophagy in OS astrocytes could be clinically exploited for the treatment of OS-associated neurodegeneration.
Results
Paraquat negatively affects basal autophagy in astrocytoma cells
We previously reported that astrocytoma U373 cells exposed to PQ-induced OS undergo a delayed, nonapoptotic cell death.36 Whether autophagy was involved in either promoting or counteracting this phenomenon remained to be elucidated. The western blotting analysis of U373 cell lysates revealed a time- and dose-dependent decrease of lipidated LC3 (LC3-II), a fundamental component of phagophore and autophagosomal membranes (Fig. 1A and B). Next, we analyzed the levels of SQSTM1, a ubiquitin-binding protein that functions as a receptor for autophagic substrates42,43 and is an abundant component of protein inclusions associated with neurodegenerative disorders.21-23 SQSTM1 levels were dose-dependently increased 24 h post-treatment (Fig. 1C and D), suggesting a reduced formation or turnover of autophagosomes. The lack of significant increase in SQSTM1 levels at 48 h was possibly due to SQSTM1 precipitation into insoluble aggregates, considering the presence of high amounts of SQSTM1 in the cell lysate fraction insoluble to 1% Igepal (Fig. 1C). The above observations were confirmed by immunofluorescence staining for autophagic structures (Fig. 2A). PQ (100 μM, 24 h) caused a more than 3–fold decrease in LC3-positive autophagic vacuoles (AVs) (Fig. 2B) and a strong reduction in AV-positive cells (Fig. 2C) compared to untreated cells. This was accompanied by a mild increase in the number of SQSTM1 granules (Fig. 2A and D) and a strong increase in the SQSTM1 staining intensity (Fig. 2A and E). In addition, PQ strongly impaired the colocalization of SQSTM1 and LC3 (Fig. 2A and F), suggesting that many SQSTM1-targeted aggregates were not engulfed within autophagosomes.
Figure 1.
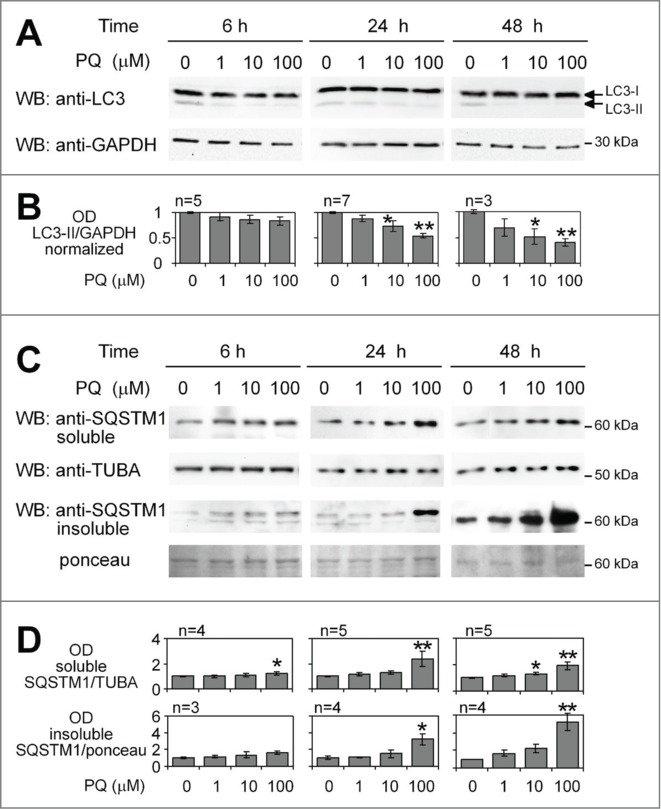
Paraquat-induced changes in LC3-II and SQSTM1 levels suggest time- and dosage-dependent inhibition of basal autophagy. (A) Time- and concentration-dependent decrease in LC3-II levels in response to PQ. U373 cells were treated with 3 different concentrations of PQ for the indicated times before lysis. LC3-II was analyzed by 15% SDS-PAGE and WB. (B) The experiment was repeated from n = 3 to 7 times as indicated. Bars represent the mean of LC3-II optical density (OD) +/− SEM normalized to vehicle-treated controls, which were set to 1. Statistical analysis was performed by ANOVA followed by Dunnett's post-test; 3 – 9 OD values for each group, 4 groups; *P < 0.05, **P < 0.01. (C) Time- and dosage-dependent accumulation of SQSTM1 in soluble and detergent-resistant fractions. The samples from the experiments described in (A) and the corresponding Igepal 1% insoluble fraction of lysates were loaded on 8% SDS-PAGE gels and analyzed for SQSTM1 expression by WB. (D) Densitometric and subsequent statistical analysis were performed as described in (A) for n experiments. TUBA/α-tubulin or ponceau staining used as loading controls, were used for OD normalization. Four–8 OD values for each group, 4 groups, *P < 0.05, **P < 0.01.
Figure 2.
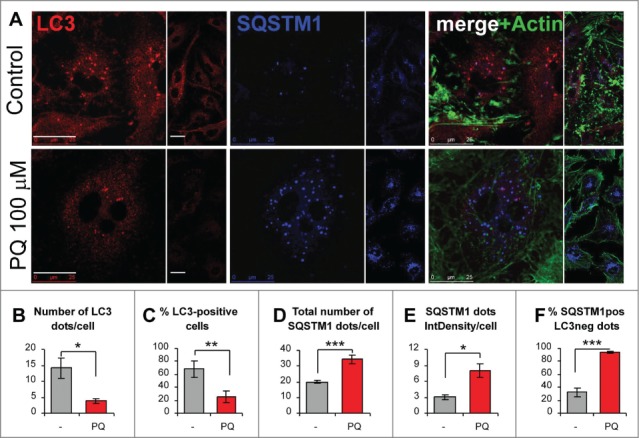
Paraquat reduces the number of autophagic vacuoles and increases the number and size of SQSTM1 aggregates in astroglial cells. (A) U373 cells growing on coverslips were treated with PBS (control) or 100 μM PQ for 24 h. Paraformaldehyde-fixed cells were stained for LC3, SQSTM1, and actin (FITC-phalloidin). Representative confocal images of middle sections from z stacks are shown (bar: 25 μm). (B-F) Statistical analysis was performed by using unpaired 2-tailed t test (t-test), for n images per treatment, at least 50 cells per treatment from 3 independent experiments. *P < 0.05, **P < 0.01., ***P < 0.001. (B) Mean number of LC3 dots per cell +/− SEM, n = 12. (C) Percentage of cells with ≥ 5 LC3 dots per cell +/− SEM, n = 12. (D) Mean number of SQSTM1 aggregates per cell +/− SEM, n = 11. (E) Mean size x intensity of SQSTM1 dots (i.e., Integrated density, IntDensity) per cell +/− SEM, n = 11. (F) Percentage of SQSTM1 dots not costaining with LC3 (SQSTM1-positive/LC3-negative dots, SQSTM1pos LC3neg) +/− SEM, n = 10.
Paraquat inhibits the formation of autophagosomes
Autophagy is a dynamic process and an apparent LC3-II downregulation may be due to scarce production or prompt consumption of autophagosomes.44 The treatment with drugs that abrogate vacuolar acidification, such as chloroquine (CQ), helps to discriminate between these 2 possibilities. To determine if the decrease of LC3 dots was the result of reduced formation or of accelerated consumption of autophagosomes, we added CQ to U373 cells during the last 4 h of PQ treatment (Fig. 3A). In cultures not exposed to PQ, CQ greatly increased the number of LC3-positive vesicles, while in PQ-treated cells this increase was modest (Fig. 3A and B). Accordingly, the levels of LC3-II protein in cells treated with 10 or 100 μM PQ along with CQ were lower than in CQ-only treated cells (Fig. 3C). We further confirmed that any treatment with CQ, ranging from 1 up to 18 h before cell lysis, resulted in a lower LC3-II accumulation in PQ-treated cells than in control cells (Figs. 4–8 and data not shown).
Figure 3.
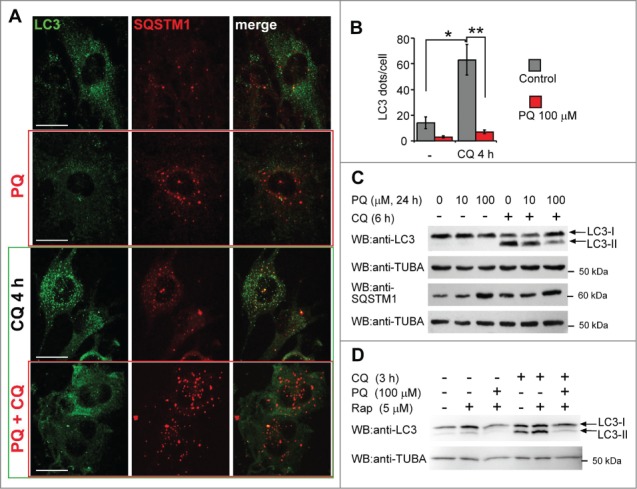
Reduction in the number of LC3 dots by PQ is not due to accelerated autophagic flux. (A) U373 cells were treated or untreated with 100 μM PQ for 24 h and with or w/o chloroquine (CQ, 25 μM) for 4 h before fixation with paraformaldehyde. A representative confocal image (extended focus of 5-μm deep stack) for each treatment and staining is shown. Bar: 25 μm. (B) Mean number of LC3 vesicles per cell +/− SEM, calculated on n = 6 images per treatment, from 3 independent experiments described in (A). Statistical analysis was performed by Mann-Whitney test, n = 6, *P < 0.05, **P < 0.01. (C) WB analysis of LC3-II and SQSTM1 in U373 cells treated as in (A) and exposed to 10 and 100 uM PQ, +/− CQ (25 μM) added 6 h before lysis. (D) PQ blocks rapamycin-induced autophagy in U373 cells. 5 μM rapamycin (Rap) +/− PQ was applied for 24 h. The autophagy flux was blocked by 25 μM CQ applied 3 h before lysis and LC3 levels were analyzed in CQ-treated and untreated cells.
Figure 4.
