

Abstract
The HOPS (homotypic fusion and protein sorting) complex functions in endocytic and autophagic pathways in both lower eukaryotes and mammalian cells through its involvement in fusion events between endosomes and lysosomes or autophagosomes and lysosomes. However, the differential molecular mechanisms underlying these fusion processes are largely unknown. Buff (bf) is a mouse mutant that carries an Asp251-to-Glu point mutation (D251E) in the VPS33A protein, a tethering protein and a core subunit of the HOPS complex. Bf mice showed impaired spontaneous locomotor activity, motor learning, and autophagic activity. Although the gross anatomy of the brain was apparently normal, the number of Purkinje cells was significantly reduced. Furthermore, we found that fusion between autophagosomes and lysosomes was defective in bf cells without compromising the endocytic pathway. The direct association of mutant VPS33AD251E with the autophagic SNARE complex, STX17 (syntaxin 17)-VAMP8-SNAP29, was enhanced. In addition, the VPS33AD251E mutation enhanced interactions with other HOPS subunits, namely VPS41, VPS39, VPS18, and VPS11, except for VPS16. Reduction of the interactions between VPS33AY440D and several other HOPS subunits led to decreased association with STX17. These results suggest that the VPS33AD251E mutation plays dual roles by increasing the HOPS complex assembly and its association with the autophagic SNARE complex, which selectively affects the autophagosome-lysosome fusion that impairs basal autophagic activity and induces Purkinje cell loss.
Keywords: autophagic SNARE complex, autophagy, Hermansky-Pudlak syndrome, HOPS complex, Purkinje cell loss, VPS33A
Abbreviations
- ACTB
β-actin
- bf
buff mutant mouse
- cKO
conditional knockout
- coIP
coimmunoprecipitation
- CTS
cathepsin
- EGFR
epidermal growth factor receptor
- GFP
green fluorescent protein
- HOPS
homotypic fusion and protein sorting
- IB
immunoblotting
- KO
knockout
- LAMP1
lysosome-associated membrane protein 1
- LRO
lysosome-related organelle
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MEF
mouse embryonic fibroblast
- RFP
red fluorescent protein
- SNARE
soluble N-ethylmaleimide-sensitive attachment protein (SNAP) receptor
- SQSTM1/p62
sequestosome 1
- STX
syntaxin
- TH
tyrosine hydroxylase
- VAMP
vesicle associated membrane protein/synaptobrevin
- VPS
vacuole protein sorting
- WT
wild type.
Introduction
The HOPS (homotypic fusion and vacuole protein sorting) complex, a tethering factor, interacts with RAB proteins and the SNARE (soluble N-ethylmaleimide-sensitive protein receptor) complex to mediate membrane fusion between endosomes and lysosomes or autophagosomes and lysosomes. It consists of 4 core subunits, VPS11, VPS16, VPS18, and VPS33A, which are called class C VPS (vacuole protein sorting) proteins, and 2 other associated proteins, VPS39 and VPS41.1 SM (Sec1/Munc18-related) proteins bind SNARE complexes to promote SNARE assembly. VPS33A is a member of the SM protein family, which is required for SNARE-mediated fusion processes.2,3 However, how VPS33A functions in SNARE-mediated fusion is largely unknown, especially in mammalian cells.
Electron microscopy combined with single-particle analysis and tomography has revealed that HOPS in yeast is an elongated and flexible complex. During the fusion process, its conformation converts from contracted to elongated to tether membranes.4 However, the molecular basis that mediates the conformational change is unknown. There is no evidence that alteration of this conformational change would affect the fusion process. In addition to its function in the maturation of lysosomes and in trafficking cargoes to lysosomes or lysosome-related organelles (LROs),5 the HOPS complex is involved in autophagosome-lysosome fusion by interacting with STX17 (syntaxin 17),6,7 one component of the autophagic SNARE complex.8
Although the function of the HOPS complex in lower eukaryotes and mammalian cells has been well studied, the underlying mechanisms in endocytic and autophagic pathways are largely unknown. An intriguing question in autophagic flux is how HOPS functions in the autophagosome-lysosome fusion. In addition, the pathophysiological roles of the HOPS complex in mammals are limited. Two mouse mutants of the HOPS complex have been characterized. One is a neural-specific Vps18-conditional knockout (cKO) mouse and the other is the buff (bf) mouse mutant, which carries a spontaneous point mutation (D251E) in Vps33a, a site that is conserved in different species.9 The Vps18 cKO mice showed severe neurodegeneration probably due to disruption of autophagy, endocytosis, and lysosomal functions. Defects in neuronal migration resulted from the upregulation of ITGB1/β1 integrin.10 The bf mutant was identified as a Hermansky–Pudlak syndrome model, exhibiting hypopigmentation, and platelet storage pool deficiency.9 It has been reported that motor deficits and Purkinje neuronal loss exist in bf mice.11 Both the Vps18-cKO mice and bf mice show neuronal loss, indicating that the HOPS complex is essential for neuronal survival. However, the underlying molecular basis is unknown.
Autophagy is a crucial process for cellular homeostasis. A variety of molecules involved in regulation of the autophagic process have been identified.12 Several studies have demonstrated that basal autophagy is essential for neuronal survival.13-15 Neurodegenerative diseases, such as Alzheimer and Parkinson diseases and schizophrenia, are associated with impaired autophagy.15,16 Here, we utilize the bf mouse with the VPS33AD251E point mutation to explore whether Purkinje cell loss is related to impaired autophagy. We found that the VPS33AD251E point mutation enhanced the interaction with the autophagic SNARE complex in both direct and indirect manners to impair the fusion process between autophagosomes and lysosomes.
Results
Bf mice exhibit motor deficits and Purkinje cell loss
Chintala et al. have reported motor dysfunction by SHIPRA protocols and Purkinje cell loss in old bf mice by immunohistochemical staining.11 To extensively assess the altered motor functions of bf mice more quantitatively, we used 2 classic methods, the open field test and the Rotarod test. There are 2 evaluation indicators in the open field test, the total distance and average speed.17,18 We tested different age groups and found that bf mice showed abnormal spontaneous locomotor activity in older age at 10 mo, but no significant changes in younger ages at 4 mo and 6 mo (Fig. 1A and B). The Rotarod test is extensively used to evaluate motor coordination and motor learning of animals.18,19 We then tested 10-mo-old bf mice by Rotarod and found that these mice displayed impaired motor learning activities (Fig. 1C). In addition, bf mice exhibited abnormal hindlimb-clasping at 16 mo of age (Fig. 1D), which is generally indicative of motor dysfunction.20 Histological analyses revealed that the gross anatomy of the brain of the bf mice at 10 mo of age was normal (Fig. 1E). Dopaminergic neurons in substantia nigra pars compacta play important roles in the regulation of motor coordination and motor learning. These neurons were specifically labeled by tyrosine hydroxylase (TH).21,22 We found no significant change in the number of TH-positive neurons in the substantia nigra pars compacta of bf mice at 10 mo of age (Fig. 1F). However, the number of Purkinje cells in the cerebellum, labeled by CALB1/calbindin, was decreased in bf mice at 6 mo of age (Fig. 1G). Purkinje cell loss has been documented in older mice at the age of approximately 13 or 14 mo.11 Taken together, the motor deficits in bf mice at the age of 10 mo may be attributable to lesions in the cerebellum as indicated by Purkinje cell loss.
Figure 1.
