

Abstract
All eukaryotic cells utilize autophagy for protein and organelle turnover, thus assuring subcellular quality control, homeostasis, and survival. In order to address recent advances in identification of human autophagy associated genes, and to describe autophagy on a system-wide level, we established an autophagy-centered gene interaction network by merging various primary data sets and by retrieving respective interaction data. The resulting network (‘AXAN’) was analyzed with respect to subnetworks, e.g. the prime gene subnetwork (including the core machinery, signaling pathways and autophagy receptors) and the transcription subnetwork. To describe aspects of evolution within this network, we assessed the presence of protein orthologs across 99 eukaryotic model organisms. We visualized evolutionary trends for prime gene categories and evolutionary tracks for selected AXAN genes. This analysis confirms the eukaryotic origin of autophagy core genes while it points to a diverse evolutionary history of autophagy receptors. Next, we used module identification to describe the functional anatomy of the network at the level of pathway modules. In addition to obvious pathways (e.g., lysosomal degradation, insulin signaling) our data unveil the existence of context-related modules such as Rho GTPase signaling. Last, we used a tripartite, image-based RNAi – screen to test candidate genes predicted to play a role in regulation of autophagy. We verified the Rho GTPase, CDC42, as a novel regulator of autophagy-related signaling. This study emphasizes the applicability of system-wide approaches to gain novel insights into a complex biological process and to describe the human autophagy pathway at a hitherto unprecedented level of detail.
Keywords: autophagy, CDC42, Cytoscape, evolution, network biology, systems biology
Abbreviations
- 3MA
3-methyladenine
- ATG
autophagy related
- AXAN
Annotated eXpanded Autophagy Network
- cAXAN
complemented Annotated eXpanded Autophagy Network
- Cvt
cytoplasm-to-vacuole targeting
- GDP
guanosine-diphosphate
- GTP
guanosine-triphosphate
- NB
neighbor
- RNAi
RNA interference
- TFs
transcription factors.
Introduction
Autophagy represents a fundamental quality control mechanism by which unwanted cytosolic components are delivered to the lytic compartment (i.e., lysosomes or the vacuole) for degradation and recycling of its constituents. Besides its pivotal function under physiological conditions (e.g., facilitating cellular homeostasis of stem cells),1 autophagy has been recognized as a decisive pathway for various human diseases such as neurodegeneration, cancer and inflammatory diseases, as well as the aging process.2,3 Underscoring the emerging interest in autophagy as a therapeutic target mechanism, several clinical trials are currently exploring the applicability of autophagy modulation in treatment of cancers (e.g., glioblastoma).4,5
Extensive research using unicellular yeasts and other model systems has led to the identification of several genes that are required for distinct steps of macroautophagy (hereafter called autophagy), from initiation and expansion of the phagophore membrane, to maturation of the resulting autophagosome, to fusion with the lysosomes (in metazoan cells) or the vacuole (in yeasts and plants). One particular group of evolutionarily ancient genes whose members are required for virtually all forms of autophagy and its subtypes is commonly called the “core machinery.”6,7 Examples of autophagy core proteins are ULK1 (a human ortholog of yeast Atg1), members of the GABARAP/MAP1LC3 family (mammalian orthologs of yeast Atg8) and representatives of the ATG4 family.8 Besides these core proteins, several signaling pathways contribute to autophagy regulation, e.g., the central MTOR pathway (which integrates most of the upstream signals), class I phosphoinositide 3-kinase (PI3K) and class III phosphatidylinositol 3-kinase (PtdIns3K), mitogen-activated protein kinases (MAPKs), and the TP53/p53 (TRP53 in mice and rats) pathway. These signaling pathways respond to external or internal stress stimuli such as amino acid depletion, limitation in growth factors, or DNA damage, and regulate autophagy by various steps of phosphorylation, protein processing, and membrane remodeling events. Another layer of regulation is represented by transcriptional regulation of key autophagy genes, e.g., by the essential transcription factor TFEB.9,10 An emerging concept is that the autophagy machinery can act nonselectively by randomly targeting bulk cytosol, but also in a tightly controlled selective mode, enabling specific degradation of damaged mitochondria, surplus peroxisomes, toxic protein aggregates, or intracellular pathogens.11-14 Selective autophagy is mediated by a group of proteins (‘autophagy receptors’), which recognize molecular tags (such as ubiquitination, phosphorylation, or acetylation) on target compartments and form the molecular link between these targets and the autophagy core machinery. Examples for autophagy receptors are Pichia pastoris Atg30 (PpAtg30) and Atg32 in yeast, and SQSTM1/p62, CALCOCO2/NDP52, as well as NBR1 in higher eukaryotes.14-16 While the vast majority of genes within the autophagy core category and most of the signaling pathways are generally believed to be evolutionarily conserved across most eukaryotic species, the evolutionary origin of autophagy receptors appears to be more complex.17 A comparative analysis of the evolutionary trends shaping different functional aspects of general and selective autophagy has not been performed yet.
Recent large scale experimental and systems biology approaches have resulted in the identification of various human genes contributing to different aspects of autophagy and its selective subtypes, thus expanding the molecular framework beyond the well-characterized core machinery and signaling pathways.18-23 While the results of these approaches hold the promise to uncover the complexity of the autophagy process on a global scale, there is very little overlap of genes found using different systems-biology strategies to date, suggesting that a more global analysis is necessary. To address this point, the work presented here describes a gene interaction network, which was constructed by combined analysis of these primary data sets and by manual curation of relevant literature. The resulting interaction network was used to describe evolutionary aspects of autophagy genes with special focus on the prime gene categories. Furthermore, we analyzed the functional architecture of the network regarding regulatory subnetworks and functional modules, and finally used network predictions to identify novel genes regulating aspects of autophagy (see flow diagram of study design, Figure S1A and Tables S1–S12).
Results
Overview of establishing AXAN
As a starting point for a global analysis of human autophagy-associated genes, we compared the primary data sets from 6 recent large-scale experimental or systems biology-based approaches.18-23 It is important to note that these source studies had different objectives and also applied different approaches (Table S1): Study I, biochemical screen for interactors of candidate proteins;19 study II, text parsing for autophagy- and lysosome-related genes;20 study III, GFP-LC3-based siRNA screen under normal nutrient conditions;18 study IV, image-based siRNA screen for selective autophagy types (virophagy, mitophagy);21 study V, siRNA screen under amino acid starvation conditions;22 study VI, genetic screen in Caenorhabditis elegans for removal of autophagy targets.23 In total, 3 studies (III, IV and V) used RNAi screening but different cultivation conditions as a discovery platform; one study (I) applied pull down of protein complexes and one (VI) performed a genetic screen in model organism C. elegans. Study II used automated text parsing of published data to analyze genes of the autophagy-lysosomal pathway and thus recapitulated data sets published before. Study VI represented a special case in that the actual genetic screen was performed in C.elegans while only the mammalian ortholog genes (n=3) of confirmed hits were further assessed experimentally. Similarly, study V described a multistep siRNA screening procedure and for functional analysis excluded known autophagy genes contained in the validated hit gene set, thus focusing on 9 novel human autophagy-associated genes. As expected, this diversity is reflected in the level of overlap of genes described in the source studies. After mapping of gene IDs, the merging of the gene sets resulted in a total set of 1170 human genes (Table S2). Study V does not have any intersection with other sources, and study VI shares one (out of 3) human genes (VMP1) with study II. 79 genes were shared between 2 analyses in each case, and 4 genes were shared by 3 studies: ATG5, ATG7, PRKAA2 (Studies I, II &III) and ATG13 (Studies I, II & IV). No common gene was found in more than 3 studies. The highest overlap was noted for studies I and II (52 genes within the binary intersection I & II) which may be explained by the text mining approach applied in study II. Intriguingly, the 2 siRNA screens studies III and IV showed an overlap of 5 genes (HILPDA, SLC25A19, FANCC, CHAF1B, and FABP1), which did not overlap with those predicted by any other analysis. The overlap of 2 studies contained 7 genes each for the intersections I & III, II & III and I & IV, and 4 genes for the intersection II & IV. The Euler diagram in Figure S1B illustrates the level of overlap between these primary gene sets. With the exception of studies V and VI (which contain relatively small number of genes as compared to the other studies) the overlap of genes shared between the data sets was higher than expected by chance. (For example, a total of 56 genes are shared by I and II while 9 would have been expected by random selection out of 20,000 human genes). In summary, this analysis emphasizes 2 main points: First, given the higher-than-random overlap between individual source data sets, the various studies seem to capture elements of a complex system with underlying biological commonality. Second, with respect to the individual functional context of each study, this merged data set can be regarded as a framework for several functional subnetworks each describing different aspects of autophagy.
Complementation of this initial gene collection with individual genes from recent literature (52 genes, Table S2) expanded the final gene collection to 1222 human genes. Within this gene set, we next defined “prime gene” categories to address different functional aspects of autophagy regulation and execution. For the purposes of the present study, 38 genes were defined as autophagy ‘core’ genes (required for virtually all subtypes of autophagy), 56 genes as autophagy ‘signaling’ molecules (mediating signal function to regulate autophagy), 9 genes as autophagy ‘receptors’ (physically linking targets of selective autophagy to the core machinery) and 5 genes as autophagy ‘mediators’ (modulating autophagy regulation under certain conditions), representing a set of 108 human ‘autophagy prime genes’ (Table S3). Notably, all 4 genes that were shared by 3 source studies (see above) represent prime genes (core, ATG5, ATG7, ATG13; and signaling, PRKAA2), thus further supporting our conceptual approach.
Next, we integrated respective interactions for all these genes, resulting in assembly of the Annotated eXpanded Autophagy Network (AXAN) with 1222 genes (nodes of the network) that are connected by 5835 interactions [edges of the network; protein-protein interactions (ppi) n = 5470 (medium confidence n = 3793; high confidence n = 1677); transcriptional regulation (transreg): n = 365, Fig. 1A and B, Table S4A]. Ten genes display pairwise interactions that are not connected to the main network (C5AR1-C5, AMH-ARL8B, TMEM39B-IL13RA1, HPS1-HPS4, CAPN7-IST1) and 236 genes represent single nodes (not connected to any other node), including the prime genes ATG4D, DRAM1, PIK3R6 and DAPK2. Intriguingly, AXAN contains almost 10 times more medium and high confidence ppi-edges than random gene sets of similar size within the entire human interactome (Table S4B). Moreover, comparison of the clustering coefficient between AXAN, 3 random networks of comparable size, and 3 randomized versions of AXAN confirmed that our network behaves in nonrandom fashion (Fig. 1C), further arguing for its role as functional biological entity. Next, we aimed to analyze cell-type specific expression of AXAN genes using the current built of the Human Protein Atlas (HPA).24 While the entire AXAN gene set did not show significant differences regarding gene expression as compared to a control gene set (not shown), AXAN prime genes displayed significant overrepresentation in several cell types (Fig. 1D, and Table S5A and B). The difference was most pronounced for cells from the germinal center, neuronal cells and macrophages, and was observable but below our significance threshold for hepatocytes and smooth muscle cells. This analysis emphasizes the pivotal role of the autophagy system in various tissues and cell types, especially in neural cells and the immune system.