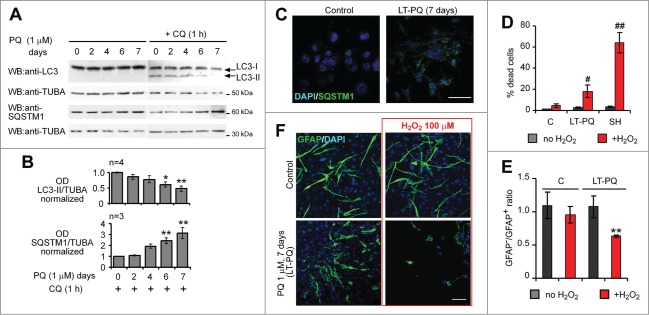
Reduction of basal autophagy by long-term pretreatment with PQ leads to functional defects in antioxidant capacity of astrocytes. (A) U373 cells were cultured in the presence of 1 μM PQ for 7, 6, 4 and 2 d, under optimal culture conditions to prevent PQ-induced cell death. On d 7, CQ (25 μM) was added 1 h before lysis. LC3 and SQSTM1 expression were analyzed by WB. (B) Densitometric analysis of data in (A); Mean +/− SEM; statistical analysis: one-way ANOVA followed by Dunnett's post-test; n, number of analyzed experiments; 5 groups, 3 to 6 samples for each group, *P < 0.05, **P < 0.01. (C) Representative confocal images (extended focus) of SQSTM1 immunostaining in control and PQ-long-term pretreated (LT-PQ) U373 cells (7 days treatment with 1 μM PQ). Bar: 100 μm. Note accumulation of SQSTM1 aggregates in LT-PQ cells. (D–F) LT-PQ U373 cells are more sensitive to H2O2 and less effectively protect dopaminergic SH-SY5Y neuroblastoma cells from H2O2-induced cell death. (D) Control (C), PQ-long-term pretreated U373 cells (LT-PQ) and SH-SY5Y (SH) cells were plated separately under standard culture conditions and 2 days later exposed to 100 μM H2O2 for the next 24 h. Cell mortality was analyzed by trypan blue exclusion assay and flow cytometry. The graph shows the mean +/− SD of a representative experiment out of 3 independent experiments, performed in triplicate. Statistical analysis: Mann-Whitney test, n = 3 in each group,#P < 0.05,##P < 0.01 indicate significant differences vs H2O2-treated C cells. (E) Control and LT-PQ U373 cells were mixed 1:1 with dopaminergic SH-SY5Y cells, plated on cover slips and cultured for 2 d under standard conditions. Next, cocultures were exposed to 100 μM H2O2 for 24 h and subsequently fixed and stained for GFAP and DAPI. The ratio of SH-SY5Y dopaminergic cells to astroglial cells was assessed by counting GFAP-negative and GFAP-positive cells from at least 1000 DAPI-positive cells. The graph shows the mean ratio of GFAP− to GFAP+ cells +/− SEM, from 3 independent experiments, performed in triplicate. Statistical analysis: Mann-Whitney test, n = 9 samples in each group, **P < 0.01. (F) Representative confocal images (extended focus) of U373-SH-SY5Y cocultures described in (E). Bar: 100 μm.
Figure 5.

Inhibition of autophagy in astroglial U373 cells and in primary astrocytes by PQ is mediated by oxidative stress. (A) ROS induction by PQ and H2O2 as detected by CA-DCF-DA in U373 cells. Live fluorescence of cell monolayers was analyzed by fluorometry 1 h after staining with CA-DCF-DA. (B) Mitochondrial ROS induction by PQ and H2O2 as detected by MitoSox staining and flow cytometry in U373 cells. Statistical analysis for (A) and (B): one-way ANOVA followed by Dunnett's post-test, n = 6 samples for each CA-DCF-DA group, 6 groups; n = 4 −5 samples for each MitoSox group, 4 groups; *P < 0.05, **P < 0.01. (C) H2O2 inhibits basal autophagy in U373 cells. The cells were treated with H2O2 (0.2 or 1 mM) for the indicated times, +/− CQ (50 μM, added 1 h before the lysis). LC3 and SQSTM1 expression was analyzed by WB after 15% and 8% SDS-PAGE, respectively. (D) Densitometric analysis of LC3-II (+CQ) and SQSTM1 bands (no CQ) from (C). Mean +/− SEM from 3 independent experiments. Statistical analysis: 2-way ANOVA followed by Bonferroni post-test, time vs concentration, n = 3 samples for each group, 5 groups for each OD analysis; *P < 0.05, **P < 0.01, compared to vehicle-treated cells. (E) Inhibition of autophagy in primary astrocytes in response to prolonged oxidative stress. Primary cultures of astrocytes isolated from mouse cortex were treated or untreated with PQ (100 μM) or H2O2 (1 mM) +/− CQ (50 μM, 3 h before lysis) and analyzed for markers of autophagy as described in (C). (F) 48 h before the treatment described in (E) the astrocytes were transfected with GFP-LC3 construct and analyzed for the presence of green dots. Representative, extended focus confocal images of GFP-LC3-expressing cells, pretreated for 3 h with CQ (50 μM), are shown. Bar: 10 μm. (G) Upregulation of SQSTM1 aggregates in primary mouse astrocytes exposed to prolonged treatment with PQ. Eight d after isolation primary cells were split and then cultured in the presence (LT-PQ) or absence (Control) of 3 μM PQ for 7 d. Primary cultures were then stained for GFAP and SQSTM1 and analyzed by confocal microscopy. Bar: 20 μM.
Figure 6.

PQ activates pro-autophagic signaling pathways in spite of its negative effect on LC3 lipidation. (A) Samples from experiments described in Fig. 1 (6 and 24 h PQ treatment) were analyzed for BECN1 expression by 8% SDS-PAGE and WB. Densitometric analysis of data (in lower panel) was performed as described in Fig. 1B (TUBA/α-tubulin was used as loading control). (B) PQ causes accumulation of insoluble BECN1 in cells exposed for 48 h to PQ. Samples of insoluble proteins in lysates were prepared and analyzed as described in Figure 1C. Statistical analysis for (A) and (B): one-way ANOVA followed by Dunnett's post-test; n indicates number of independent experiments; 4 groups, 3 – 6 OD values for each group; *P < 0.05, ***P < 0.001. (C) U373 cells were seeded on coverslips and treated or untreated with 100 μM PQ for 6 h, fixed and stained for BECN1 (green) and PIK3C3 (red). Representative confocal images of middle sections of stained cells are shown. Bar: 25 μm. (D–G) U373 cells were treated for the indicated time with 100 μM PQ and cell lysates were analyzed by NuPage 4–12% gradient gel electrophoresis followed by WB. MTOR (D), RPS6KB/p70S6 kinase) (E), MAPK8 (F) and MAPK1/3 (p44/p42) (G) phosphorylation were detected by phospho-specific antibodies. Densitometric analysis of n = 3 to 5 independent experiments was performed. Statistical analysis: one-way ANOVA for (D and E; 7 groups each experiment) and Mann-Whitney test for (F and G; 5 – 9 OD values for each control group, 3 - 5 OD values for other groups); *P < 0.05. (H) UO126 MEK1 inhibitor (0.2 μM) has little effect on basal autophagy and does not restore autophagy in PQ-treated cells. (I) Effects of UO126 used in (I) on MAPK1/3 phosphorylation in U373 cells.
Figure 7.
For figure legend, see page 1072.
Figure 8.

For figure legend, see page 1074.
The inhibitory effect of PQ was not limited to basal autophagy. In fact, the herbicide also inhibited rapamycin-induced autophagy in U373 cells, as the induction of LC3-II levels by rapamycin was potently blocked by PQ, both in the presence and absence of CQ (Fig. 3D). These data consistently demonstrate that in this cell system PQ blocks autophagy before or at the step of autophagosome formation.
Inhibition of autophagy limits the anti-oxidant and neuroprotective capabilities of astrocytes
Chronic exposure to very low concentrations of PQ likely contributes to PD pathogenesis.45,46 To mimic this condition in vitro, we chronically applied nonlethal concentrations of PQ for several days. 1 μM PQ is not lethal for at least 9 d to U373 astrocytes, if cells are maintained in daily-changed culture medium containing 4.5% glucose (data not shown). 1 µM PQ applied for 7 d (long term, LT-PQ) led to a significant decrease in LC3-II, more evident in the presence of CQ, and caused a concomitant SQSTM1 accumulation (Fig. 4A and B) and aggregation (Fig. 4C). Next, we asked if the impairment of autophagy in astrocytes could affect their viability and antioxidant capacity. After 24-h exposure to H2O2, LT-PQ cultures contained significantly less viable cells than control U373 cultures, yet LT-PQ U373 cells were more resistant to H2O2 than dopaminergic neuroblastoma SH-SY5Y cells (Fig. 4D). We have previously reported that SH-SY5Y cells subjected to similar concentrations of H2O2 rapidly activate autophagy and eventually undergo autophagy-dependent cell death.38,39 The important role of astrocytes, among others, is to protect dopaminergic neurons from oxidative insults.10,13 This antioxidant capacity of astrocytes can be assessed in vitro in coculture with neuronal cells.15 We established mixed cell cultures and found that dopaminergic neuroblastoma SH-SY5Y cells were significantly protected from H2O2 if cultured in the presence of control U373 cells, but not if cocultured with LT-PQ U373 (Fig. 4E and F). In the latter case, a decreased ratio of viable GFAP-negative cells (SH-SY5Y) compared to GFAP-positive U373 cells was observed (Fig. 4E and F). These data indicate that a chronic exposure to low concentration of PQ negatively affects autophagy and concomitantly diminishes the antioxidant and neuroprotective capacities of astrocytes.
Downregulation of LC3-II is a response to chronic, but not transient, OS in astrocytes
Astrocytes are highly resistant to OS.36,47 The lack of autophagic response to H2O2 in primary mouse astrocytes has been previously attributed to no ROS generation.48,49 To exclude such a possibility in U373 cells, we measured ROS levels in response to the herbicide and H2O2 by using the fluorescent probes CA-DCF-DA and MitoSox, which preferentially detect hydroxylradical (OH•) and mitochondrial and cytoplasmic superoxide (O2•−), respectively.39 PQ turned out to be a poor inducer of OH• compared to H2O2 (Fig. 5A), but both PQ and H2O2 caused a significant increase of O2•−, although the kinetics and the extent of its induction were different for PQ and H2O2 (Fig. 5B). In addition, the rapid induction of O2•− in U373 cells by H2O2 temporally correlated with a dose-dependent decrease of LC3-II levels at 4 h and 24 h post-treatment, in the presence or absence of CQ (Fig. 5C and D). H2O2 also induced a dose-dependent formation of insoluble SQSTM1 aggregates (Fig. 5C and D), which, however, did not accumulate with time as cell death rapidly occurred (data not shown).