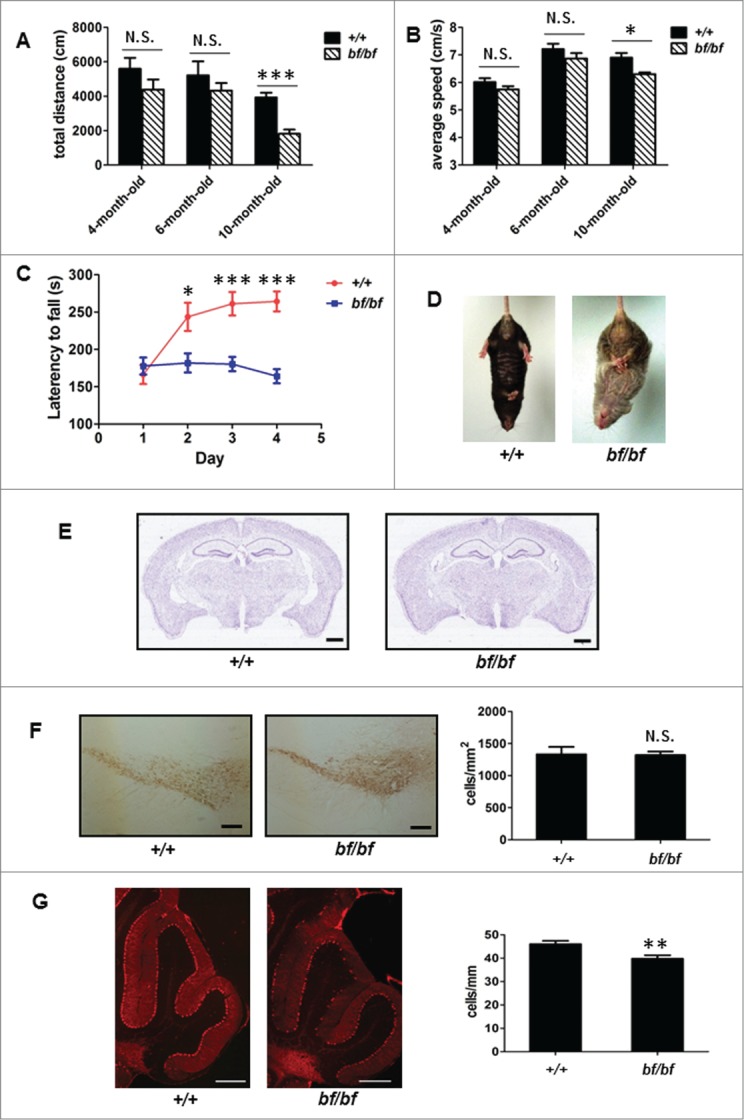
Motor deficits and Purkinje cell loss in bf mice. (A–C) Spontaneous locomotor activity and motor learning in bf mice were impaired in open field tests and Rotarod tests. The total distance (A) and the average speed (B) were decreased during 30-min test in bf mice at different ages. Bf mice exhibited defective motor learning in 10-mo-old mice (C). +/+, wild-type (B6), n = 9; bf/bf, bf mutant, n = 9. N. S., not significant (P > 0.05); *P< 0.05; ***P < 0.001 (P values for total distance: 4-mo-old, P = 0.1895; 6-mo-old, P = 0.3730; 10-mo-old, P = 0.0006. For average speed: 4-mo-old, P = 0.1547; 6-mo-old, P = 0.2367; 10-mo-old, P = 0.0318. For Rotarod test: d 1, P = 0.5147; d 2, P = 0.0175; d 3, P = 0.0004; d 4, P < 0.0001). (D) Bf mice culminated in abnormalhindlimb-clasping at 16 months of age. (E) Hematoxylin and eosin staining showed that the gross morphology of the brain was normal in bf mice (10 mo of age) compared with the wild type. Scale bar: 2 mm. (F) Immunohistochemistry by anti-TH (tyrosine hydroxylase) showed that the number of dopaminergic (DA) neurons in substantia nigra pars compacta of bf mice was not significantly changed compared with the wild-type (10 mo of age). +/+, 1333 ± 114 cells/mm2; bf/bf, 1319 ± 56 cells/mm2. Three mice were used. Scale bar: 100 μm. N.S., not significant (P = 0.9302). (G) The number of Purkinje cells labeled by CALB1/calbindin D28K was decreased in bf mice at 6 mo old. +/+, 46 ± 5 cells/mm; bf/bf, 40 ± 6 cells/mm. Three mice were used. Scale bar: 100 μm. **P < 0.01 (P = 0.0041).
The autophagic flux is impaired in bf mice
The HOPS complex is involved in endocytic and autophagic pathways. To explore the underlying mechanism of Purkinje cell loss, we first tested whether the autophagic pathway is affected in bf mice. LC3B-II is an indicator of autophagy.23 We found that the LC3B-II level was significantly increased in the cerebellum of bf mice at different ages (Fig. 2A). Similarly, the LC3B-II level in the substantia nigra was increased in 4- and 10-mo-old mice, and had a tendency to show increases (P = 0.0804) in 2-mo-old mice (Fig. 2B). However, the number of dopaminergic neurons in this region was normal in 10-mo-old mice (Fig. 1F), suggesting that Purkinje cells are more susceptible to cell death in bf mice upon the altered basal autophagy. Furthermore, electron microscopic analyses of Purkinje cells showed that autophagic vacuoles were significantly increased in bf mice (Fig. 2C). To confirm that the VPS33A mutation affects the autophagic process, we overexpressed mutant VPS33AD251E in human embryonic kidney (HEK) 293T cells, and found that the LC3B-II level was likewise increased compared with the overexpression of wild-type VPS33A (Fig. 2D). This suggests that mutant VPS33AD251E may compete with endogenous VPS33A and lead to altered autophagic activity with a dominant-negative effect. In addition, the SQSTM1/p62 (sequestosome 1) protein levels were increased in both cerebellum and substantia nigra, suggesting the autophagic flux is impaired (Fig. 2E and F). Taken together, our results demonstrate that the VPS33AD251E mutation affects the autophagic pathway in bf mice.
Figure 2.
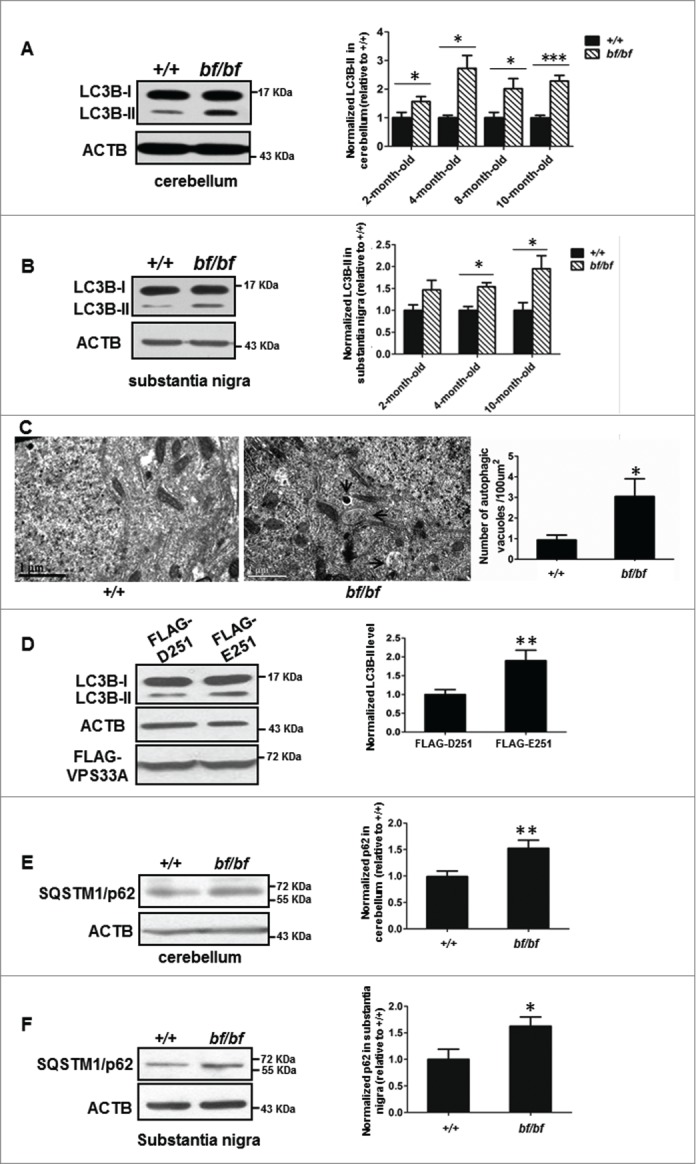
Autophagic activity was impaired in bf mice. (A) LC3B-II level was increased in the cerebellum of bf mice, and quantification data showed LC3B-II levels were increased at different ages of bf mice in the cerebellum (P values: 2-mo-old, P = 0.0387; 4-mo-old, P = 0.0005; 8-mo-old, P = 0.0291; 10-mo-old, P< 0.0001). *P < 0.05; ***P < 0.001; n = 6. (B) LC3B-II level was increased in the substantia nigra of bf mice at different age groups (P values: 2-mo-old, P = 0.0804; 4-mo-old, P = 0.0026; 10-mo-old, P = 0.0138). The representative blot was from 4-mo-old mice. N.S., not significant; *P < 0.05; **P < 0.001; n = 6. (C) Autophagic vacuoles were accumulated in Purkinje cells of bf mice at 10 mo of age as determined by electron microscopy. The arrows indicated autophagic vacuoles. Scale bar: 1 μm. Autophagic vacuoles from 20 cells were counted. *P < 0.05 (P = 0.0251). (D) LC3B-II was increased when overexpressed mutant VPS33AD251E in HEK293T cells compared with overexpressed wild-type VPS33A. **P < 0.01 (P = 0.0065), n = 4. (E) SQSTM1/p62 in the cerebellum of bf mice at 4 mo of age was increased. **P < 0.01 (P = 0.0080), n = 10. (F) SQSTM1/p62 in the substantia nigra of bf mice at 4 mo of age was increased. *P < 0.05 (P = 0.0296), n = 8. ACTB (β-actin) was used as a loading control.
Autophagosome-lysosome fusion is defective in bf MEFs
As the HOPS complex is involved in autophagosome-lysosome fusion in vitro,6 we tested whether the autolysosome formation is altered in bf cells. mRFP-GFP-LC3B is utilized to track the transitions of different stages of the autophagic process.24,25 Since GFP is sensitive to acidic condition, the green signal from GFP will disappear when autophagosomes fuse with lysosomes. Thus, yellow and red puncta represent autophagosomes and autolysosomes, respectively. Mouse embryonic fibroblasts (MEFs) transfected with mRFP-GFP-LC3B were starved for 2 h before immunofluorescence staining. We observed that there were more yellow dots accumulated in bf MEFs compared with WT MEFs (Fig. 3A). This phenomenon could result either from the impaired fusion of autophagosomes and lysosomes or from elevated lysosomal pH. To distinguish these 2 possibilities, we incubated a LysoSensor Green DND-189 with MEFs for 30 min to monitor the acidic condition of lysosomes. Our results revealed that the lysosomal acidification in bf cells was unaltered compared with WT cells (Fig. 3B). To further examine the fusion events between autophagosomes and lysosomes, we observed the localization of endogenous LC3A/B and LAMP1. MEFs were incubated with lysosomal enzyme inhibitors including pepstatin A (10 μg/ml), E-64 (10 μM) and leupeptin (1 μg/ml) for 24 h, and starved for 2 h before fixation. The colocalization of LC3A/B and LAMP1, which represented the LC3A/B fraction on autolysosomes, was significantly decreased in bf cells compared with WT cells (Fig. 3C and D). To exclude the possibility that decreased colocalization of LAMP1 and LC3A/B was likely due to the increased total LC3A/B puncta, we quantified the LC3A/B dots and the total number of LC3A/B puncta was no significant change in bf MEFs (Fig. 3D), suggesting that the LC3A/B fraction on autophagosomes is likely increased which is in agreement with the findings in Fig. 3A. Taken together, these results suggest that fusion between autophagosomes and lysosomes in bf MEFs is impaired.