Figure 1.
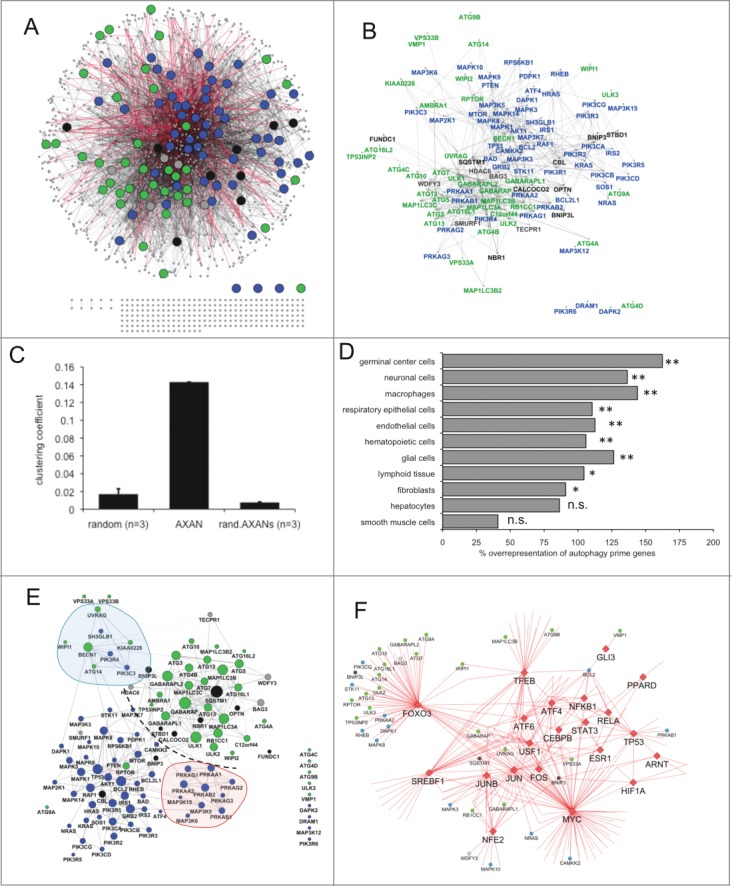
Overview of AXAN and its subnetworks. (A, B) Overview of the Annotated eXpanded Autophagy Network (AXAN) displaying autophagy core (green), signaling (blue), receptors (black) and mediators (gray), as well as additional genes (small gray circles). Gray edges: protein-protein interactions (light: medium confidence; dark: high confidence); red edges: transcriptional regulation. Singleton genes and pairwise gene interactions are displayed at the bottom. For simplicity, only edges between prime genes are displayed in (B). (C) Quantitative network parameters of AXAN, randomized versions of AXAN (rand.AXANs) and 3 random human networks of similar size. (D) Cell type-specific expression of autophagy prime genes. The bar graph displays the percentage over-representation of prime genes expressed in given cell types as compared to random nonautophagy genes. *, P < 0.05; **, P < 0.01; n.s., not significant. (E) Prime gene interaction network. All AXAN prime genes and their interactions are displayed. Node size corresponds to the degree (number of neighbors). The degree distribution across this network and the layout algorithm (spring embedded layout, weighted by edge betweenness) undescores the existence of subregions: Dotted line, spatial separation between autophagy core and signaling domains; red area, protein kinase A subregion; blue, BECN1-UVRAG subregion. (F) The transcription subnetwork within AXAN. Transcription factors (red diamonds) and their target genes within AXAN are displayed. Autophagy prime genes (large circles) are marked by color-coding as described above. Non-prime target genes within AXAN are displayed as small circles.
Subnetworks
Using prime gene categories and other functional aspects intrinsic to the network, we next aimed to describe subnetworks within AXAN. Figure 1E visualizes the interactions of all autophagy prime genes. The layout chosen to display this subnetwork (spring embedded, weighted by edge betweenness) emphasizes the architecture of this subnetwork and points to the physical separation of subregions containing core genes (upper right region) and signaling genes (lower left region). Among the core genes, the 7 members of the superfamily of mammalian orthologs of yeast ATG8 localize to the center of the region and display high degree values, emphasizing their prominent role in the autophagy process (Fig. 1E, Fig. S2A and B). Signaling genes tend to separate by pathways, e.g. the PRKA/protein kinase A subregion and the BECN1-UVRAG subregion. While some autophagy receptors (FUNDC1, BNIP3, BNIP3L, OPTN, STBD1) tend to localize to the periphery of the subnetworks due to limited number of edges and low connectivity, the prototypic receptor SQSTM1 localizes to the very center of the core subnetwork, thus underscoring its pleiotropic and prominent functional role as a molecular key player of the autophagy pathway and integration into various regulatory events. (See the supplemental materials for additional aspects of prime gene interactions: Fig. S2A and B display the autophagy core interaction subnetwork [A] and a subnetwork of autophagy receptors and their first-degree neighbors [B].)
Next, we visualized the transcription subnetwork within AXAN. We identified a total of 23 transcription factors (TFs) belonging to various families: These include members of the AP1, ATF, HIF, NFKB, TP53/p53, STAT, GLI, PPAR, and FOXO/forkhead families. Based on publicly available data, 20 of these 23 TFs regulate transcription of 204 target genes in total (see also Table S6). STAT2 (a member of the STAT family), TP73 (TP53/p53 family) and FOS (AP1 family) do not have known target genes within this network. Figure 1F displays the architecture of the AXAN transcription subnetwork. The TFs MYC, FOXO3, and TFEB are the factors with the highest number of AXAN target genes (83, 63, and 31, respectively) but among these 3 the fraction of autophagy prime genes is highest for FOXO3 (35%) and lowest for MYC (10%). Strikingly, the AP1 family factors JUN and JUNB both show a high percentage of prime genes among their targets (28% and 25%, respectively), even exceeding the percentage of TFEB (20%) that is usually considered as the key transcriptional regulator of lysosomal biogenesis and autophagy. Besides uncovering well established regulatory relationships (TFEB, FOXO3), our data set emphasizes the role of other TF groups for autophagy regulation that are related to immunomodulatory and proinflammatory pathways, such as the NFKB/NF-κB pathway (represented by NFKB1 and RELA), downstream components of the MAPK cascade (JUN, JUNB and FOS), as well as the JAK-STAT pathway (STAT3). These findings point to the recently established connection between autophagy induction and regulation of the immune system.
Assessment of evolutionary conservation of prime gene categories within AXAN
In order to describe evolutionary aspects of autophagy on a global scale, we next aimed to perform a systematic evolutionary assessment of prime gene categories and of individual prime genes. Using the InParanoid7 database for protein ortholog identification, we assessed the presence or absence of orthologs for AXAN prime genes in 100 model organisms ranging from Escherichia coli (as the prokaryotic outgroup) to Homo sapiens. The percentage of human genes having an ortholog in 2 or more species within higher order taxonomic groups was plotted for autophagy core, signaling, and receptor genes within our autophagy network to demonstrate evolutionary trends. Two thousand randomly chosen human genes served as control group. As shown in Figure 2A, approximately 90% of the core genes are conserved across all eukaryotic species analyzed in this study. TP53INP2, ATG13, and ATG14 show strong evidence for an origin during metazoan radiation, and ATG9B is restricted to the taxon Homo sapiens (see Table S7). Moreover, signaling genes are less ancient than the core genes, but more ancient than random human genes. While genes of the MAPK pathways exhibit deep conservation (with an estimated origin in the last common ancestor of all eukaryotes at least 2 billion years ago), other signaling pathway members, such as those modulated by the BCL2 family, are less conserved, likely originating in parallel with the rise of multicellularity in the last common ancestor of all Eumetazoans 650 million y ago based on these data. In contrast to core and signaling genes, autophagy receptors on average display a rather low conservation in ancient taxa, with the majority of these genes appearing to have evolved in the course of Eumetazoan evolution (estimated 650 million y ago; Fig. 2B and Fig. S4, and Table S7). In particular, only 2 of these genes (NBR1 and FUNDC1) show direct orthologs in metazoans or older taxa, while the other autophagy receptors seem to be restricted to Eumetazoans and more recent groups. SQSTM1 and CBL show at least weak evidence for older evolutionary origin. Interestingly, orthologs of STBD1 are restricted to the group of Theria within the phylum chordata and the class mammalia (Fig. 2B and Fig. S4). Figure 2C and D display core gene ATG4B and the autophagy receptor for glycogen metabolism, STBD1, as examples for ortholog tracking across all model species and taxonomic groups used within this study. As depicted by color coding, ATG4B has orthologs across the entire eukaryotic domain, while STBD1 is explicitly restricted to the subclass Theria within the class of mammals. (Details about the conservation of exemplary prime genes across all 100 model organisms can be found in Fig. S4 and S5). Importantly, none of the autophagy prime genes show orthologs in E. coli (data not shown), supporting the notion of an evolutionary origin of the autophagy pathway in the last common ancestor of all eukaryotic cells that is estimated to have evolved more than 2 billion y ago.
Figure 2.
