

Abstract
Detachment of nonmalignant intestinal epithelial cells from the extracellular matrix (ECM) triggers their growth arrest and, ultimately, apoptosis. In contrast, colorectal cancer cells can grow without attachment to the ECM. This ability is critical for their malignant potential. We found previously that detachment-induced growth arrest of nonmalignant intestinal epithelial cells is driven by their detachment-triggered autophagy, and that RAS, a major oncogene, promotes growth of detached cells by blocking such autophagy. In an effort to identify the mechanisms of detachment-induced autophagy and growth arrest of nonmalignant cells we found here that detachment of these cells causes upregulation of ATG3 and that ATG3 upregulation contributes to autophagy and growth arrest of detached cells. We also observed that when ATG3 expression is artificially increased in the attached cells, ATG3 promotes neither autophagy nor growth arrest but triggers their apoptosis. ATG3 upregulation likely promotes autophagy of the detached but not that of the attached cells because detachment-dependent autophagy requires other detachment-induced events, such as the upregulation of ATG7. We further observed that those few adherent cells that do not die by apoptosis induced by ATG3 become resistant to apoptosis caused by cell detachment, a property that is critical for the ability of normal epithelial cells to become malignant. We conclude that cell-ECM adhesion can switch ATG3 functions: when upregulated in detached cells in the context of other autophagy-promoting events, ATG3 contributes to autophagy. However, when overexpressed in the adherent cells, in the circumstances not favoring autophagy, ATG3 triggers apoptosis.
Keywords: autophagy, apoptosis, anoikis, ATG3, ATG7, extracellular matrix
Abbreviations
- ATG
autophagy-related
- BCL2L1
Bcl2-like1
- CDK4
cyclin-dependent kinase 4
- ECM
extracellular matrix
- EGFR
epidermal growth factor receptor
- GABARAP
GABA(A) receptor-associated protein
- GFP
green fluorescent protein
- LC3/MAP1LC3
microtubule-associated protein 1 light chain 3
- PE
phosphatidylethanolamine
- qPCR
quantitative polymerase chain reaction
- SD
standard deviation
- SH3GLB1/Bif1
SH3-domain GRB2-like endophilin B1
- siRNA
small interfering RNA
- UVRAG
UV radiation resistance-associated.
Introduction
Many types of epithelial cells grow in vivo attached to a form of the extracellular matrix (ECM) called the basement membrane. Detachment of such cells from the ECM causes their growth arrest and ultimately, apoptosis.1-3 The latter form of cell death is called anoikis.3 It is now believed that these phenomena preclude growth of the indicated cells outside of their original location. Unlike normal epithelial cells, cells composing carcinomas, cancers of epithelial origin, are well known to be able to survive and grow without being attached to the ECM.4-6 Such ability is thought to be critical for carcinoma progression.7,8 This notion is based on numerous experimental data. For example, the ability of cancer cells to survive and proliferate without being attached to the ECM as colonies in soft agar has served as a standard for malignant transformation for several decades.9,10 Also, the ability to grow anchorage-independently without adhesion to the ECM is conferred to cancer cells by activation of major oncoproteins, such as RAS, SRC, EGFR, and CTNNB1/β-catenin, as well as by loss of tumor suppressor genes, such as PTEN.2,11-14 Furthermore, treatments that block the ability of cancer cells to grow without attachment to the ECM, suppress their in vivo tumorigenicity and the capacity for metastasis.4,5,15-21 Finally, acquisition by the nonmalignant cells of the ability to grow anchorage-independently results in a spontaneous attainment of in vivo tumorigenicity by these cells.22,23 Thus, the ability of tumor cells to grow without adhesion to the ECM represents a potential novel target for cancer treatment. The development of such treatments has been hampered by the fact that the mechanisms by which the nonmalignant cells stop growing and die after detachment from the ECM and those by which cancer cells acquire the ability to grow anchorage-independently are understood only in part.
In an effort to understand these mechanisms we found previously that anoikis of nonmalignant intestinal epithelial cells is driven by detachment-induced downregulation of the anti-apoptotic protein BCL2L1/BclXL and detachment-induced upregulation of the pro-apoptotic molecule FASLG/Fas ligand as well as that of a cell-death promoting protein kinase CHEK2/Chk2.4,24,25 We also established that oncogenic RAS, an oncoprotein that often occurs in colorectal cancer, blocks anoikis of colon cancer cells via multiple mechanisms that involve the downregulation of pro-apoptotic proteins BAK1/Bak and CASP2/caspase-2 and the upregulation of BCL2L1 as well as that of apoptosis inhibitors BIRC3/cIAP2 and XIAP.4,5,18,26
In searching for the mechanisms by which detachment of nonmalignant intestinal epithelial cells blocks their growth we found that growth arrest of detached cells is driven by their detachment-induced autophagy. Autophagy, degradation of cellular macromolecules and organelles, begins when a membrane of unknown origin surrounds a part of the cytoplasm or an organelle.27 The resulting vesicle (autophagosome) fuses with the lysosome to form the autolysosome, and lysosomal enzymes degrade the vesicle content.28 Autophagy, whose mechanisms are understood in part, is regulated by over 20 ATG proteins.29 Autophagosome formation is begun in part by the action of class III phosphatidylinositol 3-kinase and BECN1/Beclin 1 and is then driven by a conjugate between the lipid phosphatidylethanolamine (PE) and proteins of the MAP1LC3/LC3 subfamily as well as that between PE and the GABARAP subfamily of proteins.29 MAP1LC3 lipidation starts when it's C-terminus binds the E1 ubiquitin-activating enzyme-like protein ATG7. ATG7-activated MAP1LC3 is then linked to the E2 ubiquitin-conjugating enzyme-like ATG3, and then to PE.29 GABARAP is lipidated via a similar mechanism.30,31
We found that ablation of BECN1 blocks detachment-induced autophagosome formation in the nonmalignant intestinal epithelial cells and detachment induced reduction of the fraction of these cells in the S-phase of the cell cycle without affecting their viability.1 We further observed that oncogenic RAS triggers degradation of BECN1 protein in malignant intestinal epithelial cells by activating one or more members of the calpain family of proteases.1 We found that BECN1 loss results in the suppression of detachment-induced autophagy in the RAS-transformed cells and promotes proliferation of the indicated cells without adhesion to the ECM.1
Our findings are consistent with observations indicating that autophagy can act as a tumor-suppressing mechanism. For example, loss of BECN1 and other mediators of autophagy, such as ATG5, ATG7, and SH3GLB1/Bif-1, is known to promote various types of cancer in mice.32-35 Furthermore, anti-cancer drugs, such as erlotinib, block EGFR-dependent tumor growth in vivo at least in part, by activating BECN1 and inducing autophagy of tumor cells.36 Overall, the data emerging from the literature indicate that excessive autophagy can suppress tumor growth by causing growth arrest of cancer cells and perhaps via other mechanisms.37,38 This is presumably the reason why activation of major oncogenes, such as RAS, EGFR, and AKT, or loss of tumor suppressor genes, such as TP53, restrict (but do not completely block) autophagy in cancer cells.1,36,39,40 Conversely, given that such cells often exist in a state of metabolic stress (e.g. that caused by lack of properly organized vasculature),41 some degree of autophagy is likely required by these cells for the generation of essential cellular macromolecules (that are required for cell metabolism) out of the respective monomeric components that become available after the degradation of nonessential molecules via autophagic mechanisms. This is likely the reason why complete suppression of autophagy prevents growth of various types of tumors in vivo.42,43
It has to be noted that many conclusions regarding the role of autophagy in tumor growth were made based on the susceptibility of mice lacking various mediators of autophagy to cancer.32,34,35,42,43 Furthermore, levels of some of these mediators are altered in diverse types of human tumors.44,45 Based on the changes in these levels, conclusions regarding the role of autophagy in the progression of respective cancers were made.44,45 However, some critical mediators of autophagy seem to be able to play autophagy-unrelated roles in cells. For example, ATG12, one of the important stimulators of autophagosome formation, was found to cause apoptosis via physical interactions with components of the cellular apoptotic machinery (such as the BCL2 family members) in a manner independent of its ability to trigger autophagy.46 The capacity to kill cells regardless of its involvement in autophagy has also been demonstrated for ATG3, another protein critical for autophagosome formation.47 Thus, the contribution of such proteins to cancer could, in principle, be explained by their autophagy-unrelated functions, such as the ability to control apoptosis.
Since the mechanisms by which detachment of intestinal epithelial cells from the ECM triggers autophagy (and such autophagy is critical for the suppression of anchorage-independent growth of cancer cells1) are not known, we explored these mechanisms in the present study. We found that detachment-induced upregulation of ATG3 contributes to autophagy of detached cells and the reduction of the fraction of these cells in the S-phase of the cell cycle. We established that in addition to ATG3, detachment-induced upregulation of other autophagy mediators, such as ATG7, also contributes to detachment-induced autophagy. We found that when ATG3 is overexpressed in the attached intestinal epithelial cells by itself, it does not cause autophagy but instead triggers their apoptotic death. Thus, cell-ECM interactions represent a regulator of the biological functions of ATG3: when ATG3 is upregulated in detached cells in the context of other autophagy-promoting events this protein contributes to the induction of autophagy. However, when overexpressed in the adherent cells, in the circumstances that do not favor autophagy induction, ATG3 triggers apoptosis. We also found that those (relatively few) adherent intestinal epithelial cells that adapt to the presence of increased ATG3 levels and survive despite ATG3 upregulation acquire anoikis resistance, a property that is typically displayed by the malignant cells.48
Results
Detachment of intestinal epithelial cells from the ECM triggers autophagosome formation
Previously we used electron microscopy (EM) to demonstrate that detachment from the ECM triggers autophagosome formation in the nonmalignant spontaneously immortalized rat intestinal epithelial cells IEC-18.1 Another approach for detecting this process that we used in the past was assaying the cells for autophagosome formation by counting the percentage of cells with puncta formed by green fluorescent protein (GFP)-tagged MAP1LC3B (GFP-LC3B) that was introduced into these cells by transient transfection.1 Detecting the change from diffuse cytoplasmic to punctate (characteristic of autophagy) distribution of GFP-LC3B in cells is an often used assay of autophagosome formation.49
We have now confirmed these findings by several additional approaches. First we assayed the cells before and after detachment for the number of puncta per cell formed by GFP-LC3B (that was introduced into these cells by transient transfection) which is now thought to represent a more appropriate method of detecting autophagosome formation than determining the percentage of the cells with such puncta in the total cell population.49 As shown in Figure 1A and B, detachment resulted in a substantial increase in the number of GFP-LC3B puncta per cell. To demonstrate that detachment-induced increase in these puncta reflects increased autophagosome formation, rather than decreased fusion of the autophagosomes with the lysosomes, we treated attached and detached cells with bafilomycin A1, an inhibitor of vacuolar H+-ATPases and a suppressor of autophagosome-lysosome fusion.49 We found that bafilomycin A1 treatment noticeably increased the number of GFP-LC3B puncta per cell in the attached cells (Fig. 1A and B). This indicates that IEC-18 cells undergo a significant degree of basal autophagy which can be disrupted by treatment with bafilomycin A1. However, detached bafilomycin A1-treated cells displayed a substantially higher number of GFP-LC3B puncta per cell than bafilomycin A1-treated attached cells (Fig. 1A, B). These data are consistent with a scenario, according to which detachment increases the formation of autophagic vacuoles, rather than disrupts their fusion with the lysosomes in the indicated cells.