Figure 3.
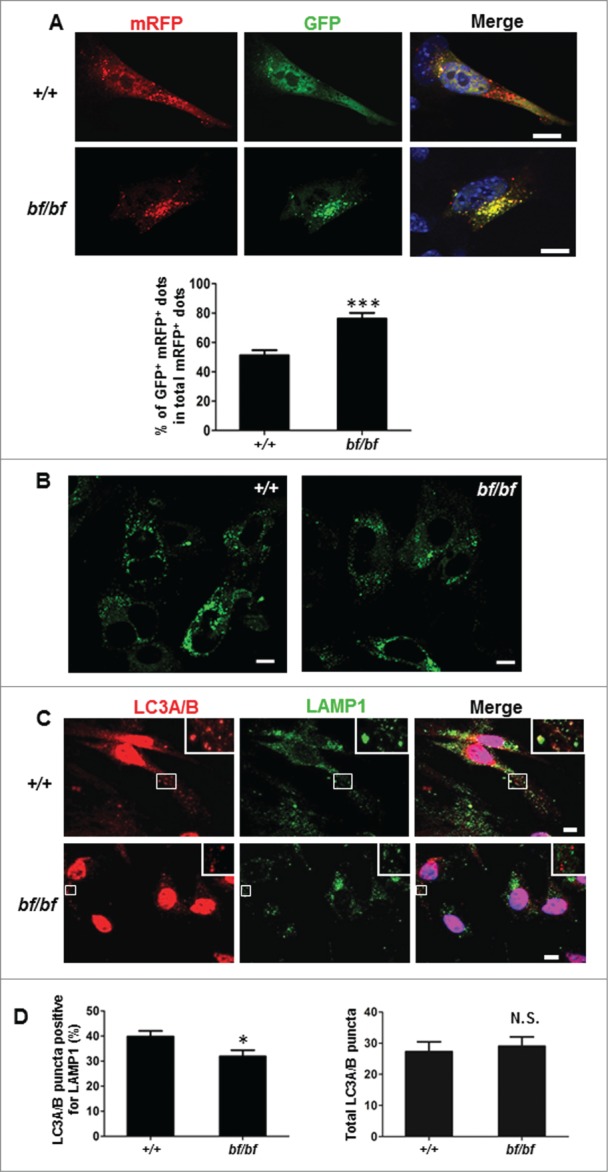
Fusion between autophagsomes and lysosomes was impaired in bf MEFs. (A) MEFs were transfected with mRFP-GFP-LC3B. After 24 h, cells were starved in DMEM without amino acids for 2 h before fixation and analyzed by confocal microscopy. Quantification data are presented as the percent of yellow puncta (autophagosomes) per total red puncta (autophagosomes and autolysosomes) from 20 randomly selected images. DAPI staining of nuclei is labeled in blue. Scale bar: 10 μm. ***P< 0.0001. (B) MEFs were stained with LysoSensor™ Green DND-189 for 30 min to observe the acidification of lysosomes. There was no apparent change in lysosomal acidification in bf MEFs compared with wild-type cells. Scale bar: 10 μm. (C) The colocalization of endogenous autophagosomal marker LC3A/B and lysosomal marker LAMP1 was reduced in bf MEFs. Cells were incubated with lysosomal inhibitors (E-64, pepstatin A and leupeptin) for 24 h and starved for additional 2 h before fixation. (D) The percentage of LC3A/B puncta colocalizing with LAMP1 relative to total LC3A/B puncta was lower in bf MEFs from 30 cells (left panel). *P < 0.05 (P = 0.0241). The total counted LC3A/B puncta were not significantly changed in bf MEFs compared with the wild-type MEFs. N.S., not significant (P = 0.7052). Scale bar: 10 μm.
VPS33A protein stability, subcellular localization, and the protein levels of other HOPS subunits in bf mice are normal
We next explored how the VPS33A point mutation disrupts its protein function to impair autophagy. We generated a polyclonal VPS33A antibody to examine the VPS33A expression pattern in mouse tissues. VPS33A was ubiquitously expressed and was highly expressed in the brain (Fig. 4A), suggesting it may play important functions in the central nervous system. We also found that the VPS33A protein levels in different brain sub-regions and the protein stability of several other HOPS subunits (VPS16, VPS18 and VPS11) were normal in bf mice compared with wild-type mice (Fig. 4B and C). Furthermore, both WT and mutant VPS33AD251E were partially colocalized with LAMP1-GFP labeled lysosomes in HeLa cells (Fig. 4D). To further confirm the localization of VPS33A in WT and bf mice, we performed subcellular fractionation by the discontinuous sucrose density gradient assay. We found that both WT and mutant VPS33A were in the same fractions, which correspond to the fractions enriched with lysosomes labeled by LAMP1 (Fig. 4E). Taken together, these results suggest that the point mutation D251E does not change the protein stability, complex assembly, and subcellular localization, which distinguishes it from the null mutation of VPS33A.
Figure 4.
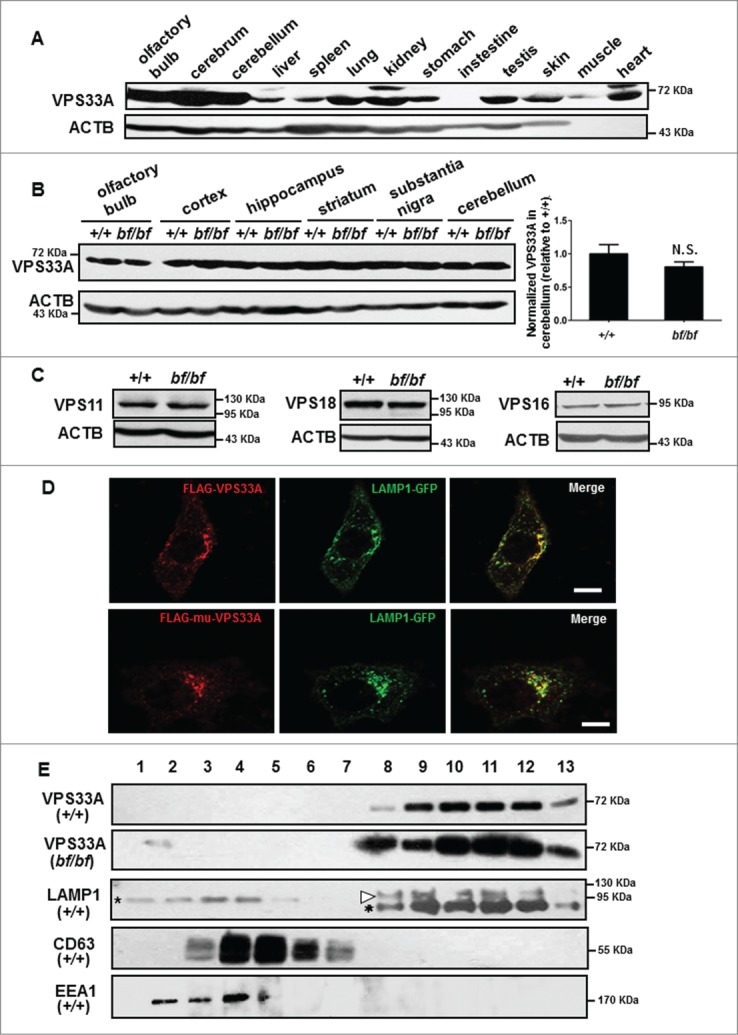
Normal steady-state levels of HOPS proteins in bf mice and unaltered subcellular localization of mutant VPS33AD251E. (A) VPS33A was ubiquitously expressed in multiple mouse tissues. Note that higher expression was shown in the central nervous system. (B) The steady-state protein level of VPS33A was normal in different brain subregions of bf mice compared with age- and sex-matched wild-type mice. The quantification data on the right demonstrated that VPS33A protein level was not significantly different in cerebellum of bf mice compared with wild-type mice. N.S., not significant (P = 0.2383), n = 7. (C) The steady-state protein levels of VPS11, VPS18 and VPS16 were not altered in the cerebellum of bf mice compared with the wild-type mice. (D) Wild-type VPS33A or VPS33AD251E (mu) was partially colocalized with LAMP1 in HeLa cells. Scale bar: 10 μm. (E) No difference in the VPS33A distribution in the cerebellum in wild-type (+/+) and bf mice by sucrose gradient analysis (fractions approximately 1 to 13). Antibodies of LAMP1, CD63 and EEA1 were used as markers for lysosomes, late endosomes/multivesicular bodies, and early endosomes, respectively. In the LAMP1 blot, Asterisks indicate nonspecific bands and arrowhead indicates the 110 kDa LAMP1 band. ACTB (β-actin) was used as a loading control.
The D251E point mutation of VPS33A enhances its interaction with the autophagic SNARE complex
To further explore the underlying mechanism of impaired autophagosome-lysosome fusion in bf cells, we examined whether the VPS33AD251E point mutation affects its interaction with the autophagic SNARE complex (STX17-SNAP29-VAMP8), which has been identified as the HOPS complex interactor to mediate autophagosome-lysosome fusion.6,8 We first tested interactions between WT or mutant VPS33A and STX17 by coimmunoprecipitation (coIP). The interaction of mutant VPS33AD251E and STX17 was enhanced compared with WT in both normal and rapamycin-treated (a drug that induces autophagy by inhibiting the MTOR pathway) conditions (Fig. 5A).