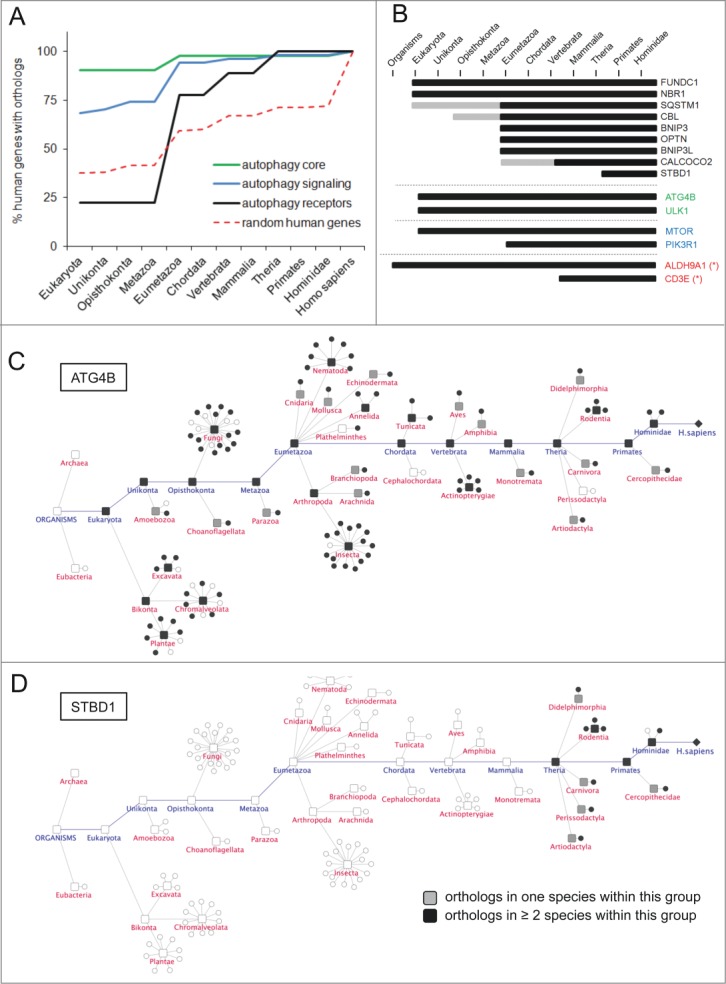
Evolutionary trends of AXAN prime genes. (A) The evolutionary conservation of human genes from functional categories within AXAN was assessed by ortholog identification in 99 eukaryotic model organisms (and Escherichia coli as outgroup) using the InParanoid7 database. Evolutionary trends were displayed as the percentage of conserved human genes in respective phylogenetic groups. Two thousand randomly chosen human genes served as control (red dotted line). (B) Evolutionary conservation of all 9 human autophagy receptors, 2 core genes, 2 signaling genes and 2 non-AXAN control genes (housekeeping enzyme ALDH9A1 and T cell receptor gene CD3E, marked by asterisks). Gray bars, ortholog in one species within this group; black bars, orthologs in at least 2 species within this group. (C and D) Evolutionary tracks of 2 exemplary AXAN genes (core gene ATG4B and receptor STBD1). Circles represent species, and boxes represent higher order taxonomic groups containing these species. Presence of an ortholog in a given species is visualized by black symbols. For higher groups, coloring displays presence of orthologs in one (gray) or at least 2 (black) species within this group. As examples, the highly conserved autophagy core gene, ATG4B, and the autophagy receptor gene, STBD1, (the most recent autophagy receptor) are displayed.
Analysis of the functional architecture of AXAN
As a next step toward global description of AXAN, we performed pathway-based module identification. Among all AXAN genes, 773 (63%) were found within the Reactome database to be associated with a subcellular pathway. After importing respective pathway interactions, GLay community clustering was performed to identify individual highly intraconnected modules. As shown in Figure 3A and B, this approach identified 21 pathway modules with node numbers ranging from 2 to 145 (Table S8 and Fig. S6). These modules represent biological entities at the process or pathway level, which in total make up the functional architecture of the autophagy network. Several of these pathway modules were expected to be present in AXAN, including those tagged by the lead terms ‘lysosome and antigen presentation’ (module M03), 'NFKB/NF-κB & MAPKs’ (M02), ‘cell stress and cell cycle’ (M04), ‘metabolic pathways’ (M11) and the prime gene enriched modules M05 and M06. Module M05 (Fig. 3C) represents the module with the highest enrichment of autophagy core genes (19 / 53 = 36% compared to 28 / 766 = 4% in all Reactome modules combined) and consequently exhibits ‘autophagy’ as the highest enriched pathway. Interestingly, it contains several smaller submodules (accentuated by red dotted lines; Fig. 3C), i.e., those containing VPS genes (involved in vacuolar protein sorting), small GTPases of the Rab family and their regulators (GDI1/2), and the HOOK gene submodule (involved in endocytic membrane trafficking and microtubule association). Module M06 also contains a high number of core genes (10%), but is mainly enriched in signaling components of the INS/insulin and MTOR signaling pathways (‘signaling’ prime genes: 18% compared to 7% in all modules combined; Fig 3D). It is noteworthy that these 2 modules (M05, M06) also harbor those 4 genes that are located in the intersection of 3 source studies. Intriguingly, our approach identified several pathway modules that were not expected to be part of AXAN. These include those tagged by lead terms ‘extracellular matrix (ECM) organization’ (M12), ‘nicotinamide metabolism’ (M10), and ‘RNA processing’ (M07). Moreover, there are several small modules (node number = 3) whose genes are not directly connected to any other module by Reactome functional interactions. These include modules characterized by the lead terms ‘secretion and absorption’ (M20), ‘fatty acid metabolism’ (M18) and ‘peroxisome’ (M16). Intriguingly, some modules appear to be linked to a specific biological context as suggested by the source studies their defining genes were identified in: While core modules such as M05 (‘autophagy’) or M03 (‘lysosome’) display contribution of source studies as expected by chance, other modules are dominated by or depleted of genes linked to a specific context (Table S8A). For example, genes from study V (siRNA screen under amino acid starvation conditions) are enriched in modules M04 (‘cell stress and cell cycle’) and M06 (‘MTOR signaling’), in accordance with their role during conditions of cellular stress by amino acid depletion. In addition, genes from source study IV (image-based screen for selective autophagy) are enriched in several modules, such as M13 (‘Rho GTPases’), M13 and M20 (‘secretion and absorption’) and M16 (‘peroxisome’) but absent from module M09 (‘protein folding’). This nonuniform allocation may reflect mechanistic aspects of autophagy types under specific environmental conditions, in this case for example the role of Rho GTPases for subcellular dynamics, vesicle trafficking and organelle distribution.25
Figure 3.
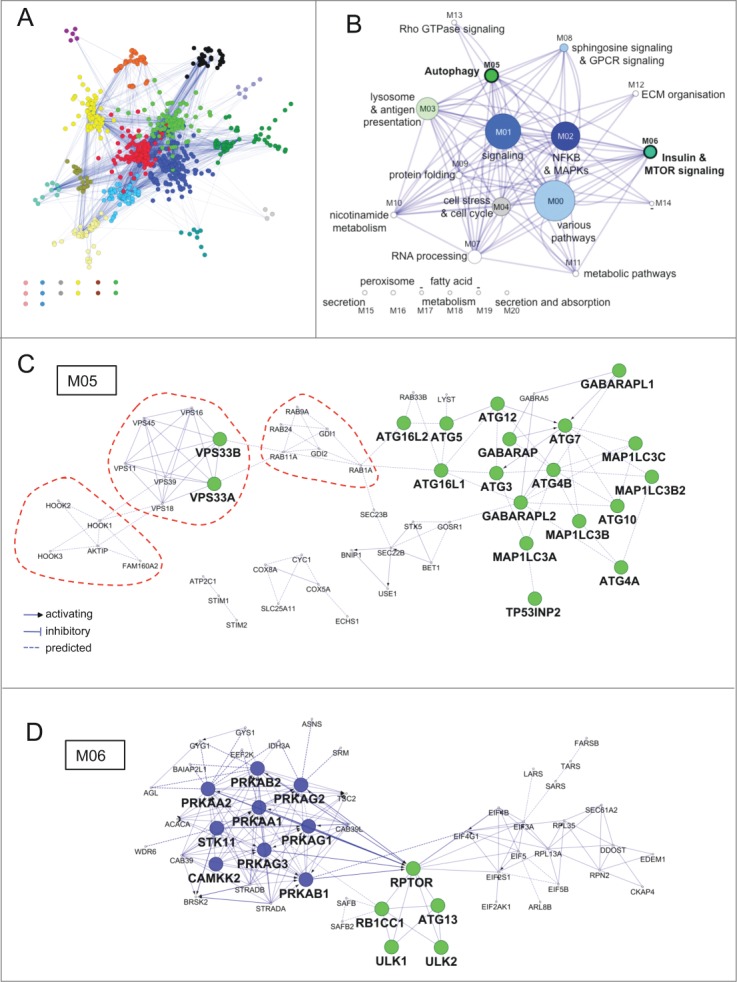
Functional architecture of AXAN. (A) AXAN genes were used to construct a pathway-based functional interaction network (using Reactome database) followed by GLay community clustering for identification of modules. The 21 individual modules are displayed by different colors. (B) The modules were transformed into metanodes with node size corresponding to the number of nodes. Coloring illustrates the fraction of autophagy core (green) and/or signaling (blue) genes. Lead terms were added based on pathway enrichment analysis. The edges between metanodes correspond to interactions between the underlying genes. Modules M05 and M06 are accentuated by bold font. (C) The ‘autophagy’ module (M05). (D) The ‘Insulin & MTOR signaling’ module (M06). In both modules prime genes are displayed according to their category (green, core; blue, signaling). The red dotted lines emphasize existence of smaller functional submodules associated with specific biological functions (see main text).
Taken together, these data are indicative of the modular nature of complex processes, such as the autophagy pathway, in which several upstream and downstream events are contributing to the entire biological execution of the process. This type of analyses can help to understand the role of given genes for the whole process and thus can lead to a deeper understanding of its biology.
Experimental assessment of network predictions
Finally, we aimed to test the potential of our network approach to assist in phrasing hypotheses that can be experimentally tested. We decided to use network interaction data as a discovery platform for novel autophagy mediating genes. As a novel concept, we focused on the subpopulation of 236 single nodes, which do not show direct interactions with any of the genes from the main network. We used protein-protein interaction data to introduce external linker nodes that are able to connect the solitary nodes to the main network by direct interaction. These linker nodes act as molecular links between subregions of the entire network and thus represent promising candidates for being novel autophagy mediators. Using this strategy, 136 of the 236 singletons were linked up to the main network by introduction of 343 linker nodes via first degree interactions, resulting in the establishment of the ‘complemented AXAN / cAXAN’ (see supplementary network file ‘cAXAN’). Interestingly, 117 of the 343 linker nodes (34%) emerged as potential candidate genes within at least one of the major source studies but were omitted from further analysis because of the respective scoring restrictions (Table S9A). In order to demonstrate exemplarily a potential functional role of some of these linker genes for the autophagy pathway, we selected 17 linker genes, based on their interaction characteristics, availability of functional annotation and accessibility to knockdown approaches (Fig. 4A and Table S9B), to be used in a focused siRNA screen applying 3 independent image-based assays for a role in autophagy pathways (see Supplemental Materials for details). In short, these assays were based on 1) delivery of a red+green RFP-EGFP tandem fluorochrome from the cytosol to the lysosome under autophagy conditions (‘lysoRG’ assay), 2) accumulation of lysine 63/K63-linked ubiquitin chains under autophagy-promoting conditions (‘UB-K63’), and 3) abundance of peroxisomal mass (as analyzed through endogenous CAT [catalase] levels) under pexophagy-promoting conditions (‘CT’) (see Figs. S7–S9 for details on the screen strategy and the assays applied, and Fig. S10 and the Movie S1 for quality control experiments). Figure 4B summarizes the main findings of this approach: After applying stringent quality control and exclusion criteria (see Table S10), 9 out of 17 genes tested passed the tests for at least 2 assays, and 4 genes (CDC42, CSTA, RHOQ, ITSN2) passed all 3 assay criteria while displaying effects in the same direction (i.e. knockdown resulting in impaired autophagy execution). Intriguingly, among these high confidence hits 2 genes belong to the Rho GTPase family (CDC42 and RHOQ). This result was especially interesting since the functional architecture of AXAN had revealed presence of a Rho GTPase module (M13) already contained in the network before importing external linker nodes (Fig. 3B).
