

Abstract
Peroxisomes are ubiquitous cell organelles essential for human health. To maintain a healthy cellular environment, dysfunctional and superfluous peroxisomes need to be selectively removed. Although emerging evidence suggests that peroxisomes are mainly degraded by pexophagy, little is known about the triggers and molecular mechanisms underlying this process in mammalian cells. In this study, we show that PEX5 proteins fused to a bulky C-terminal tag trigger peroxisome degradation in SV40 large T antigen-transformed mouse embryonic fibroblasts. In addition, we provide evidence that this process is autophagy-dependent and requires monoubiquitination of the N-terminal cysteine residue that marks PEX5 for recycling. As our findings also demonstrate that the addition of a bulky tag to the C terminus of PEX5 does not interfere with PEX5 monoubiquitination but strongly inhibits its export from the peroxisomal membrane, we hypothesize that such a tag mimics a cargo protein that cannot be released from PEX5, thus keeping monoubiquitinated PEX5 at the membrane for a sufficiently long time to be recognized by the autophagic machinery. This in turn suggests that monoubiquitination of the N-terminal cysteine of peroxisome-associated PEX5 not only functions to recycle the peroxin back to the cytosol, but also serves as a quality control mechanism to eliminate peroxisomes with a defective protein import machinery.
Keywords: peroxisome, PEX5, pexophagy, protein import, receptor recycling, selective autophagy, ubiquitin
Abbreviations
- 3-MA
3-methyladenine
- ABCD3/PMP70
ATP-binding cassette, sub-family D (ALD), member 3
- ATG
autophagy-related
- DTM
docking/translocation machinery
- EGFP
enhanced green fluorescent protein
- Hs
homo sapiens
- KR
KillerRed
- LAMP
lysosomal-associated membrane protein
- MAP1LC3A/B
microtubule-associated protein 1 light chain 3 α/β
- MEFs
mouse embryonic fibroblasts
- Mm
Mus musculus
- NBR1
neighbor of BRCA1 gene 1
- OPTN
optineurin
- PEX
peroxisomal biogenesis factor
- PMP
peroxisomal membrane protein
- PNS
postnuclear supernatant
- PtdIns3K
class III phosphatidylinositol 3-kinase
- PTS
peroxisomal targeting signal
- REM
receptor export machinery
- ROS
reactive oxygen species
- SLC25A17
solute carrier family 25 (mitochondrial carrier; peroxisomal membrane protein, 34kDa), member 17
- SQSTM1
sequestosome 1
- SV40T
SV40 large T antigen-transformed
- TPR
tetratricopeptide repeat
- Ub
ubiquitin.
Introduction
To maintain a healthy intracellular environment, cells need to eliminate excessive and dysfunctional organelles. This turnover mainly occurs within the lysosome in a process called autophagy. Currently, 3 major autophagy pathways have been characterized in eukaryotic cells: macroautophagy, microautophagy, and chaperone-mediated autophagy.1-3 During macroautophagy—hereafter simply referred to as autophagy—a cup-shaped, double membrane-bound structure, called the phagophore, is formed in the cytoplasm. This structure elongates to engulf the cargo and eventually becomes a cargo-laden short-lived organelle known as the autophagosome. The intra-autophagosomal components are, upon autophagosome-lysosome fusion, finally degraded by lysosomal hydrolases.
Autophagy is mainly mediated by AuTophaGy-related (ATG) proteins, of which at least 38 have been identified in yeast.4,5 Of these, less than half are thought to be required for canonical autophagy, which is a highly conserved process among eukaryotes.4 Autophagy occurs both selectively and nonselectively, and—in contrast to bulk autophagy—selective autophagy pathways require the additional specific action of autophagy receptors (e.g., SQSTM1/p62, NBR1, and OPTN).6,7 These receptors act independently, or concertedly, to bridge substrates targeted for degradation with the elongating phagophore via tethering of both structures. Substrate binding generally occurs through a ubiquitin-binding domain, and the binding to the phagophore via an LC3-interacting region.7 Microtubule-associated protein 1 light chain 3 α (MAP1LC3A) and its homologs are present on the phagophore convex and concave membranes, where they—among other functions—mediate the specificity of selective autophagy.7
Peroxisomes are dynamic organelles that rapidly adapt their size, protein content, and number in response to altering environmental conditions. Important functions of peroxisomes in mammals include α- and β-oxidation of fatty acids and the biosynthesis of plasmalogens and docosahexaenoic acid.8-10 Importantly, as (i) peroxisomal enzymes produce vast amounts of reactive oxygen species (ROS) as part of their catalytic cycle,11 and (ii) ROS are involved in an array of signaling pathways,12 these organelles are also increasingly being recognized as important redox signaling platforms.13,14
To perform their various functions, peroxisomes require an operational and efficient import machinery for matrix proteins. The vast majority of these proteins contain a peroxisomal targeting signal type 1 (PTS1), made up of a C-terminally located tripeptide with the consensus sequence (S/A/C)-(K/R/H)-(L/A) (in single-letter amino acid code).15,16 PTS1-containing proteins are recognized in the cytosol by the peroxisomal matrix protein import receptor PEX5, which interacts with the PTS1 via 6 tetratricopeptide repeats (TPRs) that are located in its C terminus.17 Importantly, the mammalian PEX5 transcript undergoes alternative splicing yielding 2 major isoforms, PEX5(S) (the short variant) and PEX5(L) (the long variant), the latter of which is also involved in PTS2-import.18 Upon cargo recognition, the protein complex is transported to the peroxisomal membrane where PEX5 docks on the docking/translocation machinery (DTM), consisting of the peroxins PEX13, PEX14, and the 3 RING proteins PEX2, PEX10, and PEX12.19,20 The PEX5-cargo protein complex is then inserted into the DTM with the concomitant translocation of the cargo protein into the organelle matrix. Finally, DTM-inserted PEX5 is monoubiquitinated on an evolutionarily conserved cysteine residue (in humans and mice at amino acid position 11) and subsequently extracted from the peroxisomal membrane in an ATP-dependent manner by the receptor export machinery.21,22 This machinery, often called 'REM'23 or 'exportomer',24 consists in mammals of the core proteins PEX1 and PEX6, 2 AAA+ ATPases that can interact and form a heterohexameric ring complex.25,26
Over the past decades, it has become increasingly clear that mammalian peroxisomes are degraded via selective autophagy (a process known as 'pexophagy').27-32 This is perhaps best illustrated by the observation that proliferated rat peroxisomes are rapidly turned over in an autophagy-dependent manner upon removal of the proliferation stimulus.27,28 Unfortunately, little is currently known about the physiological triggers and molecular mechanisms underlying mammalian pexophagy. However, since (i) peroxisomes—like mitochondria—produce large amounts of ROS as part of their metabolism,11 (ii) excessive organelle-specific ROS-generation causes mitochondria- and endoplasmic reticulum-selective degradation in mammalian cells,33-35 and (iii) highly oxidized peroxisomes can be degraded via pexophagy in Hansenula polymorpha and Arabidopsis thaliana,36,37 it is tempting to speculate that mammalian pexophagy can be triggered via ROS-related mechanisms.
Finally, accumulating evidence points toward an integral role of ubiquitin in the targeting of mammalian peroxisomes to autophagosomes.30,32,38 For example, ectopic expression of peroxisomal membrane proteins (PMPs) attached to cytosolically exposed ubiquitin triggers pexophagy in a SQSTM1-dependent manner.38 In addition, overexpression of both NBR1 and PEX3 has been demonstrated to induce pexophagy in a ubiquitin-dependent fashion.30,32 However, endogenous PMPs that are ubiquitinated during pexophagy have not yet been identified. Nevertheless, a proposed candidate is PEX5,30,39 which—as mentioned above—is known to be monoubiquitinated on a cysteine residue (Cys11 in human and mouse PEX5) at the peroxisomal membrane during its normal import cycle.21 In addition, it has recently been shown that cytosolic PEX5 can also be ubiquitinated at Lys527, probably the result of a yet uncharacterized quality control process.40 In this study, we provide evidence that expression of Cys11-monoubiquitinatable but export-deficient variants of PEX5 trigger peroxisome degradation in SV40 large T antigen-transformed (SV40T) mouse embryonic fibroblasts (MEFs). This finding strongly indicates that the amount of Cys11-monoubiquitinated PEX5 at the peroxisomal membrane may function as a quality control mechanism to eliminate peroxisomes with a defective/jammed protein import machinery.