Figure 5.
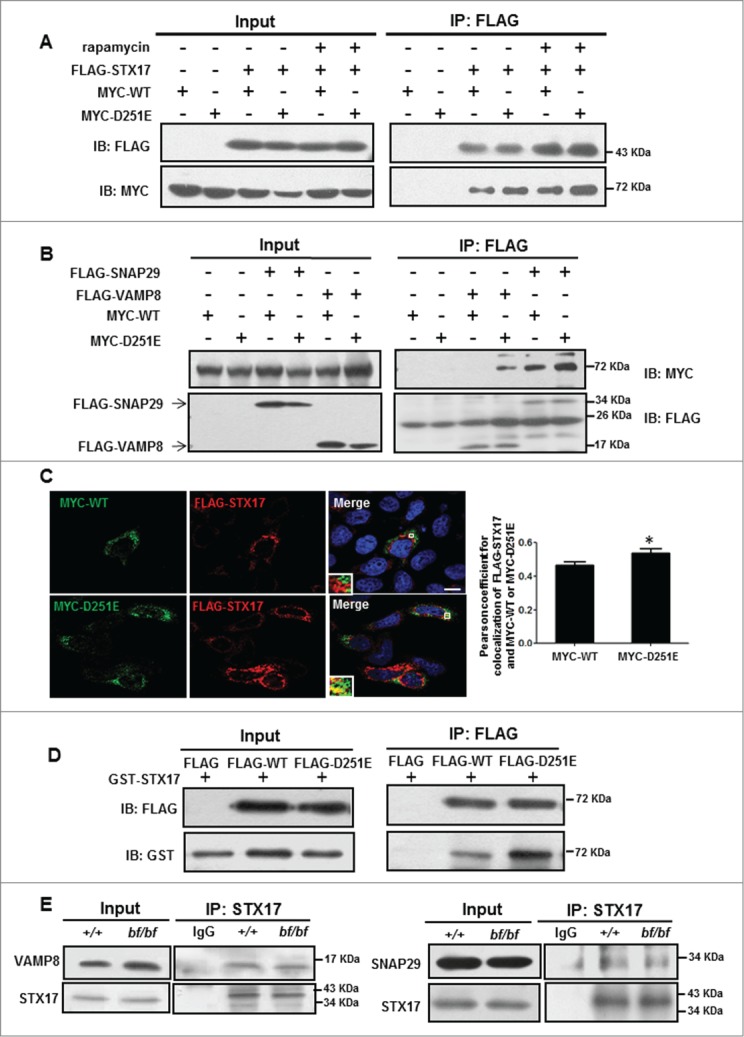
The interactions of mutant VPS33A and autophagic SNARE complex were increased. (A) MYC-VPS33AWT or VPS33AD251E and FLAG-STX17 were cotransfected in HEK293T cells. Cells were treated with DMSO or rapamycin (100 nM) for 4 h before being harvested, immunoprecipitated with FLAG beads, and followed by anti-MYC immunoblotting. FLAG-STX17 precipitated more mutant MYC-VPS33A with or without rapamycin treatment as shown in the blots. (B) MYC-VPS33A WT or VPS33AD251E and FLAG-SNAP29 or VAMP8 were cotransfected in HEK293T cells. After 24 h, the cells were harvested for immunoprecipitation analyses. FLAG-SNAP29 or VAMP8 precipitated more mutant MYC-VPS33A as shown in the blots. (C) COS7 cells were cotransfected with MYC-VPS33A (WT or D251E) and FLAG-STX17 for 24 h. Cells were cultured in DMEM without amino acids for 2 h before fixation. Then cells were fixed, made permeable in 0.1% saponin, stained with anti-MYC and anti-FLAG antibodies, and finally examined under a confocal microscope. Scale bars: 10 µm. Quantification of colocalization represented by Pearson correlation coefficient showed that STX17 colocalized more with the mutant VPS33A than the wild-type. /P< 0.05 (P = 0.0419), n = 20 cells. (D) FLAG alone, FLAG-VPS33AWT or FLAG-VPS33AD251E were expressed in HEK293T cells, and immunoprecipitated with FLAG beads. Then the FLAG beads were incubated with bacterially purified GST-STX17. Samples were immunoblotted with anti-GST antibody. FLAG-D251E binds more GST-STX17 as shown in the blots. (E) STX17 pulled down almost equal amounts of endogenous VAMP8 and SNAP29 in both wide-type and bf MEFs. IB, immunoblotting; IP, immunoprecipitation.
We then tested whether the mutant VPS33A also bind stronger to SNAP29 and VAMP8. Indeed, the mutant VPS33AD251E likewise enhanced interactions with SNAP29 and VAMP8 by coIP assays (Fig. 5B). We confirmed the increased interaction of mutant VPS33AD251E with STX17 by immunofluorescence. After 24 h cotransfection of MYC-VPS33A (wild type [WT] or D251E) and FLAG-STX17 in cultured cells and starvation for 2 h before fixation, we found that the colocalization ratio (Pearson coefficient) of mutant VPS33A and STX17 was significantly increased (Fig. 5C).
Inferred from the structure of VPS33, the D251 residue is localized in the domain 3a region, which is predicted as the interacting interface between VPS33 and syntaxins.26 We hypothesized that the direct interaction between VPS33AD251E and STX17 is enhanced. To test this, FLAG alone, FLAG-VPS33AWT or FLAG-VPS33AD251E were expressed in HEK293T cells, and immunoprecipitated using FLAG beads. Then the FLAG beads were incubated with bacterially purified GST-STX17. We showed that the direct interaction of VPS33AD251E and STX17 was increased (Fig. 5D).
Furthermore, as the SM protein VPS33 has been reported to protect trans-SNARE complex from disassembly by Sec18 in vitro,27 we tested whether the VPS33AD251E mutation affects the autophagic SNARE complex formation. By endogenous IP, we found that STX17 formed a complex with VAMP8 and SNAP29 properly in bf cells (Fig. 5E), suggesting that the impaired autophagosome-lysosome fusion efficiency does not result from the abnormal formation of the autophagic SNARE complex.
The D251E point mutation of VPS33A enhances interactions with other subunits of the HOPS complex except VPS16
As the steady-state levels of mutant VPS33A and HOPS complex were unaffected (Fig. 3B and C), we then tested whether the point mutation (D251E) interferes with its interactions with other subunits of the HOPS complex. In our coIP assays, the VPS33AD251E mutation increased interactions with VPS39, VPS18, VPS11 or VPS41, but not with VPS16 (Fig. 6). To confirm these results, we generated different mutations, including converting D251 to other residues (L (leucine, Leu), K (Lysine, Lys), N (Asparagine, Asp)) and deleting D251 (D251Δ). All the amino acid substitutions revealed enhanced interactions with VPS39, but not with VPS16 (Fig. S1A and B). Similar to the D251E mutation, D251Δ resulted in enhanced interactions with VPS39 or VPS18, but not with VPS16 (Fig. S1C and D).
Figure 6.
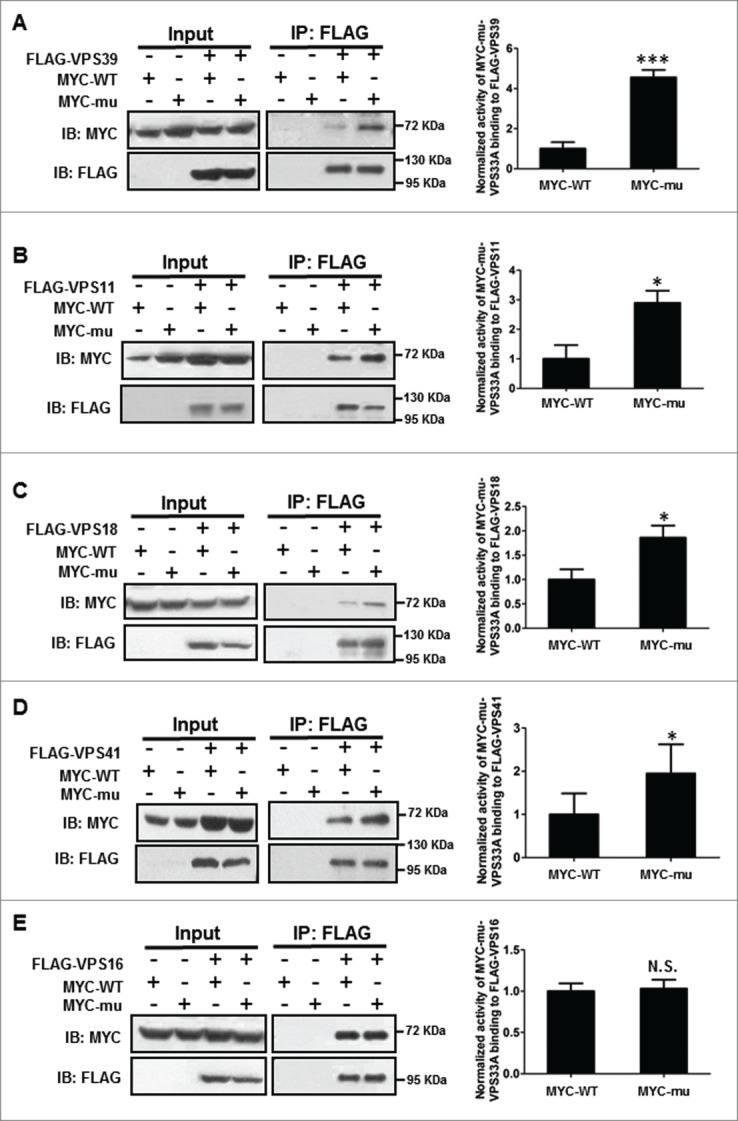
Interactions were increased between mutant VPS33A and other subunits of HOPS complex except VPS16 by coimmunoprecipitation assays. (A–E) MYC-WT (wild-type) or VPS33AD251E (mu) and FLAG-VPS39, -VPS11, -VPS18, -VPS41 or -VPS16 were cotransfected in HEK293T cells, respectively. After 24 h, the cells were harvested, FLAG-tagged HOPS subunits immunoprecipitated with FLAG beads and samples were immunoblotted for detecting MYC-VPS33A. In the blots, the amount of FLAG-tagged proteins was mostly equalized, in order to show the increase of immunoprecipitated proteins in the mutant VPS33AD251E lanes. Compared with the wild-type VPS33A, the mutant VPS33AD251E increased the interactions with (A) VPS39 (P = 0.0004), (B) VPS11 (P = 0.0373), (C) VPS18 (P = 0.0295) and (D) VPS41 (P = 0.0477), but did not affect the interaction with (E) VPS16 (P = 0.8325). *P < 0.05; ***P < 0.001; N.S., not significant; n = 5. IB, immunoblotting; IP, immunoprecipitation.