We further addressed if the negative effect of PQ and H2O2 on basal autophagy in U373 cells was an unusual feature of this cell line or a common response to prolonged OS in astrocytes. Primary mouse astrocytes were expanded for 10 d after isolation before exposing the cells to OS. 100 μM PQ and 1 mM H2O2 applied for 16 h had little effect on LC3 lipidation, but a longer exposure (48 h) to pro-oxidants led to a significant decrease in LC3-II levels (Fig. 5E) and in LC3 dots (Fig. 5F). This effect occurred days before any signs of cell death, as primary astrocytes showed very low mortality until d 4 and d 3 post-treatment in response to 100 μM PQ (Fig. 7H) and 1 mM H2O2 (data not shown), respectively. In addition, primary astrocytes exposed for 7 d to a low concentration of PQ (3 μM) also accumulated SQSTM1 aggregates in contrast to astrocytes cultured under the same conditions, but without PQ (Fig. 5G).
PQ triggers upstream pro-autophagic signals
To find the mechanism underlying the PQ-induced block of basal autophagy we measured the levels of BECN1 upon PQ treatment, as downregulation of this protein inhibits basal and starvation-induced autophagy.50 Unexpectedly, we found a dose-dependent induction of BECN1 in cells exposed to different concentrations of PQ (Fig. 6A). Interestingly, BECN1 accumulated in the insoluble fraction of lysates at the 48 h time point (Fig. 6B), suggesting that it was part of a membrane-associated oligomeric complex. This interpretation was supported by immunofluorescence staining for BECN1, which showed increased intensity and colocalized with PIK3C3/VPS34 upon 16 h treatment with PQ (Fig. 6C).
We investigated other possible signaling pathways regulating autophagy in response to PQ. MTOR activation leads to autophagy suppression in the majority of cell systems.17 However, 100 μM PQ did not induce significant activation of MTOR at early time points, whereas it strongly inhibited MTOR after 24 h (Fig. 6D), when autophagy was clearly suppressed in our system. Accordingly, RPS6KB/p70S6 kinase phosphorylation, which reflects MTOR activity, was inhibited or unchanged up to 6 h after PQ treatment and strongly suppressed after 24 h (Fig. 6E). These data suggest that the negative regulation of autophagy by PQ does not involve MTOR. Starvation-induced autophagy and MPP+-induced autophagy are mediated by the activation of MAPK8/JNK1 (mitogen-activated protein kinase 8).50,51 Similarly, a transient increase in MAPK1/ERK2-MAPK3/ERK1 activity correlates with autophagy induction and is required for MPP+-50 and zVAD-induced autophagic cell death.52 PQ led to a significant elevation of MAPK8 phosphorylation within 1 h, followed by a very slow decrease back to control levels (Fig. 6F). Phosphorylation of MAPK1/3 was similarly modulated by PQ-induced stress, but a significant increase in p-MAPK1/3 was still detectable 24 h post-treatment (Fig. 6G). Sustained MAPK1/3 activation blocks the maturation of autophagosomes in cancer cell lines exposed to the pesticide Lindane.53 Remarkably, the MEK1 inhibitor UO126 had little effect on basal LC3-II levels in control cells and further reduced LC3-II in PQ-treated cells (Fig. 6H), although it strongly decreased MAPK1/3 phosphorylation in PQ-treated and control cells (Fig. 6I). These data indicate that MAPK1/3 activation is not an inhibitory signal elicited by PQ.
Inhibition of OS by a NQO2 inhibitor, but not by N-acetylcysteine, enhances autophagy in U373 cells and in primary astrocytes exposed to PQ
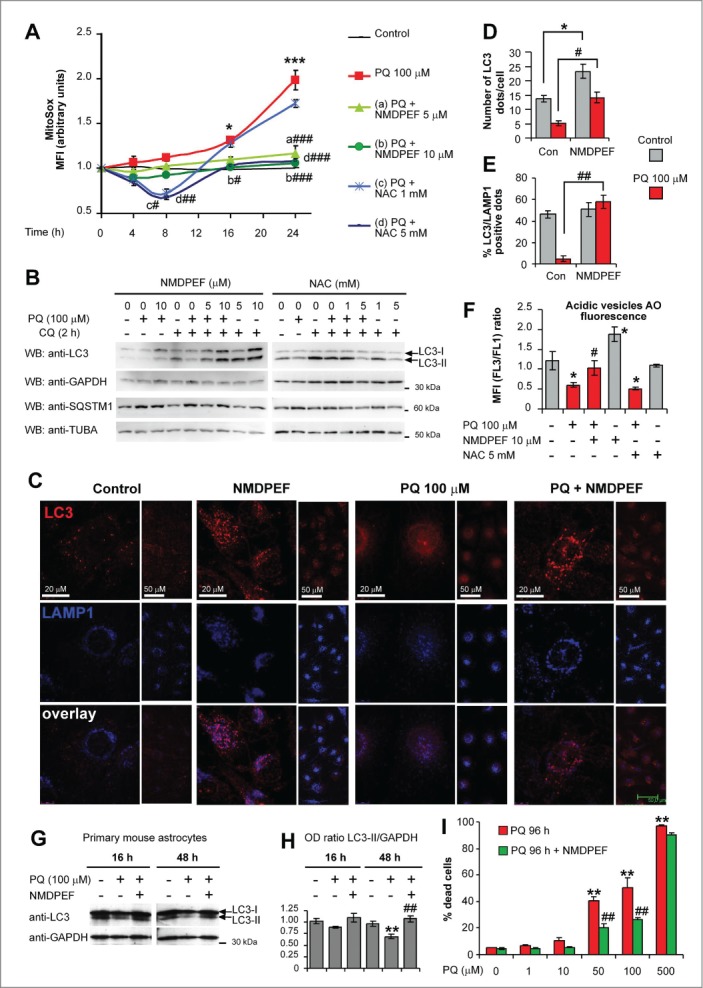
To examine the causal role of OS in PQ-induced downregulation of basal autophagy we used N-acetylcystein (NAC), a thiol antioxidant, and NMDPEF (N-[2-(2-methoxy-6H-dipyrido[2,3-a:3,2-e]pyrrolizin-11-yl)ethyl]-2-furamide), a pharmacological inhibitor of NQO2 activity and a powerful PQ-specific antioxidant in U373 cells.36 According to our previous observations,36 10 μM NMDPEF strongly reduced PQ-induced ROS, as detected by MitoSox (Fig. 7A). 5 mM NAC showed comparable effects on mitochondrial ROS, but the effects of the NQO2 inhibitor were more stable over time compared to NAC (Fig. 7A). Remarkably, the addition of NMDPEF to U373 cells for 24 h potently enhanced LC3-II processing (Fig. 7B) and increased the number of AV dots both in PQ-treated and untreated cells (Fig. 7C and D). In contrast, cotreatment with NAC did not restore LC3-II levels in PQ-treated astrocytoma cells, but it dose-dependently decreased LC3-II in control cells (Fig. 7D). Of note, NMDPEF not only increased the number of LC3-positive dots, but it restored the normal percentage of LC3-LAMP1 double-positive autolysosomes, which were markedly reduced in PQ-treated cells compared to control cells (Fig. 7C and E), suggesting that NQO2 inhibition stimulates autophagy flux. This observation was confirmed by acridine orange staining and flow cytometry used to assess quantitatively acidic vesicles such as autolysosomes and lysosomes.54 NMDPEF increased, whereas NAC reduced, the number of acidic vesicles, in control and PQ-treated cells (Fig. 7F).
The pro-autophagic effect of the NQO2 inhibitor was further confirmed in primary mouse astrocytes. NMDPEF restored LC3-II levels in primary cultures treated with PQ (Fig. 7G and H), which correlated with a significant protective effect of this agent against PQ-induced cell death (Fig. 7I), in accordance to what we previously observed in U373 cells.36 Thus, a NQO2-specific antioxidant, but not a generic inhibitor of PQ-induced OS, can prevent deleterious effects of PQ on LC3 lipidation and autolysosome formation.
NQO2 mediates the negative regulation of LC3-lipidation by PQ
We further investigated the active involvement of NQO2 in the negative regulation of basal autophagy induced by PQ. NMDPEF dose-dependently and time-dependently induced autophagy, as shown by the increase in LC3 lipidation and BECN1 (Fig. 8A). This effect was not due to inhibition of the autophagic flux, since the upregulation of LC3-II in the presence of NMDPEF and CQ was much higher than in vehicle-treated cells in the presence of CQ (Fig. 7B). NQO2 is well expressed in primary astrocytes and U373 cells, although very low levels of NQO2 can be detected in mouse brain total lysates, suggesting an astrocyte-specific expression of this enzyme (Fig. 8B). Since NQO2 activity contributes to OS in astrocytes,36 it may play a role in the negative regulation of autophagy by chronic OS. To test this hypothesis, we stimulated NQO2 activity in U373 cells with the NQO2 substrate menadione (K3). Stimulation of NQO2 activity rapidly inhibited LC3 lipidation at 1 and 6 h, although at later time points LC3-II accumulated most likely due to inhibition of the autophagic flux (Fig. 8C). This effect correlated with NQO2 activity. 10 μM K3 led to 3- to 4-fold activation of NQO2 at 1 and 6 h, respectively, while PQ stimulated 2-fold NQO2 activation within 20 h (Fig. 8D). Next, we addressed the role of NQO2 in autophagy by genetic modulation of its expression. NQO2 was silenced by transient transfection of different NQO2-specific siRNAs or overexpressed by a stable expression of the human NQO2-coding lentiviral construct (Fig. 8E). NQO2 knockdown prevented PQ-induced reduction in LC3 lipidation and partially reduced PQ-dependent accumulation of SQSTM1 (Fig. 8F and G). In addition, NQO2 silencing markedly reduced the LC3 response to NMDPEF alone, but augmented the response to NMDPEF in the PQ-treated cells (Fig. 8F and G). We further analyzed LC3 processing in U373 cells stably overexpressing NQO2. NQO2 overexpression reproducibly led to a stronger reduction of LC3 lipidation than in control vector-expressing cells in response to PQ (Fig. 8H and I). Strikingly, this correlated with a significantly higher sensitivity of NQO2-transfected cells to PQ-induced toxicity, since these cells massively underwent cell death 48 h after the treatment with PQ in contrast to control vector-transfected cells, which were still alive at this time point (Fig. 8J). The increased mortality was mediated by an increased NQO2 activity in NQO2-overexpressing U373 cells, since its inhibition by the NQO2 inhibitor NMDPEF efficiently protected these cells against cell death. In addition, cotreatment with the autophagy inhibitor 3-methyladenine and PQ increased the percentage of dead cells in NQO2-overexpressing and in control cells (Fig. 8J). Thus, the residual autophagy in PQ-treated cells has a protective function in astroglial cells, and its stimulation with the NQO2 inhibitor protects from PQ-induced cell death.
