

Abstract
Aging was recently described as a life event programmed by the hypothalamus, a key brain region that is crucial for the neuroendocrine interaction between the central nervous system and the periphery. Autophagy impairment is a hallmark of aging, contributing to the aging phenotype and to the aggravation of age-related diseases. Since hypothalamic autophagy decreases with age, strategies to promote autophagy in the hypothalamus may be relevant for control of the aging process. NPY (neuropeptide Y) is an endogenous neuropeptide mainly produced by the hypothalamus. We recently reported, for the first time, that NPY stimulates autophagy in rodent hypothalamus and mediates caloric restriction-induced autophagy in hypothalamic neurons. Moreover, we observed that NPY acts through NPY1R (neuropeptide Y receptor Y1) or NPY5R activation involving a concerted action of different signaling pathways. Since both hypothalamic autophagy and NPY levels decrease with age, modulation of NPY levels could provide new putative therapeutic tools to ameliorate age-related deteriorations and extend longevity.
Keywords: autophagy, caloric restriction, hypothalamus, neuropeptide Y
The hypothalamus, the brain region that regulates development, growth, and metabolism, has gained increased attention for its key role in the progression of whole-body aging. Aging is characterized by autophagy impairment, which leads to loss of cellular proteostasis. Moreover, autophagy mediates, in part, the beneficial effects of caloric restriction (CR) on life-span extension and delaying the progression of age-related diseases. At the same time, CR increases hypothalamic NPY levels, the front line neurotransmitter of the hypothalamic arcuate nucleus response to low energy availability. This observation, in addition to the fact that aging is controlled by the hypothalamus, and autophagy is impaired in the aged hypothalamus, raised the question, does NPY mediate CR-induced autophagy in the hypothalamus? Our study demonstrates that CR stimulates autophagic flux in hypothalamic neurons, and that NPY1R, NPY2R, or NPY5R antagonists block this effect. These results suggest that endogenous NPY mediates CR-induced autophagy in hypothalamic neurons. We then questioned the effect of NPY per se in autophagy regulation. We show that NPY not only induces autophagy, increasing the number of autophagosomes, as shown by the rapid accumulation of LC3B-II, but also increases autophagic flux in hypothalamic neurons. In both hypothalamic neuronal in vitro models, rat hypothalamic neural cell primary cultures, and a mouse hypothalamic cell line, NPY1R, NPY2R, or NPY5R antagonists inhibit the conversion of LC3B-I into LC3B-II, blocking therefore autophagy induction. However, while the inhibition of NPY1R or NPY5R blocked autophagy flux induced by NPY in both hypothalamic neuronal models, the blockage of NPY2R only prevents the autophagic flux induced by NPY in rat differentiated hypothalamic neural cells, a culture constituted mainly by neurons but also with other cell types, namely astrocytes and microglia. This observation suggests that NPY2R may be relevant for autophagy regulation in other hypothalamic neural cells. Although NPY2R is not involved in the increase of autophagic flux in mHypoN42 hypothalamic neurons, the activation of this receptor could contribute to the first step of autophagy induction. Further studies need to be performed to clarify the contribution of this receptor in NPY regulation of autophagy.
We then characterize the mechanisms downstream of NPY receptor activation. The inhibition of mechanistic target of rapamycin (serine/threonine kinase) complex 1 (MTORC1) is one of the molecular switches for autophagy induction. We observed that NPY does not alter the phosphorylation status of MTOR, or its substrate ribosomal protein S6 kinase, 70 kDa (RPS6KB), suggesting that NPY does not induce autophagy through the inhibition of MTORC1. This was not unexpected, given that MTOR signaling responds to nutrient availability, and autophagy regulation depends on the metabolic state of the cell and nutrient availability; as the cell culture models used are under nutrient-rich conditions, MTOR kinase is active and it is possible that NPY may induce autophagy through a MTOR-independent pathway. Indeed, other signaling pathways regulate autophagy in mammalian cells independent of canonical MTOR inhibition. We observed that the class I phosphoinositide 3-kinase (PI3K), MAPK1/ERK2-MAPK3/ERK1 and PKA signaling pathways mediate NPY-induced autophagy in hypothalamic neurons. An in-depth mechanistic analysis revealed that NPY1R activates both AKT and PKA signaling pathways in a PI3K-dependent manner, and NPY5R activation increases both MAPK1/3 and PKA phosphorylation. Moreover, PI3K or PKA inhibitors block NPY1R-induced autophagic flux whereas PI3K, MAPK1/3, or PKA inhibitors inhibit NPY5R-induced autophagic flux. Collectively, these results show that NPY induces autophagic flux through NPY1R and NPY5R activation, and this involves a concerted action of the PI3K, MAPK1/3, and PKA pathways (Fig. 1).