As the exogenous VPS33A competes for the endogenous VPS33A in complex formation, we next tested whether the direct interactions between these protein pairs exist in a cell-free system. We expressed the VPS18-VPS33A pair in the E. coli and performed affinity isolation assays. We confirmed the enhanced interaction of mutant VPS33A with VPS18 (Fig. 7A). In addition, we confirmed the enhanced interactions of mutant VPS33A with VPS18 or VPS39 in the extracts of cerebellum by endogenous IP assays using the anti-VPS33A antibody (Fig. 7B). These results suggest that the D251 residue is critical for mediating VPS33A interactions with other subunits of the HOPS complex except for VPS16.
Figure 7.
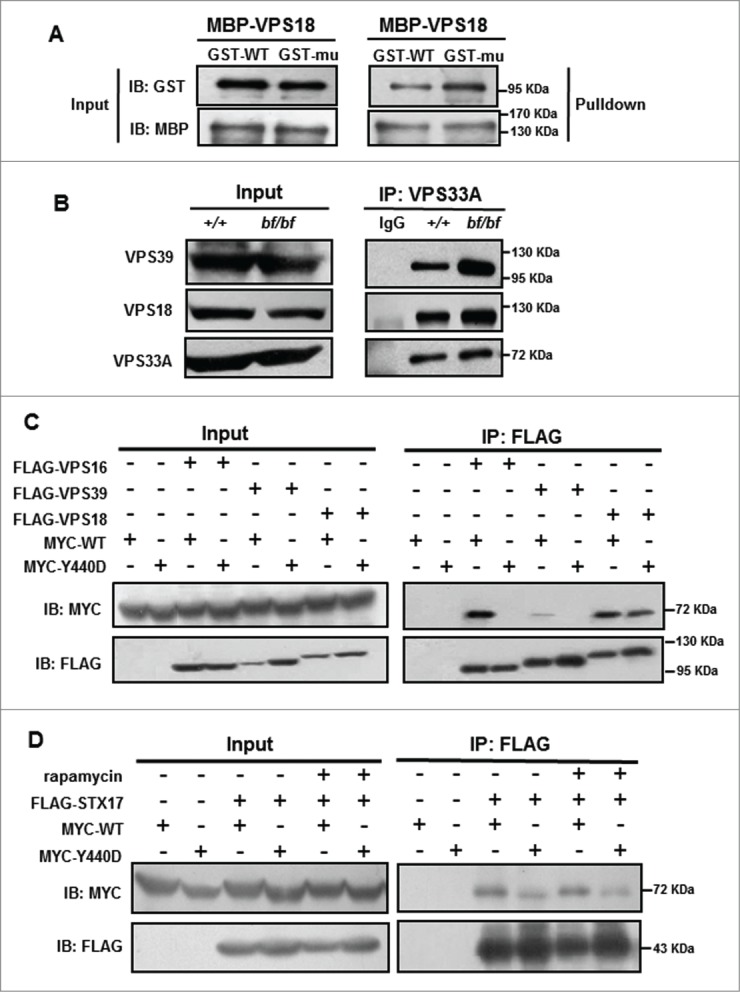
Increased interactions with HOPS subunits (VPS18 and VPS39) by affinity isolation assays and disruption of interactions between VPS33AY440D and HOPS subunits (VPS16, VPS18 and VPS39) or STX17. (A) VPS18 pulled down increased amounts of mutant VPS33AD251E (mu) by the maltose binding protein (MBP)-affinity isolation assay compared with the wild-type VPS33A (WT). (B) The cerebellum lysates were incubated with IgG or anti-VPS33A antibody, and immunoprecipitated using protein A beads. VPS33A pulled down increased amounts of endogenous VPS39 or VPS18 in bf mice. IgG serves as a negative control. (C) MYC-WT or VPS33AY440D and FLAG-VPS39, -VPS18, or -VPS16 were cotransfected in HEK293T cells. After 24 h, the cells were harvested, immunoprecipitated with FLAG beads and followed by anti-MYC immunoblotting. The interactions between mutant VPS33AY440D and VPS16, VPS39, or VPS18 were greatly reduced compared with the wild-typeVPS33A. (D) MYC-WT or VPS33AY440D and FLAG-STX17 were cotransfected in HEK293T cells. Cells were treated with DMSO or rapamycin (100 nM) for 4 h, then harvested and immunoprecipitated with FLAG beads and followed by anti-Myc immunoblotting. The Y440D mutation decreased its interaction with STX17 both in normal and rapamycin-treated conditions compared with wild-type VPS33A. The experiments in (A–D) were repeated at least twice. IB, immunoblotting; IP, immunoprecipitation.
We then explored whether the enhanced HOPS interactions result in altered interaction with STX17. Inferred from the crystal structure of human VPS33A, the Tyr438-to-Asp (Y438D) mutation is expected to disrupt its interaction with VPS16.28 We made a homologous Y440D mutation in mouse VPS33A. Indeed, we found that the Y440D mutation greatly reduced the interactions with VPS16, VPS39 or VPS18 by coIP assays (Fig. 7C). Similarly, the Y440D mutation decreased its interaction with STX17 both in normal and rapamycin-treated conditions (Fig. 7D). In addition, the Y440 residue is not located in the interaction interface between VPS33A and STX17.26 Thus it is unlikely to interfere with the direct interaction with STX17. Thus we propose that the enhanced interactions of VPS33AD251E with other HOPS subunits may indirectly affect its interaction with STX17 or the autophagic SNARE complex.
The endocytic pathway and lysosome function are normal in bf mice
The HOPS complex also functions in the endosome-lysosome fusion process.1,29 If this endocytic pathway is impaired, it may affect the lysosomal functions and lead to impaired autolysosomal formation. For example, lack of VPS18 caused the disruption of autophagy, endocytosis, and lysosomal function.10 We therefore tested whether the endocytic pathway and lysosomal function in bf mice are affected. EGFR (epidermal growth factor receptor) is a typical cargo protein that is transported to the lysosomes through the endocytic pathway. After EGF stimulation for indicated time points, cells were harvested for immunobloting. EGFR trafficking is normal in bf MEFs compared with WT MEFs (Fig. 8A and B). We then examined the endocytic compartments in MEFs by electron microscopy (Fig. 8C and D). We adopted definitions for autophagosomes, endolysosomes, and lysosomes as previously described.30 We found that the number of endolysosomes as well as the number of lysosomes was not significantly changed in bf cells. However, the number of autophagosomes was significantly increased in bf MEFs (Fig. 8E), which is consistent with the data obtained from Purkinje cells of bf mice (Fig. 2C).
Figure 8.