PQ and NMDPF cotreatment, but not PQ alone, stimulates NFE2L2 activity independently from SQSTM1
Finally, we asked whether NFE2L2 was involved in PQ-dependent inhibition of autophagy. NFE2L2 is a ubiquitously expressed transcription factor that regulates the expression of genes bearing an antioxidant response element (ARE) in the promoter region, including NQO2.55 Importantly, autophagic activity inversely correlates with NFE2L2-mediated antioxidant responses, due to SQSTM1-dependent disruption of KEAP1-NFE2L2 binding on the one hand56,57 and due to NFE2L2-dependent upregulation of SQSTM1 on the other.58,59 First, we assessed the expression at protein level of NFE2L2, NQO2, and SQSTM1 in U373 cells exposed to PQ, NMDPF, or PQ+NMDPF for up to 24 h (Fig. 9A). PQ induced the accumulation of SQSTM1 at 24 h, as expected, but did not alter NFE2L2 levels. Though not statistically significant, a transient upregulation of NQO2 protein level was observed in PQ-treated cells. NMDPF alone had no effects on the expression of these 3 proteins. However, when the cells were cotreated with PQ and NMDPF, NFE2L2 levels significantly increased at 24 h (Fig. 9A). Next, NFE2L2 transcriptional activity was determined by luciferase reporter assay monitoring ARE induction in control, PQ, NMDPEF, and PQ+NMDPEF treatment conditions. The cotreatment with PQ and NMDPEF robustly induced ARE-dependent transcription (Fig. 9B), which is consistent with the upregulation of NFE2L2 under these conditions (Fig. 9A). Finally, we asked whether SQSTM1 could be involved in NFE2L2 expression and activity. To this end, we repeated the experiments in siRNA-SQSTM1-silenced cells. Data shown in Figs 9C and D confirm that PQ alone does not induce NFE2L2 and also demonstrate that transcriptional activation of NFE2L2 (as probed by luciferase activity) in PQ+NMDPF-treated cells occurred independently from SQSTM1.
Figure 9.

PQ and NMDPF cotreatment, but not PQ alone, stimulate NFE2L2 activity independently from SQSTM1. (A) WB analysis of NFE2L2, SQSTM1 and NQO2 protein levels treated with PQ (100 μM), NMDPEF (5 μM) or PQ (100 μM) + NMDPEF (5 μM) for the indicated times. Graphs show the OD analysis (ratio to GAPDH, normalized to vehicle-treated controls) as means +/− SEM of n = 3 blots from 3 independent experiments. Statistical analysis: one-way ANOVA followed by Dunnett post-test; 5 groups, n = 3 - 6 OD values in each group; *P < 0.05, **P < 0.01. (B) NFE2L2 activity is activated by PQ plus NMDPEF. U373 cells were transfected with ARE-Firefly and Renilla luciferase reporter plasmids and 2 days later treated with 100 μM PQ or/and 5 μM NMDPEF for 6 or 24 h. The ratio of Firefly to Renilla luciferase activity was determined twice in triplicates. The graph shows means +/− SEM of a representative experiment out of 3 independent experiments that yielded similar results. Statistical analysis: one-way ANOVA followed by Dunnett post-test; n = 6 determinations in each group, 5 groups; *P < 0.05, **P < 0.01. (C) U373 cells were transfected with a control siRNA or a SQSTM1-specific siRNA, treated as indicated and analyzed by WB for the expression of SQSTM1, NFE2L2 and NQO2. GAPDH was used as loading control. (D) U373 cells were transfected with reporter plasmids as described in (B) together with 1 μg of control siRNA or SQSTM1-targeting siRNA. The cells were then treated as indicated for 24 h and subsequently assayed for luciferase activity as described in (B). Statistical analysis: one-way ANOVA followed by Bonferroni post-test; *P < 0.05, **P < 0.01, when compared to control cells; #P < 0.05, when compared to matched group expressing control siRNA. Note that NFE2L2-dependent luciferase activity is not suppressed by SQSTM1 silencing.
Discussion
The data presented in this paper led to several original and rather unexpected conclusions. We showed for the first time that prolonged OS reduces the basal autophagy rate in primary astrocytes and U373 astrocytoma cells exposed to PQ or H2O2, as evidenced by decreased LC3 lipidation correlating with impaired autophagosome formation and concomitant accumulation of SQSTM1 aggregates. Paradoxically, this occurs in spite of an apparent activation of the majority of pro-autophagic pathways by PQ. This inhibitory effect on autophagy is mediated by the quinone reductase NQO2 and can be prevented by pharmacological and genetic modulation of its activity. Prolonged inhibition of basal autophagy by low-doses of PQ has functional consequences on the antioxidant capacity of astrocytes and negatively affects their neuroprotective capability toward dopaminergic cells exposed to OS.
These findings are unexpected for several reasons. First, numerous previous reports documented the causal role of OS induced by H2O2 in the activation of autophagy in different cell systems,39,48,49,60,61 implicating O2•− as the main messenger of this response. Second, in neuroblastoma cell lines62,63 and in Drosophila synapse development64 OS induced by PQ was reported to induce autophagy. Third, MPPT/MPP+, which is structurally related to PQ, excessively stimulates an autophagic response, leading to cell death in different neuronal cells.41,50,54 This led to the concept that nigrostriatal degeneration induced by these toxins arises from a pathological enhancement of autophagy in dopaminergic neurons. Furthermore, OS has been implicated in the induction of autophagy under different experimental conditions in vitro and in vivo, such as amino acid starvation in cancer cell lines65 or ischemia/reperfusion in cardiomyocytes.66 Finally, thiol antioxidants such as NAC or vitamin E inhibit basal and rapamycin-induced autophagy.67 Accordingly, we also observed such a negative effect of NAC in our system (Fig. 7B). In contrast, the specific NQO2 inhibitor NMDPEF, that apparently shows a similar antioxidant activity, elicited an increase of the autophagic activity in response to OS (Fig. 7). This discrepancy suggests that the effect of an antioxidant on cell physiology depends on its target(s) and its way of action. Similarly, the source, type and time of action of OS may have opposite effects on the autophagy machinery, as also emerges from some recent findings and from more careful analysis of the literature data.17 ROS generated in response to different stimuli seem to both induce and inhibit upstream signaling pathways regulating autophagy and may also negatively influence lysosomal function.54,68,69 For example, it has been reported that rotenone inhibits autophagic flux via inhibition of lysosomal degradation, as substantiated by increased levels of SQSTM1 and SNCA in dopaminergic SH-SY5Y cells.70 The suppression of autophagic flux was also observed in hepatoma cells in response to H2O2 and TNF/TNFα71 and in PRNP (prion protein)-deficient hippocampal primary neurons, but not in normal hippocampal neurons exposed to H2O2, suggesting a possible role of a functional PRNP in regulation of the autophagic response to OS.72
In our experimental conditions, PQ-induced OS exerted a dual effect on the autophagy machinery: on the one hand, it activated autophagy-inducing signals including MAPK1/3 and MAPK8 pathways, inhibited MTOR and upregulated BECN1 (Fig. 6) and, on the other hand, it impaired LC3 processing, AV formation (Figs. 1–3) and led to a reduced number of autolysosomes (Fig. 7C and E). The negative regulation of basal autophagy by PQ (as well as by H2O2) was time- and dose-dependent. The effect was hardly detectable after short exposure or with low concentration (1 μM) of PQ (Fig. 1A). However, the prolonged exposure to low concentrations or the acute exposure to higher doses of PQ invariably led to the impairment of basal autophagy.
At low concentrations of PQ positive and negative signals are balanced and no detectable response in terms of autophagy induction is observed, in spite of a toxic accumulation of oxidation products. In the long run, such a situation potentially leads to astroglial degeneration. Indeed, astroglial cells survived up to 7 d under nonlethal concentrations of PQ (1 μM), but eventually showed a significant reduction in LC3-II levels associated with a significant accumulation of insoluble SQSTM1 aggregates (Fig. 4A–C), with a concurrent reduced resistance to OS and neuroprotective capacity toward dopaminergic cells (Fig. 4D–F). This observation can explain why PQ chronically administered to rodents causes accumulation of protein inclusions, similar to Lewy bodies in SN and other brain regions, before any loss of dopaminergic neurons becomes apparent.73 Accordingly, a recent report addressing the autophagic response to PQ in vivo reported appreciable modulation of autophagic markers in different regions of the brain, suggesting a possible inhibition of autophagy in vivo by PQ, but not by maneb, another herbicide implicated in PD etiology.30 However, the interpretation of the results obtained in rodent models is hampered by the difficulty in assessing the rate of autophagic flux in vivo.
Our in vitro system made it possible to show for the first time an inhibitory effect of PQ on the autophagy machinery in astrocytes. In addition, we could demonstrate that such a negative effect is mediated by the oxidoreductase NQO2. Several data support this conclusion: (i) pharmacological inhibition of NQO2 by NMDPEF potently upregulates LC3-II levels and autophagosome formation, as well as autophagosome-lysosome fusion and cell survival in the presence of PQ; (ii) NQO2 activation by menadione in astroglial cells leads to a rapid decrease in LC3-II levels; (iii) silencing of NQO2 expression by siRNA or shRNA reduces, whereas NQO2 overexpression (iv) augments, the inhibitory effect of PQ on LC3 processing. Induction of NQO2 activity and SQSTM1 accumulation by PQ did not correlate with NFE2L2 activation. Intriguingly, in PQ-NMDPEF cotreated cells NFE2L2 expression and activity were upregulated. Astrocytes have been reported to possess high basal expression of NFE2L2,13 and apparently PQ cannot further induce its expression, unless NQO2 is inhibited. This complex regulation definitely deserves a more in depth investigation. However, considering the lack of correlation between NFE2L2 and SQSTM1 levels (Fig. 9A) and no effect of SQSTM1 silencing on NFE2L2 activity and levels (Fig. 9C and D), we can conclude that PQ+NMDPEF-induced NFE2L2 upregulation is SQSTM1 independent in U373 astroglial cells.
Another interesting finding presented here is that primary astrocytes and astroglial cell lines express much higher NQO2 protein levels compared to other brain cells (Fig. 8B). Consistently, we found that PQ exerted its inhibitory effect on autophagy mainly in astrocytes and to a lesser extent in other cell types (Janda et al., unpublished observations).