Figure 1.

Detachment from the ECM triggers autophagosome formation in intestinal epithelial cells. (A, B) Nonmalignant intestinal epithelial cells IEC-18 cells were transfected with the GFP-LC3B expression vector, cultured attached to or detached from the ECM for 24 h in the absence (−) or in the presence (+) of 50 nM bafilomycin A1 (BafA1) and green puncta per cell were counted. (A) Representative fluorescence microscopy images of the attached and detached cells. (B) Quantification of the number of green puncta per cell. The numbers represent the average of the number of puncta per cell observed for 30–40 cells plus the SE. This experiment was repeated 3 times with similar results. (C–E) IEC-18 cells were transduced with the mCherry-GFP-LC3-encoding virus, cultured attached to (att) or detached from (det) the ECM for the indicated times and green, red, or yellow puncta were counted. (C) Representative fluorescence microscopy images of the attached and detached cells. (D, E) Quantification of the number of yellow (D) and red (E) puncta per cell performed within the same experiment. The numbers represent the average of the number of puncta per cell observed for 30–40 cells plus the SE. This experiment was repeated twice with similar results. (F) IEC-18 cells were cultured attached (att) to or detached from (det) the ECM for 20 h and assayed for MAP1LC3B expression by western blot. Positions of MAP1LC3B-I and MAP1LC3B-II on the gel are indicated. (G, H) IEC-18 cells were cultured attached (att) to or detached from the ECM (det) for the indicated times and in the presence of 50 nM bafilomycin A1 (BafA1) and assayed for MAP1LC3B expression by western blot. (I) IEC-18 cells were transfected with the GFP-LC3 expression vector, cultured attached to or detached from the ECM for 24 h and assayed by western blot by using an anti-GFP antibody. Positions of GFP-LC3B and free GFP on the gel are indicated. CDK4 was used as a loading control in (F) and ACTB in (G–I). *, p < 0.05.
To verify these results by a complementary method we infected IEC-18 cells with a tandem mCherry-GFP-tagged LC3-encoding Moloney murine leukemia virus. GFP does not generate fluorescence at an acidic pH, and mCherry-GFP-LC3 can only yield green puncta outside of the lysosomes (the lumen of which is acidic). 49 In contrast, mCherry can give rise to red fluorescence both at the cytosolic and the lysosomal pH, and thus mCherry-GFP-LC3 can promote the formation of the red puncta both inside and outside of the lysosomes.49 We found that mCherry-GFP-LC3-producing cells displayed an increased number of the yellow puncta (formed due to simultaneous fluorescence of both mCherry and GFP) after being detached for 7 h, and that the number of such puncta was significantly reduced after the cells were detached for 30 h (Fig. 1C, D). Conversely, the number of the red puncta was increased after the cells were detached for 7 h and remained increased after the cells were detached for 30h (Fig. 1C, E). These data are consistent with a scenario according to which detachment triggers increased formation of the autophagic vacuoles (signified by the presence of yellow dots) which later fuse with the lysosomes (and this results in the disappearance of the yellow dots due to GFP inactivation at the lysosomal pH). Since mCherry is stable both at the cytosolic and lysosomal pH, the number of autophagic vacuoles generating red fluorescence increased after cell detachment and remained high even when these vacuoles fused with the lysosomes.
We further found that detachment of IEC-18 cells resulted in a significant decrease in the amount of the nonlipidated MAP1LC3B (often called MAP1LC3B-I) in these cells and a strong increase in the amount of the lipidated MAP1LC3B (often referred to as MAP1LC3B-II), a transition that represents one of the hallmarks of autophagosome formation (Fig. 1F).49 Detachment did not change the amount of the MAP1LC3B mRNA in these cells (Fig. S1A). To confirm that the increase in the amount of MAP1LC3B-II in detached cells reflects increased MAP1LC3B lipidation, rather than decreased autophagy-dependent degradation of MAP1LC3B-II (as occurs during autophagy)49 we measured such lipidation before and after treatment of attached and detached cells with bafilomycin A1 (Fig. 1G, H). Attached cells treated with this drug displayed a strong increase in the amount of MAP1LC3B-II compared to that of MAP1LC3B-I (under these conditions MAP1LC3B-I could only be detected after a long exposure of the respective western blot (Fig. S1B, C)). This increase is consistent with the possibility that bafilomycin A1 treatment disrupted the basal autophagy in the attached cells which prevented MAP1LC3B-II degradation and caused its accumulation in these cells. Importantly, the amount of MAP1LC3B-II in bafilomycin A1-treated detached cells was even higher than that in the attached cells treated with this drug (Fig. 1G, H). Hence, detachment likely promotes increased MAP1LC3B synthesis and lipidation, rather than blocks MAP1LC3B-II autophagy-dependent degradation.
When GFP-LC3B is delivered to the lysosome as a part of the autophagosome, the inner autophagosomal membrane (and the LC3 component of the fusion protein) is degraded, while the relatively more degradation-resistant GFP component remains intact and emerges as free GFP on a western blot. This free GFP emergence represents another way to monitor autophagy.49 We observed in this regard that detachment triggered a significant increase of free GFP in GFP-LC3B-transfected IEC-18 cells (Fig. 1I). Collectively, the data shown in Figure 1 indicate that detachment triggers increased autophagy of intestinal epithelial cells.
We also confirmed that similar to what we observed before, detachment of intestinal epithelial cells triggers a significant reduction of the fraction of the cells in the S phase of the cell cycle (Fig. 2A) (and a concomitant increase in the G1-phase, Fig. S2A) and apoptosis (detected by the ability to bind ANXA5, a characteristic property of apoptotic cells) (Fig. 2B; Fig. S2B).1,50
Figure 2.
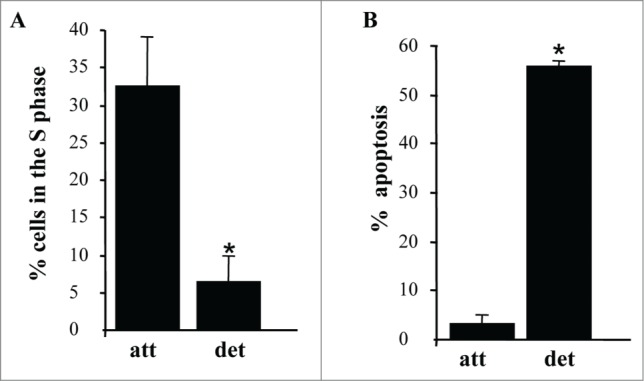
Detachment from the ECM triggers growth arrest and apoptosis of intestinal epithelial cells. (A) IEC-18 cells were cultured attached to or detached form the ECM for 15 h and assayed for the distribution of the cells in phases of the cell cycle by flow cytometry. Percentage of the cells in the S phase of the cell cycle is shown. The numbers represent the average of 2 independent experiments plus the SD. (B) The cells were cultured as in (A), stained with propidium iodide (PI) and ANXA5 and assayed for ANXA5 and PtdIns binding by flow cytometry. Percent apoptosis was calculated as the sum of the percentages of ANXA5-positive-PI-negative cells (undergoing early stages of apoptosis) and ANXA5-positive-PtdIns-positive cells (undergoing late stages of apoptosis). The numbers represent the average of 2 independent experiments plus the SD. * Indicates that the p-value was less than 0.05.
Detachment of intestinal epithelial cells from the ECM triggers ATG3 and ATG7 upregulation
In an effort to understand the mechanisms of detachment-induced autophagy of intestinal epithelial cells we found that detachment of IEC-18 cells does not alter the expression of autophagy mediators, such as UVRAG, SH3GLB1/Bif-1, ATG16L1, and ATG10, in these cells (Fig. 3A–D). We also found that all of ATG5 is present in a complex that most likely represents ATG5 conjugated with ATG12 in both attached and detached cells (Fig. 3E). The fact that all cellular ATG5 can be conjugated with ATG12 even in the absence of autophagy has been described by several groups.49
Figure 3.
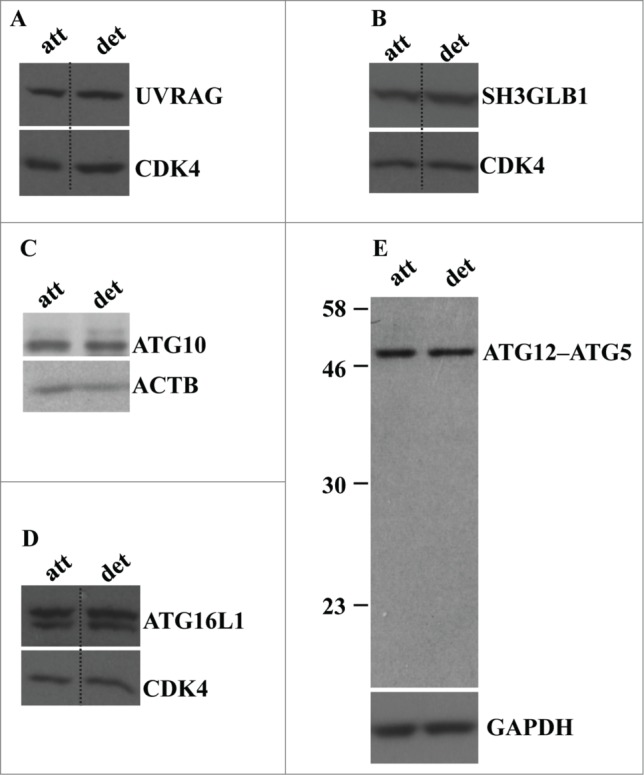
Detachment from the ECM does not alter the levels of UVRAG, SH3GLB1, ATG16L1, ATG10, and ATG12-ATG5 conjugate in intestinal epithelial cells. (A–D) IEC-18 cells were cultured attached to (att) or detached (det) from the ECM for 20 h and assayed for the expression of the indicated proteins by western blot. CDK4 was used as a loading control in (A, B, D), ACTB in (C) and GAPDH (glyceraldehyde 3-phosphate dehydrogenase) in (E).