Figure 4.
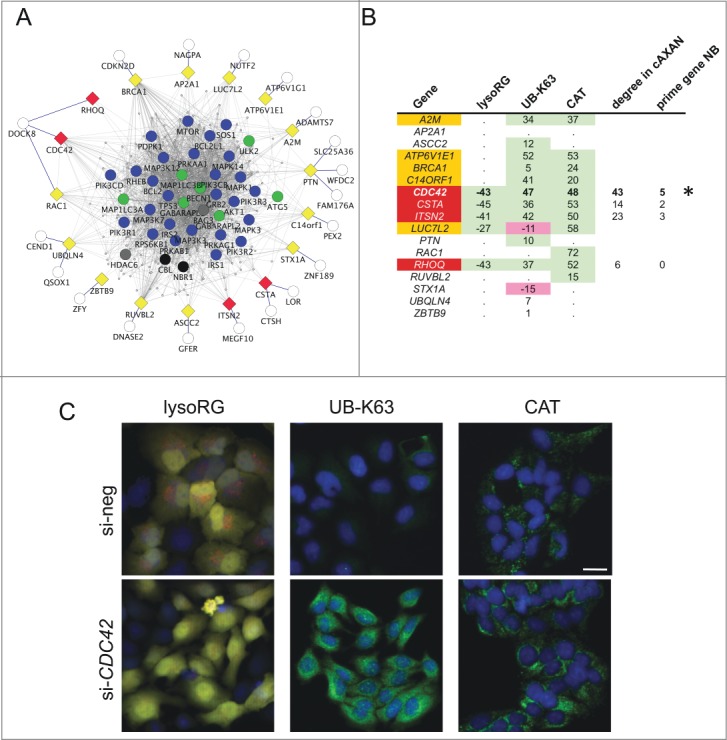
Functional assessment of network predictions. (A) External linker nodes (diamonds) were introduced to connect hitherto solitary nodes (white circles) to AXAN main network (color coding of prime genes as described above). Only those linker nodes chosen for functional siRNA-screening and their first-degree neighbors are shown. Yellow, candidate linker genes that did not pass all test criteria; red, hit genes from functional screen. (B) Summary of the results from a tripartite image-based siRNA screen aimed to assess the role of linker nodes for autophagy pathways. Three independent assays were used: delivery of the cytosolic RFP-EGFP fusion protein to the lysosome (lysoRG) as a measure of autophagy, quantification of endogenous Lys63/K63-linked ubiquitin (UB-K63) levels and quantification of peroxisomal mass (using CAT/catalase as an endogenous marker) under pexophagy conditions (CT, as a measure of pexophagy). Effect strength is displayed using si-negCTRL normalized values. Purple, gene knockdown decreases autophagy effect; green, knockdown increases effect. Of 17 candidate genes tested, 5 passed 2 of the 3 tests with effects in the same direction (orange), and 4 passed all 3 tests in the same direction (red). CDC42 (marked by asterisk) was chosen for further analyses, based on effect strength, its degree in cAXAN and the number of prime genes among its direct neighbors (NB). (C) Examples of how knockdown of Rho GTPase, CDC42, affects the 3 image-based readouts (Scale bar: 10 μm).
Next we aimed to focus on functional validation of one exemplary high confidence hit gene. We analyzed the degree of hit genes within cAXAN and the number of prime genes among direct neighbors (Fig 4B). CDC42 displayed the highest degree and the highest number of prime genes as first-degree neighbors. This observation, the consistent strength of the effect on the 3 independent functional assays and the intriguing presence of a Rho GTPase module within the pathway analysis prompted us to further analyze the role of CDC42 for autophagy. As shown in Figure 4C, knockdown of CDC42 resulted in significantly impaired delivery of the cytosolic reporter protein, RFP-EGFP, to the lysosomal compartment, increased levels of the autophagy target K63-linked ubiquitin, and elevated levels of CAT under pexophagy conditions. In addition, knockdown of CDC42 (or control gene ATG5) impaired the formation of LAMP1-associated red-only structures in Huh7 cells transfected with ptfLC3 and starved for 4 h in EBSS (Fig. 5, and Figs. S10 and S11). These results point to a role of CDC42 as positive regulator of autophagy pathways.
Figure 5.
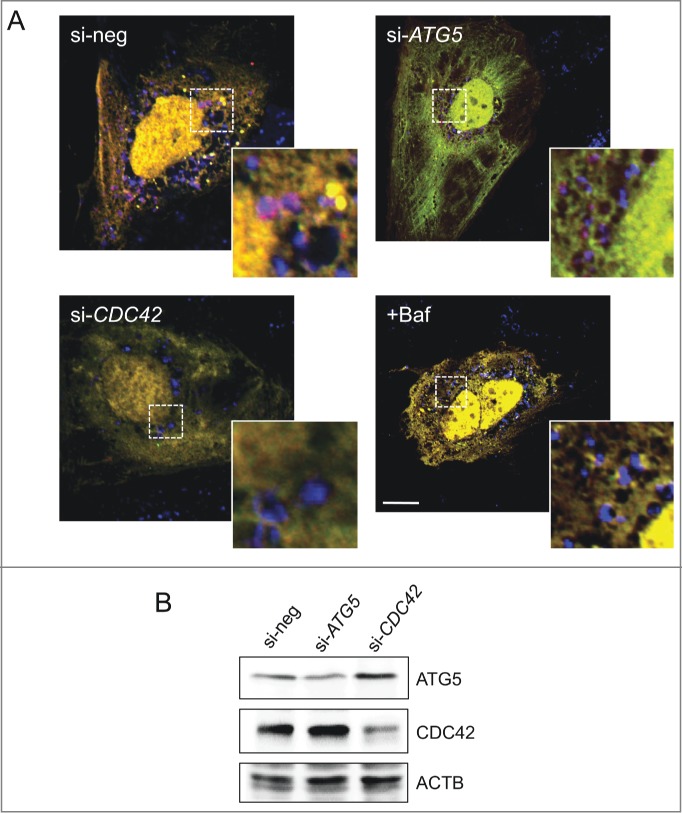
Effect of CDC42 on autophagy pathways. (A) Knockdown of CDC42 (or ATG5 as positive control) impairs formation of red-only structures in ptfLC3-transfected Huh7 cells (4 h starvation in EBSS). Colocalization of these structures with lysosomal marker LAMP1 (blue) only appears in cells transfected with negative control siRNA (si-neg). Treatment with lysosome inhibitor bafilomycin A1 (Baf, 20 nM) served as positive control. Scale bar: 5 μm. (B) Knockdown of ATG5 and CDC42 was analyzed by western blotting. ACTB/β-actin served as loading control.
Finally, we aimed to use network-derived information to generate a hypothesis about the mechanism underlying CDC42 function in autophagy regulation. As shown in Figure 6A, 4 of the 5 prime genes interacting directly with CDC42 represent signaling molecules involved in either RPS6KB signaling, the RRAS-MAPK cascade or the TP53/p53/DNA damage pathway. Twenty seven of the 43 first-degree neighbors were assigned to pathway modules (Fig. 3B) with 11 genes located within M01 (signaling) and 5 genes located within M02 (NFKB and MAPKs) (Table S12). Intriguingly, none of these CDC42 interactors was contained in the data set from source study IV (which focused on selective types of autophagy), arguing for a general role of this CDC42-centered subnetwork for basal autophagy under varying nutrient conditions. From these network-intrinsic data we hypothesized that, in the context of basal autophagy regulation, CDC42 may play a role in modifying regulatory signaling events mediated by ribosomal S6 kinase and/or MAPK cascades. As demonstrated in Figure 6B, our data revealed that CDC42 knockdown decreased signaling activity of components of these pathways, thus strongly supporting our hypothesis. Since both signaling cascades are pivotally involved in various aspects of transcriptional/translational regulation of the autophagy system, we next assessed the effect of CDC42 on LC3 abundance and processing in our system. As shown in Figure 6C, knockdown of CDC42 resulted in reduction not only of processed LC3-II but also of LC3-I, both after rapamycin treatment and under basal growth conditions, thus supporting a role of this Rho GTPase for regulating the abundance of the key proteins for autophagosome formation. These data confirm the role of CDC42 as positive regulator of autophagy-mediating signaling events.
Figure 6.
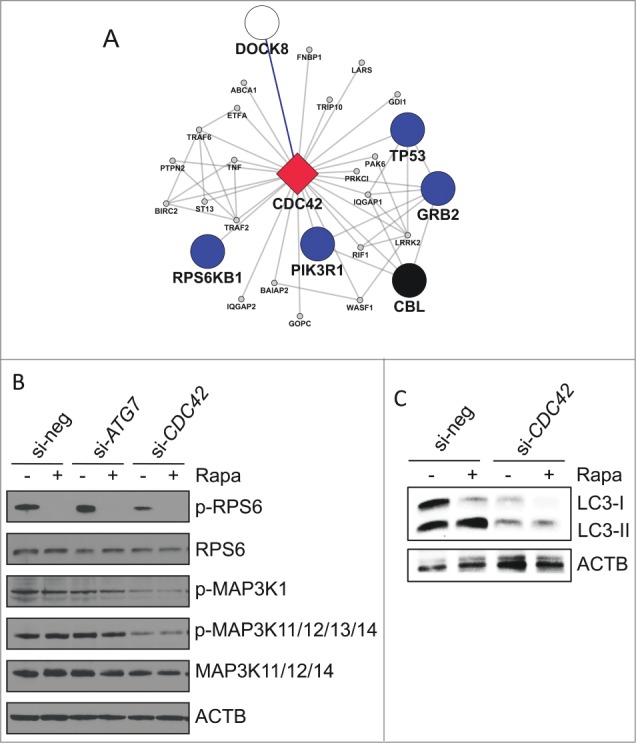
Functional role of CDC42 on autophagy-associated pathways. (A) Subnetwork of first-degree neighbors of CDC42 within the complemented AXAN (cAXAN). CDC42 (red diamond), prime genes (blue, signaling; black, receptors) and the formerly singleton gene DOCK8 (white) are displayed with large symbols, other AXAN genes with small symbols. (B) Effect of knockdown of ATG7 (control) and CDC42 on signaling pathways induced by autophagy activator rapamycin (Rapa). ACTB/β-actin served as loading control. (C) Effect of knockdown of CDC42 on LC3 abundance and lipidation induced by rapamycin (Rapa) in Huh7 cells.