We have recently demonstrated that NQO2 clearly contributes to OS induced by PQ36 likely because its catalytic activity is intrinsically linked to ROS generation.74 Here, we confirm these findings and show that overexpression of NQO2 further sensitizes astroglial cells to PQ-induced cell death (Fig. 8J), an effect associated with impairment of autophagy. Most importantly, we show that NQO2-mediated inhibition of autophagy in astroglial cells has pathophysiological consequences since it limits the intrinsic ability of these cells to protect dopaminergic cells from OS.
The mechanism involved in free radical production by NQO2 is unknown and difficult to envisage in consideration of the fact that NQO2 catalyzes 2-electron reductions thought to be free of ROS byproducts.37 In addition, the toxifying effects of NQO2 activity are difficult to reconcile with a proposed detoxifying function of NQO2 in oxidative dopamine metabolism.75 Nevertheless, since NQO2 causes toxic activation of certain substrates (menadione, mitomycin)37 and contributes to ROS production by PQ,36 it cannot be excluded that similar ROS generating events accompany dopamine quinone reactions mediated by NQO2.
Thus, under certain conditions, NQO2 can lead to chronic OS and autophagy impairment, which may be prevented by pharmacological inhibition of its activity. The upregulation of basal autophagy associated with the inhibition of NQO2 activity in the absence of exogenous OS (Fig. 8A) suggests that ROS-independent mechanisms are also involved. The exact molecular mechanism of NQO2-mediated inhibition of basal autophagy is presently under investigation. Based on the differential effects exerted by the antioxidants NAC (more general) and NMDPEF (specific to NQO2-related ROS), we envisage a role for the localized NQO2-dependent production of O2•−, which might in turn affect the machinery for LC3 processing.
Regardless of the molecular mechanisms implicated in NQO2-dependent regulation of autophagy, the identification of this novel regulator of autophagy in oxidatively stressed cells has important practical implications. NQO2 can be easily modulated pharmacologically by several natural and synthetic inhibitors, including NMDPEF and certain flavonoids such as quercetin and resveratrol. These findings will foster future studies to define if pharmacological targeting of NQO2 in vivo can prevent SQSTM1-positive protein inclusions and pathological changes present in PD and other neurodegenerative disorders.
Material and Methods
Cell culture
Astrocytoma U373 cells (kind gift of Michela Pollicita, University Tor Vergata, Rome, Italy) were cultured as previously described.10 After thawing, cells were expanded in DMEM 4.5% glucose (LONZA, 12-604) and then routinely cultured in standard medium: DMEM 1% glucose (LONZA, 12-707), supplemented with 2 mM l-glutamine (PAA Laboratories, M11-004), 10% fetal calf serum, 1 mM sodium pyruvate (Gibco, 11360-070), 100 units/ml penicillin, 100 μg/ml streptomycin (Pen/Strep, Gibco, 15070-063), in standard 5% CO2 conditions. The medium was regularly changed every 48 h.
Isolation of primary astrocytes
Brains of 5 newborn pups (mouse strain Bl6/129, postnatal d 0–1) were isolated immediately after decapitation as previously described.47 Briefly, the brains were placed on plastic dishes containing sterile phosphate-buffered saline (PBS; Lonza, 4984) plus Pen/Strep (1:100). Cortices were dissected, meninges carefully removed and the tissue was cut into small pieces with scissors. All the cortex tissue was then collected, resuspended in 10 ml of DMEM, containing trypsin and DNAase (Sigma-Aldrich, T4799, P4513) and digested for 30 min at 37°C with occasional shaking. The digested cortex was washed and plated on 5 culture dishes (10 cm) in standard medium. The next day the medium was changed and then renewed twice a wk. At confluence (usually at d 7–9 after plating) the culture was washed 3 times with PBS, trypsinized in 0.05% trypsin for 5–8 min and plated 1:3. When confluent, the primary cells were split again and plated for required experiments. All experiments were performed before 4 weeks after isolation. The percentage of astrocytes in primary culture was assessed on the basis of GFAP positivity upon staining with rabbit anti-GFAP (Thermo Scientific, OPA-06100), followed by anti-rabbit Alexa Cy5 (Molecular Probes, A-21070). The animal care and procedure for sacrifice complied with the recommendation of the local ethics committee.
Reagents and antibodies
Unless otherwise specified, all reagents were purchased from Sigma-Aldrich: paraquat (M2254; 100 mM stock in PBS), chloroquine (C6628; 25 mM stock in PBS), 3-methyladenine (M9281; 10 mg/ml, 95% ethanol), DMSO (D4540), NAC (A9165; 1 M stock in PBS), were kept in aliquots at −80°C and thawed shortly before each treatment. Rapamycin (37094) was kept as 5 mM stock at −20°C. H2O2 (95321) was diluted from a 33% stock into 1 M aliquots and stored at −20°C. NMDPEF (a kind gift of Philipe Delagrange, Servier, France) was stored as a 20 mM stock in DMSO at −20°C.
The following antibodies (Abs) used for western blotting (WB) were purchased from MBL International Corporation: rabbit anti-LC3 (PM036; used 1:4000 and occasionally 1:1000), rabbit anti-BECN1 (PD017; 1:2000), anti-SQSTM1/p62, mouse (M162-3; 1:1000) or rabbit (PM045; 1:2000). In some experiments, the following Santa Cruz Biotechnology Abs were used: rabbit anti-BECN1 (sc-11427; 1:200), goat anti-NQO2 (sc-18574; 1:200), rabbit anti-NFE2L2/NRF2 (sc-722, C-20; 1:1000), anti-TUBA/α-tubulin (sc-23948; 1:800) and rabbit anti-GAPDH (clone FL335, sc-25778; 1:1000). Other Abs used were from Sigma-Aldrich: rabbit anti-NQO2 (HPA332; 1:1000), anti-ACTB/β-actin (clone A40; 1:5000) and mouse anti-GFAP (G4546; 1:1000). The following Abs were from Cell Signaling Technology (used at 1:2000 or 1:1000), rabbit anti-LC3B (used only for lysates from primary astrocytes; 2775), anti-p-MTOR (Ser2448; 2971) and anti-MTOR (clone 7C10; 2983S), rabbit anti-p-RPS6KB/p70S6 kinase (Thr389; 9205S) and anti-total RPS6KB/p70S6 kinase (2708S), anti-p-MAPK1/3 (P-p44/42, T202/Y204, clone 197G2, 4377S) and anti-MAPK1/3 (p44/42, clone 3A7, 9107), anti-RPS6KB (p-SAPK/JNK, T183/Y185, 4668). The Abs used for confocal immunofluorescence analysis were: rabbit anti-PIK3C3/VPS34 (RayBiotech, RB-08-0037; 1:200), mouse anti-BECN1 (MBL, PM015; 1:100), mouse anti-LAMP1 (BD Biosciences, 611042; 1:100). Secondary antibodies anti-rabbit (Alexa Fluor 568, A-11011 and Alexa Fluor 488, A21206) and anti-mouse (Alexa Fluor 633, A-21052) were used at 1:800 dilution and were purchased from Molecular Probes. Fluorescein (FITC) phalloidin (Molecular Probes, F432; 1:400) was used to detect actin.
Treatments and experimental setup for western blotting analysis
U373 cells were seeded on plastic dishes 24 h before treatment at the density 2 × 104 cells/cm2 or 1 × 104 cells/cm2 and lysed 24 or 48 h after initial treatment, respectively. All treatments were performed in freshly renewed medium and the compound (PQ, NMDPEF, H2O2, NAC) was added for the indicated time (usually 6, 24, 48 h) before the end of the experiment, so that control and treated cells were all lysed at the same time. CQ (25 µM or 50 µM) was added 1 to 6 h before the end of the experiment, as specified; the time of exposure to CQ was identical for each sample in the same experiment. For prolonged incubation, U373 cells were seeded at low density (5 × 104/cm2) and cultured in 4.5% glucose DMEM for 7 d. A half volume of the culture medium was renewed each day. Cells were exposed to 1 µM PQ for 7, 6, 4 and 2 d before the lysis and lysed at the same time on day 7. CQ (25 μM) was added 1 h before lysis. For signaling experiments the cells were treated for 15 and 30 min, and 1, 3, 6 and 24 h.
Western blotting
Cell monolayers were washed once with PBS and scraped in cold Lysis buffer 1 (LB1) containing 50 mM Tris-HCl, pH 7.3, 150 mM NaCl and the following reagents from Sigma-Aldrich: 1% Igepal (I7771), 1 mM EDTA (E5134), 1 mM phenylmethylsulfonyl fluoride (P7626), phosphatase inhibitors (1mM NaF [S1504], 0.01 mM sodium orthovanadate [S6508]) and protease inhibitor cocktail (Complete Mini, Roche Diagnostic, 11836153001). The lysate was placed on ice for 10 min and then centrifuged at 4°C, 15000 g for 10 min. The supernatant fraction was then mixed (3:1) with Laemmli Sample buffer (4 ×: 100 mM Tris-HCl, pH 6.8, 4% SDS [Sigma-Aldrich, L4390], 16% glycerol, 20% 2-mercaptoethanol [Carlo Erba Reagents, 460691]), boiled 15 min at 90°C and then stored at −80°C until analysis. Samples were boiled again for 3 min at 90°C before loading onto 15% or 8% polyacrylamide SDS (SDS-PAGE) gels. Protein concentration was quantified by BCA protein assay (Thermo Scientific, 23227). Alternatively, for analysis of detergent-resistant fractions of the lysates, cell monolayers were lysed in LB1 as above. After centrifugation (4°C, 15000 g for 10 min), the pellets were washed once in 200 μl cold LB1 buffer and then lysed in 30 μl Laemmli Sample buffer (4x) by heating 10 min at 90°C. Next LB1 buffer (30 μl) was added and samples were heated again 10 min at 90°C. For loading on gels, it was assumed that protein concentration in the pellet fraction was proportional to the protein concentration in the supernatant fraction.