Results
Expression of PEX5 proteins fused to a bulky C-terminal tag triggers peroxisome removal in SV40T-MEFs
To gain more insight into potential triggers for peroxisome degradation in mammalian cells, we first tested the hypothesis that these organelles can be selectively removed upon oxidative damage. To generate oxidative stress in the peroxisomal matrix or at the peroxisomal membrane in a temporally controlled manner, we designed a set of KillerRed (KR)-fusion proteins. KR is a genetically-encoded photosensitizer that generates ROS upon green light illumination.41 Intriguingly, although we were unable to show an increase in peroxisome turnover upon photoactivation of peroxisomal matrix-targeted KR,42 we found that overexpression of nonphotoactivated human (Homo sapiens, Hs) PEX5(L)-KR in SV40T-MEFs already resulted in a partial or complete disappearance of peroxisomes in a large number of cells, as detected by immunofluorescence with anti-PEX14 antibodies (Fig. 1A). As such a phenotype was not discernible in cells overexpressing only PEX5(L) or KR (Fig. 1B and C, respectively), we next investigated whether or not overexpression of other PEX5-fusion proteins could also induce peroxisome removal. Note that to facilitate the identification of transfected cells, the indicated test plasmids were routinely cotransfected with a plasmid encoding mitochondria-targeted EGFP. In addition, to quantify and compare the results in an easy and reliable way, the number of peroxisomes in each transfected cell was counted and cataloged as more than 50 (no or moderate reduction in peroxisome number), between one and 50 (strong reduction in peroxisome number), or none (complete absence of peroxisomes).
Figure 1.
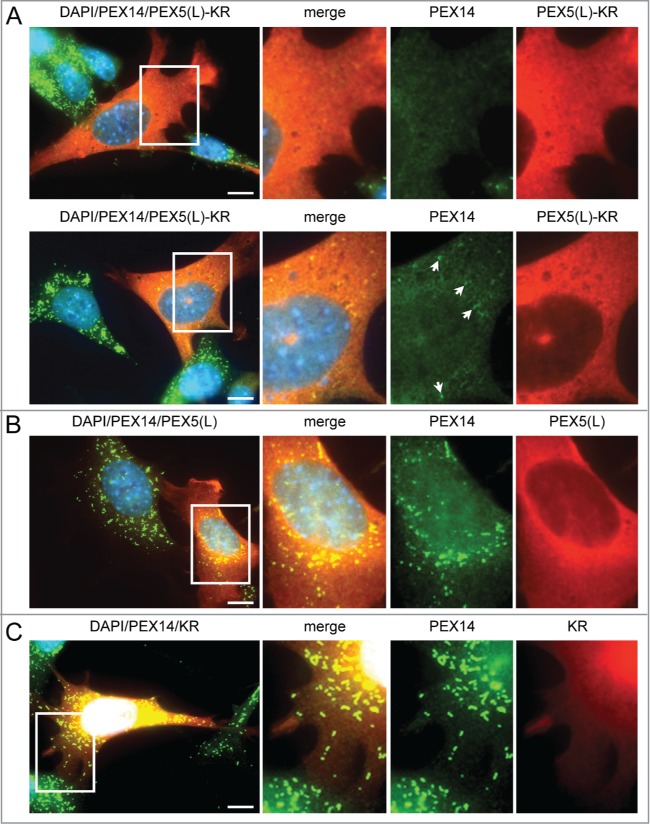
Expression of PEX5-KR triggers the removal of peroxisomes. SV40T-MEFs were transfected with plasmids encoding either (A) PEX5(L)-KR, (B) PEX5(L), or (C) KR. One day later, the cells were fixed, counterstained with DAPI, and processed for immunofluorescence with anti-PEX5 and/or anti-PEX14 antibodies followed by TxRed- and/or Alexa Fluor 488-conjugated secondary antibodies. (A) Upper and lower panels show a transfected cell where all, or most, peroxisomes are absent, respectively. Arrows indicate some of the remaining peroxisomes. Scale bar: 10 µm.
A first set of experiments, in which peroxisome number was analyzed with ABCD3/PMP70-antibodies (Fig. 2A), revealed that (i) also PEX5(S)-EGFP—but not EGFP-PEX5(S) or PEX5(S)-FLAG—could trigger peroxisome removal, (ii) the PEX5-EGFP-mediated removal process of peroxisomes was PEX5 splice-variant independent, and (iii) this process could be triggered by both human and mouse PEX5(L) (Fig. 2B). Co-expression of mt-EGFP had no effect on the outcome of the experiment (Fig. S1). To eliminate the possibility that overexpression of PEX5-EGFP resulted in the masking of the PEX14 and ABCD3 epitopes or the selective degradation of these PMPs, we also carried out immunostainings with antibodies recognizing either CAT/catalase or a mix of peroxisomal matrix proteins (ab-MF16). The results of these experiments clearly showed that also peroxisomal matrix proteins disappeared upon PEX5(L)-EGFP expression (Fig. S2), thereby confirming and extending our initial observations.
Figure 2.
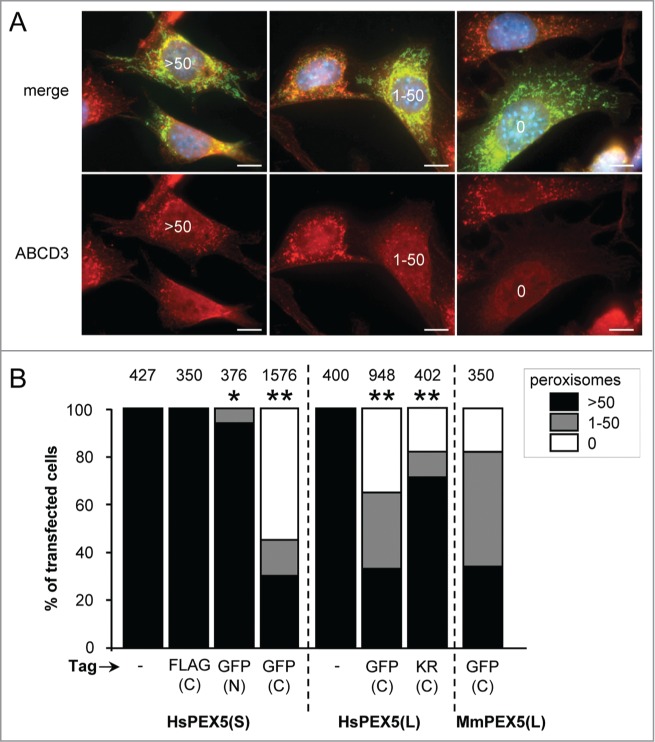
Expression of PEX5 proteins fused to a bulky C-terminal tag promotes a decrease in peroxisome number. SV40T-MEFs were cotransfected with plasmids encoding mitochondria-targeted EGFP (mt-EGFP; green color; marker for transfected cells) and either HsPEX5(S), HsPEX5(S)-FLAG, EGFP-HsPEX5(S), HsPEX5(S)-EGFP, HsPEX5(L), HsPEX5(L)-EGFP, HsPEX5(L)-KR, or mouse (Mus musculus, Mm) PEX5(L)-EGFP. One day later, the cells were fixed, counterstained with DAPI, and processed for immunofluorescence with anti-ABCD3 antibodies followed by TxRed- or Alexa Fluor 488-conjugated secondary antibodies. The number of peroxisomes in each transfected cell was counted and cataloged as more than 50 (>50 ), between 1 and 50 (1–50), or none (0). (A) Images of cells co-expressing mt-EGFP and PEX5(S)-EGFP with >50 (left panels), 1–50 (middle panels), or 0 (right panels) remaining peroxisomes are shown (these images depict representative examples of all phenotypes observed). Scale bar: 10 µm. (B) The percentage of transfected cells displaying each phenotype is plotted. The values above each bar represent the number of transfected cells analyzed per condition. A compilation of the results of at least 3 independent experiments (see Fig. S21) is shown. The “>50 peroxisomes” values from the “HsPEX5(S)” and “HsPEX5(L)” subpanels were statistically compared with the value from the corresponding control (−) condition (* p < 0.05; ** p < 0.01).
In a subsequent series of experiments, we obtained evidence that the degree of peroxisome removal could be correlated with the expression levels of PEX5-EGFP (Fig. S3) and that this process steadily increased up to 20 h post-transfection, after which no further increase was observed (Fig. S4). Finally, as KR is a dimeric protein and EGFP has a weak tendency to dimerize,43 we also checked the peroxisome removal capacity of PEX5-mCherry and PEX5-HaloTag (mCherry and HaloTag tags are strictly monomeric tags) and observed that also these PEX5-fusion proteins could induce peroxisome removal (Fig. S5). Taken together, these data clearly show that the presence of a bulky tag at the C terminus of PEX5 can trigger peroxisome degradation in SV40T-MEFs in a time- and expression level-dependent manner.