In summary, our data emphasize the applicability of network based strategies for hypothesis generation, as exemplified by the identification of several novel autophagy regulating human genes, and point to a role of CDC42 as modifier of autophagy related signaling.
Discussion
Usability of meta-analyses on functional data sets
In this study we present a global description of the Annotated eXpanded Autophagy Network (AXAN), which was constructed by merging individual data sets which focus on selected aspects of autophagy, and by complementation of these primary data sets with additional layers of data (i.e., interactions, expression profiles, subcellular localization, evolutionary conservation, pathway information). We consider the entire resulting data set as the global framework for individual subnetworks describing aspects of autophagy in a given biological context (e.g., general and selective types, varying nutritional conditions). In addition, we demonstrate that the global network contains more information than its individual components, and we exemplify the utility of this approach by testing the predictive character of our network. This concept goes along with recent efforts in the field of genetics where meta-analyses of various large scale genetic data sets are analyzed in its entirety, successfully identifying genetic variations linked to specific phenotypes that were not identified by any individual study. One prominent example is the dissection of genetic susceptibility factors for complex human diseases such as inflammatory bowel diseases revealing a multilayered interplay between host organism and microbial genetics.26 This conceptual approach benefits from the increase in statistical power due to utilization of multiple primary data sets and the broad availability of quantitative data that were generated by similar technological platforms. In contrast, functional data generated in different laboratories by applying various types of screening technologies, high throughput methodology or systems biology approaches usually are much less consistent and coherent. The large variability of cell types, conditions and technologies used may thus explain the relatively low overlap in data shared by more than one of our primary data sets, but it also reflects the variant nature of autophagy, its subtypes and individual facets. While our study may represent the first attempt to perform a meta-analysis of multiplatform functional data, pointing to limitations and opportunities of this concept, we believe that similar strategies applied in the fields of cell biology and functional genomics may in the future open the avenues for a deeper understanding of complex biological processes. In this context, the discipline of systems biology offers a plethora of valuable strategies, e.g. by establishing a universal language for computational biological experiments27 or for description and presentation of biochemical reactions.28 The establishment of a common set of parameters and descriptors to be contained in experimental documentation is thus expected to be of utmost importance for future functional meta-analysis approaches.
Shedding light onto the evolution of autophagy
Using protein ortholog identification, we aimed to describe evolutionary trends within AXAN with a focus on prime gene categories. Our data support the generally accepted idea that the evolutionary origin of the core autophagy system lies in the last common ancestor of all eukaryotic cells.29 This is emphasized by the significantly high conservation of autophagy core and signaling genes in eukaryota as compared to random human gene sets. The fact that none of the prime genes shows orthologs in the prokaryotic outgroup (E. coli) supports the exclusively eukaryotic nature of the autophagy system. Indeed, it has been proposed that autophagy originates from various modifications of the endomembrane system in the last common ancestor of all eukaryotic cells29 alongside other membrane remodeling mechanisms such as endocytosis (which in itself is mechanistically linked to autophagy). In contrast to the core genes, autophagy receptors are significantly less conserved as a gene group while showing a large variance of evolutionary conservation among the individual group members. Kraft and colleagues have discussed the phylogenetic history of selectivity factors for autophagy and conclude that NBR1-type receptors resemble ancient autophagy mediators closely related to the yeast cytoplasm-to-vacuole targeting (Cvt) receptor, ATG19, that have been lost in the genomes of most invertebrate species.30 In contrast, the report describes SQSTM1-type receptors to have more recent origin mainly in the metazoan group, a view that is in line with our own observations. Based on our data, the gene coding for the autophagy receptor, STBD1, can be regarded as the most recent representative of this group. STBD1 gene expression is most prominent in liver and muscle where it has been demonstrated to be involved in selective autophagy of the energy reserve glycogen (‘glycophagy’).31,32 Recently, the function of this protein in glycogen handling has been linked to gender-specific differences in development of cardiac metabolic stress.33 It is tempting to speculate that future studies will unravel even more mediators of selective autophagy with a precise cargo binding profile and restricted tissue and cell type specificity. It is noteworthy to mention that the restriction of some autophagy receptor groups to higher phylogenetic taxa does not mean that selective autophagy is a feature of recent evolutionary origin. Indeed, molecular analyses using yeast and other simple model systems emphasize the notion that selectivity of autophagy is present in even very distant taxa to a significant level, whereas the recognition of selective cargos is achieved by different proteins other than orthologs of human autophagy receptors. Examples for this concept include the yeast mitophagy receptor Atg32, the Cvt receptor Atg19, and pexophagy receptors Atg36 (Saccharomyces cerevisiae) and Atg30 (Pichia pastoris).16,34-36 In this context it is important to emphasize the differential role of molecular tags for selective autophagy receptors: While a subgroup of mammalian receptors (including SQSTM1, NBR1 and CALCOCO2) utilize ubiquitin recognition on the cargo to physically link it to LC3 family members, this requirement for ubiquitin recognition is not evident in ancient receptor systems and is also not relevant for all mammalian receptors (e.g., BNIP3L, STBD1). Interestingly, recent data point to remarkable similarities between ubiquitin-dependent selective autophagy (‘ubiquitinophagy’) mediated by NBR1- and SQSTM1-type receptors and the Cvt pathway in yeast.30 The complexity of these processes is further exemplified by recent studies describing that members of the tripartite motif-containing protein (TRIM) family selectively mediate various different aspects of autophagy, ranging from assembly of the ULK1-BECN1 signaling platform to actual activity as selective autophagy receptors.37 In conclusion, while the autophagy core and signaling machinery represent eukaryote-specific ancient system components, selectivity of autophagy is achieved by different, but probably related, mechanisms that may or may not have shared evolutionary origin. Knowledge about evolutionary conservation of single genes, groups of genes or mechanistic modules can help in establishing new biological model systems for validating results from established ones. We anticipate that the data presented here and similar approaches will support the interpretation of data sets across a plethora of taxonomic groups, thus speeding up the process of assessing autophagy biology in various organisms.
Transcriptional regulation of autophagy genes
Our approach to describe functionally relevant subnetworks within AXAN has unraveled the complex nature of the transcription network within the autophagy system. While the role of transcriptional regulation of autophagy was previously underestimated, recent work has pointed to the significant contribution of specific transcription factors for the fine-tuning of autophagic responses. One prominent example is the basic-helix-loop-helix, leucine-zipper transcription factor, TFEB, which controls both lysosomal biogenesis and autophagy-linked genes to coordinate cellular homeostasis.9,10 This vital role of TFEB is supported by our analysis. Moreover, our data highlight the even more prominent role of the forkhead transcription factor FOXO3, which is known to coordinate proteostasis (protein homeostasis) by both autophagic and proteasomal degradation pathways.38 Interestingly, allelic variants of FOXO3 have been linked to human longevity.39,40 Evolutionary conservation of this salient finding has been demonstrated by studies performed in the lab of Thomas Bosch reporting on the essential role of FOXO3 for long term survival of the simple metazoan Hydra.41,42 Because autophagy is recognized as a longevity-promoting pathway in various model organisms,43,44 it is tempting to speculate that the life prolonging role of FOXO3 family members may at least partially be attributed to its pivotal role in regulating autophagy prime genes. In addition to these well-described regulatory systems, our subnetwork approach highlights the contribution of other TFs that are involved in multiple mechanisms linked directly or indirectly to autophagy-mediated cellular and organismal homeostasis. These processes include proinflammatory immune responses (NFKB, JUN-FOS, STAT3), DNA damage sensing (TP53), development and tumorigenesis (MYC, GLI3), and glucose and lipid handling (SREBF1, PPARD, see also recent findings on nutrient-sensing nuclear receptors and autophagy).45 Our study thus supports the key role of autophagy beyond its subcellular housekeeping function46-48 and may inspire future studies to assess the detailed transcriptional programs induced by, and regulating, autophagy under varying environmental conditions and cellular context.
Rho GTPAse CDC42 involved in autophagy regulation
In order to identify novel genes contributing to autophagy regulation, we used the introduction of external linker genes for unconnected (‘singleton’) nodes as a novel conceptual strategy. The result from our tripartite functional screen, which identifies and confirms at least 4 high-confidence-hit genes, underscores the validity and applicability of this approach. The exemplary hit gene that was chosen for further analysis, CDC42, represents a member of the Rho GTPase family. These proteins are RRAS-related signaling molecules that switch between a GDP-bound inactive state to GTP-bound active state, and which are involved in regulation of the cytoskeleton, cell polarity, cell cycle, and gene expression.49 CDC42 is well described for its pleiotropic role in cell cycle regulation, cytokinesis, mitosis, neuronal development, stem cell aging, and neurodegeneration.50,51 Our data indicate that in the context of autophagy regulation, CDC42 plays a role in regulation of upstream signaling events (MAPK and RPS6KB signaling cascades) and thus may represent a molecular regulator for fine tuning autophagic responses to environmental conditions. While it is well established that CDC42 is complexing with and regulating the activity of RPS6KB52 and contributes to MAPK activation downstream of RRAS and other stimuli,53 an intriguing aspect is that protein synthesis and activity of CDC42 (and other small GTPases such as RHOA and RAC1) are negatively regulated by rapamycin in some tissues.54 We could confirm this effect in our system (Fig. S13), supporting the concept of complex regulatory circuits including positive and negative feedback mechanisms shaping context-specific regulation of autophagy. It is noteworthy that Chung and colleagues have reported about an autophagy-independent role of microtubule-associated LC3-II in the regulation of CDC42 activity.55 This mechanism appears to play an important role in bone-resorption by osteoclasts and represents another example for the pleiotropic nature of multifunctional proteins such as GTPases or LC3 family members apart from their supposed key functions. Interestingly, among the high confidence hit genes, we also identified RHOQ as another Rho GTPase to regulate autophagic responses. This is in line with our results from pathway module identification efforts, which emphasize the role of Rho GTPases as a functional unit within the entire AXAN.