For analysis of serine-threonine phosphorylations (p-MTOR, p-RPS6KB, p-MAPK1/3, p-MAPK8/JNK) cell monolayers were washed in PBS containing NaF (1 mM) and lysed as described previously.76 In general, 10–20 μg of protein lysate was loaded on 15% or 8% SDS-PAGE gels or 4–12% Bis-Tris gradient NuPAGE gels (Invitrogen, NP0323). For NQO2 expression analysis, 50 μg of protein lysate was loaded on 10% gels. After electrophoresis, polypeptides were electrophoretically transferred to PVDF membranes (Bio-Rad, 162–0177), which were thereafter saturated with 5% milk (Panrea AppliChem, A0830) in TTBS for 1 h at room temperature (RT). The primary antibody was usually incubated overnight, followed by 2-h incubation with secondary antibody. Blots were developed with the enhanced chemiluminescence (ECL) procedure, using ECL reagents (ImmunoStarTM, Bio-Rad, 170–5070 or Santa Cruz Biotechnology, sc-2040) and digitally acquired using a ChemiDoc XRS imaging system (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Transient and lentiviral silencing of NQO2
Nonsilencing (control, All-stars, 1027280) siRNA and a set of human NQO2-targeting siRNA were purchased from Qiagen (1027416). Transfection of U373 cells was performed using Lipofectamine 2000 (Life Technologies, Invitrogen, 11668027) and Optimem (Invitrogen, 51985026) according to the manufacturer's instructions. Cells were incubated in Opitmem containing siRNA-lipofectamine complexes for 6 h, washed with DMEM and then incubated in culture medium containing 4.5% glucose for 20 h before treatments with PQ or NMDPEF for 24 h. The nonsilencing siRNA was used as a negative control. The silencing with lentiviruses coding for human NQO2-targeting shRNA was performed as previously described.36 Four d after infection with lentiviral supernatant fractions U373 cells were plated at standard conditions; 24 h later cells were treated with PQ +/− NMDPEF for 24 h, lysed and assayed by WB for autophagy markers and NQO2 expression. Sixty μg of total protein lysate were usually necessary to detect NQO2 with goat anti-NQO2 (Santa Cruz Biotechnology) or less, if rabbit anti-NQO2 Ab (Sigma-Aldrich) was used.
Preparation of NQO2-overexpresing cells
NQO2-overexpressing U373 cells were generated by infection with a lentivirus coding for human NQO2 pLenti6.2# × 2044;V5-DEST™ Gateway®. This plasmid was obtained by clonase-mediated recombination (Gateway LR Clonase Mix, Life Technologies, Invitrogen, 11791-020) between the destination vector and pENTRY221-NQO2 (clone ID IOH6580) according to the manufacturer's indications. Lentiviral particles were generated in HEK293-FT cells (Invitrogen, R700-07) from pLenti6.2#x2044;V5-DEST™ NQO2 plasmids and control pLenti6.2/GW/emGFP (empty) vectors and used to infect U373 cells as described previously.36 After 6 d selection in blasticidin (Invitrogen, R210-01) and 4 d recovery in the absence of the drug, NQO2 overexpression was verified and NQO2-overexpressing and control vector cells were used for experiments.
Immunofluorescence staining
For immunofluorescent staining, U373 cells were seeded (20–30 × 103 cells/cm2) on cover slips and the next day treated as indicated, washed in PBS, fixed in 4% paraformaldehyde (Sigma-Aldrich, 158127) in PBS (pH 7.4) for 5 min and then blocked for 30 min at RT with PBS (pH 7.4) containing 1% fetal bovine serum, 0.1% Tween 20 (Sigma-Aldrich, P-1379) (blocking buffer). For immunodetection of antigens, the cells were incubated for 1 h at RT with the primary antibody; afterward the coverslips were washed 3 × in blocking buffer and then incubated for 45 min with the secondary antibody. Antibody dilutions were made in blocking buffer. After washing once in blocking buffer and 2 times in PBS, the cover slips were finally mounted on glass slides with 4 μl Slow-fade Gold mounting reagent (Life Technologies, S36936). A negative control was performed by omitting the primary antibody.
Confocal microscopy and analysis of images
Intracellular localization of LC3-positive vesicles, SQSTM1 dots and other markers of interest was assessed by confocal microscopy. Images were collected on a Leica TCS sp5X confocal microscope (LAS AF V2.2 DMI 6000 Argon Me Ne 543, Me Ne 633) with a 40 × ApoPLA oil immersion objective, magnification 5 ×, and 60 µm aperture (Leica Biosystems Inc., Buffalo Grove, IL, USA). Z stacks of images were collected as described previously.76 Extended focus projection were used to generate the images used for counting of total number of LC3 vesicles and SQSTM1 dots per cell. The colocalization analysis of LAMP1 and LC3 or SQSTM1 and LC3 was performed on a single central plane confocal image showing 2 to 5 cells. Counting of dots was performed manually and by ImageJ software. The parameters for ImageJ analysis were adjusted suitably to obtain similar results to manual counting. Several independent images with at least 50 cells were collected for each experiment. The mean of dots per cell was first calculated for single image and then the mean for the entire experiment was calculated by adding the single image means divided by the total number of images.
Live imaging of GFP-LC3 positive vesicles
Primary mouse astrocytes (12 d after isolation) were plated at the density 2 × 104 × cm−2 on coverslips in 12-well plates and the next day transfected with 0.75 μg of GFP-LC3 plasmid (kind gift of Dr E. Bampton, Leichester, UK) per well with 1.5 μl Lipofectamine 2000 (Invitrogen, 11668027) according to the manufacturer's instructions. 48 h after transfection, cells were treated as required and 24 h later live cell images were recorded on an A1r Nikon inverted confocal microscope equipped with resonant scanner for fast imaging and motorized XY stage by using a 63X CFI Plan APO VC 1.4 DT:0.13mm immersion oil objective (Nikon AG Egg/ZH, Switzerland). For confocal z-axis stacks, 25 images separated by 0.2 μm along the z-axis were acquired.
Detection of acidic vesicles by flow cytometry
U373 cells were seeded and cultured on 24-well plates as for WB experiments, but at the end of the 24-h treatment the cells were incubated for 15 min with 10 μg/ml acridine orange (Sigma-Aldrich, 318337). Next, the cells were washed once in PBS, trypsinized for 3 min, collected with 1 ml 10% fetal calf serum culture medium in conical tubes (for flow cytometry analysis), centrifuged at 800 g for 5 min and suspended in 0.4 ml PBS containing 1% BSA (Santa Cruz Biotechnology, 9048-46-8) and 0.1% EDTA (Sigma-Aldrich, E5134). Cells were acquired in the far red (>650 nm, FL3) channel and in the green channel (510–530 nm, FL1) by FACSCanto II (BD Biosciences, Erenbodgem, Belgium). Mean fluorescence intensity (MFI) for FL1 and FL3 channels was determined on the entire viable cell population by BD FACSDiva software. The ratio of MFI (FL3)/MFI (FL1) was calculated for each sample and data analyzed by Microsoft Excel.
Mitochondrial ROS detection by MitoSox
To detect ROS in mitochondria U373 cells were treated or untreated with PQ (200 μM) or H2O2 (200 μM) before analysis. MitoSox (Molecular Probes, M36008) staining and flow cytometry were performed as previously described.36
NQO2 activity assay
NQO2 acitivity in U373 cells was measured as previously described36 with a few modifications. Briefly, U373 cells plated and cultured under standard conditions were treated with 10 μM menadione (K3, Supelco, 4-7775; 10 mM stock in ethanol), or PQ and then homogenized on ice with a glass tissue grinder (Wheaton, 357538) in reaction buffer (50 mM Tris-HCl, pH 7.5, 1 mM n-octyl-β-D-glucopyranoside [Sigma-Aldrich, O8001-100 MG]). 5 μg of freshly prepared cell homogenates were added to a reaction mixture containing 150 μl of reaction buffer and 10 μM BNAH (Santa Cruz Biotechnology, 208609) and no K3. The decrease in BNAH fluorescence was monitored for 30 min at RT and normalized to a spontaneous decay in BNAH fluorescence in the absence of cell homogenate.
ROS detection by a DCF (dichlorofluorescein) derivative
U373 cells were plated at standard conditions (2 × 104/cm2) in 96-well plates. PQ (100 μM) or H2O2 (200 μM) was added at 24, 6 or 3 h or 60, 30 and 15 min before analysis (all treatments were performed 6 times). Cell staining with cytoplasmic ROS fluorescent indicator 5(6)-carboxy-2′,7′ dichlorofluorescein diacetate (CA-DCF-DA; Sigma-Aldrich, 21884) was performed 1.5 h before analysis. For this purpose, the conditioned medium +/− PQ or H2O2 was temporarily removed from cells (kept in the incubator in the meantime) and the cells were incubated with prewarmed basal DMEM containing 20 μM CA-DCF-DA or vehicle (DMSO) for 30 min. Stained cells were gently rinsed in warm basal DMEM and incubated again for 1 h with conditioned medium. Where indicated, PQ or H2O2 was added for short-term treatments. Fluorescent signal detection of live cell monolayers was performed on multilabel fluorimeter Victor 2 (Perkin-Elmer, Singapore) with fluorescein filter settings. Background readings (cells stained with DMSO) were subtracted for analysis of fluorescence of DCF-stained cells.
Transfection of siRNA and cDNA and luciferase reporter assay
Transfection of reporter plasmids and siRNA was performed with Lipofectamine 2000 (Invitrogen, 11668027), which was used according to the manufacturer's instructions. SQSTM1 siRNA (EHU027651) and control siRNA (EHUEGFP) were purchased from Sigma-Aldrich. Reporter assay plasmids (ARE–firefly luciferase and thymidine kinase (TK)-Renilla luciferase (internal control) were purchased as Cignal reporter kit (Qiagen, CCS-5020L) and used according to the manufacturer's instructions. In particular, 1.5 to 2 µg of reporter DNA and 1 µg of siRNA per 35-mm plate were used for the transfection of subconfluent U373 cells. Luciferase activities were measured by using the Dual-Luciferase Reporter Assay system (Promega, E1910) in 96-well plates on a Multilabel luminometer/fluorimeter Victor 2 (Perkin-Elmer, Singapore). The experiment was run in triplicate and each measurement was repeated 3 times.
Data analysis and statistical procedures
Statistical analysis of all the experiments was performed using GraphPad Prism software (version 3.0). The data were presented as means ±SEM (95% confidence interval) of 3 or more independent experiments, unless otherwise specified. The statistical significance of the differences between treatments was evaluated by analysis of variance test ANOVA, one-way or 2-way depending on the number of variable parameters, followed by Dunnett's or Bonferroni multiple comparison tests, respectively. Mann-Whitey or unpaired 2-tailed t test test were used for 2 small-size (n < 10) or 2 middle-size (n ≥ 10) group comparisons.