PEX5-EGFP-induced peroxisome removal is dependent on autophagy
As autophagy is thought to be the responsible mechanism for most, if not all, peroxisome turnover in mammalian cells,29,31 we next investigated the potential involvement of this pathway in PEX5-EGFP-mediated peroxisome degradation. For this purpose, we employed SV40 large T antigen-transformed atg5−/− MEFs as well as control MEFs treated with the autophagy inhibitors 3-methyladenine (3-MA) or LY294002: ATG5 is essential for efficient MAP1LC3-lipidation, a crucial step in the formation of canonical autophagosomes;44 and 3-MA and LY294002 are inhibitors of class I phosphoinositide 3-kinases and class III phosphatidylinositol 3-kinases (PtdIns3Ks) that suppress autophagosome formation via inhibition of the class III enzyme.45 Importantly, as both PtdIns3K inhibitors (Fig. 3A) as well as ATG5 inactivation (Fig. 3B) interfered with PEX5-EGFP-induced peroxisome removal, the observed phenotype is highly likely to be autophagy dependent. In this context, it is also interesting to mention that—despite the fact that peroxisome removal was not completely blocked in atg5−/− cells—PEX5-EGFP behaved similarly to SLC25A17-Ub, a non-natural protein already reported to selectively trigger peroxisome removal in mammalian cells in an autophagy-dependent manner (Fig. 3C).38 Taken together, these data strongly point toward autophagy as the major mechanism for PEX5-EGFP-induced peroxisome removal.
Figure 3.
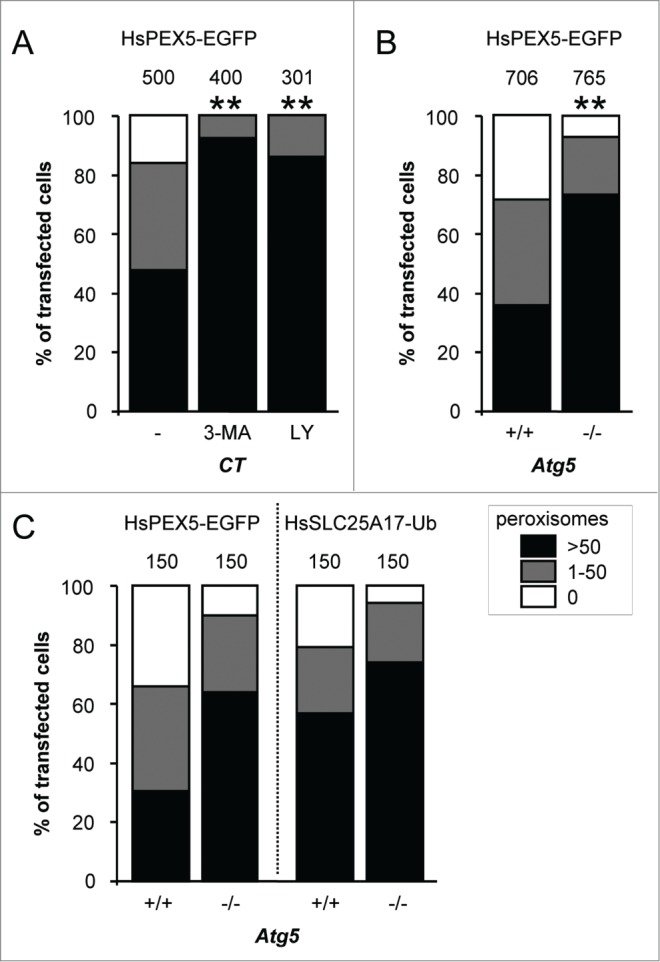
PEX5-EGFP-induced peroxisome removal is dependent on autophagy. Control (CT), Atg5+/+ or atg5−/− SV40T-MEFs were co-transfected with plasmids encoding mitochondria-targeted EGFP (marker for transfected cells) and a plasmid encoding either PEX5-EGFP or FLAG-SLC25A17-Ub in the absence (−) or presence of 10 mM 3-methyladenine (3-MA) or 10 µM LY294002 (LY). One day later, the cells were fixed and processed for immunofluorescence with either anti-ABCD3 or anti-PEX14 antibodies. Peroxisome degradation was quantified and plotted as in Figure 2B. The values above each bar represent the number of transfected cells analyzed per condition. (A, B) A compilation of the results of at least 3 independent experiments (see Fig. S22) is shown. The “>50 peroxisomes” values from the different (sub)panels were statistically compared with the value from the corresponding control condition (**, p < 0.01). (C) The results of a single experiment are shown.
Nevertheless, and despite repeated efforts, we were unable to colocalize peroxisomes with endogenous LC3 or LAMP1 (lysosomal-associated membrane protein 1) nor with recombinant EGFP-LC3, LAMP1-EGFP, or LAMP2A-EGFP, not even in the presence of chloroquine (a lysosomal lumen alkalizer) or protease inhibitor mixtures of N-(trans-epoxysuccinyl)-L-leucine 4 guanidinobutylamide (E-64), pepstatin A and/or leupeptin (data not shown). Potential explanations for these negative results may be that the percentage of (GFP-)LC3 involved in PEX5-EGFP-induced peroxisome removal and the amount of peroxisomal markers trapped within the lysosomal compartment is simply below the detection limit. Regarding the latter, it is important to note that, as (i) the peroxisomal volume in mammalian cells is ≤1% of the total cellular volume,46 and (ii) the PEX5-EGFP-induced removal of the peroxisome population was spread over 20 h (Fig. S4), the presence of even a minor residual protease activity may be sufficient to prevent the detection of peroxisomal marker proteins in lysosomes. In addition, as even low concentrations of chloroquine (e.g.,, 20 µM) cause a dramatic expansion of the lysosomal compartment, signal dilution effects need also to be taken into account.
To rule out the possibility that the observed decrease in peroxisome number was not a direct result of high basal turnover rates in combination with a reduction in peroxisome formation, we also performed a series of pulse-chase labeling experiments to estimate the basal turnover rate of peroxisomes in SV40T-MEFs (peroxisomes are continually formed and degraded, and the actual number of these organelles within a cell depends on the kinetics of both processes). However, as could be expected from similar studies performed in Chinese hamster ovary cells,47 the basal turnover rate of peroxisomes was not high enough to consider a reduction in peroxisome formation as the causal factor for the PEX5-EGFP-induced phenotype (Fig. S6). In this context, it is also worth noting that, under basal conditions, the number of peroxisomes was not statistically different (p < 0.01) between any of the SV40T-MEFs under study (Fig. S7).
Finally, we also investigated whether or not PEX5-EGFP expression activated general autophagy. As these studies revealed that such expression did not lead to an increase in the average number of MAP1LC3B puncta per cell (data not shown) or to differences in the amount of LC3-II between samples in the presence or absence of the vacuolar-type ATPase inhibitor bafilomycin A1 (Fig. S8; for reasons that will become clear later, PEX5C11A-EGFP was included as a negative control), PEX5-EGFP expression did not seem to affect the overall autophagic flux. This idea was further corroborated by the observation that expression of this protein selectively eliminated peroxisomes and had no effect on the normal distribution and morphology of the endoplasmic reticulum and mitochondria (Fig. S9).
The N-terminal cysteine residue that marks PEX5 for recycling is crucial for PEX5-EGFP-induced pexophagy
As (i) it has been hypothesized that ubiquitination of endogenous proteins at the peroxisomal membrane may trigger peroxisome removal in mammalian cells,30,32,38 and (ii) PEX5 export requires monoubiquitination of the protein at Cys11,21 we next examined whether or not 2 N-terminally truncated (PEX5(S)ΔN16 and PEX5(S)ΔN110) variants of PEX5 lacking this cysteine residue could still induce peroxisome degradation. Since these experiments clearly showed that a deletion of the first 16 amino acid residues in PEX5 is sufficient to disrupt PEX5-EGFP-induced peroxisome removal (Fig. 4A), we performed another series of experiments in which we tested the capability of PEX5(L)C11K-EGFP, PEX5(L)C11S, PEX5(L)C11S-EGFP, PEX5(L)C11A, and PEX5(L)C11A-EGFP to trigger peroxisome removal. Note that (i) PEX5(L)C11K is a monoubiquitinatable and fully functional variant of PEX5 in which Cys11 has been substituted by a lysine,48 (ii) PEX5(L)C11S is an export-incompetent PEX5 mutant in which Cys11 has been replaced by a serine, an amino acid residue that can be slowly ubiquitinated under specific conditions,49,50 and (iii) PEX5(L)C11A is also an export-incompetent PEX5 mutant in which Cys11 has been exchanged for an alanine, a nonubiquitinatable amino acid.48,51 As shown in Fig. 4B, expression of PEX5(L)C11K-EGFP, PEX5(L)C11S-EGFP, and PEX5(L)C11A-EGFP respectively caused a strong, moderate, or no pexophagy phenotype in most transfected cells, strongly indicating that the presence of a ubiquitinatable residue at amino acid position 11 of PEX5 is crucial for PEX5-EGFP-induced peroxisome removal. Note that further analysis of the key variants of PEX5-EGFP demonstrated that these proteins were all partially localized to peroxisomes (Fig. S10) and expressed to a similar extent (Fig. S11). Interestingly, expression of nontagged PEX5(L)C11S, but not PEX5(L)C11A, also triggered peroxisome degradation in a small number of cells (Fig. 4B). Although the reason for this phenomenon is not yet clear (but see Discussion), this result further supports the importance of a ubiquitinatable residue at amino acid position 11 of PEX5 to trigger peroxisome removal.