We further found that detachment of IEC-18 cells resulted in a significant upregulation of autophagy mediators ATG3 and ATG7 (Fig. 4A, B). These events were not unique to IEC-18 cells as we also observed upregulation of both ATG3 and ATG7 in the case of nonmalignant Hkh-2 human intestinal epithelial cells (Fig. 4C, D). These cells were derived from human colon carcinoma cells HCT-116 (that carry one oncogenic KRAS allele) by targeted deletion of the activated KRAS allele via homologous recombination.51 Hkh-2 cells are non-tumorigenic in mice and are incapable of growing without adhesion to the ECM.23,51 Furthermore, we found previously that detachment from the ECM significantly enhances autophagosome formation in these cells.1 We also found that detachment of IEC-18 cells causes a significant upregulation of the ATG3 mRNA (Fig. 4E) but not that of the ATG7 mRNA (Fig. 4F). Hence, detachment-induced ATG3 upregulation could occur due to increased transcription of the ATG3 gene or increased ATG3 mRNA stability. Conversely, it is possible that detachment-dependent upregulation of ATG7 is the consequence of increased ATG7 protein synthesis or enhanced ATG7 protein stability.
Figure 4.
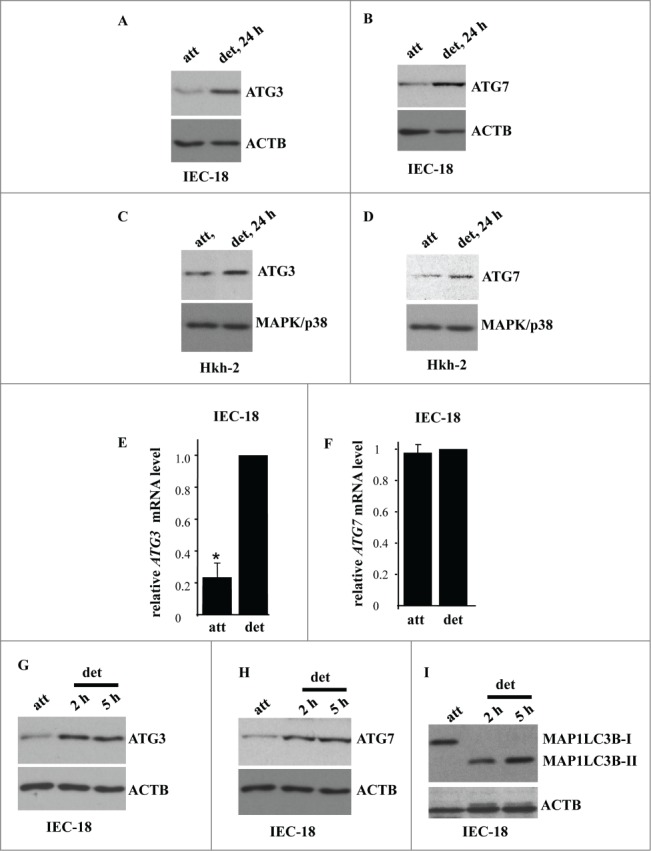
Detachment from the ECM triggers ATG3 and ATG7 upregulation in intestinal epithelial cells. (A, B) IEC-18 cells were cultured attached (att) to or detached from (det) the ECM for the indicated times and assayed for ATG3 (A) or ATG7 (B) protein expression by western blot. (D, E) Human nonmalignant intestinal epithelial Hkh-2 cells were cultured attached (att) to or detached (det) from the ECM for the indicated times and assayed for ATG3 (C) or ATG7 (D) protein expression by western blot. (E, F) IEC-18 cells were cultured attached to or detached from the ECM for 24 h and assayed for ATG3 (E) or ATG7 (F) mRNA levels by qPCR. The observed ATG3 and ATG7 mRNA levels were normalized by the levels of RNA18S/18S rRNA, which were also determined by qPCR. The resulting levels of ATG3 (E) or ATG7 (F) mRNA in the detached cells were arbitrarily designated as 1.0. The data represent the average of 4 independent experiments plus the SD in (E) and 2 independent experiments in (F). * indicates that the p-value was less than 0.05. (G–I) IEC-18 cells were cultured attached to or detached from the ECM for the indicated times and assayed for ATG3 (G), ATG7 (H) or MAP1LC3B (I) protein expression by western blot. Positions of MAP1LC3B-I and MAP1LC3B-II on the gel are indicated. ACTB was used as a loading control in (A, B, G–I) and MAPK/p38 in (C, D).
We further observed that detachment-induced ATG3 and ATG7 upregulation occurs at the same time as MAP1LC3B lipidation (Fig. 4G-I). These events were detected by us as early as at 2 h post-detachment. We published previously that detachment of IEC-18 cells increases the number of autophagic vacuoles per cell (detected by EM) from approximately 1 in the attached cells to 5 at 5 h post-detachment and further, to 12 at 17 h post-detachment.1 Thus, as might have been expected, ATG3 and ATG7 upregulation and MAP1LC3B lipidation is followed by the formation of autophagic vacuoles.
ATG3 upregulation is required for autophagosome formation in detached intestinal epithelial cells
To test whether detachment-induced ATG3 upregulation is required for autophagy of detached intestinal epithelial cells we ablated ATG3 in detached IEC-18 cells by transfecting them with 2 separate ATG3-specific small interfering (si)RNAs (Fig. 5A). We observed that siRNA-transfected detached IEC-18 cells displayed ATG3 levels comparable to those observed by us in the attached IEC-18 cells (Fig. 5A). We then assayed attached and detached cells for autophagosome formation by counting the number of puncta formed per cell by GFP-LC3B (after transient transfection of these cells with the GFP-LC3B expression vector). We found that ATG3 ablation significantly reduced autophagosome formation in the detached cells (Fig. 5B). We also observed that ATG3 loss noticeably reduced the levels of MAP1LC3B-II (Fig. 5C) in detached IEC-18 cells. We further established that ATG3 ablation blocked lipidation of GABARAP (Fig. 5E).52 Covalent GABARAP binding to PE represents another marker of autophagy.52 We found that attached IEC-18 cells carried almost exclusively unlipidated GABARAP (often referred to as GABARAP-I) whereas detached cells displayed a noticeable increase in the presence of the lipidated form (GABARAP-II) (Fig. 5D).53 We also observed that detachment did not alter the level of GABARAP mRNA (Fig. S3A) in IEC-18 cells. We established that ATG3 loss significantly reduced the amount of GABARAP-II in the detached cells (Fig. 5E). Collectively, our data indicate that the reversal of detachment-induced ATG3 upregulation blocks detachment-induced autophagy of intestinal epithelial cells.
Figure 5.

For figure legend,see page 1238.
We further found that enforced ATG3 downregulation significantly increased the fraction of detached cells in the S-phase of the cell cycle (Fig. 5F; Fig. S3B). Given that further to detachment and growth arrest, IEC-18 cells undergo apoptosis, we measured such apoptosis of these cells before and after ATG3 ablation.2,4 We found that ATG3 loss did not reduce apoptosis of the detached cells (Fig. 5G; Fig. S3C).
Since at least some autophagy mediators are known to play autophagy-unrelated roles in cells, we decided to confirm the role of autophagy in detachment-induced reduction in the fraction of intestinal epithelial cells in the S phase of the cell cycle by ablating a different autophagy mediator in these cells.46,47 Since, according to our data (Fig. 5E), GABARAP likely mediates the effect of ATG3 on autophagy, we tested the effect of GABARAP ablation on the cell cycle progression of detached IEC-18 cells. Similar to what was observed in case of ATG3 loss, we found that GABARAP ablation by siRNAs (Fig. 6A) triggered a significant increase in the fraction of detached IEC-18 cells in the S phase of the cell cycle (Fig. 6B; Fig. S4). We conclude that detachment-induced ATG3 upregulation and subsequent autophagy is necessary for detachment-induced reduction in the fraction of intestinal epithelial cells in the S phase of the cell cycle.
Figure 6.
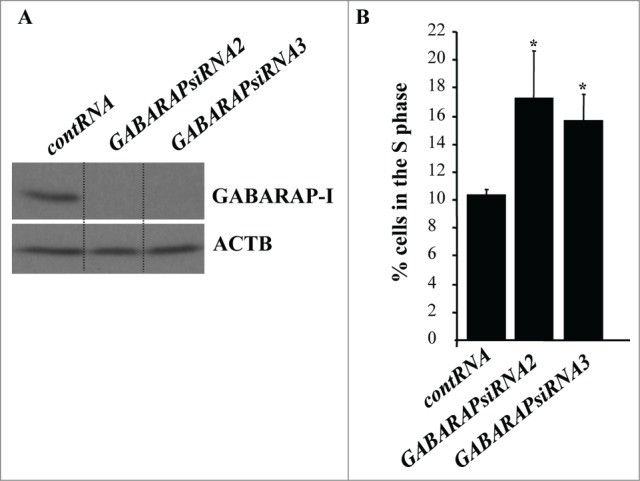
GABARAP ablation increases the fraction of detached intestinal epithelial cells in the S-phase of the cell cycle. (A) IEC-18 cells were transfected with 100 nM control RNA (contRNA) or GABARAP-specific small interfering RNA (GABARAPsiRNA)2 or 3 and assayed for GABARAP expression by western blot. ACTB was used as a loading control. (B) IEC-18 cells were treated as in (A), detached from the ECM for 14h and assayed for the distribution of the cells in phases of the cell cycle by flow cytometry. Percentage of the cells in the S phase of the cell cycle is shown. The numbers represent the average of 3 independent experiments plus the SD. * indicates that the p-value was less than 0.05.