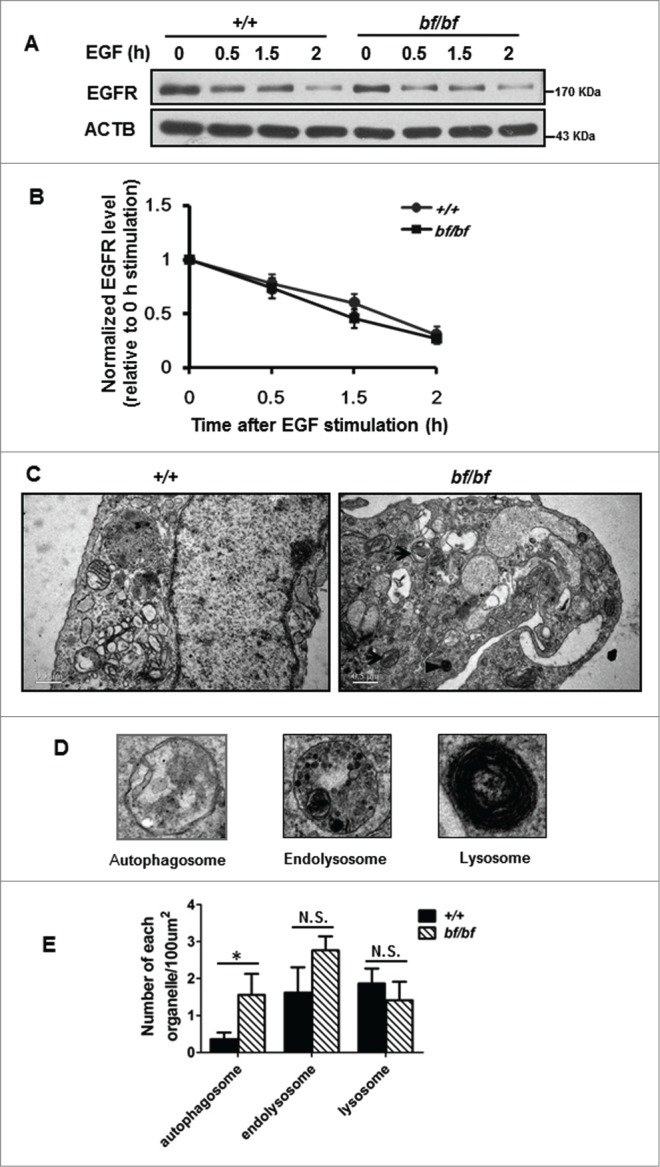
The endocytic pathway was normal in bf cells. (A) MEFs isolated from bf mice were starved overnight and stimulated with 100 ng/ml EGF. Cells were harvested at indicated time points and the protein levels of EGFR were determined. ACTB (β-actin) was used as a loading control. (B) EGFR degradation was quantified by using the protein level at 0 min as control. There was no significant difference between the wild-type (+/+) and bf MEFs. n = 5. (C) Ultrastructural analyses of various endocytic or autophagic organelles in MEFs by electron microscopy. The arrows indicate autophagosomes and the arrowhead indicates a lysosome. Scale bar: 500 nm. (D) Representative structures of the autophagosome, endolysosome and lysosome. (E) The numbers of autophagosomes (P = 0.0367), endolysosomes (P = 0.2091) and lysosomes (P = 0.4889) were quantified from 80 randomly selected areas. *P < 0.05; N.S., not significant.
We have shown that the lysosomal acidification was normal (Fig. 3B). To further assess whether the function of the lysosomes is normal, we measured the maturation of lysosomal proteases in bf mice. CTSB (cathepsin B) and CTSD are 2 key lysosomal proteases. They are cleaved into mature form in the lysosomes from premature form. We found that both the immature and mature forms of CTSB and CTSD were normal in the cerebellum of bf mice (Fig. S2A). We then incubated the cells for 1.5 h with Magic Red-CTSB, which is a membrane-permeable probe and which is cleaved by CTSB into a fluorescent product.29 We confirmed that the activity of CTSB was normal in bf MEFs (Fig. S2B). All these results suggest that the endocytic pathway and lysosomal functions are normal in bf tissues and cells.
Discussion
It has been reported that the HOPS complex is involved in endosome-lysosome fusion (endocytic pathway), autophagosome-lysosome fusion (autophagic pathway), and the biogenesis of lysosome-related organelles (LRO pathway). The intriguing point is how the HOPS complex distinguishes and functions in these 3 pathways. Additional protein complexes may be required for this selectivity. For example, AP-3 interacts with HOPS in the LRO pathway,31 while the endocytic trans-SNARE (STX7, STX8, Vti1b, and VAMP7), RAB7 and PLEKHM1 interact with HOPS in endosome-lysosome fusion,32,33 and the autophagic SNARE (STX17, SNAP29, and VAMP8) interacts with HOPS in autophagosome-lysosome fusion.6,7 In the bf mice, the LRO pathway defect is rescued by the transfection of wild-type VPS33A in the melanocytes,9 suggesting a loss-of-function effect. In this study, we did not detect an effect on the endocytic pathway. In the autophagic pathway, we detected a gain-of-function effect on the interactions with autophagic SNARE and several other HOPS subunits. However, the effect on autophagosome-lysosome fusion was impaired. In this regard, it has a loss of function in autolysosome formation. These findings suggest that the HOPS complex does not function in a conserved mechanism in different pathways and the bf mutation likely affects the LRO pathway and autophagic pathway in different mechanisms.
The pathogenesis of Purkinje cell loss in the bf mutant is intriguing.9 Likewise, Purkinje cell loss and an increased level of LC3B-II in the cerebellum were observed in the Vps18-cKO mice. Both multivesicular bodies and autophagosomes were increased, suggesting that the endocytic pathway and autophagic flux are both affected due to the loss of VPS18.10 However, in bf mice, the endocytic pathway and lysosomal functions are apparently normal. The point mutation in VPS33AD251E did not alter its protein's stability or subcellular localization. Notably, unlike the knockdown or knockout of a HOPS core subunit, the point mutation VPS33AD251E did not destabilize other HOPS subunits. This suggests that the bf mutant is a unique tool in the study of VPS33A functions in membrane fusion. The VPS33AD251E mutation is distinct but hypomorphic to the loss of VPS18. It only compromises the autophagosome- lysosome fusion process without disrupting the endosome-lysosome fusion. This explains why bf mice are viable and less severely affected whereas the Vps18−/− mice are not viable.9,10
Inferred from the crystal structure of C. thermophilum VPS33, the D251 residue is localized in the domain 3a region, which is predicted as the interacting interface between VPS33 and syntaxins.26 Our results support that the D251-containing domain 3a likely mediates the direct interaction with STX17. In addition, the crystal structure of human VPS33A revealed that domain 3a is unlikely to interact directly with VPS16.28 Indeed, our data showed that the interaction between mutant VPS33AD251E and VPS16 was unaffected.
In bf cells, the autophagic SNARE complex forms properly, suggesting that the D251E mutation has little or no effect on SNARE complex formation. The enhanced interactions of VPS33AD251E with the autophagic SNARE complex and the HOPS complex may have profound effects on the autophagosome-lysosome fusion. Membrane fusion is dependent on the conformational transition of the SNARE complex from the assembly state of trans-SNARE to the disassembly state of cis-SNARE. SEC18 promotes the disassembly of cis-SNARE,27,34,35 whereas the HOPS complex facilities the assembly of trans-SNARE.36 Recently, it has been reported that VPS33A directly protects the SNARE complex from Sec18-mediated disassembly in the membrane fusion process.27 Except for VPS16, the VPS33AD251E mutation enhanced interactions with other subunits of the HOPS complex (Fig. 6), suggesting that the conformation of the HOPS complex was likely changed from an elongated form to a contracted status.4 In addition, the altered HOPS conformation induced by the VPS33AD251E mutation may cause enhanced interaction with an autolysosomal SNARE component STX17, which in turn may preclude the transition from trans-SNARE to cis-SNARE during membrane fusion. Thus, the bf mutation likely affects the flexibility of HOPS complex or SNARE trans-to-cis transition during the autophagic fusion process due to the enhanced protein interactions. However, the stability of HOPS complex (Fig. 4C) and the assembly of autophagic SNARE (Fig. 5E) were not affected. Nevertheless, this hypothesis awaits further investigation by structural biology or in vitro reconstitution SNARE fusion assays. We propose that the increased interaction of VPS33AD251E with STX17 could be resulted from their enhanced direct interaction, as well as the indirect consequence of enhanced HOPS assembly. Therefore, the VPS33AD251E mutation plays dual roles by increasing the HOPS complex assembly and its association with the autophagic SNARE complex, which indirectly and directly affects the autophagome-lysosome fusion (a model in Fig. S2C). The impaired autolysosome formation eventually leads to Purkinje cell loss in bf mice. These findings suggest that the HOPS complex does not function in a conserved mechanism in different fusion events and the bf mutation selectively affects the autophagic pathway, but not the endocytic pathway. This provides insights into the molecular machinery underlying autophagosome-lysosome fusion.
Interestingly, the Purkinje cell loss was noted in bf mice at 6 mo of age (Fig. 1G) and the number of TH+ neurons in the substantia nigra was normal at least at 10 mo of age (Fig. 1F), suggesting that the Purkinje cells are more vulnerable to cell death upon altered basal autophagic activity. Consistently, in Atg5 or Atg7 cKO mice, different neuron types have variable degrees of cell death. Purkinje cells are among the most vulnerable neurons and exhibit cell loss within 12 postnatal wk,13,14 whereas midbrain dopaminergic neurons are reduced until 9 mo of age.37 It has been speculated that different brain regions may require different basal autophagic activities to maintain neuronal survival. Therefore, impaired autophagy may have different consequences on different neuronal types although the underlying mechanisms are unknown.38 In addition, motor deficits are more severe in older mice (Fig. 1A to D). It is likely that other neuronal cell loss may occur with aging, but the Purkinje cell loss to be the most prominent in earlier stage. The bf mutant provides a good model in further understanding the impact of basal autophagy, particularly the late autophagic pathway, in neuronal survival and neurodegeneration.
Materials and Methods
Mice
The bf mutant mice (bf/bf )9 and control C57BL/6J (wild-type, WT) mice were originally obtained from Dr. Richard T. Swank's laboratory and bred in the animal facility of the Institute of Genetics and Developmental Biology (IGDB), Chinese Academy of Sciences. All experimental procedures were approved by the Committee on Animal Health and Care of IGDB.
Plasmids
Full-length cDNAs encoding mouse VPS33A (both wild-type and the D251E mutation), VPS11, VPS16, VPS18, VPS41, VPS39, VPS41, STX17, SNAP29, and VAMP8 were amplified by PCR from the total cDNA of mouse brain and subcloned into the pCMV-tag2B vector (with FLAG-tag) or pCMV-tag3B vector (with a MYC-tag) as indicated in the figures. Other mutant mouse VPS33A constructs used in coimmunoprecipitation assays were generated by a QuickChange mutagenesis kit (Stratagene, La Jolla, CA, USA). mRFP-GFP-LC3B and LAMP1-GFP plasmids were gifts from Dr. Li Yu (Tsinghua University, Beijing, China).