Figure 4.
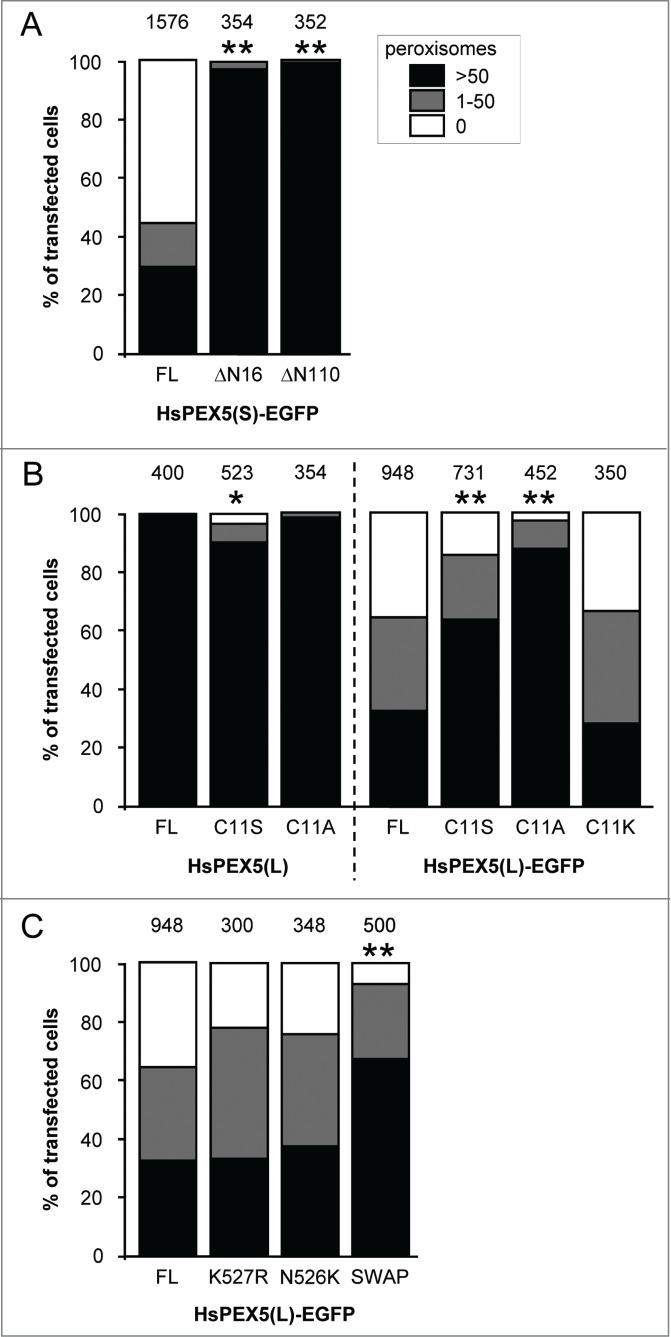
The N-terminal cysteine residue that marks PEX5 for recycling is crucial for PEX5-EGFP-induced pexophagy. SV40T-MEFs were cotransfected with plasmids encoding mitochondria-targeted EGFP (marker for transfected cells) and either (A) full-length PEX5(S)-EGFP (FL), PEX5(S)ΔN16-EGFP (ΔN16), or PEX5(S)ΔN110-EGFP (ΔN110), (B) PEX5(L) (FL), PEX5(L)C11K (C11K), PEX5(L)C11S (C11S), or PEX5(L)C11A (C11A), with or without a C-terminally fused EGFP-tag, or (C) PEX5(L)-EGFP (FL) PEX5(L)K527R-EGFP (K527R), PEX5(L)N526K-EGFP (N526K), or PEX5(S)ΔC299-PEX5(L)/PEX5RΔN326-EGFP (SWAP). For clarity reasons, the PEX5(L)-EGFP data are presented in panels (B and C). One day later, the cells were fixed and processed for immunofluorescence with anti-ABCD3 antibodies. Peroxisome degradation was quantified and plotted as in Figure 2B. The values above each bar represent the number of transfected cells analyzed per condition. A compilation of the results of at least 3 independent experiments (see Fig. S23) is shown. The “>50 peroxisomes” values from the (sub)panels were statistically compared with the value from the corresponding control (FL) condition (* p < 0.05; ** p < 0.01).
Finally, we also tested the ability of PEX5(L)N526K-EGFP, PEX5(L)K527R-EGFP, and PEX5(S)ΔC299-PEX5(L)/PEX5RΔN326-EGFP to induce peroxisome removal in our experimental setup. PEX5(L)N526K and PEX5(L)K527R are PEX5 mutants that are incapable of binding PTS1 proteins or undergo lysine-linked monoubiquitination, respectively,17,40 and PEX5(S)ΔC299-PEX5L/PEX5RΔN324-EGFP is a chimeric protein composed of the N-terminal 298 amino acids of PEX5(S) and the C-terminal TPR-containing domain (amino acids 325 to 624) of PEX5L/PEX5R (peroxisomal biogenesis factor 5-like), a PEX5-related PTS1-binding protein.52 As the PEX5(L)N526K and PEX5(L)K527R mutants did not lose their capacity to trigger PEX5-mediated peroxisome degradation (Fig. 4C), it can be concluded that neither PTS1 binding nor monoubiquitination of PEX5(L) at Lys527 were essential for this process. In addition, from the results obtained with the chimeric PEX5(S)ΔC299-PEX5L/PEX5RΔN326-EGFP protein (Fig. 4C), it was clear that—although the primary amino acid sequence of the TPRs clearly influenced the peroxisome removal phenotype—the TPRs of PEX5 were not essential for PEX5-EGFP-triggered peroxisome removal. In summary, these data provide direct evidence that the N-terminal cysteine residue that marks PEX5 for recycling is crucial for PEX5-mediated peroxisome removal.
C-terminal tagging with EGFP renders PEX5 export-incompetent and leads to the accumulation of Ub-PEX5-EGFP at the peroxisomal membrane
As previous work has pointed out that (i) the accumulation of monoubiquitinated membrane proteins on the cytosolic surface of peroxisomes can cause pexophagy,38,39 and (ii) Cys11-ubiquitination of PEX5 regulates its ATP-dependent export from peroxisomes back to the cytosol,21,51 we here investigated the dynamics and topology of PEX5(L)-EGFP at the peroxisomal membrane by employing a previously described in vitro import/export assay.21 In short, we incubated radiolabeled PEX5(L), PEX5(L)-EGFP, PEX5(L)C11S-EGFP, and PEX5(L)C11A-EGFP with a (peroxisome-containing) rat liver postnuclear supernatant fraction supplemented with ATP or AMP-PNP and either Ub or GST-Ub. Organelle and/or supernatant fractions were then treated or not with proteinase K, processed for SDS-PAGE under nonreducing or reducing conditions, and assayed by autoradiography. Recall that (i) during its transient passage through the peroxisomal membrane, PEX5 adopts a transmembrane topology, only exposing a protease-accessible N-terminal domain of approximately 2 kDa to the cytosol, (ii) monoubiquitination of PEX5 exclusively occurs when the receptor is embedded in the DTM, (iii) substitution of Cys11 by Ser or Ala results in PEX5 proteins that are still functional in docking and membrane insertion but are largely or completely incompetent in the ubiquitination/export process, respectively, and (iv) AMP-PNP, a nonhydrolyzable analog of ATP, blocks the export of monoubiquitinated PEX5 back into the cytosol.48,53
In a first series of experiments, we found that PEX5(L)-EGFP entered the DTM and became monoubiquitinated similar to nontagged PEX5(L) (Fig. 5A, left panels, compare the AMP-PNP conditions). However, we also observed that Ub-PEX5(L)-EGFP, in contrast to Ub-PEX5(L), encountered difficulties in leaving the DTM (same panels, compare the ATP conditions), a property that can be better appreciated using a 2-step import/export assay (Fig. 5B). In these assays, radiolabeled PEX5(L) and PEX5(L)-EGFP were first imported in the presence of AMP-PNP. The organelles were then sedimented and the supernatant fraction (containing nonimported PEX5 proteins) was discarded. Finally, the organelles were resuspended in fresh ATP-containing import buffer and incubated for 5 min at 37°C to promote export of the monoubiquitinated species. As shown in Fig. 5B, the vast majority of monoubiquitinated PEX5(L)-EGFP remained in the organelle fraction, in contrast to PEX5(L), which was recovered mainly in the soluble fraction. PEX5(L)C11S-EGFP and PEX5(L)C11A-EGFP could also efficiently enter the DTM (Fig. 5A, right panels). Nonetheless, as these molecules are respectively poor substrates or not substrates for monoubiquitination, they became trapped at the DTM, even in the presence of ATP. Note that, as (i) PEX5(L)C11S-EGFP and PEX5(L)C11A-EGFP were not posttranslationally modified at the peroxisomal membrane (Fig. 5A, right panels), (ii) replacement of Ub with GST-Ub resulted in higher molecular weight species of PEX5(L) and PEX5(L)-EGFP (Fig. 5C, upper panels), and (iii) treatment of the protein samples with DTT destroyed the thioester bond between (GST-)Ub and PEX5(L)(-EGFP) (Fig. 5C, lower panels; see also Fig. S12), it is clear that the PEX5(L) modifications in our assays represent Cys11-dependent monoubiquitination events.