Optical density of western blots was assessed by Quantity One image software (Bio-Rad) for each individual blot and the respective loading control. The main OD values were divided by loading control OD and normalized to OD values in untreated cells. The latter were assigned an arbitrary value of “1” or 100%. All experiments were replicated at least 3 times.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Giovanni Porciatti and Nuria Vadalà for excellent technical support. NQO2 inhibitor NMDPEF was a kind gift of Philippe DeLagrange, Servier Pharmaceuticals, Paris, France. We thank Luca Scorrano's laboratory for providing the biological material, necessary tools, and instructions for the isolation and culture of primary murine astrocytes.
Funding
This work was financially supported by a Research and Competitiveness PON3a-00359 grant and by Environmental Protection Agency Calabria (ARPACal). CI was financially supported by Compagnia S.Paolo (Project Neuroscience; Torino, Italy) and Comoli, Ferrari & SpA (Novara, Italy).
References
- 1.Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res 2005; 81:302-13; PMID:15954124; http://dx.doi.org/ 10.1002/jnr.20562 [DOI] [PubMed] [Google Scholar]
- 2.McGeer PL, McGeer EG. Glial reactions in Parkinson's disease. Mov Disord 2008; 23:474-83; PMID:18044695; http://dx.doi.org/ 10.1002/mds.21751 [DOI] [PubMed] [Google Scholar]
- 3.Rappold PM, Tieu K. Astrocytes and therapeutics for Parkinson's disease. Neurotherapeutics 2010; 7:413-23; PMID:20880505; http://dx.doi.org/ 10.1016/j.nurt.2010.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vila M, Jackson-Lewis V, Guegan C, Wu DC, Teismann P, Choi DK, Tieu K, Przedborski S. The role of glial cells in Parkinson's disease. Curr Opin Neurol 2001; 14:483-9; PMID:11470965; http://dx.doi.org/ 10.1097/00019052-200108000-00009 [DOI] [PubMed] [Google Scholar]
- 5.Le W, Rowe D, Xie W, Ortiz I, He Y, Appel SH. Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson's disease. J Neurosci 2001; 21:8447-55; PMID:11606633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inyushin MY, Huertas A, Kucheryavykh YV, Kucheryavykh LY, Tsydzik V, Sanabria P, Eaton MJ, Skatchkov SN, Rojas LV, Wessinger WD. L-DOPA Uptake in Astrocytic Endfeet Enwrapping Blood Vessels in Rat Brain. Parkinsons Dis 2012; 2012:321406; PMID:22888467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meiser J, Weindl D, Hiller K. Complexity of dopamine metabolism. Cell Commun Signal 2013; 11:34; PMID:23683503; http://dx.doi.org/ 10.1186/1478-811X-11-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 2011; 14:724-38; PMID:22152301; http://dx.doi.org/ 10.1016/j.cmet.2011.08.016 [DOI] [PubMed] [Google Scholar]
- 9.Gates MA, Dunnett SB. The influence of astrocytes on the development, regeneration and reconstruction of the nigrostriatal dopamine system. Restor Neurol Neurosci 2001; 19:67-83; PMID:12082230 [PubMed] [Google Scholar]
- 10.Janda E, Visalli V, Colica C, Aprigliano S, Musolino V, Vadala N, Muscoli C, Sacco I, Iannone M, Rotiroti D, et al.. The protective effect of tianeptine on Gp120-induced apoptosis in astroglial cells: role of GS and NOS, and NF-kappaB suppression. Br J Pharmacol 2011; 164:1590-9; PMID:21175585; http://dx.doi.org/ 10.1111/j.1476-5381.2010.01172.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desagher S, Glowinski J, Premont J. Astrocytes protect neurons from hydrogen peroxide toxicity. J Neurosci 1996; 16:2553-62; PMID:8786431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mena MA, Casarejos MJ, Carazo A, Paino CL, Garcia de Yebenes J. Glia conditioned medium protects fetal rat midbrain neurones in culture from L-DOPA toxicity. Neuroreport 1996; 7:441-5; PMID:8730801; http://dx.doi.org/ 10.1097/00001756-199601310-00016 [DOI] [PubMed] [Google Scholar]
- 13.Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 2003; 23:3394-406; PMID:12716947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devenish RJ, Klionsky DJ. Autophagy: mechanism and physiological relevance ‘brewed’ from yeast studies. Front Biosci (Schol Ed) 2012; 4:1354-63; PMID:22652877; http://dx.doi.org/ 10.2741/S337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 2011; 8:108-17; PMID:22187000; http://dx.doi.org/ 10.1038/nrneurol.2011.200 [DOI] [PubMed] [Google Scholar]
- 16.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931-7; PMID:17712358; http://dx.doi.org/ 10.1038/nrm2245 [DOI] [PubMed] [Google Scholar]
- 17.Janda E, Isidoro C, Carresi C, Mollace V. Defective autophagy in Parkinson's disease: role of oxidative stress. Mol Neurobiol 2012; 46:639-61; PMID:22899187; http://dx.doi.org/ 10.1007/s12035-012-8318-1 [DOI] [PubMed] [Google Scholar]
- 18.Jiang P, Mizushima N. Autophagy and human diseases. Cell Res 2014; 24:69-79; PMID:24323045; http://dx.doi.org/ 10.1038/cr.2013.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braak H, Sastre M, Del Tredici K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson's disease. Acta Neuropathol 2007; 114:231-41; PMID:17576580; http://dx.doi.org/ 10.1007/s00401-007-0244-3 [DOI] [PubMed] [Google Scholar]
- 20.Wakabayashi K, Hayashi S, Yoshimoto M, Kudo H, Takahashi H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson's disease brains. Acta Neuropathol 2000; 99:14-20; PMID:10651022; http://dx.doi.org/ 10.1007/PL00007400 [DOI] [PubMed] [Google Scholar]
- 21.Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 2002; 160:255-63; PMID:11786419; http://dx.doi.org/ 10.1016/S0002-9440(10)64369-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arai T, Nonaka T, Hasegawa M, Akiyama H, Yoshida M, Hashizume Y, Tsuchiya K, Oda T, Ikeda K. Neuronal and glial inclusions in frontotemporal dementia with or without motor neuron disease are immunopositive for p62. Neurosci Lett 2003; 342:41-4; PMID:12727313; http://dx.doi.org/ 10.1016/S0304-3940(03)00216-7 [DOI] [PubMed] [Google Scholar]
- 23.Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 2001; 12:2085-90; PMID:11447312; http://dx.doi.org/ 10.1097/00001756-200107200-00009 [DOI] [PubMed] [Google Scholar]
- 24.Cheung ZH, Ip NY. Autophagy deregulation in neurodegenerative diseases - recent advances and future perspectives. J Neurochem 2011; 118:317-25; PMID:21599666; http://dx.doi.org/ 10.1111/j.1471-4159.2011.07314.x [DOI] [PubMed] [Google Scholar]
- 25.Dagda RK, Das Banerjee T, Janda E. How Parkinsonian toxins dysregulate the autophagy machinery. Int J Mol Sci 2013; 14:22163-89; PMID:24217228; http://dx.doi.org/ 10.3390/ijms141122163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Episcopo FL, Tirolo C, Testa N, Caniglia S, Morale MC, Marchetti B. Reactive astrocytes are key players in nigrostriatal dopaminergic neurorepair in the MPTP mouse model of Parkinson's disease: focus on endogenous neurorestoration. Curr Aging Sci 2013; 6:45-55; PMID:23895521; http://dx.doi.org/ 10.2174/1874609811306010007 [DOI] [PubMed] [Google Scholar]
- 27.Gupta K, Patani R, Baxter P, Serio A, Story D, Tsujita T, Hayes JD, Pedersen RA, Hardingham GE, Chandran S. Human embryonic stem cell derived astrocytes mediate non-cell-autonomous neuroprotection through endogenous and drug-induced mechanisms. Cell Death Differ 2012; 19:779-87; PMID:22095276; http://dx.doi.org/ 10.1038/cdd.2011.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokoyama H, Uchida H, Kuroiwa H, Kasahara J, Araki T. Role of glial cells in neurotoxin-induced animal models of Parkinson's disease. Neurol Sci 2011; 32:1-7; PMID:21107876; http://dx.doi.org/ 10.1007/s10072-010-0424-0 [DOI] [PubMed] [Google Scholar]
- 29.Duty S, Jenner P. Animal models of Parkinson's disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol 2011; 164:1357-91; PMID:21486284; http://dx.doi.org/ 10.1111/j.1476-5381.2011.01426.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wills J, Credle J, Oaks AW, Duka V, Lee JH, Jones J, Sidhu A. Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS One 2012; 7:e30745; PMID:22292029; http://dx.doi.org/ 10.1371/journal.pone.0030745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drolet RE, Cannon JR, Montero L, Greenamyre JT. Chronic rotenone exposure reproduces Parkinson's disease gastrointestinal neuropathology. Neurobiol Dis 2009; 36:96-102; PMID:19595768; http://dx.doi.org/ 10.1016/j.nbd.2009.06.017 [DOI] [PubMed] [Google Scholar]
- 32.Dranka BP, Zielonka J, Kanthasamy AG, Kalyanaraman B. Alterations in bioenergetic function induced by Parkinson's disease mimetic compounds: lack of correlation with superoxide generation. J Neurochem 2012; 122:941-51; PMID:22708893; http://dx.doi.org/ 10.1111/j.1471-4159.2012.07836.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drechsel DA, Patel M. Differential contribution of the mitochondrial respiratory chain complexes to reactive oxygen species production by redox cycling agents implicated in parkinsonism. Toxicol Sci 2009; 112:427-34; PMID:19767442; http://dx.doi.org/ 10.1093/toxsci/kfp223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cristovao AC, Choi DH, Baltazar G, Beal MF, Kim YS. The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid Redox Signal 2009; 11:2105-18; PMID:19450058; http://dx.doi.org/ 10.1089/ars.2009.2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castello PR, Drechsel DA, Patel M. Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J Biol Chem 2007; 282:14186-93; PMID:17389593; http://dx.doi.org/ 10.1074/jbc.M700827200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janda E, Parafati M, Aprigliano S, Carresi C, Visalli V, Sacco I, Ventrice D, Mega T, Vadala N, Rinaldi S, et al.. The antidote effect of quinone oxidoreductase 2 inhibitor against paraquat-induced toxicity in vitro and in vivo. Br J Pharmacol 2013; 168:46-59; PMID:22289031; http://dx.doi.org/ 10.1111/j.1476-5381.2012.01870.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vella F, Ferry G, Delagrange P, Boutin JA. NRH:quinone reductase 2: an enzyme of surprises and mysteries. Biochem Pharmacol 2005; 71:1-12; PMID:16253210; http://dx.doi.org/ 10.1016/j.bcp.2005.09.019 [DOI] [PubMed] [Google Scholar]