ATG3 upregulation triggers apoptosis, rather than autophagosome formation and growth arrest, in the attached intestinal epithelial cells
We further tested whether ATG3 upregulation is sufficient for the induction of autophagy and subsequent growth arrest of attached intestinal epithelial cells. To achieve ATG3 upregulation in the attached IEC-18 cells we infected them with an ATG3-encoding Moloney murine leukemia virus carrying a puromycin resistance gene. We found that about 90% of control cells that were not transduced with the virus died within the first 24 h of puromycin treatment (not shown). We also observed that after the infection of IEC-18 cells with the control virus the ratio of the number of colonies formed by these cells in monolayer culture after puromycin treatment to that formed by the cells infected with the same amount of the control virus but not treated with puromycin was approximately 0.7 (not shown). Thus, the efficiency of viral transduction under these conditions was approximately 70%. We further established that when infected with the ATG3-encoding virus and treated with puromycin for 24 h, the adherent IEC-18 cells produced ATG3 at levels that were significantly higher than those in the cells infected with the same number of the control viral particles and comparable to those observed in the detached uninfected IEC-18 cells (Fig. 7A). Thus, we have found conditions under which ATG3 could be produced in the attached cells at the level similar to that induced by cell detachment.
Figure 7.
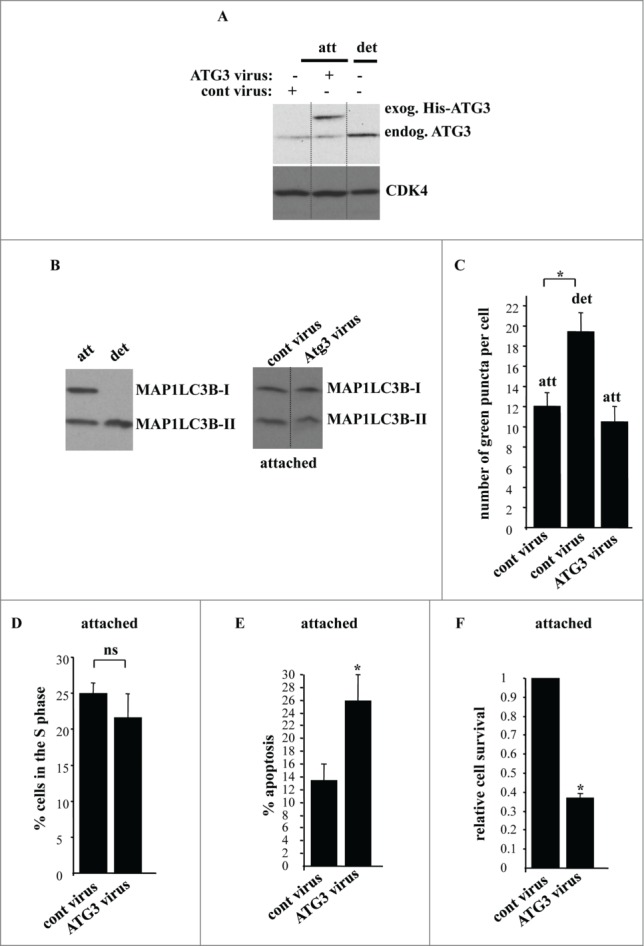
ATG3 upregulation in attached intestinal epithelial cells does not trigger their autophagy or cell cycle arrest but promotes apoptosis. (A) IEC-18 cells were infected with 4 × 104 viral particles per cell of the control retrovirus (cont virus) or ATG3-encoding retrovirus (ATG3 virus), treated with 2 μg/ml puromycin for 24 h and assayed for ATG3 expression by western blot along with IEC-18 cells that were detached for 20 h. The positions of endogenous ATG3 (endog. ATG3) and exogenous His-tagged ATG3 (exog. ATG3) on the gel are indicated. CDK4 was used as a loading control. (B, left) IEC-18 cells were cultured attached (att) to or detached (det) from the ECM for 20 h and assayed for MAP1LC3B expression by western blot. Positions of MAP1LC3B-I and MAP1LC3B-II on the gel are indicated. (B, right) IEC-18 cells infected with the indicated viruses as in (A) were cultured attached to the ECM for an additional 20 h and analyzed as in (B, left). (C) IEC-18 cells infected with the indicated viruses as in (A) were transiently transfected with a GFP-LC3B expression vector and plated attached to or detached from the ECM for 20 h and analyzed for the presence of GFP-LC3B puncta as in Figure 1. The results represent the average of the number of GFP-LC3B puncta per cell observed for 30–45 cells plus the SE. (D) IEC-18 cells infected with the indicated viruses as in (A) were cultured attached to the ECM for an additional 24 h and assayed for the distribution of the cells in phases of the cell cycle by flow cytometry. Percentage of the cells in the S phase of the cell cycle is shown. The numbers represent the average of 3 independent experiments plus the SD. (E) IEC-18 cells infected with the indicated viruses as in (A) were cultured attached to the ECM for an additional 24 h and assayed for ANXA5 binding. The numbers represent the average of 2 independent experiments plus the SD. (F) IEC-18 cells infected with the indicated viruses as in (A) were cultured attached to the ECM for 7–10d. Cell colonies were then stained and counted. The numbers represent the average of the triplicates plus the SD. This experiment was repeated twice with similar results. Relative cell survival was calculated as the number of colonies formed by the ATG3-expressing cells relative to that formed by the control cells which was arbitrarily designated as 1.0. * indicates that the p-value was less than 0.05.
We subsequently assayed the control and ATG-3 expressing cells (generated after infection of IEC-18 cells with respective viruses and a 24 h-long selection with puromycin) for autophagy by detecting the conversion of MAP1LC3B from the nonlipidated form to the PE-conjugated form. Whereas, as expected, detachment of IEC-18 cells resulted in a quantitative conversion of MAP1LC3B-I to MAP1LC3B-II (Fig 7B, left), we observed no change in MAP1LC3B lipidation in the adherent IEC-18 cells in 20h after the introduction of ATG3 in the cells (Fig. 5B, right), nor in 12 h or 48 h after generating ATG3-overproducing cells (not shown). Likewise, similar to what is shown above (Fig. 1B), we found that detachment of IEC-18 cells resulted in a substantial increase in the number of GFP-LC3B puncta per cell, whereas the introduction of ATG3 in these cells has no impact on GFP-LC3B distribution in these cells when they were attached to the ECM (Fig. 7C). Furthermore, unlike what was observed after cell detachment (Fig. 2A), overexpression of ATG3 in the adherent cells did not change the fraction of these cells in the S-phase of the cell cycle (Fig. 7D). Thus, it appears that ATG3 upregulation is not sufficient for the induction of autophagy and growth arrest in attached intestinal epithelial cells.
We further found that when the cells producing ectopic ATG3 were generated (in the process of a 24 h-long selection with puromycin) and placed in monolayer culture for an additional 20 h, a significant fraction of these cells displayed a typical feature of apoptosis, such as the ability to bind ANXA5 (Fig. 7E). When cultured in the presence of exogenous ATG3 for an extended period of time (7–10 d) a significant fraction of these cells (60–70%) lost viability as was evident from their inability to form colonies (Fig. 7F). Thus, upregulation of ATG3 in the adherent intestinal epithelial cells is insufficient for the induction of autophagy in these cells and triggers their apoptotic death.
Conjugation of ATG3 to ATG12 is not required for the ability of ATG3 to kill attached intestinal epithelial cells
Others found that ATG3 can cause apoptosis via covalent binding to ATG12 via lysine 243 (of ATG3).47 It was proposed that the ATG12–ATG3 conjugate can increase the susceptibility of cells to various apoptotic stimuli by altering mitochondrial homeostasis. This apoptosis is thought to be mediated by mechanisms that are unrelated to autophagy.47 It was demonstrated in this regard that an ATG3 variant in which lysine 243 is mutated to arginine (ATG3KR) is capable of causing autophagy but does not promote apoptosis.47 We thus used this mutant to test the role of ATG12–ATG3 conjugation in ATG3-dependent death of adherent intestinal epithelial cells. We found that the ATG3KR mutant when expressed in IEC-18 cells (as described above for the wild-type ATG3) triggered loss of the adherent cells with the same efficiency as the wild-type ATG3 (Fig. 8). Thus, ATG12 conjugation to ATG3 is unlikely required for the ability of ATG3 to kill attached intestinal epithelial cells.
Figure 8.
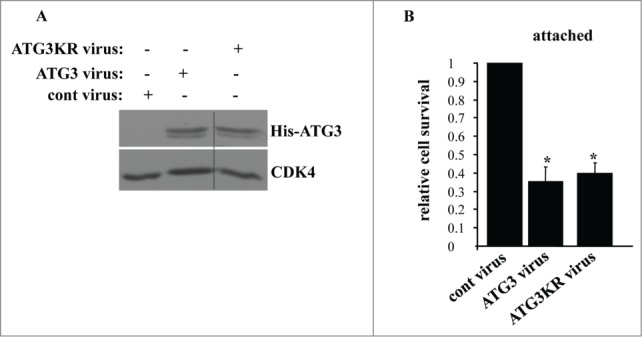
Lysine 243 of ATG3 is not required for the ability of ATG3 to kill attached intestinal epithelial cells. (A) IEC-18 cells were infected (+) or not infected (−) with 4 × 104 viral particles per cell of the control retrovirus (cont virus) or ATG3-encoding retrovirus (ATG3 virus), or the virus encoding ATG3 in which K243 was mutated to arginine (ATG3KR virus), treated with 2 μg/ml puromycin for 24 h and assayed for ATG3 expression by western blot. CDK4 was used as a loading control. (B) IEC-18 cells treated as in (A) were assayed for survival as in Figure 5F. The numbers represent the average of triplicates plus the SD. This experiment was repeated twice with similar results. * indicates that the p-value was less than 0.05.
Detachment-induced upregulation of ATG7 is required for detachment-induced autophagosome formation in intestinal epithelial cells
Since ATG3 upregulation cannot by itself induce autophagosome formation in intestinal epithelial cells we speculated that, in addition to such upregulation, other autophagy-promoting events occur in the detached cells. In addition to ATG3, detachment of intestinal epithelial cells triggered upregulation of ATG7 (Fig. 4B, D, H). We reasoned that such upregulation (possibly in concert with other yet unknown detachment-dependent events) could also contribute to detachment-induced autophagy. We therefore tested the role of ATG7 in autophagosome formation in detached cells.