Figure 5.
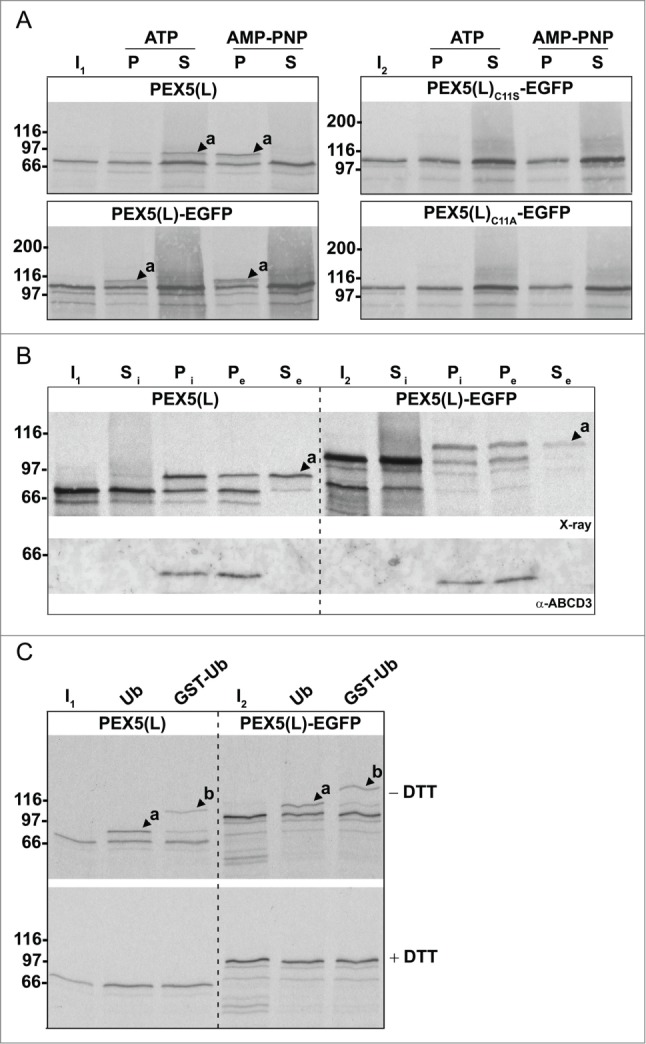
For figure legend, see page 1349.
In a second series of experiments, we found that organelle-bound (Ub-)PEX5(L) and (Ub-)PEX5(L)-EGFP displayed a similar topology, as assessed by protease-protection assays. Indeed, in the presence of ATP or AMP-PNP, we respectively detected (i) small amounts of PEX5(L) and PEX5(L)-EGFP exposing the majority of their mass in the peroxisomal matrix (Fig. 6, upper panel, lanes 2 and 5, arrowheads labeled 'a'), and (ii) large amounts of DTM-embedded Ub-PEX5(L) and Ub-PEX5(L)-EGFP (Fig. 6, upper panel, lanes 3 and 6, arrowheads labeled 'b'). At first sight, this finding may be counterintuitive given our earlier observation that, in the presence of ATP, Ub-PEX5(L)-EGFP was more abundant in the organellar fraction than Ub-PEX5(L) (Fig. 5A, compare lane 2 in the 2 upper panels). However, a careful analysis of the autoradiographs that are shown in Fig. 6 revealed that a major fraction of Ub-PEX5(L)-EGFP obtained in the presence of ATP was accessible to proteinase K, whereas the one obtained in the presence of AMP-PNP was not. Indeed, in the condition with ATP, we observed 3 additional protease-resistant PEX5(L)-EGFP fragments around 35 kDa (Fig. 6, see asterisks). Note that these fragments contained the EGFP moiety of the chimeric protein because they were recognized by an anti-EGFP antibody (see Fig. S13). In addition, as they could not be observed for export-incompetent PEX5(L)C11A-EGFP (Fig. 6, upper and lower panels, 2 last lanes), our findings strongly indicate that the REM is capable of extracting PEX5-EGFP partially out of the DTM, but most likely becomes jammed when it encounters a tightly folded domain (e.g., EGFP or KR).
Figure 6.
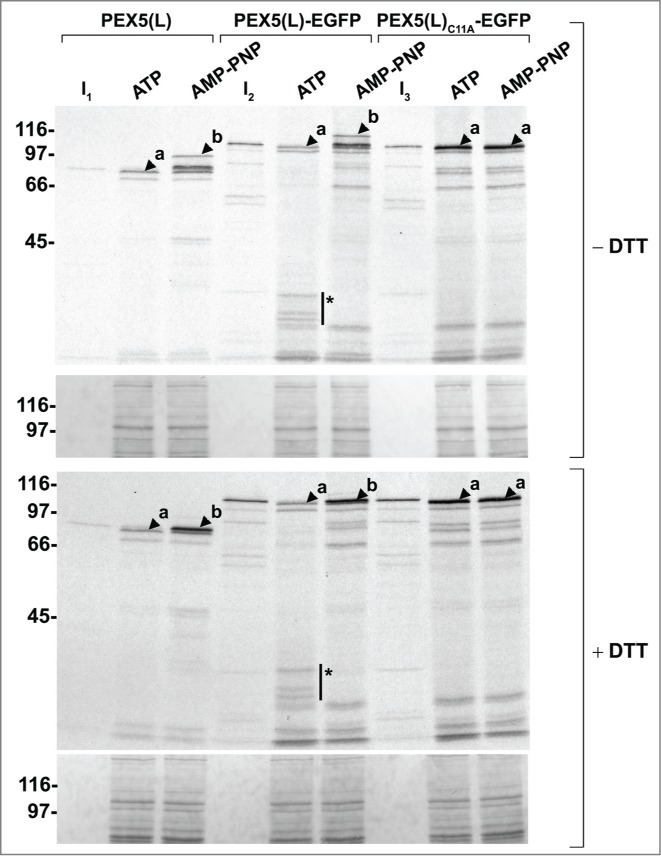
Monoubiquitinated PEX5-EGFP trapped at the DTM in the presence of ATP is only partially protease protected. Radiolabeled PEX5(L), PEX5(L)-EGFP and PEX5(L)C11A-EGFP were subjected to in vitro import assays in the presence of Ub aldehyde and either ATP or AMP-PNP, as indicated. After incubation at 37°C, organelle suspensions were treated with proteinase K. NEM-treated organelles were then isolated, and subjected to SDS-PAGE under reducing (+ DTT) and non-reducing (− DTT) conditions. The autoradiographs (upper panels) and a section of the corresponding Ponceau S-stained membranes (lower panels) are shown. a and b represent DTM-inserted PEX5(L) exposing 2 kDa of its N terminus to the cytosol and DTM-embedded monoubiquitinated PEX5(L), respectively. 21 The asterisks mark a set of PEX5(L)-EGFP-derived fragments that are protease resistant. Lanes I1, I2, I3, 5% of the radiolabeled protein used in the assays.
PEX5-EGFP is monoubiquitinated in cellulo in a Cys11-dependent manner
To find evidence that PEX5-EGFP is also monoubiquitinated at Cys11 when expressed in SV40T-MEFs, whole lysates of cells expressing PEX5C11A-EGFP or PEX5-EGFP were subjected to SDS-PAGE under nonreducing and reducing conditions and processed for immunoblotting with antibodies against PEX5. From these experiments, it is clear that a small portion of PEX5-EGFP was posttranslationally modified upon expression in these cells (Fig. S14). In addition, as (i) this modification is Cys11-dependent, DTT-sensitive, and caused a molecular shift of approximately 8 kDa (Fig. S14), (ii) this behavior mimics that of PEX5-EGFP in vitro (Figs. 5 and S12), and (iii) previous studies have shown that, in mammals, the DTT-sensitive form (with monoubiquitination at the conserved cysteine residue) is associated with peroxisomes and the DTT-insensitive form (with unknown modification) is located in the cytosol,40,48;51,54 it is reasonable to conclude that the posttranslationally-modified form of PEX5-EGFP represents peroxisome-associated ubiquitinated PEX5-EGFP.