Network analyses as a vital tool set for current biology
In this study we use strategies from network biology to describe the autophagy pathway from a global perspective beyond the well-described core machinery. Network biology is one of the approaches to investigate molecular interactions within a cell. The first step of this approach is to collect interactions which represent the edges in the network by linking up nodes (i.e. genes or proteins). For collecting novel interactions, many high-throughput experimental methods such as yeast 2-hybrid or tandem affinity purification are well established. These huge amounts of interactions as well as curated interactions are available through public databases. Various methodologies for the subsequent steps have been developed. They include those for integration of interactions,56-58 visualizing and exploring them,59,60 and analyzing them in various ways such as pathway mappings,61 functional enrichment analysis,62 node degree analysis,63 network motif extraction,64 and functional module prediction.65 Especially Cytoscape59 was employed as a useful software platform to conduct many of these network analyses. These analytical strategies can assist in describing the functional properties of a biological system. Importantly, network analyses have been useful for understanding complex biological processes66 such as the complex interplay between host and viral factors during influenza and HIV virus infection.67,68
Moreover, recent advances have highlighted the emerging properties revealed by analysis of network behavior under varying conditions, thus emphasizing the importance of differential as opposed to static networks.69 It is important to note that our network represents a static model without the capacity to visualize or analyze the dynamics of the underlying biological process. It will be a major challenge for future studies to implement the changing behavior of systems over time and also in response to various perturbations into existing network models. In this context, our network or its derivatives may form the basis for a computational approach to use mathematical modeling for quantitative predictions of the system's behavior in response to external stimuli.
Materials and Methods
Establishing AXAN
Gene sets were retrieved from 6 recent large scale studies,18-22 several individual studies, and our own data (see Table S1), in total accounting for the identification of 1211 human genes involved in aspects of autophagy. Gene IDs were mapped using DAVID lab tool (http://david.abcc.ncifcrf.gov/). Transcription factors were identified using the Transcriptional Regulatory Element Database (TRED, http://rulai.cshl.edu/cgi-bin/TRED/). Interactions were retrieved from the following resources: protein-protein interactions (PPI) from the human-specific protein interaction collection HIPPIE (http://cbdm.mdc-berlin.de/tools/hippie/)70 using the following cut-offs: HIPPIE score ≤0.62: low confidence (edges omitted); 0.63 ≤ HIPPIE score ≤0.72: medium confidence; ≥0.73: high confidence. Interactions describing transcriptional regulation were retrieved from the TRED and from the ToppGene server.71 The resulting network and all derivatives were visualized and analyzed using Cytoscape.59 In order to complement AXAN by connecting the primary single nodes (‘singletons’) with the AXAN main network we used the following strategy: We introduced ‘external linker nodes’ by querying the HIPPIE interactome for first degree neigbors of singletons that also represent direct nigbors of AXAN main nodes. Only medium and high confidence interactions were allowed for connecting singleton and AXAN main genes. The final network containing the initial AXAN plus linker nodes was dubbed ‘complemented AXAN’ (cAXAN).
Analysis of gene expression and protein localization profiles
To describe cell type specific expression profiles of autophagy prime genes, we used information provided by the Human Protein Atlas (HPA, http://www.proteinatlas.org/).24 Genes expressed in a given tissue or cell type were defined as those that have medium or high expression as supported by evidence from HPA. We have calculated rates of expression for 108 autophagy prime genes and 17,434 nonautophagy genes. Nonautophagy genes are defined as those which are not included in the list of entire AXAN genes but have UniProt IDs assigned. Data were expressed as percentage overrepresentation of autophagy prime genes compared to nonautophagy genes. The differences in the rates were tested using the Fisher exact test after the Bonferroni correction. The main subcellular localization of AXAN proteins (Table S5, Fig. S3) was established by combining information from the Mammalian Protein Localization database (LOCATE; http://locate.imb.uq.edu.au/), gene ontology terms (geneontology.org) and manual curation of gene-specific literature from the UniProtKB/Swiss-Prot summary at GeneCards (http://www.genecards.org, as soon as subcellular protein localization is explicitly referred to).
Assessment of network characteristics of AXAN
In order to analyze whether or not AXAN displays characteristics of a random network, we generated 3 random human gene sets of equal size (n = 1222 genes) within the HIPPIE interactome data set (http://cbdm.mdc-berlin.de/tools/hippie/) and retrieved medium and high confidence interactions (HIPPIE score >0.62). In addition, 3 randomized versions of AXAN were created by keeping the nodes and freely shuffling the same number of edges using the RandomNetworks plugin for Cytoscape (https://sites.google.com/site/randomnetworkplugin/). AXAN itself, randomized AXANs and the 3 random human networks were analyzed with respect to medium and high HIPPIE-edges using the NetworkAnalyzer plugin for Cytoscape. The clustering coefficient and average degree were used as parameters describing network characteristics. The clustering coefficient of a node quantifies how close its neighbors are to forming an enclosed group. The degree quantifies the number of direct neighbors for each node (idential for AXAN and randomized AXANs).
Assessment of evolutionary conservation of human genes
The InParanoid7 database (http://InParanoid.sbc.su.se) uses pairwise assessment of protein sequence similarity to identify orthologs of genes across 99 species of eukaryotic model organisms and Escherichia coli as prokaryotic outgroup. In its current version the database contains collectively 1.3 million proteins organized into 42.7 million pairwise ortholog groups. We used the human genome as query list and extracted presence or absence of orthologs across these species for all AXAN genes or 2000 random human genes as a control set. The phylogenetic group that encompasses Homo sapiens and the respective model organism was used to estimate the evolutionary origin of each query gene. Presence of an ortholog in one species that is represented by one higher order phylogenetic group was regarded as “evidence,” while presence of orthologs in at least 2 species belonging to a given group were regarded as “strong evidence.” For analysis of evolutionary trends, orthologs in at least 2 species of a taxonomic group were used as inclusion criterion. For each gene set, the percentage of human genes having orthologs in at least 2 species of higher taxonomic group was displayed.
Analyzing the network architecture of AXAN
For functional dissection of AXAN in terms of cellular pathways and processes, we used the Reactome FI database, which imports functional interactions of various types (complex formation, enzymatic reactions, transcriptional regulation) for a set of query genes and thus creates a pathway-focused interaction network. The resulting network was analyzed for modules (distinct areas that form enclosed functional domains) based on GLay community cluster analysis.72 These modules were visualized as metanodes and were analyzed for pathway enrichment using gene sets from several pathway databases. Enriched terms were used as “theme terms” for annotation of the modules.
Plasmid construction, cell culture, and transfection
The plasmid ptfLC3 encoding tandem fluorochrome RFP-EGFP-LC3 (Addgene plasmid 21074, provided by Tamotsu Yoshimori) has been described before.73 pAT012 encoding cytosolic tandem fluorochrome reporter protein mRFP-EGFP was constructed by deleting the coding sequence of LC3B from ptfLC3 using restriction enzymes BglII and BamHI. The principle of detection of autophagy using this assay is based on the differential stability of the 2 fluorochromes under varying pH conditions (as described by Kimura and colleagues73) with red-only structures indicating lysosomal delivery of the reporter protein. HeLa (human cervical carcinoma) cells were grown in DMEM medium supplemented with 10% fetal calf serum and antibiotics. Stable transfection of pAT012 was achieved by selection in G418 (1000 μg/ml; Sigma-Aldrich, A1720) for 3 wk. Transfection of siRNA was performed using the On Target Plus Smart Pool series of pooled siRNAs (4 individual siRNAs per gene, Dharmacon, see Table S11) and Lipofectamine RNAiMAX (Invitrogen, 13778100). In short, per well of a 96-well plate, exponentially growing cells at 60% confluency were transfected with 0.15 μl of siRNA (10 μM) and 0.25 μl Lipofectamine RNAiMAX in 25 μl of serum-free medium. Transfection was performed one day after seeding on 96-well glass bottom plates, and cells were treated as described for 72 h after transfection. Next, cells were fixed in 4% paraformaldehyde/phosphate-buffered saline (PFA/PBS; Affymetrix, 19943–1LT) and used for automated image acquisition (see below). For the confirmatory run, cells were stained for a prominent target of autophagy (Lys63/K63-linked ubiquitin chains) using a specific antibody (Millipore, 05–1308) and secondary antibody, anti-rabbit Alexa Fluor 488 (Abcam, ab150077). Nuclei were stained using DAPI (Sigma Aldrich, D9542–5MG). To induce autophagy at a high level, we first blocked autophagosomal turnover by 3-methyladenine (3MA; Sigma-Aldrich, M9281) at 10 mM for 18 h, followed by release of this block using nitrogen-free EBSS (Sigma-Aldrich, E2888–500ML). This treatment resulted in effective induction of autophagy within 4 h as assessed by formation of red-only structures due to lysosomal quenching and degradation of the EGFP portion of the tandem fluorochrome protein. For confirmatory and follow-up experiments, Huh7 (human hepatoma) and primary human glioblastoma cells (pGBMs, case 135Z)74 were used. Huh7 were transfected with siRNA using RNAiMAX (Invitrogen, 13778100) and with plasmid DNA using Trans-IT-LT1 (MirusBio, MIR 2300). pGBMs were transfected with reporter plasmids using Lipofectamine 2000 (Invitrogen, 11668–027). Where indicated, the lysosomal ATPase inhibitor Bafilomycin A1 (Calbiochem, 196000) was used at 20 nM for 6 to 12 h.
Automated image analysis
For the tripartite screen, images were automatically acquired using an Opera High Content Screening System (PerkinElmer, Waltham, MA, USA). Quantitative image analysis was performed using Cellprofiler75 and a hand-made analysis pipeline. In short, the pipeline consisted of the following steps (see Figures S7–S9): 1) identification of nuclei; 2) filtering of images based on nuclei number (5 < n < 200 ); 3) identification of cells; 4) quantification of red/green ratio (primary screen, n = 3), or fluorescence intensity (for secondary and tertiary screen pass), for each cell. Average and standard deviation for each set was calculated, and significance of differences between test genes and negative control (noncoding siRNA mix) were performed using the Student t test with a critical α level of 0.05, and with correction for multiple testing (Benjamini-Hochberg correction). Genes were defined as hits if they passed the primary screen and at least one of the confirmatory screen passes, and as high confident hits when they passed all criteria in all 3 screening passes with effects in the same direction.