To support our data on the NPY effect on autophagy regulation, we set up an in vivo study where, by gene transfer technology, we overexpressed NPY in mouse hypothalamic arcuate nucleus. After 4 wk, we observed larger LC3B puncta accumulation in hypothalamic arcuate nucleus of hypothalamic NPY-overexpressing mice. Moreover, a decrease in the protein content of both LC3B-II and SQSTM1 in the hypothalamus of hypothalamic NPY-overexpressing mice was observed, indicating an increase of the autophagic flux.
Altogether, the results show that NPY induces autophagy stimulation in the hypothalamus. Since autophagy is a key mechanism underlying CR's beneficial effects and considering that it is difficult to implement and sustain a CR regimen in humans, increasing research interest has focused on identifying agents that can mimic the beneficial effects of CR. Increasing evidence, including our study, suggest that NPY is a putative CR mimetic candidate.
In addition, because aging is associated with reduced levels of NPY in the hypothalamus and a basal decreased autophagic activity, the stimulatory effect of NPY on autophagy may contribute to delay aging. Moreover, age-related hypothalamic autophagy impairment might contribute to the metabolic dysregulation observed with age, so it is tempting to speculate that autophagy stimulation by NPY in hypothalamic neurons could restore the metabolic homeostasis, and therefore, the role of hypothalamic NPY on food intake regulation might depend on autophagy activation. Conversely, as NPY has a well-established neuroprotective effect, autophagy activation by NPY could be a relevant potential strategy to investigate in neurodegenerative diseases characterized by impaired autophagy.
Figure 1.
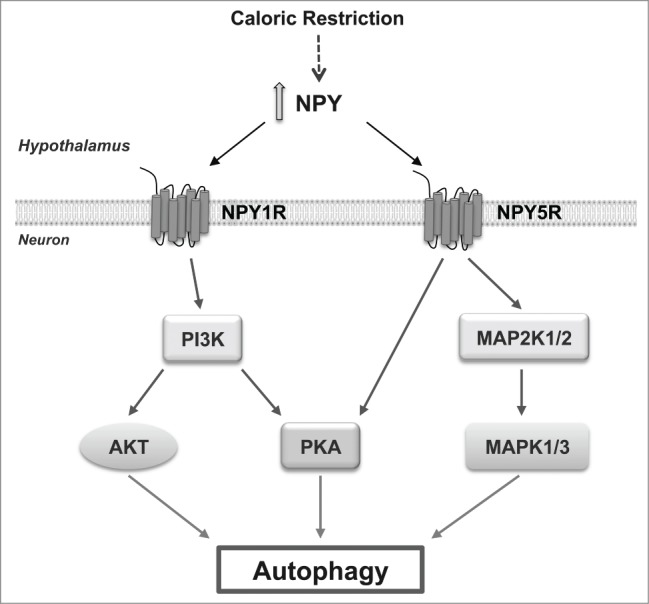
Caloric restriction increases NPY in the hypothalamus that in turn induces autophagy in hypothalamic neurons through NPY1R or NPY5R activation through a combination of intracellular pathways. NPY1R activates both AKT and PKA signaling pathways in a PI3K-dependent manner and NPY5R activation increases both MAPK/ERK and PKA phosphorylation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was funded by FEDER funds through the Operational Program Competitiveness Factors - COMPETE and national funds by FCT - Foundation for Science and Technology under the projects (PTDC/SAU-FCF/099082/2008, SFRH/BPD/73942/2010, SFRH/BD/73004/2010, SFRH/BPD/78424/2011; Projeto Mais Centro, “Aging, Stress And Chronic Diseases: From Mechanisms To Therapeutics” (CENTRO-07-ST24-FEDER-002006), and strategic project UID / NEU/04539 /2013.