Next, as expression of PEX5-EGFP is expected to lead to an accumulation of Ub-PEX5-EGFP at the peroxisomal membrane, we also checked whether or not PEX5-EGFP-expressing cells contain ubiquitin-positive peroxisomes. Unfortunately, despite the fact that we used 2 different anti-ubiquitin antibodies, including one that was already successfully used by others to visualize ubiquitin-positive peroxisome clusters in PEX3-HA2-overexpressing cells,32 no ubiquitin-positive peroxisomes could be detected, not even upon treatment of the cells with 3-MA (data not shown). However, (i) the ubiquitin-moieties in DTM-embedded PEX5-EGFP molecules may be shielded by potential interaction partners (e.g.,, PEX1, or PEX6), (ii) in contrast to what happens in PEX3-HA2 overexpressing cells, the fluorescence intensity of putative ubiquitin-positive peroxisomes is not enhanced because the organelles do not cluster upon expression of PEX5-EGFP, and (iii) ubiquitin is also ligated to a great number of endogenous proteins, the threshold for detection of ubiquitin-positive peroxisomes may be below the limit needed to visualize such structures above background fluorescence.
The PEX5-EGFP-induced phenotype is cell type-specific
We also expressed PEX5-EGFP in other mammalian cell types (e.g., human skin fibroblasts, rat embryonic fibroblasts, and a mouse oligodendrocyte cell line) and unexpectedly found that this protein could not trigger peroxisome removal in these cells (data not shown). In addition, we obtained empirical evidence that the PEX5-EGFP-induced peroxisome removal phenotype in MEFs can be directly linked to the SV40 large T antigen-induced immortalization of these cells. Indeed, expression of PEX5-EGFP quickly resulted in the disappearance of peroxisomes in SV40T-cells (e.g.,, “homemade” control MEFs,55 Atg5+/+ MEFs,56 and Perk+/+ MEFs,57), but not in primary control MEFs (passage <5)55 and spontaneously transformed Sqstm1+/+ MEFs.58 Note that, as the expression levels of pEGFP-N1-encoded proteins are comparable in spontaneously transformed- and SV40T-cells, at least within the time scale of the experiments (Fig. S15), these differences in phenotype are caused by other factors than differences in PEX5-EGFP expression levels (see Discussion).
We still considered the possibility that PEX5-EGFP displays an export defect in vivo only in SV40T-cells and that the protein is actually functional in other cells. To test this possibility, PEX5-deficient human fibroblasts and spontaneously-transformed MEFs were subjected to transfection experiments to assess whether or not PEX5-EGFP has complementing or dominant-negative activity in peroxisomal matrix protein import. Appropriate controls (non-tagged PEX5WT, PEX5C11K, PEX5C11S, and PEX5C11A) were included to discriminate between different outcomes (PEX5WT and PEX5C11K, but not PEX5C11S and PEX5C11A, restore PTS1 protein import in PEX5-deficient cells; and PEX5C11S and PEX5C11A, but not PEX5WT and PEX5C11K, exert a dominant-negative activity on the same process in control cells).48,51 From these experiments, it is clear that PEX5(L)-EGFP cannot restore peroxisomal matrix protein import in PEX5-deficient human fibroblasts (Fig. S16A) and interfered with PTS1 protein import upon expression in spontaneously transformed control MEFs (Fig. S16B).
Downregulation of PEX1, SQSTM1, or NBR1 expression does not interfere with PEX5-EGFP-induced peroxisome removal
To investigate the potential role of the PEX5 export machinery in PEX5-EGFP-induced peroxisome removal, we used Dicer substrate RNAs (DsiRNAs) to downregulate the expression level of PEX1, an essential REM component.16 Note that, under the employed conditions (for details, see Materials and Methods), virtually all cells were transfected (as confirmed by a fluorescently-labeled scrambled control RNA duplex) (Fig. S17). Despite the fact that we could knock down PEX1 (Fig. S18A and B), no effect could be observed on the level of the PEX5-EGFP-induced phenotype (Fig. 7A). In addition, such treatment did not affect peroxisome number in cells overexpressing HsPEX5 (Fig. 7B). Unfortunately, as additional experiments revealed that the residual amounts of PEX1 were sufficient to retain a functional PTS1 import machinery (Fig. S18C), no reliable conclusions can be drawn regarding the potential role of PEX1 in the process under study.
Figure 7.
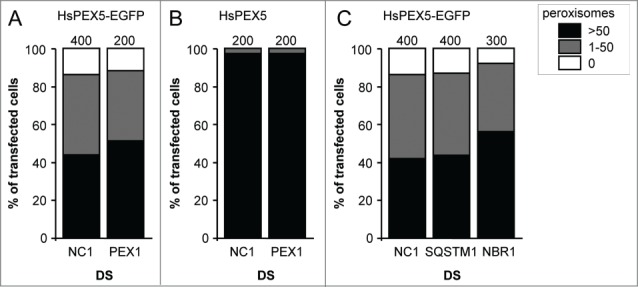
Downregulation of PEX1, SQSTM1, or NBR1 does not influence PEX5-EGFP-induced peroxisome removal. SV40T-MEFs were sequentially transfected with scrambled (NC1), PEX1-, SQSTM-, or NBR1-specific duplex siRNAs (DS) in combination or not with plasmids encoding mitochondria-targeted EGFP (marker for transfected cells) and (A, C) HsPEX5-EGFP or (B) HsPEX5 (for details, see Materials and Methods). One day later, the cells were fixed and processed for immunofluorescence microscopy with anti-PEX14 antibodies. Peroxisome degradation was quantified and plotted as in Figure 2B. The values above each bar represent the number of transfected cells analyzed per condition. A compilation of the results of at least 2 independent experiments (see Fig. S24) is shown. The “>50 peroxisomes” values from the (sub)panels were statistically compared with the value from the corresponding control (NC1) condition and found not to be statistically different.
To investigate the potential role of the ubiquitin-binding selective autophagy receptors SQSTM1 and NBR1, a similar approach was used. Also, here we could observe a significant downregulation of the expression levels of SQSTM1 (Fig. S19A and C) and, albeit to a lesser extent, NBR1 (Fig. S19B and D). Unfortunately, once again, we could not observe any effect on the level of PEX5-EGFP-induced peroxisome removal (Fig. 7C). The interpretation of these data is presented below.
Discussion
In recent years, the phenomenon of selective organelle degradation has attracted increasing attention.59-61 The main reason for this is that an accumulation of dysfunctional organelles contributes to developmental abnormalities, aging, inflammation, cancer, and other diseases.6 The pexophagy field has also gained much interest. However, despite rapid and considerable progress in our understanding of how peroxisomes are selectively degraded in (Saccharomyces cerevisiae and methylotrophic) yeasts,29,62-65 little is known about how this process is regulated in mammalian cells.31 This is perhaps best illustrated by the fact that, although it has been demonstrated that ectopic expression of PMPs attached to cytosolically exposed ubiquitin can trigger pexophagy,38 endogenous PMPs that are ubiquitinated during pexophagy have not yet been identified. In this study, we show that expression of a monoubiquitinatable but export-incompetent variant of PEX5, a naturally monoubiquitinated protein, results in the accumulation of Ub-PEX5 at the peroxisomal membrane and triggers peroxisome removal in SV40T-MEFs. These observations provide the first strong evidence in favor of the recent hypothesis that alterations in PEX5 and ubiquitin dynamics on peroxisome membranes can regulate mammalian pexophagy.30,39 The potential underlying molecular mechanisms of these findings and their implications for future research are discussed in the following paragraphs.