Functional follow-up studies
Follow up studies were performed with Huh7 cells and primary glioblastoma cells (pGBMs). In order to minimize off-target effects and exclude experimental bias due to the choice of our initial RNAi agents, follow-up gene knockdown was performed using the siGenome system (Dharmacon): CDC42, M005057–01; ATG5, M-004374–04–0005; ATG12, D-050953–03; nontargeting siRNA 3 (si-neg), D-001210–03–05. Colocalization of lysosomal marker LAMP1 with red-only structures resulting from autolysosomal delivery of the RFP-EGFP fusion protein (‘lysoRG assay’) was performed using rabbit polyclonal antibody from Abcam (ab24170). In short, Huh7 cells were grown on glass cover slips and treated as indicated. After treatment, cells were fixed in 4% paraformaldehyde and washed 3× with PBS. The fixed cells were blocked and permeabilized for 1 h in blocking buffer (3% BSA [Sigma-Aldrich, A9418–5G], 5% normal goat serum [Pierce, 31872], and 0.1% Triton X-100 [TX-100, Sigma-Aldrich, X-100] in PBS), followed by immunostaining with respective primary antibody (diluted in binding buffer; 3% BSA, 0.05% TX-100 in PBS) overnight at 4°C. The cells were washed 3× with PBS and then stained with respective secondary antibodies diluted in binding buffer for 1 h. The secondary antibodies used for immunofluorescence were Donkey-anti-Rabbit IgG conjugates from Life Technologies (Rockford, IL, USA): Alexa Fluor488 (A-21206), Alexa Fluor594 (A-21207), and Alexa Fluor647 (A-31573). The cells were washed 3× with PBS and mounted using ProLong Gold antifade mounting reagent (Molecular Probes/Invitrogen, P-36931). Images were visualized under a 100× oil objective using an Olympus FluoView 1000 (Olympus USA, Melville, NY) confocal microscope. Where indicated cells were treated with 500 nM rapamycin for 6 h. pGBMs grown on glass cover plates and transfected with reporter plasmid pAT012 were treated with 3MA overnight or left in full medium, followed by washing in PBS and replacing of medium with either 3MA containing medium, full medium or EBSS for another 4 h. After fixation and DAPI staining cells were used for confocal imaging.
Western blotting
Protein lysates were prepared and separated by SDS-PAGE as described before.76 Antibodies were purchased from following resources: Anti-ubiquitin Lys63/K63-specific (rabbit; Millipore, 05–1308); anti-LC3B (D11, LC3-II-specific, rabbit; Cell Signaling Technology, 3868), anti-ATG7 (rabbit; Epitomics, 2054–1); anti-LAMP1 (rabbit; Abcam, ab24170); anti-ATG5 (rabbit, polyclonal; Novus Biologicals, NB110–53818); anti-SQSTM1/p62 (rabbit; MBL, PM045); anti-CT/catalase (rabbit; EMD Millipore, 219010); anti-ACTB/β-actin (mouse; Sigma-Aldrich, A1978–200UL); anti-ACBD5 (rabbit; Sigma, HPA011861–100UL); anti-CDC42 (mouse; Abcam, ab41429); anti-phospho-RPS6 (Ser235/236, rabbit; Cell Signaling Technology, 4858P); anti-RPS6 (total protein, rabbit; Cell Signaling Technology, 2217S); anti-phospho-MAP3K1/MEKK1 (rabbit; Santa Cruz Biotechnology, sc-130202); anti-phospho-MAPK11/12/13/14 (phospho-p38, rabbit; Cell Signaling Technology, 4511P); anti-MAPK11/12/14 (total p38, rabbit; Cell Signaling Technology, 9212). Horseradish peroxidase (HRP) – coupled secondary antibodies for western blotting were Goat Anti-Rabbit IgG-HRP Conjugate (BioRad, 170–6515) and Goat Anti-Mouse IgG-HRP Conjugate (BioRad, 172–1011).
Concluding remarks
In summary, our approach describes the autophagy pathway from a hitherto unprecedented systematic perspective and illustrates the applicability of network biology for streamlining research efforts. It also holds the premise and supports the idea that computational approaches, especially those implementing network and systems biology, will represent indispensable assets for the interpretation of multiplatform and multicenter, large-scale functional studies in the future.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Sumit Chanda (Sanford Burnham Medical Research Institute (SBMRI), LaJolla, CA) for helpful discussions on the topic of functional screens. We also thank Susanne Heyne-Gehlen and Anton Cheltsov (both SBMRI) for support with automated image acquisition. Pedro Aza-Blanc and Paul de Jesus (both SBMRI) are gratefully acknowledged for their expert assistance on RNAi. We thank the entire Cytoscape team at University of California San Diego (UCSD) and beyond for their support. The authors are also grateful to the entire Subramani lab at UCSD for support and helpful discussions.
Funding
This work was supported by the NIH P50 grant #GM085764 through the San Diego Center for Systems Biology (SDCSB, http://sdcsb.org, to AT, TI and SS), by grants from the German Research Association (Deutsche Forschungsgemeinschaft DFG; Ti 640 1–1 and Ti 640 2–1 to AT), by grant GM069373 (to SS) and by the German Excellence Initiative “Inflammation at Interfaces” (to AT). BS is funded by the Lichtenberg program of the VW foundation, MG and BS are supported by the Federal Ministry of Education and Research, Germany (BMBF, VIP initiative, FKZ 03V0785). TI is funded by the National Resource for Network Biology (P41 GM103504) and the San Diego Center for Systems Biology (P50 GM085764). AS is supported by National Institutes of Health Grants; AI085087, DK077704, and DK08379.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Guan JL, Simon AK, Prescott M, Menendez JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A, Zhang J. Autophagy in stem cells. Autophagy 2013; 9:830-49; PMID:23486312; http://dx.doi.org/ 10.4161/auto.24132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/ 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaza N, Kohli L, Roth KA. Autophagy in brain tumors: a new target for therapeutic intervention. Brain Pathol 2012; 22:89-98; PMID:22150924; http://dx.doi.org/ 10.1111/j.1750-3639.2011.00544.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palumbo S, Comincini S. Autophagy and ionizing radiation in tumors: the “survive or not survive” dilemma. J Cell Physiol 2013; 228:1-8; PMID:22585676; http://dx.doi.org/ 10.1002/jcp.24118 [DOI] [PubMed] [Google Scholar]
- 6.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717-21; PMID:11099404; http://dx.doi.org/ 10.1126/science.290.5497.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102-9; PMID:17909521; http://dx.doi.org/ 10.1038/ncb1007-1102 [DOI] [PubMed] [Google Scholar]
- 8.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124-31; PMID:20034776; http://dx.doi.org/ 10.1016/j.ceb.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al.. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; http://dx.doi.org/ 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al.. A gene network regulating lysosomal biogenesis and function. Science 2009; 325:473-7; PMID:19556463 [DOI] [PubMed] [Google Scholar]
- 11.Sumpter R Jr., Levine B. Selective autophagy and viruses. Autophagy 2011; 7:260-5; PMID:21150267; http://dx.doi.org/ 10.4161/auto.7.3.14281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Till A, Lakhani R, Burnett SF, Subramani S. Pexophagy: the selective degradation of peroxisomes. Int J Cell Biol 2012; 2012:512721; PMID:22536249; http://dx.doi.org/ 10.1155/2012/512721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res 2010; 70:3431-4; PMID:20424122; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-4027 [DOI] [PubMed] [Google Scholar]
- 14.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259-69; PMID:19450525; http://dx.doi.org/ 10.1016/j.molcel.2009.04.026 [DOI] [PubMed] [Google Scholar]
- 15.Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 2009; 17:87-97; PMID:19619494; http://dx.doi.org/ 10.1016/j.devcel.2009.06.013 [DOI] [PubMed] [Google Scholar]
- 16.Farre JC, Manjithaya R, Mathewson RD, Subramani S. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev Cell 2008; 14:365-76; PMID:18331717; http://dx.doi.org/ 10.1016/j.devcel.2007.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meijer WH, van der Klei IJ, Veenhuis M, Kiel JA. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy 2007; 3:106-16; PMID:17204848; http://dx.doi.org/ 10.4161/auto.3595 [DOI] [PubMed] [Google Scholar]
- 18.Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, Liu X, Eisenberg J, Liu J, Blenis J, et al.. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell 2010; 18:1041-52; PMID:20627085; http://dx.doi.org/ 10.1016/j.devcel.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68-76; PMID:20562859; http://dx.doi.org/ 10.1038/nature09204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jegga AG, Schneider L, Ouyang X, Zhang J. Systems biology of the autophagy-lysosomal pathway. Autophagy 2011; 7; PMID:21293178; http://dx.doi.org/ 10.4161/auto.7.5.14811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orvedahl A, Sumpter R Jr., Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, et al.. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011; 480:113-7; PMID:22020285; http://dx.doi.org/ 10.1038/nature10546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKnight NC, Jefferies HB, Alemu EA, Saunders RE, Howell M, Johansen T, Tooze SA. Genome-wide siRNA screen reveals amino acid starvation-induced autophagy requires SCOC and WAC. EMBO J 2012; 31:1931-46; PMID:22354037; http://dx.doi.org/ 10.1038/emboj.2012.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, et al.. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010; 141:1042-55; PMID:20550938; http://dx.doi.org/ 10.1016/j.cell.2010.04.034 [DOI] [PubMed] [Google Scholar]
- 24.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al.. Proteomics. Tissue-based map of the human proteome. Science 2015; 347:1260419; PMID:25613900; http://dx.doi.org/ 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 25.Harris KP, Tepass U. Cdc42 and vesicle trafficking in polarized cells. Traffic 2013; 11:1272-9; http://dx.doi.org/ 10.1111/j.1600-0854.2010.01102.x [DOI] [PubMed] [Google Scholar]
- 26.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al.. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491:119-24; PMID:23128233; http://dx.doi.org/ 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waltemath D, Adams R, Bergmann FT, Hucka M, Kolpakov F, Miller AK, Moraru II, Nickerson D, Sahle S, Snoep JL, et al.. Reproducible computational biology experiments with SED-ML–the Simulation Experiment Description Markup Language. BMC Syst Biol 2011; 5:198; PMID:22172142; http://dx.doi.org/ 10.1186/1752-0509-5-198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hucka M, Finney A, Sauro HM, Bolouri H, Doyle JC, Kitano H, Arkin AP, Bornstein BJ, Bray D, Cornish-Bowden A, et al.. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 2003; 19:524-31; PMID:12611808; http://dx.doi.org/ 10.1093/bioinformatics/btg015 [DOI] [PubMed] [Google Scholar]