We found that ablation of ATG7 by 2 different siRNAs (Fig. 9A, left, center), significantly reduced ATG7 levels in detached IEC-18 cells. ATG7 levels in detached cells transfected with one of these siRNAs were similar to those in the attached cells whereas transfection with the second siRNA resulted in somewhat lower ATG7 levels in detached cells compared to the attached ones (Fig. 9A, right). However, transfection with each siRNA resulted in a similar (substantial) degree of reduction in the number of GFP-LC3B puncta per cell (the GFP-LC3B vector was introduced in cells via transient transfection) in detached IEC-18 cells (Fig. 9B). These data indicate that once detached cells lose the ability to upregulate ATG7 above a certain level (e.g., that observed in the attached cells), autophagosome formation cannot be induced in the detached cells. We did not observe consistent inhibition of MAP1LC3B lipidation in the detached cells after enforced ATG7 downregulation, possibly due to the fact that the amount of the remaining ATG7 was sufficient to promote such lipidation. However, the amount of ATG7 remaining in the cells transfected with ATG7-specific siRNAs was clearly not sufficient for the lipidation of GABARAP since we found that ATG7 loss caused a significant inhibition of GABARAP lipidation in the detached cells (Fig. 9C).52 Hence, upregulation of ATG7 contributes to detachment-induced autophagy of intestinal epithelial cells. These data are consistent with a notion that detachment-induced upregulation of ATG3 stimulates autophagy of detached cells together with other detachment-induced autophagy-promoting events, such as, for example, detachment-induced upregulation of ATG7.
Figure 9.
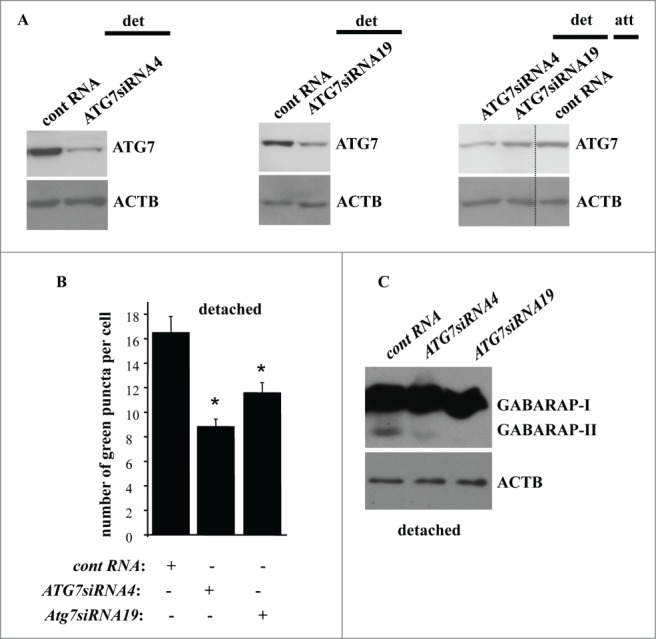
Detachment-induced upregulation of ATG7 contributes to autophagosome formation in detached intestinal epithelial cells. (A) IEC-18 cells IEC-18 cells were transfected with 100 nM control RNA (cont RNA) or ATG7-specific small interfering RNA (ATG7siRNA)4 (left) or 19 (center) and cultured detached from the ECM for 20 h (left, center). IEC-18 cells transfected as above were cultured attached to the ECM for 20 h in case of the control RNA or detached from the ECM for 20 h in case of ATG7siRNAs 4 and 19 and assayed for ATG7 expression by western blot. (B) IEC-18 cells were transfected (+) or not transfected (−) as in (A), then transiently transfected with a GFP-LC3B expression vector, detached from the ECM for 12 h and assayed for the presence of GFP-LC3B puncta as in Figure 1. The results represent the average of the number of GFP-LC3B puncta per cell observed for 60–120 cells plus the SE. This experiment was repeated twice with similar results. * indicates that the p-value was less than 0.05. (C) IEC-18 cells were transfected as in (A), cultured for 16 h detached from the ECM and assayed for GABARAP expression by western blot. Positions of GABARAP-I and GABARAP-II on the gel are indicated. ACTB was used as a loading control in (A and C).
Adherent intestinal epithelial cells that survive after ATG3 upregulation display anoikis resistance
In the course of our studies we noticed that even though the majority of adherent intestinal epithelial cells die in response to the transduction with ATG3-encoding retrovirus, some of these cells survive (Fig. 7F). Furthermore, when expanded in the presence of puromycin (resistance to which is encoded by the respective viral vector) these cells displayed substantial levels of constitutively produced exogenous ATG3 (Fig. 10A). Thus, it appears that a fraction of IEC-18 cells are capable of resisting ATG3-induced apoptosis, and the total cell population is replaced with these apoptosis-resistant cells after ATG3 upregulation. Interestingly, when detached from the ECM, the cells overproducing ATG3 in a stable manner turned out to be significantly more viable than the control cells (Fig. 10B). Hence, these cells are resistant to detachment-induced apoptosis (a phenomenon called anoikis), a property that is thought to be essential for the malignant potential of cancer cells.3-5,7,8,17,18,23,55 We further found that the cells stably producing ectopic ATG3 did not display increased autophagosome formation compared to the control cells neither when attached to the ECM nor when detached for 12 h (Fig. 10C) or 17 h (not shown). Thus, their anoikis resistance does not seem to be related to the ability of ATG3 to promote autophagy.
Figure 10.
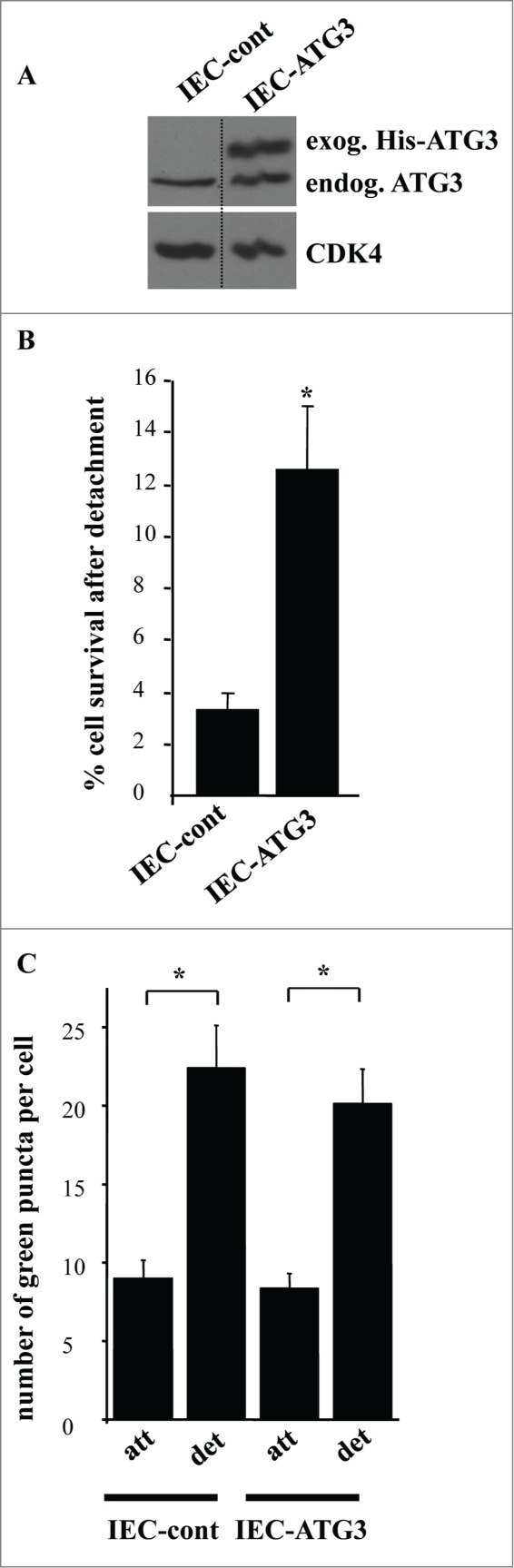
Intestinal epithelial cells that are resistant to apoptosis induced by ATG3 are also resistant to apoptosis triggered by their detachment from the ECM. (A) IEC-18 cells were infected with 4 × 104 viral particles per cell of the control retrovirus or ATG3-encoding retrovirus and cultured in the presence of 2 μg/ml puromycin until the respective control cell line (IEC-cont) and ATG3-overexpressing cell line (IEC-ATG3) were established. Cells were then assayed for ATG3 expression by western blot as in Figure 7A. (B) The indicated cell lines were plated in monolayer culture either immediately or after being cultured without adhesion to the ECM for 16 h. Cell colonies were allowed to form for 7–10 d. The colonies were then stained and counted. “% cell survival after detachment” was calculated as the percentage of the colonies formed by the cells re-plated after detachment relative to that formed by the attached cells. The results represent the average of triplicates plus the SD. This experiment was repeated twice with similar results. * indicates that the p-value was less than 0.05. (C) The indicated cell lines were transiently transfected with a GFP-LC3B expression vector, cultured attached to (att) or detached from (det) the ECM for 12 h and assayed for the presence of GFP-LC3B puncta as in Figure 1. The results represent the average of the number of GFP-LC3B puncta per cell observed for 24–40 cells plus the SE.
In summary, we have identified in this study a novel mechanism of autophagosome formation in intestinal epithelial cells induced by their detachment from the ECM. We found before that this mechanism contributes to the blockade of anchorage-independent proliferation of nonmalignant intestinal epithelial cells. We also found in the past that RAS oncogene promotes growth of detached cell by blocking their autophagy (and the ability to grow without adhesion to the ECM is thought to be critical for the malignant potential of tumor cells).1,7,8 We have found now that detachment-induced autophagosome formation in the nonmalignant intestinal epithelial cells is mediated by more than one signal, including those triggered by detachment-induced upregulation of ATG3 and ATG7. We also observed that cell-ECM interactions can determine the biological functions of ATG3: when ATG3 is upregulated in detached cells in the context of other autophagy-promoting events (e.g. detachment-induced ATG7 upregulation) ATG3 contributes to the induction of autophagy. However, when overexpressed in the adherent cells, in circumstances that do not favor autophagy induction, ATG3 triggers apoptosis. Those cells that can adapt to the presence of ATG3 and survive become anoikis-resistant, i.e. they display features of malignant cells.