In our initial experiments aimed at clarifying whether or not peroxisomes can be selectively removed upon oxidative damage, we accidentally found that expression of PEX5 proteins fused to a bulky C-terminal tag (e.g., KR, EGFP, HaloTag, or mCherry) caused the disappearance of peroxisomes in SV40T-MEFs (Figs. 1A and 2B; Fig. S5). In addition, we observed that this process required the presence of the N-terminal cysteine residue that marks PEX5 for recycling (Fig. 4B). As these observations, combined with the finding that PEX5 Cys11 monoubiquitination only takes place at the peroxisomal membrane,21 strongly indicated that the addition of a bulky tag to the C terminus of PEX5 interferes with the export step of the cycling receptor from the peroxisome to the cytosol, we employed a previously established in vitro assay21 to study the import/export kinetics of PEX5-EGFP at the peroxisomal membrane. These experiments clearly demonstrated that PEX5-EGFP can enter the DTM and become monoubiquitinated at Cys11 as is the nontagged wild-type PEX5 (Fig. 5). However, in contrast to Ub-PEX5, Ub-PEX5-EGFP remained largely associated with the organelle pellet in the presence of ATP (Fig. 5A, B), clearly showing that the existence of a tightly folded domain at the C terminus of PEX5 interfered with its export back into the cytosol (Fig. 5A, B). Importantly, Ub-PEX5-EGFP was still a substrate for the REM. Indeed, this protein became partially accessible to proteinase K under conditions where the REM was active (i.e., when ATP but not AMP-PNP was used in the in vitro assays) yielding a set of organelle-associated protease-resistant fragments of approximately 35 kDa that comprise the bulky EGFP moiety (Figs. 6 and S13). The behavior of Ub-PEX5-EGFP can be explained in 2 ways. On the one hand, it is possible that PEX5 exits the DTM via a REM-dependent threading mechanism, and that—by analogy to some other AAA+ ATPases66—tightly folded proteins such as EGFP (or a cargo protein that cannot be released from PEX5) cannot pass through a hole that may be present in the PEX1-PEX6 complex. Alternatively, it is conceivable that the DTM acts as a trap for globular proteins, letting them in but preventing them from getting out. Regardless of the mechanism, and although there is currently compelling evidence suggesting that the cargo release step occurs prior to monoubiquitination of PEX5,19,67,68 the results presented here provide experimental evidence to support the concept that the peroxisomal export machinery may also participate in cargo release.69 According to this idea, the ATP used by the REM to extract Ub-PEX5 from the DTM could also provide the energy necessary to disrupt the PEX5-cargo protein interaction, e.g., by unfolding the PTS1-binding domain of PEX5. Further data are necessary to clarify these important mechanistic aspects of the peroxisomal protein import machinery.
As it is well-known that the insertion of PEX5 into the peroxisomal membrane is a cargo-dependent process,69 the observation that both PEX5(L)N526K–EGFP and PEX5(S)ΔC299-PEX5L/PEX5RΔN324-EGFP trigger peroxisome removal may be difficult to reconcile with the suggested model that ubiquitinated versions of these proteins accumulate at the peroxisomal membrane. However, here it is important to mention that (i) the N526K mutation in PEX5(L) (and the corresponding N489K mutation in PEX5(S)) causes conformational alterations at the N-terminal half of PEX5 mimicking the ones induced by binding of a PTS1-containing peptide to the normal peroxin,49 and (ii) also C-terminally truncated versions of PEX5 have been reported to function as substrates for the peroxisomal DTM.71 In this context, it is interesting to mention that also PEX5(S)N489K-EGFP, a version of PEX5 that lacks both its PTS1 and PEX7-binding sites, can trigger peroxisome removal in SV40T-MEFs (data not shown). Another intriguing observation is that non-tagged PEX5C11S, but not PEX5C11A, triggered peroxisome removal in a small number of cells (Fig. 4B). Although PEX5C11S can be monoubiquitinated at the DTM,21 this process occurs at a very slow rate explaining why this protein accumulates at the peroxisome. While further data are necessary to explain the peroxisome removal phenotype induced by PEX5C11S, it is tempting to speculate that PEX5 molecules retained for a long time at the peroxisomal membrane (e.g., in case the cargo protein cannot be released from PEX5) become strongly (or even covalently) linked to some DTM component(s) (e.g., by ROS-promoted mechanisms). In such a scenario, the subsequent monoubiquitination of these molecules would trigger their REM-dependent extraction from the DTM. However, this process would not be completed due to the strong PEX5-DTM interaction, thus leading to the accumulation of partially exposed Ub-PEX5C11S species at the peroxisome limiting membrane, similar to the ubiquitinatable PEX5-EGFP species used in this work.
As our data indicate that the addition of a bulky tag to the C terminus of PEX5 can result in the accumulation of partially extracted Ub-PEX5 on the peroxisomal surface, it is conceivable to envisage that this will eventually result in the recruitment of the autophagic machinery to the organelle. However, despite the fact that PEX5-mediated peroxisome removal depends on ATG5 and can be blocked by 3-MA and LY294002 (Fig. 3), we were repeatedly unable to colocalize peroxisomes with EGFP-MAP1LC3B, even when culturing the cells in medium supplemented with bafilomycin A1 (Fig. S20) or chloroquine (data not shown). Nevertheless, here it should be mentioned that such colocalization could easily be observed upon expression of SLC25A17-Ub, even in the absence of autophagy inhibitors (Fig. S20, upper panels). In addition, unlike SLC25A17-Ub-, NBR1-, or PEX3-induced pexophagy, where peroxisomes cluster prior to degradation,30,32,38 we did not observe any clustering of peroxisomes upon PEX5-KR expression (Fig. S20, compare the upper and lower panels). Note that this may also impede the likeliness of finding peroxisomes within autophagosomes, which have an approximate half-life of only 10 min.44
Regarding the observations that (i) the PEX5-EGFP-induced peroxisome removal phenotype in MEFs can be directly linked to the SV40 large T antigen-induced immortalization of these cells (data not shown), and (ii) SV40 large T antigen does not directly influence the expression levels of PEX5-EGFP (Fig. S15), it is important to note that SV40T-cells display cell type-specific global changes in gene expression (including some components of the cellular ubiquitination/deubiquitination machinery).72,73 These observations suggest that the PEX5-EGFP-induced phenotype depends on a critical balance of multiple factors that remain to be determined and may even be cell type-specific (e.g., the promptness with which the Ub-moiety is recognized by the pexophagy machinery, the amount of ubiquitin at the peroxisomal membrane, the kinetics of PEX5 ubiquitination/deubiquitination, and other potential quality control mechanisms, such as proteasomal removal of [poly]ubiquitinated PEX5). For example, although PEX5 accumulates at the peroxisomal membrane in aging human fibroblasts, the number of peroxisomes in these cells is profoundly increased.74 However, here it is important to know that (i) cellular aging is associated with an increase in the GSSG (oxidized glutathione)/GSH (reduced glutathione) ratio,75 and (ii) exposure of human PEX5 to GSSG results in a ubiquitination-deficient PEX5 molecule.76
In conclusion, this work presents the first experimental evidence that addition of a bulky tag to the C terminus of PEX5 interferes with the export of monoubiquitinated PEX5 from the DTM, and that this in turn can trigger peroxisome removal in SV40T-MEFs. These findings strongly support the idea that peroxisome-associated monoubiquitinated PEX5 may act as a key surveillance factor for the selective removal of dysfunctional peroxisomes in mammalian cells.30,39 In this context, we hypothesize that the bulky tag may mimic a cargo protein that cannot be released from PEX5. However, as the study of mammalian pexophagy is still in its infancy, this and many other intriguing questions remain. For example, like in other studies that applied ectopic expression of ubiquitinated PMPs or PEX3 as a pexophagy trigger,30,32 PEX5-EGFP-induced pexophagy could only be partially blocked in conditions in which macroautophagy was inhibited. Although this finding may suggest that some peroxisomes can be removed by alternative degradation pathways,77,78 this needs further investigation. Also the potential involvement of the PEX5 export machinery, the specific autophagy receptor proteins (e.g., SQSTM1, NBR1, OPTN, CALCOCO2, HDAC6, etc.), and the mammalian Atg8 orthologs (i.e., the MAP1LC3 and GABARAP subfamilies)79 need further investigation. Indeed, as we currently cannot exclude the possibility that—upon siRNA knockdown—the remaining amounts of PEX1, SQSTM1, or NBR1 are still sufficient to sustain PEX5-EGFP-mediated peroxisome removal, the role of these proteins in this process should ideally be studied in SV40T-knockout cell lines (and that are currently not available). However, in case the triggering factor for peroxisome removal is not accumulation of Ub-PEX5 at the DTM but rather accumulation of a partially dislocated Ub-PEX5 at the DTM/REM, a complete inactivation of PEX1 would not result in an enhanced peroxisome removal phenotype. Also, in the absence of a functional PEX5 export machinery, a ubiquitin-dependent quality control pathway—called RADAR (receptor accumulation and degradation in the absence of recycling)80—may be activated (assuming there is one in mammalian cells) thereby leaving peroxisomes intact. Finally, it is also not yet clear why overexpression of SLC25A17-Ub, NBR1, or PEX3 induces peroxisomal clustering,30,32,38 whereas we do not observe this phenotype during PEX5-EGFP-triggered pexophagy. Here it is tempting to speculate that the clustering phenotype may represent a situation in which excessive peroxisomes are massively removed, while the mechanism underlying PEX5-EGFP-induced pexophagy may mimic a condition where dysfunctional organelles are individually degraded. Also, given that SQSTM1 and NBR1 play a role in PMP-Ub- and PEX3-induced peroxisome clustering and that this event precedes their targeting to autophagosomes and lysosomes,30,32 it may well be that SQSTM1 and/or NBR1 are not involved in PEX5-EGFP-induced peroxisome removal.