- 29.Hughes T, Rusten TE. Origin and evolution of self-consumption: autophagy. Adv Exp Med Biol 2007; 607:111-8; PMID:17977463 [DOI] [PubMed] [Google Scholar]
- 30.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 2010; 12:836-41; PMID:20811356; http://dx.doi.org/ 10.1038/ncb0910-836 [DOI] [PubMed] [Google Scholar]
- 31.Jiang S, Wells CD, Roach PJ. Starch-binding domain-containing protein 1 (Stbd1) and glycogen metabolism: Identification of the Atg8 family interacting motif (AIM) in Stbd1 required for interaction with GABARAPL1. Biochem Biophys Res Commun 2011; 413:420-5; PMID:21893048; http://dx.doi.org/ 10.1016/j.bbrc.2011.08.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang S, Heller B, Tagliabracci VS, Zhai L, Irimia JM, DePaoli-Roach AA, Wells CD, Skurat AV, Roach PJ. Starch binding domain-containing protein 1/genethonin 1 is a novel participant in glycogen metabolism. J Biol Chem 2011; 285:34960-71; PMID:20810658; http://dx.doi.org/ 10.1074/jbc.M110.150839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reichelt ME, Mellor KM, Curl CL, Stapleton D, Delbridge LM. Myocardial glycophagy – a specific glycogen handling response to metabolic stress is accentuated in the female heart. J Mol Cell Cardiol 2013; 65:67-75; PMID:24080183; http://dx.doi.org/ 10.1016/j.yjmcc.2013.09.014 [DOI] [PubMed] [Google Scholar]
- 34.Scott SV, Guan J, Hutchins MU, Kim J, Klionsky DJ. Cvt19 is a receptor for the cytoplasm-to-vacuole targeting pathway. Mol Cell 2001; 7:1131-41; PMID:11430817; http://dx.doi.org/ 10.1016/S1097-2765(01)00263-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 2009; 17:98-109; PMID:19619495; http://dx.doi.org/ 10.1016/j.devcel.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Motley AM, Nuttall JM, Hettema EH. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J 2012; 31:2852-68; PMID:22643220; http://dx.doi.org/ 10.1038/emboj.2012.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mandell MA, Jain A, Arko-Mensah J, Chauhan S, Kimura T, Dinkins C, Silvestri G, Münch J, Kirchhoff F, Simonsen A, et al.. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell 2014; 30:394-409; PMID:25127057; http://dx.doi.org/ 10.1016/j.devcel.2014.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472-83; PMID:18054316; http://dx.doi.org/ 10.1016/j.cmet.2007.11.004 [DOI] [PubMed] [Google Scholar]
- 39.Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, Masaki KH, Willcox DC, Rodriguez B, Curb JD. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A 2008; 105:13987-92; PMID:18765803; http://dx.doi.org/ 10.1073/pnas.0801030105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flachsbart F, Caliebe A, Kleindorp R, Blanche H, von Eller-Eberstein H, Nikolaus S, Schreiber S, Nebel A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci U S A 2009; 106:2700-5; PMID:19196970; http://dx.doi.org/ 10.1073/pnas.0809594106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boehm AM, Khalturin K, Anton-Erxleben F, Hemmrich G, Klostermeier UC, Lopez-Quintero JA, Oberg HH, Puchert M, Rosenstiel P, Wittlieb J, et al.. FoxO is a critical regulator of stem cell maintenance in immortal Hydra. Proc Natl Acad Sci U S A 2012; 109:19697-702; PMID:23150562; http://dx.doi.org/ 10.1073/pnas.1209714109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nebel A, Bosch TC. Evolution of human longevity: lessons from Hydra. Aging (Albany NY) 2012; 4:730-1; PMID:23241851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008; 4:176-84; PMID:18059160; http://dx.doi.org/ 10.4161/auto.5269 [DOI] [PubMed] [Google Scholar]
- 44.Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet 2008; 4:e24; PMID:18282106; http://dx.doi.org/ 10.1371/journal.pgen.0040024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014; 516(7529):112-5; PMID:25383539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res 2014; PMID:24459182 [DOI] [PubMed] [Google Scholar]
- 47.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013; 13:722-37; PMID:24064518; http://dx.doi.org/ 10.1038/nri3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15:713-20; PMID:23817233; http://dx.doi.org/ 10.1038/ncb2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stengel K, Zheng Y. Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cell Signal 2011; 23:1415-23; PMID:21515363; http://dx.doi.org/ 10.1016/j.cellsig.2011.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Geiger H, Zheng Y. Regulation of hematopoietic stem cell aging by the small RhoGTPase Cdc42. Exp Cell Res 2014; 329:214-9; PMID:25220425; http://dx.doi.org/ 10.1016/j.yexcr.2014.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chircop M. Rho GTPases as regulators of mitosis and cytokinesis in mammalian cells. Small GTPases 2014; 5:pii: e29770; PMID:24988197; http://dx.doi.org/ 10.4161/sgtp.29770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chou MM, Blenis J. The 70 kDa S6 kinase complexes with and is activated by the Rho family G proteins Cdc42 and Rac1. Cell 1996; 85:573-83; PMID:8653792; http://dx.doi.org/ 10.1016/S0092-8674(00)81257-X [DOI] [PubMed] [Google Scholar]
- 53.Lopez-Ilasaca M. Signaling from G-protein-coupled receptors to mitogen-activated protein (MAP)-kinase cascades. Biochem Pharmacol 1998; 56:269-77; PMID:9744561; http://dx.doi.org/ 10.1016/S0006-2952(98)00059-8 [DOI] [PubMed] [Google Scholar]
- 54.Liu L, Luo Y, Chen L, Shen T, Xu B, Chen W, Zhou H, Han X, Huang S. Rapamycin inhibits cytoskeleton reorganization and cell motility by suppressing RhoA expression and activity. J Biol Chem 2010; 285:38362-73; PMID:20937815; http://dx.doi.org/ 10.1074/jbc.M110.141168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chung YH, Yoon SY, Choi B, Sohn DH, Yoon KH, Kim WJ, Kim DH, Chang EJ. Microtubule-associated protein light chain 3 regulates Cdc42-dependent actin ring formation in osteoclast. Int J Biochem Cell Biol 2012; 44:989-97; PMID:22465708; http://dx.doi.org/ 10.1016/j.biocel.2012.03.007 [DOI] [PubMed] [Google Scholar]
- 56.Bar-Joseph Z, Gerber GK, Lee TI, Rinaldi NJ, Yoo JY, Robert F, Gordon DB, Fraenkel E, Jaakkola TS, Young RA, et al.. Computational discovery of gene modules and regulatory networks. Nat Biotechnol 2003; 21:1337-42; PMID:14555958; http://dx.doi.org/ 10.1038/nbt890 [DOI] [PubMed] [Google Scholar]
- 57.Yeger-Lotem E, Sattath S, Kashtan N, Itzkovitz S, Milo R, Pinter RY, Alon U, Margalit H. Network motifs in integrated cellular networks of transcription-regulation and protein-protein interaction. Proc Natl Acad Sci U S A 2004; 101:5934-9; PMID:15079056; http://dx.doi.org/ 10.1073/pnas.0306752101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelley R, Ideker T. Systematic interpretation of genetic interactions using protein networks. Nat Biotechnol 2005; 23:561-6; PMID:15877074; http://dx.doi.org/ 10.1038/nbt1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13:2498-504; PMID:14597658; http://dx.doi.org/ 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suzuki H, Saito R, Kanamori M, Kai C, Schonbach C, Nagashima T, Hosaka J, Hayashizaki Y. The mammalian protein-protein interaction database and its viewing system that is linked to the main FANTOM2 viewer. Genome Res 2003; 13:1534-41; PMID:12819152; http://dx.doi.org/ 10.1101/gr.956303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, Anwar N, Bader GD, Sander C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res 2011; 39:D685-90; PMID:21071392; http://dx.doi.org/ 10.1093/nar/gkq1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005; 21:3448-9; PMID:15972284; http://dx.doi.org/ 10.1093/bioinformatics/bti551 [DOI] [PubMed] [Google Scholar]
- 63.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature 2000; 407:651-4; PMID:11034217; http://dx.doi.org/ 10.1038/35036627 [DOI] [PubMed] [Google Scholar]
- 64.Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Network motifs: simple building blocks of complex networks. Science 2002; 298:824-7; PMID:12399590; http://dx.doi.org/ 10.1126/science.298.5594.824 [DOI] [PubMed] [Google Scholar]
- 65.Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003; 4:2; PMID:12525261; http://dx.doi.org/ 10.1186/1471-2105-4-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ideker T, Sharan R. Protein networks in disease. Genome Res 2008; 18:644-52; PMID:18381899; http://dx.doi.org/ 10.1101/gr.071852.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Chiang CY, Tu BP, De Jesus PD, Lilley CE, et al.. Human host factors required for influenza virus replication. Nature 2010; 463:813-7; PMID:20027183; http://dx.doi.org/ 10.1038/nature08699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, et al.. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008; 135:49-60; PMID:18854154; http://dx.doi.org/ 10.1016/j.cell.2008.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ideker T, Krogan NJ. Differential network biology. Mol Syst Biol 2012; 8:565; PMID:22252388; http://dx.doi.org/ 10.1038/msb.2011.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schaefer MH, Fontaine JF, Vinayagam A, Porras P, Wanker EE, Andrade-Navarro MA. HIPPIE: Integrating protein interaction networks with experiment based quality scores. PLoS One 2012; 7:e31826; PMID:22348130; http://dx.doi.org/ 10.1371/journal.pone.0031826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009; 37:W305-11; PMID:19465376; http://dx.doi.org/ 10.1093/nar/gkp427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Su G, Kuchinsky A, Morris JH, States DJ, Meng F. GLay: community structure analysis of biological networks. Bioinformatics 2010; 26:3135-7; PMID:21123224; http://dx.doi.org/ 10.1093/bioinformatics/btq596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; http://dx.doi.org/ 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- 74.Glas M, Rath BH, Simon M, Reinartz R, Schramme A, Trageser D, Eisenreich R, Leinhaas A, Keller M, Schildhaus HU, et al.. Residual tumor cells are unique cellular targets in glioblastoma. Ann Neurol 2010; 68:264-9; PMID:20695020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al.. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 2006; 7:R100; PMID:17076895; http://dx.doi.org/ 10.1186/gb-2006-7-10-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nazarko TY, Ozeki K, Till A, Ramakrishnan G, Lotfi P, Yan M, Subramani S. Peroxisomal Atg37 binds Atg30 or palmitoyl-CoA to regulate phagophore formation during pexophagy. J Cell Biol 2014; 204:541-57; PMID:24535825; http://dx.doi.org/ 10.1083/jcb.201307050 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.