Discussion
In this study we have identified novel mechanisms of autophagy of intestinal epithelial cells induced by their detachment from the ECM. The importance of identification of these mechanisms stems from our earlier findings indicating that detachment-induced autophagy triggers growth arrest of such cells.1 The ability of cancer cells to grow without adhesion to the ECM is thought to represent a critical prerequisite for tumor progression.4,5,10-14 We found that oncogenic RRAS promotes proliferation of intestinal epithelial cells in the absence of adhesion to the ECM by blocking their autophagy.1 The mechanisms by which RAS exerts this effect were examined by us in the past. We found that RAS blocks detachment-induced autophagy by triggering calpain-dependent degradation of BECN1.1 In agreement with our findings, others observed that inhibition of detachment-dependent autophagy enhances proliferation of breast epithelial cells carrying an oncogenic mutant of the PI3-kinase when these cells are placed in a 3-dimensional culture.56 So far, the mechanisms by which detachment of the nonmalignant intestinal epithelial cells from the ECM promotes autophagy have remained unknown. We have shown now that such autophagy is triggered by detachment-induced upregulation of ATG3 and ATG7.
Our results indicate that detachment-induced upregulation of ATG3 stimulates autophagy of detached cells together with other detachment-induced autophagy-promoting events, such as, for example detachment-induced upregulation of ATG7. When upregulated in the adherent cells by itself, ATG3 does not trigger autophagy but induces apoptosis instead. These data are consistent with findings made by others that ATG3 and other mediators of autophagy, such as ATG12, can trigger apoptosis via mechanisms unrelated to their ability to promote autophagosme formation.46,47 Others proposed that in some cases the ability of ATG3 to trigger apoptosis requires the ability of this protein to conjugate with ATG12.47 According to our data, ATG3-induced apoptotic death of attached intestinal epithelial cells occurs regardless of whether or not ATG3 can conjugate with ATG12. Understanding the mechanisms of ATG3-induced apoptosis represents the subject of our ongoing research.
Whether or not detachment-induced upregulation of ATG3 and ATG7 are the only events that contribute to detachment-induced autophagy of intestinal epithelial cells is presently not known. It was demonstrated that detachment of other types of epithelial cells (e.g., those of the breast) blocks several major metabolic pathways in such cells.57 Such metabolic stress could contribute to the induction of autophagy. In addition, it cannot be excluded that other yet unidentified autophagy-promoting changes in the expression of the regulators of autophagy are triggered by detachment of intestinal epithelial cells. Such events, together with detachment-dependent upregulation of ATG3 and ATG7 could in principle contribute to autophagy of detached intestinal epithelial cells. Hence, which combination of detachment-induced autophagy-promoting signals would be sufficient for the induction of autophagy in the adherent cells remains to be investigated. Studies aimed at addressing these questions are currently underway in our lab.
One potentially important physiological implication of our studies is that inhibition of autophagy due to inactivation or loss of ATG3 partners involved in this process can create a situation when ATG3 is no longer needed for autophagosome formation and instead of participating in this process triggers apoptosis. It is now thought in this regard that when autophagy is increased beyond a certain level it blocks tumor growth, and this is presumably the reason why oncogenes, such as RAS, ERBB2, AKT, and others have the potential to reduce autophagy in cancer cells.1,36,39 However, some level of autophagy in malignant cells is thought to be required for tumor growth as complete inhibition of autophagy achieved by, for example deletion of genes coding for autophagy mediators in mice (e.g. that of the Atg7 gene) prevents tumor growth.42 One possible interpretation of these data is that cancer cells often experience disruption of metabolism due to their inadequate supply with blood and require basal levels of autophagy for the degradation of the nonessential cellular macromolecules and utilization of the resulting monomeric components of these molecules for the restoration of defective metabolic pathways.42 However, it is conceivable that loss of ATG7 or other autophagy-related proteins creates a situation when ATG3 which normally acts as an ATG7 partner in the induction of autophagy can no longer be involved in facilitating autophagy (since autophagosome formation is blocked due to the absence of ATG7) and promotes apoptosis of tumor cells instead.29 Thus, lack of basal autophagy and ATG3-induced apoptosis could in principle cooperate with each other in suppressing tumor growth in mice lacking ATG7 or other autophagy-promoting proteins.
We have demonstrated here that those adherent intestinal epithelial cells that do not succumb to ATG3-induced apoptosis become anoikis-resistant. What are the possible mechanisms of such resistance? We found previously that anoikis of intestinal epithelial cells is driven by multiple mechanisms mediated by detachment-induced changes in the expression of various regulators of apoptosis.4,18,24,25 Importantly for this study, we and others observed previously that a small fraction of nonmalignant intestinal epithelial cells, such as IEC-18 and Hkh-2 used in this study, remains viable after being cultured without adhesion to the ECM and if expanded displays pronounced and well reproducible resistance to anoikis 22,23 (the nature of this cellular heterogeneity is presently not known). These data indicate that intestinal epithelial cells contain a cell population in which at least some of the anoikis-promoting mechanisms are blocked. It is possible that apoptosis induced by ATG3 is driven by mechanisms that overlap with those triggered by cell detachment. Thus, conceivably, unlike the majority of the cells which succumb to ATG3-induced apoptosis after ATG3 upregulation, the cells composing the anoikis-resistant fraction and hence, lacking the indicated apoptosis-promoting mechanisms, do not die in response to ATG3 upregulation.
What is the possible physiological relevance of the ability of the cells to survive and become anoikis-resistance in response to the upregulation of ATG3? It was found in this regard that ATG3 is upregulated, at least in some types of cells, under ischemia, a condition that frequently occurs in tumors due to the irregularities of the tumor vasculature.58-60 Ischemia induces apoptosis, and it is possible that such apoptosis is mediated at least in part by ATG3 upregulation.60 Our data indicate that cells that remain viable despite ATG3 upregulation become anoikis-resistant. On the other hand, the fact that cancer cells can acquire resistance to ischemia-induced apoptosis is well documented.61 Thus, it is conceivable that cancer cells that do not die by ischemia-induced apoptosis and are hence selected for resistance to ATG3-induced apoptosis are at the same time selected for increased anoikis resistance. Anoikis resistance of cancer cells is thought to substantially contribute to their tumorigenicity.4,5,10-14 Hence, the ischemia-resistant cells would be expected to have a higher malignant potential than those before the onset of ischemia. Perhaps not by coincidence, tumors formed by cells that survive during hypoxia (the major consequence of ischemia) are particularly aggressive.61
ATG3 is upregulated in colorectal cancer cells during photodynamic therapy (PDT), a treatment that kills cancer cells by promoting their apoptosis.62 It was previously established that malignant cells that survive after PDT can form more aggressive tumors than those formed by the untreated cells.63 It cannot be excluded that ATG3 contributes to PDT-induced apoptosis of cancer cells and that those cells that survive after such treatment (and are hence selected for resistance to ATG3-dependent apoptosis) possess increased anoikis resistance. Therefore tumors formed by such cells can be expected to display increased aggressiveness.
Levels of expression of some of the autophagy mediators in various human cancers were compared to those in respective normal cells in several studies, and used to assess the possible role of autophagy in the progression of these malignancies.37,44,45 To our knowledge, whether or not ATG3 levels change in the course of progression of any cancers has not yet been explored. Our data indicate that if changes in ATG3 expression are found in the future in human cancers, these changes could reflect the contribution of not only autophagy but also that of ATG3-dependent apoptosis in the course of the respective diseases.
In summary, we have found in this study that intestinal epithelial cell-ECM adhesion can switch the biological function of ATG3: ATG3 contributes to autophagy of detached cells but kills those attached to the ECM.
Materials and Methods
Cell culture
Cells were cultured as described previously.4 For suspension cultures cells were plated above a layer of 1% sea plaque agarose (lONZA, 50100) polymerized in α-minimal essential medium (MEM; Invitrogen, 11900) in case of IEC-18 cells and Dulbecco's modified essential medium (DMEM; Invitrogen, 12800) in case of Hkh-2 cells.
Western blot
This assay was performed as described previously.4 The following antibodies were used in the study. Anti-ATG3 (Cell Signaling Technology, 3415), anti-ATG7 (Cell Signaling Technology, 2631), anti-ATG16L1 (ProSci, 4425), anti-MAP1LC3B (Cell Signaling Technology, 3868), anti-GABARAP (Sigma, SAB2100873), anti-SH3GLB1 (ProSci, 45–143), anti-UVRAG (Abcam, ab72073), anti-ATG5 (Proteintech, 10181–2-AP), anti-ATG10 (Prosci 43990), anti-GFP (Clontech, 632381), anti-CDK4 (Santa Cruz Biotechnology, sc-601) and anti-AKTB (Santa Cruz Biotechnology, sc-130656).
RNA interference
RNAi was performed as described previously.26 The sequences of the siRNAs were as follows. ATG3 siRNA1: UGUACAUCACUUACGACAA; ATG3 siRNA2: CGGAUGAAUUG GAGG CUAU; ATG7 siRNA4: GGCACGAACUGACCCAGAA; ATG7 siRNA19: AGUGAAUGCCA GCGGGUUC; GABARAP siRNA2: AUGAAAGCGUCUACGGUCU; GABARAP siRNA3: ACAAAGAGGAGCAUCCGUU.
Quantitative polymerase chain reaction (qPCR)
This procedure was performed as described previously.18 Sequences of the primers used for the amplification of ATG3 mRNA were GCAAACAAGAACCTATGACCTG and GTCTTCATACATGTGCTCAACTG, sequences of the primers used for the amplification of ATG7 mRNA were GCTGGGAGAAGAACCAGAAA and GAGATTCAGATCCACGGATGAC, sequences of the primers used for the amplification of MAP1LC3B mRNA were TCTTTGTAAGGGCG GTTCTG and TCACAAGCATGGCTCTCTTC, and sequences of the primers used for the amplification of GABARAP mRNA were TCAGGCTCAAACACCATCTC and GCGGCTGC ATCTACCATTA.