Antibodies and reagents
Polyclonal VPS33A antibody was generated in New Zealand White rabbits against the full-length mouse Vps33a gene with a GST-tag. Affinity-purified antibody was produced by using nitrocellulose membranes with the full-length VPS33A antigen. Other antibodies used in this study were: rabbit polyclonal anti-LC3B (Sigma-Aldrich, L7543) or anti-LC3A/B (Novus Biologicals, NB100-2331), anti-tyrosine hydroxylase (Chemicon, AB152), anti-c-Myc (Sigma-Aldrich, C3956), anti-EGFR (Fitzgerald, 20R-ER004), anti-GST (Santa Cruz Biotechnology, SC-138), anti-CTSB/cathepsin B (Abcam, ab33538), anti-TUBB/β-tubulin (Santa Cruz Biotechnology, sc-9104); goat polyclonal anti-VPS11 (Santa Cruz Biotechnology, sc-100893), anti-VPS16 (Santa Cruz Biotechnology, sc-86939), anti-VPS18 (Santa Cruz Biotechnology, sc-100890), anti-VPS39 (Santa Cruz Biotechnology, sc-104761), anti-CTSD/cathepsin D (Santa Cruz Biotechnolgy, sc-6486); rat monoclonal anti-LAMP1 (BD Biosciences, CD107); mouse monoclonal anti-CALB1/calbindin-D-28K (Sigma-Aldrich, C9848), anti-FLAG (Sigma-Aldrich, F3165), anti-CD63 (Millipore, CBL553), anti-ACTB/β-actin (Sigma-Aldrich, A5441), anti-EEA1 (BD Biosciences, 610457), anti-VAMP8 (Novus Biologicals, EP2629Y), anti-STX17/syntaxin 17 (Sigma-Aldrich, HPA001204), anti-SNAP29 (Abcam, ab138500), anti-SQSTM1/p62 (MBL, PM045). All Alexa Fluor-conjugated secondary antibodies were from Invitrogen (donkey anti-Mouse IgG (H+L), Alexa Fluor 594 conjugate (R37115); donkey anti-Rabbit IgG (H+L), Alexa Fluor 488 conjugate (R37118); donkey anti-Rabbit IgG (H+L), Alexa Fluor 594 conjugate (A-21209); goat anti-rat IgG (H+L), Alexa Fluor 488 conjugate (A-11006)). Reagents used in this study were: E-64 (Sigma-Aldrich, E3132), pepstatin A (Sigma-Aldrich, P5318), leupeptin (Sigma-Aldrich, L2884), rapamycin (Sigma-Aldrich, R0395), saponin (Sigma-Aldrich, S7900), LysoSensor™ probes (Invitrogen, L7535). The Magic Red™ Cathepsin Detection Kit was from Immunochemistry Technologies (#937).
Spontaneous locomotor activity
A single mouse was placed in a chamber (40 cm length × 40 cm width × 35 cm height) for 30 min. Parameters including total distance and average speed were measured by Anilab software (AniLab, Ningbo, China). After each test, the chambers were cleaned with water and 75% alcohol. At least 6 mice at different age groups were used in these experiments.
Rotarod test
The experiment was performed as described previously.39 In brief, for each test, the Rotarod apparatus (Ugo Basile, Comerio, Italy) was set to accelerate from 4 to approximately 40 r.p.m. within 5 min. A 10-min break was granted to each mouse between the tests. The experiment lasted for 4 d with 4 tests per d.
Cell culture
HeLa cells, COS7 cells, HEK293T cells, and MEFs were cultured in regular Dulbecco's modified Eagle's medium (DMEM) from HyClone (SH30022.01B) supplemented with 10% fetal bovine serum (GIBCO, 10099-141), 100 μg/ml penicillin and streptomycin in a 5% CO2 incubator. For starvation treatment, cells were washed with phosphate-buffered saline (HyClone, SH30256.01) twice and incubated in amino acid-free DMEM without serum (starvation medium) for 2 h. Cells were transiently transfected using Lipofectamine 2000 (Invitrogen, 11668-019).
Histological studies
For Nissel staining or immunohistochemical staining, 20-µm frozen sections of mouse brain were prepared. The procedures were followed by standard protocols. The results were captured by a TS100 microscope (Nikon, Tokyo, Japan).
Electron microscopy
To observe the autophagic vacuoles in Purkinje cells of cerebellum, mice were anesthetized and perfused with 2% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. The cerebellum was removed and cut into several cubes with 1-mm3 size. Then the cubes were postfixed overnight at 4°C with 2% paraformaldehyde, 2.5% glutaraldehyde, 0.5% tannic acid. The cubes were rinsed and treated with 1% osmium tetroxide for 2 h at 4°C. After washing, the cubes were dehydrated in an ascending series of dilutions of acetone and impregnated in ERL-4206 Spur (NSA (SPI-CHEM, 02829-AF), ERL 4221 (SPI-CHEM, 02815-NA), DER 736 (SPI-CHEM, 02830-AA), DMAE (SPI-CHEM, 02824-AA)) at 70°C for 12 h. We observed the organelles in MEFs with a JEM1400 electron microscope (JEOL, Japan) by following the procedures previously described.30
Immunofluorescence confocal imaging
Cells were grown on glass coverslips. Under indicated experimental conditions, the cells were washed with phosphate-buffered saline and fixed in 4% paraformaldehyde for 15 min. All procedures were previously described.40 To label lysosomes, LysoSensor Green DND-189 (1:1000) was incubated with MEFs for 30 min. Magic Red CTSB substrate was utilized as described in kit protocols. All the confocal images were captured with C2 laser-scanning microscope (Nikon, Japan).
Immunoblotting
Cell lysates prepared in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 [aMReSCO, 0694]), and tissue lysates prepared in RIPA buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% DOC [aMReSCO, 0613], 0.1% SDS [aMReSCO, 0227], 1% NP-40 [Abcam, ab142227]) were subjected to 10–15% SDS polyacrylamide gel electrophoresis, and followed by regular procedures.40
Sucrose gradient centrifugation
Mouse cerebellum was homogenized in sample buffer (250 mM sucrose, 20 mM Tris-HCl, pH 7.4, 1 mM EDTA) followed by centrifugation at 1000 × g for 15 min. The supernatant fraction was placed onto the top of a 1.5 ml discontinuous sucrose gradient from 0.42 M to 1.38 M. The gradient was centrifuged at 100,000 × g for 6.5 h. Thirteen fractions were collected and analyzed by immunoblotting.
Coimmunoprecipitation and pulldown assays
Transfected HEK293T cells were harvested and lysed in cell lysis buffer with protease inhibitor cocktail (Sigma-Aldrich, P8340) for 1 h at 4°C. All the procedures for coimmunoprecipitation were as described previously.30 For cell endogenous IP, MEFs were lysed, precipitated with STX17 antibody (rabbit) and eluted by protein A beads (Cell Signaling Technology, 17-0780-01). Rabbit IgG was added as negative control. For tissue endogenous IP, cerebellum extracts were sequentially lysed, precipitated with VPS33A antibody, eluted by Protein A beads and immunoblotted with antibodies of other HOPS subunits. For affinity isolation assays, GST-WT or mu-VPS33A and maltose binding protein (MBP)-VPS18 were purified by binding to glutathione Sepharose 4B (GE Healthcare, 17-0756-01) and amylose resin (New England Biolabs, E8021S) and eluted with glutathione and maltose, respectively. The purified proteins were mixed and incubated overnight at 4°C, immunoprecipitated with amylose resin. The immunoprecipitates were immunoblotted with the indicated antibodies.
EGFR degradation assay
MEFs isolated from buff and wild-type mice were starved in DMEM containing 10 mM HEPES, pH 7.4 and 0.2% BSA (Roche, A6020) overnight. The procedures were performed as described previously.30
Statistics
For all experiments, statistical significance was determined by the Student t test. In the figures, all data represent mean ± SEM.
Disclosure of Potential Conflicts of Interest
There were no potential conflicts of interest to be disclosed.
Acknowledgments
We are in debt to Dr. Richard T. Swank who provided the mouse mutants for this study and proofread this manuscript. We thank Dr. Lin Yang for technical assistance in using the electron microscope. We are grateful for critical comments from Drs. Judith Klumperman, Hong Zhang, and Chonglin Yang.