- 38.Castino R, Bellio N, Follo C, Murphy D, Isidoro C. Inhibition of PI3k class III-dependent autophagy prevents apoptosis and necrosis by oxidative stress in dopaminergic neuroblastoma cells. Toxicol Sci 2010; 117:152-62; PMID:20525898; http://dx.doi.org/ 10.1093/toxsci/kfq170 [DOI] [PubMed] [Google Scholar]
- 39.Castino R, Fiorentino I, Cagnin M, Giovia A, Isidoro C. Chelation of lysosomal iron protects dopaminergic SH-SY5Y neuroblastoma cells from hydrogen peroxide toxicity by precluding autophagy and Akt dephosphorylation. Toxicol Sci 2011; 123:523-41; PMID:21742779; http://dx.doi.org/ 10.1093/toxsci/kfr179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Higgins GC, Devenish RJ, Beart PM, Nagley P. Autophagic activity in cortical neurons under acute oxidative stress directly contributes to cell death. Cell Mol Life Sci 2011; 68:3725-40; PMID:21437645; http://dx.doi.org/ 10.1007/s00018-011-0667-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong AS, Lee RH, Cheung AY, Yeung PK, Chung SK, Cheung ZH, Ip NY. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson's disease. Nat Cell Biol 2011; 13:568-79; PMID:21499257; http://dx.doi.org/ 10.1038/ncb2217 [DOI] [PubMed] [Google Scholar]
- 42.Geetha T, Vishwaprakash N, Sycheva M, Babu JR. Sequestosome 1/p62: across diseases. Biomarkers 2012; 17:99-103; PMID:22296116; http://dx.doi.org/ 10.3109/1354750X.2011.653986 [DOI] [PubMed] [Google Scholar]
- 43.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al.. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062-75; PMID:19524509; http://dx.doi.org/ 10.1016/j.cell.2009.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartlett RM, Holden JE, Nickles RJ, Murali D, Barbee DL, Barnhart TE, Christian BT, DeJesus OT. Paraquat is excluded by the blood brain barrier in rhesus macaque: An in vivo pet study. Brain Res 2009; 1259:74-9; PMID:19135428; http://dx.doi.org/ 10.1016/j.brainres.2008.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang A, Costello S, Cockburn M, Zhang X, Bronstein J, Ritz B. Parkinson's disease risk from ambient exposure to pesticides. Eur J Epidemiol 2011; 26:547-55; PMID:21505849; http://dx.doi.org/ 10.1007/s10654-011-9574-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Irace C, Scorziello A, Maffettone C, Pignataro G, Matrone C, Adornetto A, Santamaria R, Annunziato L, Colonna A. Divergent modulation of iron regulatory proteins and ferritin biosynthesis by hypoxia/reoxygenation in neurones and glial cells. J Neurochem 2005; 95:1321-31; PMID:16135072; http://dx.doi.org/ 10.1111/j.1471-4159.2005.03449.x [DOI] [PubMed] [Google Scholar]
- 48.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci 2007; 120:4155-66; PMID:18032788; http://dx.doi.org/ 10.1242/jcs.011163 [DOI] [PubMed] [Google Scholar]
- 49.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 2008; 15:171-82; PMID:17917680; http://dx.doi.org/ 10.1038/sj.cdd.4402233 [DOI] [PubMed] [Google Scholar]
- 50.Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol 2007; 170:75-86; PMID:17200184; http://dx.doi.org/ 10.2353/ajpath.2007.060524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/ 10.1016/j.molcel.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen SY, Chiu LY, Maa MC, Wang JS, Chien CL, Lin WW. zVAD-induced autophagic cell death requires c-Src-dependent ERK and JNK activation and reactive oxygen species generation. Autophagy 2011; 7:217-28; PMID:21127402; http://dx.doi.org/ 10.4161/auto.7.2.14212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corcelle E, Nebout M, Bekri S, Gauthier N, Hofman P, Poujeol P, Fenichel P, Mograbi B. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res 2006; 66:6861-70; PMID:16818664; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3557 [DOI] [PubMed] [Google Scholar]
- 54.Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci 2010; 30:12535-44; PMID:20844148; http://dx.doi.org/ 10.1523/JNEUROSCI.1920-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal 2010; 13:1713-48; PMID:20446772; http://dx.doi.org/ 10.1089/ars.2010.3221 [DOI] [PubMed] [Google Scholar]
- 56.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al.. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010; 12:213-23; PMID:20173742 [DOI] [PubMed] [Google Scholar]
- 57.Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 2010; 30:3275-85; PMID:20421418; http://dx.doi.org/ 10.1128/MCB.00248-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 2010; 285:22576-91; PMID:20452972; http://dx.doi.org/ 10.1074/jbc.M110.118976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Riley BE, Kaiser SE, Shaler TA, Ng AC, Hara T, Hipp MS, Lage K, Xavier RJ, Ryu KY, Taguchi K, et al.. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol 2010; 191:537-52; PMID:21041446; http://dx.doi.org/ 10.1083/jcb.201005012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castino R, Lazzeri G, Lenzi P, Bellio N, Follo C, Ferrucci M, Fornai F, Isidoro C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J Neurochem 2008; 106:1426-39; PMID:18489716; http://dx.doi.org/ 10.1111/j.1471-4159.2008.05488.x [DOI] [PubMed] [Google Scholar]
- 61.Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 2009; 16:1040-52; PMID:19407826; http://dx.doi.org/ 10.1038/cdd.2009.49 [DOI] [PubMed] [Google Scholar]
- 62.Gonzalez-Polo R, Niso-Santano M, Moran JM, Ortiz-Ortiz MA, Bravo-San Pedro JM, Soler G, Fuentes JM. Silencing DJ-1 reveals its contribution in paraquat-induced autophagy. J Neurochem 2009; 109:889-98; PMID:19425177; http://dx.doi.org/ 10.1111/j.1471-4159.2009.06020.x [DOI] [PubMed] [Google Scholar]
- 63.Gonzalez-Polo RA, Niso-Santano M, Ortiz-Ortiz MA, Gomez-Martin A, Moran JM, Garcia-Rubio L, Francisco-Morcillo J, Zaragoza C, Soler G, Fuentes JM. Inhibition of paraquat-induced autophagy accelerates the apoptotic cell death in neuroblastoma SH-SY5Y cells. Toxicol Sci 2007; 97:448-58; PMID:17341480; http://dx.doi.org/ 10.1093/toxsci/kfm040 [DOI] [PubMed] [Google Scholar]
- 64.Milton VJ, Jarrett HE, Gowers K, Chalak S, Briggs L, Robinson IM, Sweeney ST. Oxidative stress induces overgrowth of the Drosophila neuromuscular junction. Proc Natl Acad Sci U S A 2011; 108:17521-6; PMID:21987827; http://dx.doi.org/ 10.1073/pnas.1014511108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J 2007; 26:1749-60; PMID:17347651; http://dx.doi.org/ 10.1038/sj.emboj.7601623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell Death Differ 2009; 16:31-8; PMID:19008922; http://dx.doi.org/ 10.1038/cdd.2008.163 [DOI] [PubMed] [Google Scholar]
- 67.Underwood BR, Imarisio S, Fleming A, Rose C, Krishna G, Heard P, Quick M, Korolchuk VI, Renna M, Sarkar S, et al.. Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum Mol Genet 2010; 19:3413-29; PMID:20566712; http://dx.doi.org/ 10.1093/hmg/ddq253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boya P. Lysosomal function and dysfunction: mechanism and disease. Antioxid Redox Signal 2012; 17:766-74; PMID:22098160; http://dx.doi.org/ 10.1089/ars.2011.4405 [DOI] [PubMed] [Google Scholar]
- 69.Walls KC, Ghosh AP, Franklin AV, Klocke BJ, Ballestas M, Shacka JJ, Zhang J, Roth KA. Lysosome dysfunction triggers Atg7-dependent neural apoptosis. J Biol Chem 2010; 285:10497-507; PMID:20123985; http://dx.doi.org/ 10.1074/jbc.M110.103747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mader BJ, Pivtoraiko VN, Flippo HM, Klocke BJ, Roth KA, Mangieri LR, Shacka JJ. Rotenone inhibits autophagic flux prior to inducing cell death. ACS Chem Neurosci 2012; 3:1063-72; PMID:23259041; http://dx.doi.org/ 10.1021/cn300145z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jiang P, Huang Z, Zhao H, Wei T. Hydrogen peroxide impairs autophagic flux in a cell model of nonalcoholic fatty liver disease. Biochem Biophys Res Commun 2013; 433:408-14; PMID:23537653; http://dx.doi.org/ 10.1016/j.bbrc.2013.02.118 [DOI] [PubMed] [Google Scholar]
- 72.Oh JM, Choi EK, Carp RI, Kim YS. Oxidative stress impairs autophagic flux in prion protein-deficient hippocampal cells. Autophagy 2012; 8:1448-61; PMID:22889724; http://dx.doi.org/ 10.4161/auto.21164 [DOI] [PubMed] [Google Scholar]
- 73.Manning-Bog AB, McCormack AL, Purisai MG, Bolin LM, Di Monte DA. Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. J Neurosci 2003; 23:3095-9; PMID:12716914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reybier K, Perio P, Ferry G, Bouajila J, Delagrange P, Boutin JA, Nepveu F. Insights into the redox cycle of human quinone reductase 2. Free Radic Res 2011; 45:1184-95; PMID:21762045; http://dx.doi.org/ 10.3109/10715762.2011.605788 [DOI] [PubMed] [Google Scholar]
- 75.Fu Y, Buryanovskyy L, Zhang Z. Quinone reductase 2 is a catechol quinone reductase. J Biol Chem 2008; 283:23829-35; PMID:18579530; http://dx.doi.org/ 10.1074/jbc.M801371200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Janda E, Palmieri C, Pisano A, Pontoriero M, Iaccino E, Falcone C, Fiume G, Gaspari M, Nevolo M, Di Salle E, et al.. Btk regulation in human and mouse B cells via protein kinase C phosphorylation of IBtkgamma. Blood 2011; 117:6520-31; PMID:21482705; http://dx.doi.org/ 10.1182/blood-2010-09-308080 [DOI] [PubMed] [Google Scholar]