Expression vectors
pBabe puroWTATG3, and pBabe puroKRATG3 retroviral vectors were from Addgene (26926 and 26927, respectively). pBabe mCherry-GFP-LC3 vector was provided by Dr. N. Ridgway (Dalhousie University, Canada). pBabe as well as pHIT and pVSVG vectors (components of the retroviral packaging system) were provided by Dr. P. Lee (Dalhousie University, Canada). GFP-LC3B expression vector was provided by Dr. T. Yoshimori (Osaka University, Japan).
Production of the retroviruses
3 × 106 293T cells were plated on a 100 mm tissue culture dish, grown overnight and transfected with 10 μg of either pBabe or pBabepuroWTAtg3 or pBabepuroKR Atg3 DNA, 5 μg of pHIT and 5 μg of pVSVG in the presence of 12 μl of Lipofectamine 2000 (Invitrogen, 11668–019) in 6 ml of OPTI-MEM medium (Gibco, 31985–070). The medium was changed 4 h later to DMEM containing 10% fetal calf serum. The medium was collected 48 h later and filtered through a 0.45-micron filter unit. Viral concentration in the resulting solution was measured by use of the Retrovirus Quantitation Kit (Cell Biolabs Inc., VPK-120) according to the manufacturer's instructions.
Retroviral transduction
5 × 105 IEC-18 cells were plated in a 100 mm tissue culture dish and grown overnight. 4 × 104 viral particles per cell were then added to the cells in the presence of 8 μg/ml polybrene (Sigma, 107689) and the cells were cultured for 24 h. Medium was then changed to fresh medium and the cells were cultured for 24 h in the presence of 2 μg/ml puromycin. In the case of transduction of IEC-18 cells with the mCherry-GFP-LC3B virus, 6 ml of the virus-containing culture medium prepared as described above was added to 5 × 105 IEC-18 cells in the presence of 8 μg/ml polybrene and the cells were further cultured for 24 h. Medium was then changed to fresh medium and the cells were cultured for an additional 24 h.
Detection of GFP-LC3B puncta in cells
In the case of the cells infected with retroviruses the cells were first transduced with the indicated viruses as described above and then transfected with 3 μg GFP-LC3B expression vector in the presence of 6 μl Lipofectamine 2000 in 6 ml of OPTI-MEM medium. Four h later the medium was changed to regular growth medium not containing puromycin. Twenty-four h later the cells were harvested by trypsinization, washed with phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4), and fixed with 4% paraformaldehyde for 20 min at room temperature. Images of the cells were captured by use of a Carl Zeiss Axiovert 200M microscope. In the case of the ATG3-deficient cells the cells were first transfected with the GFP-LC3B vector1 and then with the control RNA and ATG3-specific siRNAs26 and assayed as described above. In the case of the ATG7-deficient cells, the cells were initially transfected with respective RNAs26 and then with the GFP-LC3B vector.1 8 h after the transfection ATG7-deficient cells were plated in monolayer or suspension.
Detection of mCherry-GFP-LC3 puncta in cells
Cells transduced with the mCherry-GFP-LC3-encoding virus as described above were analyzed by the use of a Carl Zeiss Axiovert 200M microscope for the presence of green, red, or yellow puncta.
Flow cytometry
This assay was performed as described previously.1 Cells infected with retroviruses were cultured for 24 h in the absence of puromycin prior to being harvested for the assay.
Detection of apoptosis
Apoptosis was detected by use of ANXA5 as described previously.25 Cells infected with retroviruses were cultured for 24 h in the absence of puromycin prior to being harvested for the assay.
Detection of clonogenic cell survival
Cell colonies were stained with Crystal Violet.24 Cells infected with retroviruses were maintained in the presence of 2 μg/ml puromycin for 7–10 d while the cell colonies were allowed to form.
Statistical analysis
Statistical analysis of the data in Figures 1A, 2A, 3C, 4C, 5C, F, 6B, 7H, 8B, and C was performed by the unpaired Student's t-test. Statistical analysis of the data in Figures 1C, D and 5E, was performed by the chi-square test for goodness-of-fit.
Western blot images
Western blot images were generated by Photoshop. When lanes were removed from these images, and separate parts of the same image were joined together, a vertical dotted line was used to indicate where the image was cut.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Drs. Yoshimori, Ridgway and Lee for the materials provided for this study.
Funding
This study was supported by the Canadian Institutes of Health Research Nova Scotia Regional Partnership Program (CIHR/NS RPP) operating grant 125109 and the CIHR/NS RPP New Investigator award held by Kirill Rosen. Byong Yoo was a recipient of the IWK Health Centre Research Associateship. Yongling Li was a recipient of the IWK Health Centre Pos-Doctoral Fellowship.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Yoo BH, Wu X, Li Y, Haniff M, Sasazuki T, Shirasawa S, Eskelinen EL, Rosen RV. Oncogenic ras-induced down-regulation of autophagy mediator Beclin-1 is required for malignant transformation of intestinal epithelial cells. J Biol Chem 2010; 285:5438-49; PMID:19778902; http://dx.doi.org/ 10.1074/jbc.M109.046789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rak J, Mitsuhashi Y, Erdos V, Huang SN, Filmus J, Kerbel RS. Massive programmed cell death in intestinal epithelial cells induced by three-dimensional growth conditions: suppression by mutant c-H-ras oncogene expression. J Cell Biol 1995; 131:1587-98; PMID:8522614; http://dx.doi.org/ 10.1083/jcb.131.6.1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 1994; 124:619-626; PMID:8106557; http://dx.doi.org/ 10.1083/jcb.124.4.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosen K, Rak J, Leung T, Dean NM, Kerbal RS, Filmus J. Activated Ras prevents downregulation of Bcl-X(L) triggered by detachment from the extracellular matrix. A mechanism of Ras-induced resistance to anoikis in intestinal epithelial cells. J Cell Biol 2000; 149:447-56; PMID:10769035; http://dx.doi.org/ 10.1083/jcb.149.2.447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosen K, Rak J, Jin J, Kerbel RS, Newman MJ, Filmus J. Downregulation of the pro-apoptotic protein Bak is required for the ras-induced transformation of intestinal epithelial cells. Curr Biol 1998; 8:1331-4; PMID:9843689; http://dx.doi.org/ 10.1016/S0960-9822(07)00564-7 [DOI] [PubMed] [Google Scholar]
- 6.Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J 1997; 16:2783-93; PMID:9184223; http://dx.doi.org/ 10.1093/emboj/16.10.2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacks T, Weinberg RA. Taking the study of cancer cell survival to a new dimension. Cell 2002; 111:923-5; PMID:12507419; http://dx.doi.org/ 10.1016/S0092-8674(02)01229-1 [DOI] [PubMed] [Google Scholar]
- 8.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature 2001; 411:342-8; PMID:11357141; http://dx.doi.org/ 10.1038/35077213 [DOI] [PubMed] [Google Scholar]
- 9.Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell 1974; 3:355-9; PMID:4442124; http://dx.doi.org/ 10.1016/0092-8674(74)90050-6 [DOI] [PubMed] [Google Scholar]
- 10.Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, Der CJ, Counter CM. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell 2005; 7:533-45; PMID:15950903; http://dx.doi.org/ 10.1016/j.ccr.2005.04.030 [DOI] [PubMed] [Google Scholar]
- 11.Coll ML, Rosen K, Ladeda V, Filmus J. Increased Bcl-xL expression mediates v-Src-induced resistance to anoikis in intestinal epithelial cells. Oncogene 2002; 21:2908-13; PMID:11973652; http://dx.doi.org/ 10.1038/sj.onc.1205388 [DOI] [PubMed] [Google Scholar]
- 12.Rosen K, Coll M, Li A, Filmus J. Transforming growth factor-alpha prevents detachment-induced inhibition of c-Src kinase activity, Bcl-XL down-regulation, and apoptosis of intestinal epithelial cells. J Biol Chem 2001; 276:37273-9; PMID:11487584; http://dx.doi.org/ 10.1074/jbc.M106424200 [DOI] [PubMed] [Google Scholar]
- 13.Li H, Ray G, Yoo BH, Erdogan M, Rosen KV. Down-regulation of death-associated protein kinase-2 is required for beta-catenin-induced anoikis resistance of malignant epithelial cells. J Biol Chem 2009; 284:2012-22; PMID:18957423; http://dx.doi.org/ 10.1074/jbc.M805612200 [DOI] [PubMed] [Google Scholar]
- 14.Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX, Davies MA, Khan H, Furui T, Mao M, et al.. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene 1999; 18:7034-45; PMID:10597304; http://dx.doi.org/ 10.1038/sj.onc.1203183 [DOI] [PubMed] [Google Scholar]
- 15.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. EphA2: a determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene 2004; 23:1448-56; PMID:14973554; http://dx.doi.org/ 10.1038/sj.onc.1207247 [DOI] [PubMed] [Google Scholar]
- 16.Scotlandi K, Maini C, Manara MC, Benini S, Serra M, Cerisano V, Strammiello R, Baldini N, Lollini PL, Nanni P, et al.. Effectiveness of insulin-like growth factor I receptor antisense strategy against Ewing's sarcoma cells. Cancer Gene Ther 2002; 9:296-307; PMID:11896447; http://dx.doi.org/ 10.1038/sj.cgt.7700442 [DOI] [PubMed] [Google Scholar]
- 17.Frankel A, Rosen K, Filmus J, Kerbel RS. Induction of anoikis and suppression of human ovarian tumor growth in vivo by down-regulation of Bcl-X(L). Cancer Res 2001; 61:4837-41; PMID:11406560 [PubMed] [Google Scholar]
- 18.Yoo BH, Wang Y, Erdogan M, Sasazuki T, Shirasawa S, Corcos L, Sabapathy K, Rosen KV. Oncogenic ras-induced down-regulation of pro-apoptotic protease caspase-2 is required for malignant transformation of intestinal epithelial cells. J Biol Chem 2011; 286:38894-903; PMID:21903589; http://dx.doi.org/ 10.1074/jbc.M111.290692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. CEACAM6 gene silencing impairs anoikis resistance and in vivo metastatic ability of pancreatic adenocarcinoma cells. Oncogene 2004; 23:465-73; PMID:14724575; http://dx.doi.org/ 10.1038/sj.onc.1207036 [DOI] [PubMed] [Google Scholar]