Funding
This work was partially supported by grants from National Natural Science Foundation of China (Nos. 31230046; 91332116), and from Chinese Academy of Sciences (KJZD-EW-L08).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Balderhaar HJ, Ungermann C. CORVET and HOPS tethering complexes - coordinators of endosome and lysosome fusion. J Cell Sci 2013; 126:1307-16; PMID:23645161; http://dx.doi.org/ 10.1242/jcs.107805 [DOI] [PubMed] [Google Scholar]
- 2.Hong W, Lev S. Tethering the assembly of SNARE complexes. Trends Cell Biol 2014; 24:35-43; PMID:24119662; http://dx.doi.org/ 10.1016/j.tcb.2013.09.006 [DOI] [PubMed] [Google Scholar]
- 3.Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science 2009; 323:474-7; PMID:19164740; http://dx.doi.org/ 10.1126/science.1161748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brocker C, Kuhlee A, Gatsogiannis C, Balderhaar HJ, Honscher C, Engelbrecht-Vandre S, Ungermann C, Raunser S. Molecular architecture of the multisubunit homotypic fusion and vacuole protein sorting (HOPS) tethering complex. Proc Natl Acad Sci U S A 2012; 109:1991-6; PMID:22308417; http://dx.doi.org/ 10.1073/pnas.1117797109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei AH, Li W. Hermansky-Pudlak syndrome: pigmentary and non-pigmentary defects and their pathogenesis. Pigment Cell Melanoma Res 2013; 26:176-92; PMID:23171219; http://dx.doi.org/ 10.1111/pcmr.12051 [DOI] [PubMed] [Google Scholar]
- 6.Jiang P, Nishimura T, Sakamaki Y, Itakura E, Hatta T, Natsume T, Mizushima N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell 2014; 25:1327-37; PMID:24554770; http://dx.doi.org/ 10.1091/mbc.E13-08-0447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takats S, Pircs K, Nagy P, Varga A, Karpati M, Hegedus K, Kramer H, Kovacs AL, Sass M, Juhasz G. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol Biol Cell 2014; 25:1338-54; PMID:24554766; http://dx.doi.org/ 10.1091/mbc.E13-08-0449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012; 151:1256-69; PMID:23217709; http://dx.doi.org/ 10.1016/j.cell.2012.11.001 [DOI] [PubMed] [Google Scholar]
- 9.Suzuki T, Oiso N, Gautam R, Novak EK, Panthier JJ, Suprabha PG, Vida T, Swank RT, Spritz RA. The mouse organellar biogenesis mutant buff results from a mutation in Vps33a, a homologue of yeast vps33 and Drosophila carnation. Proc Natl Acad Sci U S A 2003; 100:1146-50; PMID:12538872; http://dx.doi.org/ 10.1073/pnas.0237292100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng C, Ye J, Yan S, Kong S, Shen Y, Li C, Li Q, Zheng Y, Deng K, Xu T, et al.. Ablation of vacuole protein sorting 18 (Vps18) gene leads to neurodegeneration and impaired neuronal migration by disrupting multiple vesicle transport pathways to lysosomes. J Biol Chem 2012; 287:32861-73; PMID:22854957; http://dx.doi.org/ 10.1074/jbc.M112.384305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chintala S, Novak EK, Spernyak JA, Mazurchuk R, Torres G, Patel S, Busch K, Meeder BA, Horowitz JM, Vaughan MM, et al.. The Vps33a gene regulates behavior and cerebellar Purkinje cell number. Brain Res 2009; 1266:18-28; PMID:19254700; http://dx.doi.org/ 10.1016/j.brainres.2009.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24-41; PMID:24366339; http://dx.doi.org/ 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al.. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/ 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- 14.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al.. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/ 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- 15.Yuan Y, Wang H, Wei Z, Li W. Impaired autophagy in hilar mossy cells of the dentate gyrus and its implication in schizophrenia. J Genet Genomics 2015; 42:1-8; PMID:25619597; http://dx.doi.org/ 10.1016/j.jgg.2014.12.001 [DOI] [PubMed] [Google Scholar]
- 16.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983-97; PMID:23921753; http://dx.doi.org/ 10.1038/nm.3232 [DOI] [PubMed] [Google Scholar]
- 17.Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci 1992; 15:133-9; PMID:1374971; http://dx.doi.org/ 10.1016/0166-2236(92)90355-C [DOI] [PubMed] [Google Scholar]
- 18.Graybiel AM. The basal ganglia. Trends Neurosci 1995; 18:60-2; PMID:7537409; http://dx.doi.org/ 10.1016/0166-2236(95)80019-X [DOI] [PubMed] [Google Scholar]
- 19.Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc Am Pharm Assoc (Baltim) 1957; 46:208-9; PMID:13502156; http://dx.doi.org/ 10.1002/jps.3030460322 [DOI] [PubMed] [Google Scholar]
- 20.Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, et al.. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet 1999; 8:397-407; PMID:9949199; http://dx.doi.org/ 10.1093/hmg/8.3.397 [DOI] [PubMed] [Google Scholar]
- 21.Da Cunha C, Wietzikoski S, Wietzikoski EC, Miyoshi E, Ferro MM, Anselmo-Franci JA, Canteras NS. Evidence for the substantia nigra pars compacta as an essential component of a memory system independent of the hippocampal memory system. Neurobiol Learn Mem 2003; 79:236-42; PMID:12676522; http://dx.doi.org/ 10.1016/S1074-7427(03)00008-X [DOI] [PubMed] [Google Scholar]
- 22.Pioli EY, Meissner W, Sohr R, Gross CE, Bezard E, Bioulac BH. Differential behavioral effects of partial bilateral lesions of ventral tegmental area or substantia nigra pars compacta in rats. Neuroscience 2008; 153:1213-24; PMID:18455318; http://dx.doi.org/ 10.1016/j.neuroscience.2008.01.084 [DOI] [PubMed] [Google Scholar]
- 23.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/ 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- 24.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; http://dx.doi.org/ 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- 25.Sarkar S, Korolchuk V, Renna M, Winslow A, Rubinsztein DC. Methodological considerations for assessing autophagy modulators: a study with calcium phosphate precipitates. Autophagy 2009; 5:307-13; PMID:19182529; http://dx.doi.org/ 10.4161/auto.5.3.7664 [DOI] [PubMed] [Google Scholar]
- 26.Baker RW, Jeffrey PD, Hughson FM. Crystal Structures of the Sec1/Munc18 (SM) Protein Vps33, Alone and Bound to the Homotypic Fusion and Vacuolar Protein Sorting (HOPS) Subunit Vps16/. PLoS One 2013; 8:e67409; PMID:23840694; http://dx.doi.org/ 10.1371/journal.pone.0067409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lobingier BT, Nickerson DP, Lo SY, Merz AJ. SM proteins Sly1 and Vps33 co-assemble with Sec17 and SNARE complexes to oppose SNARE disassembly by Sec18. Elife 2014; 3:e02272; PMID:24837546; http://dx.doi.org/ 10.7554/eLife.02272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graham SC, Wartosch L, Gray SR, Scourfield EJ, Deane JE, Luzio JP, Owen DJ. Structural basis of Vps33A recruitment to the human HOPS complex by Vps16. Proc Natl Acad Sci U S A 2013; 110:13345-50; PMID:23901104; http://dx.doi.org/ 10.1073/pnas.1307074110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pols MS, ten Brink C, Gosavi P, Oorschot V, Klumperman J. The HOPS proteins hVps41 and hVps39 are required for homotypic and heterotypic late endosome fusion. Traffic 2013; 14:219-32; PMID:23167963; http://dx.doi.org/ 10.1111/tra.12027 [DOI] [PubMed] [Google Scholar]
- 30.Zhang A, He X, Zhang L, Yang L, Woodman P, Li W. Biogenesis of Lysosome-related Organelles Complex-1 Subunit 1 (BLOS1) Interacts with Sorting Nexin 2 and the Endosomal Sorting Complex Required for Transport-I (ESCRT-I) Component TSG101 to Mediate the Sorting of Epidermal Growth Factor Receptor into Endosomal Compartments. J Biol Chem 2014; 289:29180-94; PMID:25183008; http://dx.doi.org/ 10.1074/jbc.M114.576561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Angers CG, Merz AJ. HOPS interacts with Apl5 at the vacuole membrane and is required for consumption of AP-3 transport vesicles. Mol Biol Cell 2009; 20:4563-74; PMID:19741093; http://dx.doi.org/ 10.1091/mbc.E09-04-0272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rawet-Slobodkin M, Elazar Z. PLEKHM1: a multiprotein adaptor for the endolysosomal system. Mol Cell 2015; 57:1-3; PMID:25574946; http://dx.doi.org/ 10.1016/j.molcel.2014.12.022 [DOI] [PubMed] [Google Scholar]
- 33.Collins KM, Wickner WT. Trans-SNARE complex assembly and yeast vacuole membrane fusion. Proc Natl Acad Sci U S A 2007; 104:8755-60; PMID:17502611; http://dx.doi.org/ 10.1073/pnas.0702290104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jun Y, Xu H, Thorngren N, Wickner W. Sec18p and Vam7p remodel trans-SNARE complexes to permit a lipid-anchored R-SNARE to support yeast vacuole fusion. Embo j 2007; 26:4935-45; PMID:18007597; http://dx.doi.org/ 10.1038/sj.emboj.7601915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ungermann C, Nichols BJ, Pelham HR, Wickner W. A vacuolar v-t-SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J Cell Biol 1998; 140:61-9; PMID:9425154; http://dx.doi.org/ 10.1083/jcb.140.1.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu H, Jun Y, Thompson J, Yates J, Wickner W. HOPS prevents the disassembly of trans-SNARE complexes by Sec17p/Sec18p during membrane fusion. EMBO J 2010; 29:1948-60; PMID:20473271; http://dx.doi.org/ 10.1038/emboj.2010.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR, Yue Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J Neurosci 2012; 32:7585-93; PMID:22649237; http://dx.doi.org/ 10.1523/JNEUROSCI.5809-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto A, Yue Z. Autophagy and its normal and pathogenic states in the brain. Annu Rev Neurosci 2014; 37:55-78; PMID:24821313; http://dx.doi.org/ 10.1146/annurev-neuro-071013-014149 [DOI] [PubMed] [Google Scholar]
- 39.Cheever TR, Li B, Ervasti JM. Restricted morphological and behavioral abnormalities following ablation of β-actin in the brain. PLoS One 2012; 7:e32970; PMID:22403730; http://dx.doi.org/ 10.1371/journal.pone.0032970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z, Hao CJ, Li CG, Zang DJ, Zhao J, Li XN, Wei AH, Wei ZB, Yang L, He X, et al.. Mutation of SLC35D3 causes metabolic syndrome by impairing dopamine signaling in striatal D1 neurons. PLoS Genet 2014; 10:e1004124; PMID:24550737; http://dx.doi.org/ 10.1371/journal.pgen.1004124 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.