- 20.Jiang K, Sun J, Cheng J, Djeu JY, Wei S, Sebti S. Akt mediates Ras downregulation of RhoB, a suppressor of transformation, invasion, and metastasis. Mol Cell Biol 2004; 24:5565-76; PMID:15169915; http://dx.doi.org/ 10.1128/MCB.24.12.5565-5576.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berezovskaya O, Schimmer AD, Glinskii AB, Pinilla C, Hoffman RM, Reed JC, Glinsky GV. Increased expression of apoptosis inhibitor protein XIAP contributes to anoikis resistance of circulating human prostate cancer metastasis precursor cells. Cancer Res 2005; 65:2378-86; PMID:15781653; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-2649 [DOI] [PubMed] [Google Scholar]
- 22.Rak J, Mitsuhashi Y, Sheehan C, Krestow JK, Florenes VA, Filmus J, Kerbel RS. Collateral expression of proangiogenic and tumorigenic properties in intestinal epithelial cell variants selected for resistance to anoikis. Neoplasia 1999; 1:23-30; PMID:10935467; http://dx.doi.org/ 10.1038/sj.neo.7900001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Derouet M, Wu X, May L, Hoon Yoo B, Sasazuki T, Shirasawa S, Rak J, Rosen RV. Acquisition of anoikis resistance promotes the emergence of oncogenic K-ras mutations in colorectal cancer cells and stimulates their tumorigenicity in vivo. Neoplasia 2007; 9:536-45; PMID:17710156; http://dx.doi.org/ 10.1593/neo.07217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosen K, Shi W, Calabretta B, Filmus J. Cell detachment triggers p38 mitogen-activated protein kinase-dependent overexpression of fas ligand. A novel mechanism of anoikis of intestinal epithelial cells. J Biol Chem 2002; 277:46123-30; PMID:12356751; http://dx.doi.org/ 10.1074/jbc.M207883200 [DOI] [PubMed] [Google Scholar]
- 25.Yoo BH, Berezkin A, Wang Y, Zagryazhskaya A, Rosen KV. Tumor suppressor protein kinase Chk2 is a mediator of anoikis of intestinal epithelial cells. Int J Cancer 2012; 131:357-66; PMID:21834075; http://dx.doi.org/ 10.1002/ijc.26368 [DOI] [PubMed] [Google Scholar]
- 26.Liu Z, Li H, Derouet M, Filmus J, LaCasse EC, Korneluk RG, Kerbel RS, Rosen RV. ras Oncogene triggers up-regulation of cIAP2 and XIAP in intestinal epithelial cells: epidermal growth factor receptor-dependent and -independent mechanisms of ras-induced transformation. J Biol Chem 2005; 280:37383-92; PMID:16115895; http://dx.doi.org/ 10.1074/jbc.M503724200 [DOI] [PubMed] [Google Scholar]
- 27.Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer 2005; 5:886-97; PMID:16239905; http://dx.doi.org/ 10.1038/nrc1738 [DOI] [PubMed] [Google Scholar]
- 28.Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy 2005; 1:1-10; PMID:16874026; http://dx.doi.org/ 10.4161/auto.1.1.1270 [DOI] [PubMed] [Google Scholar]
- 29.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ 2005; 12(Suppl 2):1542-52; PMID:16247502; http://dx.doi.org/ 10.1038/sj.cdd.4401765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanida I, Komatsu M, Ueno T, Kominami E. GATE-16 and GABARAP are authentic modifiers mediated by Apg7 and Apg3. Biochem Biophys Res Commun 2003; 300:637-44; PMID:12507496; http://dx.doi.org/ 10.1016/S0006-291X(02)02907-8 [DOI] [PubMed] [Google Scholar]
- 31.Tanida I, Tanida-Miyake E, Komatsu M, Ueno T, Kominami E. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J Biol Chem 2002; 277:13739-44; PMID:11825910; http://dx.doi.org/ 10.1074/jbc.M200385200 [DOI] [PubMed] [Google Scholar]
- 32.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen E-L, Mizushima N, Ohsumi Y, et al.. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809-20; PMID:14638851; http://dx.doi.org/ 10.1172/JCI20039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003; 100:15077-82; PMID:14657337; http://dx.doi.org/ 10.1073/pnas.2436255100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 2011; 25:795-800; PMID:21498569; http://dx.doi.org/ 10.1101/gad.2016211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, et al.. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007; 9:1142-51; PMID:17891140; http://dx.doi.org/ 10.1038/ncb1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et al.. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013; 154:1269-84; PMID:24034250; http://dx.doi.org/ 10.1016/j.cell.2013.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672-6; PMID:10604474; http://dx.doi.org/ 10.1038/45257 [DOI] [PubMed] [Google Scholar]
- 38.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, et al.. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51-64; PMID:16843265; http://dx.doi.org/ 10.1016/j.ccr.2006.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012; 338:956-9; PMID:23112296; http://dx.doi.org/ 10.1126/science.1225967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126:121-34; PMID:16839881; http://dx.doi.org/ 10.1016/j.cell.2006.05.034 [DOI] [PubMed] [Google Scholar]
- 41.Yu JL, Rak JW, Coomber BL, Hicklin DJ, Kerbel RS. Effect of p53 status on tumor response to antiangiogenic therapy. Science 2002; 295:1526-8; PMID:11859195; http://dx.doi.org/ 10.1126/science.1068327 [DOI] [PubMed] [Google Scholar]
- 42.Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, et al.. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 2014; 4:914-27; PMID:24875857; http://dx.doi.org/ 10.1158/2159-8290.CD-14-0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al.. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013; 504:296-300; PMID:24305049; http://dx.doi.org/ 10.1038/nature12865 [DOI] [PubMed] [Google Scholar]
- 44.Zhao H, Yang M, Zhao J, Wang J, Zhang Y, Zhang Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med Oncol 2013; 30:475; PMID:23371253; http://dx.doi.org/ 10.1007/s12032-013-0475-1 [DOI] [PubMed] [Google Scholar]
- 45.Li BX, Li CY, Peng RQ, Wu XJ, Wang HY, Wan DS, Zhu XF, Zhang XS. The expression of beclin 1 is associated with favorable prognosis in stage IIIB colon cancers. Autophagy 2009; 5:303-6; PMID:19066461; http://dx.doi.org/ 10.4161/auto.5.3.7491 [DOI] [PubMed] [Google Scholar]
- 46.Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell 2011; 44:698-709; PMID:22152474; http://dx.doi.org/ 10.1016/j.molcel.2011.10.014 [DOI] [PubMed] [Google Scholar]
- 47.Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, Debnath J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010; 142:590-600; PMID:20723759; http://dx.doi.org/ 10.1016/j.cell.2010.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol 2001; 13:555-62; PMID:11544023; http://dx.doi.org/ 10.1016/S0955-0674(00)00251-9 [DOI] [PubMed] [Google Scholar]
- 49.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Engeland M, Nieland LJ, Ramaekers FC, Schutte B, Reutelingsperger CP. Annexin V-affinity assay: a review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 1998; 31:1-9; PMID:9450519; http://dx.doi.org/ 10.1002/(SICI)1097-0320(19980101)31:1%3c1::AID-CYTO1%3e3.0.CO;2-R [DOI] [PubMed] [Google Scholar]
- 51.Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 1993; 260:85-8; PMID:8465203; http://dx.doi.org/ 10.1126/science.8465203 [DOI] [PubMed] [Google Scholar]
- 52.Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010; 29:1792-802; PMID:20418806; http://dx.doi.org/ 10.1038/emboj.2010.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805-12; PMID:15169837; http://dx.doi.org/ 10.1242/jcs.01131 [DOI] [PubMed] [Google Scholar]
- 54.Wirth M, Joachim J, Tooze SA. Autophagosome formation–the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol 2013; 23:301-09; PMID:23727157; http://dx.doi.org/ 10.1016/j.semcancer.2013.05.007 [DOI] [PubMed] [Google Scholar]
- 55.Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. CA Nature 2004; 430:1034-9; PMID:15329723; http://dx.doi.org/ 10.1038/nature02765 [DOI] [PubMed] [Google Scholar]
- 56.Chen N, Eritja N, Lock R, Debnath J. Autophagy restricts proliferation driven by oncogenic phosphatidylinositol 3-kinase in three-dimensional culture. Oncogene 2013; 32: 2543-54; PMID:22777351; http://dx.doi.org/ 10.1038/onc.2012.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schafer Z T, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie H Y, Gao S, Puigserver P, Brugge J S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009; 461: 109-13; PMID:19693011; http://dx.doi.org/ 10.1038/nature08268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu BX, Darden AG, Laser M, Li Y, Crosson CE, Hazard ES III, Ma J-X. The rat Apg3p/Aut1p homolog is upregulated by ischemic preconditioning in the retina. Mol Vis 2006; 12:1292-302; PMID:17110912 [PubMed] [Google Scholar]
- 59.Hammond EM, Giaccia AJ. The role of p53 in hypoxia-induced apoptosis. Biochem Biophys Res Commun 2005; 331:718-25; PMID:15865928; http://dx.doi.org/ 10.1016/j.bbrc.2005.03.154 [DOI] [PubMed] [Google Scholar]
- 60.Rak J, Yu JL. Oncogenes and tumor angiogenesis: the question of vascular “supply” and vascular “demand”. Semin Cancer Biol 2004; 14:93-104; PMID:15018893; http://dx.doi.org/ 10.1016/j.semcancer.2003.09.014 [DOI] [PubMed] [Google Scholar]
- 61.Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996; 379:88-91; PMID:8538748; http://dx.doi.org/ 10.1038/379088a0 [DOI] [PubMed] [Google Scholar]
- 62.Wei MF, Chen MW, Chen KC, Lou PJ, Lin SY, Hung SC, Hsiao M, Yao CJ, Shieh MJ. Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy 2014; 10:1179-92; PMID:24905352; http://dx.doi.org/ 10.4161/auto.28679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gilaberte Y, Milla L, Salazar N, Vera-Alvarez J, Kourani O, Damian A, Rivarola V, Roca MJ, Espada J, Gonzalez S. Cellular intrinsic factors involved in the resistance of squamous cell carcinoma to photodynamic therapy. J Invest Dermatol 2014; 134:2428-37; PMID:24717244; http://dx.doi.org/ 10.1038/jid.2014.178 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.