In summary, this study provides strong evidence that monoubiquitinated PEX5 can serve as a quality control mechanism to eliminate peroxisomes. In addition, it paves the way for further investigations aimed at elucidating the molecular basis underlying peroxisome degradation in mammalian cells, an essential prerequisite to understand how defects in this process may be linked to clinically relevant disease phenotypes.
Materials and Methods
DNA manipulations and plasmids
Polymerase chain reactions were routinely performed using Pfx DNA polymerase (Invitrogen, 11708039). Oligonucleotides and RNAi duplex oligonucleotides used in this study were synthesized by Integrated DNA Technologies and are listed in Tables S1 and S2, respectively. The Escherichia coli strain TOP10F' (Invitrogen, C3030–03) was used for all DNA manipulations. Restriction enzymes were purchased from TaKaRa. The mammalian expression vectors pEGFP-N1, pCMV-Tag 2B, pKillerRed-dMito, and pHT2 were commercially obtained from Clontech (6085–1), Stratagene (211172), Evrogen (FP964), and Promega (G8241), respectively. A detailed description of the noncommercial plasmids used in this study is available in the supplementary information (Materials S1). All new plasmids were verified by DNA sequencing (LGC Genomics).
Cell culture, immunofluorescence, and live-cell microscopy
SV40 large T-antigen transformed Atg5+/+, atg5−/−, and Perk+/+ MEFs were kindly provided by Dr. P. Agostinis (KU Leuven, Belgium).56,57 PEX5−/− human fibroblasts, spontaneously transformed Sqstm1+/+ MEFs, and the murine oligodendrocyte cell line (158N) were generous gifts from Dr. G. Dodt (University of Tübingen, Germany), Dr. T. Yanagawa (Niigata University, Japan), and Dr. S. Ghandour (University of Strasbourg, France), respectively.58,76,81 Control primary MEFs (C57BL/6) and rat embryonic fibroblasts (Sprague-Dawley) were generated as before.76 All cells were cultured at 37°C in a humidified 5% CO2 incubator in minimum essential medium Eagle α (Lonza, BE12–169F) supplemented with 10% (v/v) fetal bovine serum superior (Biochrom AG, BCHRS0615), 2 mM ultraglutamine-1 (Lonza, BE17–605E/U1/12), and 0.2% (v/v) Mycozap (Lonza, VZA-2012). MEFs were transfected using Invitrogen´s Neon Transfection System (1350 V, 30 ms pulse width, 1 pulse).82 To knock down the expression of target genes, the cells were first electroporated with the appropriate Dicer-substrate RNAs (DsiRNAs), and—2 d later—co-electroporated with the same DsiRNAs and the plasmid encoding the protein under study. The final concentrations in the 10-µl microporator tip were 2 µM for individual DsiRNAs; 0.66 µM per DsiRNA for TriFECTa RNAi kit duplex combinations, and 1 µg of plasmid. The transfected cells were either processed for indirect immunofluorescence or lysed for SDS-PAGE and immunoblot analysis (for sample analysis under nonreducing conditions, cell pellets were first treated with 10 mM N-ethylmaleimide (NEM; Across Organics, 156100100) to block deubiquitinases and any endogenous nucleophilic groups that may attack the Ub-PEX5-EGFP thioester). Samples for immunofluorescence microscopy were fixed and processed as described before.47 The rabbit polyclonal antiserum against human PEX14,83 the mouse polyclonal antiserum against bovine CT/catalase84 and the antibodies raised against a mixture of peroxisomal matrix proteins (ab-MF16)85 have been described elsewhere. DAPI (Roche, 10236276001), Hoechst 33258 (Sigma, 14530), the rabbit anti-ABCD3 antibodies (Sigma, P0497), the mouse anti-FLAG antibodies (Stratagene, 200472–21), the rabbit anti-HaloTag antibodies (Promega, G9281), the mouse anti-LAMP1 antibodies (BD Pharmingen, 553792), the rabbit (Cell Signaling Technology, 2775) and mouse (Nanotools, 0231–100/LC3–5F10) anti-LC3B antibodies, the rabbit (Proteintech, 16004–1-AP) and mouse (Abcam, ab55474) anti-NBR1 antibodies, the rabbit anti-PEX1 antibodies (Bio-Connect, 13669–1-AP), the rabbit anti-SQSTM1 antibodies (Sigma, P0067), the rabbit anti-ubiquitin antibodies (Cell Signaling Technology, 3933), the mouse anti-mono/polyubiquitin monoclonal antibody (Enzo, FK2, BML-PW8810), the Alexa Fluor 350- (Invitrogen, A11069), Alexa Fluor 488- (Invitrogen, A11017 and A11070) or Texas Red- (TxRed; Calbiochem, 401355 and 401230) conjugated secondary antibodies were commercially obtained. To interfere with the autophagic process, the cells were cultivated in the presence of 10 mM 3-MA (Sigma, M9281), 100 nM bafilomycin A1 (Sigma, B1793), 10 µM LY294002 (Sigma, L-9908), 20 µM chloroquine (Sigma, C6628), 10 µM E-64 (MP Biomedicals, 152846), 10 µM pepstatin A (Sigma, P-4265), and/or 100 µM leupeptin hemisulfate (Fluka, 62070). Cells for live-cell imaging were seeded and imaged in FluoroDish cell culture dishes (World Precision Instruments, FD35–100). Where indicated, cells were treated for 1 h with 500 nM MitoTracker® Red CM-H2Xros (Life Technologies, M7513) or ER-TrackerTM Blue-White DPX (Life Technologies, E-12253) in regular cell culture medium, and washed once with the same medium immediately before imaging. The sequential labeling of live cells expressing HaloTag-HsHAO2 (hydroxyacid oxidase 2 [long chain]) was done for the specified period of time with 250 nM HaloTag TMR (Promega, G8251) and 10 nM HaloTag R110Direct (Promega, G3221) ligands as described elsewhere.86 After the first and second labeling reactions, the cells were washed 6 times and once, respectively, with standard growth medium. Fluorescence was evaluated on a motorized inverted IX-81 microscope (Olympus), controlled by Cell-M software (Olympus) and equipped with a temperature-, humidity-, and CO2-controlled incubation chamber. The technical specifications of the objectives, excitation and emission filters, and digital camera have been described elsewhere.42 The Cell-M software was used for quantitative image analysis.
In vitro import/export assays
Rat liver postnuclear supernatant (PNS) for in vitro assays was prepared in SEM buffer (0.25 M sucrose, 20 mM MOPS-KOH, pH 7.2, 1 mM EDTA-NaOH, pH 7.2) supplemented with 2 µg/ml E-64 (Sigma, E3132), as described before.70 [35S]-labeled proteins were synthesized in vitro using the TNT® T7 Quick Coupled Transcription/Translation System (Promega, L1170) in the presence of [35S]methionine (specific activity >1000 Ci/mmol; PerkinElmer Life Sciences, NEG709A001MC). In the in vitro import reactions (100 µl final volume), 1 µl of the relevant 35S-labeled protein was added to 600 µg of PNS protein that had been primed for import (incubation for 5 min at 37°C in import buffer (0.25 M sucrose, 50 mM KCl, 20 mM MOPS-KOH, pH 7.2, 3 mM MgCl2, 20 µM methionine, and 2 µg/ml E-64) containing 0.3 mM ATP (Sigma, A2383).22,67 Import assays also contained 2 mM glutathione (Sigma, G4251), 3 μM ubiquitin aldehyde53 and, where indicated, ATP (3 mM), AMP-PNP (3 mM; Sigma, A2647), bovine ubiquitin (15 μM; Sigma, U6253) or GST-Ub21 (15 μM). After incubation for 30 min at 37°C, samples were treated with 20 mM NEM (Sigma, E3876) on ice for 5 min, as described before.48 To separate organelles from soluble proteins, the in vitro import reactions were diluted with ice-cold SEMK buffer (SEM buffer containing 80 mM KCl) and centrifuged at 16,000 × g for 20 min at 4°C. Samples were subjected to trichloroacetic acid precipitation and processed for SDS-PAGE under reducing or nonreducing conditions, as specified. Protease protection assays were done using proteinase K (400 μg/ml final concentration; Sigma, P2308) for 40 min on ice.53 The 2-step in vitro import/export assay was exactly done as described before.68 For the immunoprecipitation assays, protease-treated organelles from import assays were solubilized for 30 min at 4°C in immunoprecipitation buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA-NaOH, pH 8.0, 0.1% [w/v] SDS [Merck, 1137601000], 1% [w/v] Triton X-100 [Sigma, T9284], 0.5% [w/v] sodium deoxycholate [Sigma, D5670], 500 µg/ml phenylmethylsulfonyl fluoride [Sigma, P7626] and 1/200 [v/v] mammalian protease inhibitor mixture [Sigma, P8346]). After removing the insoluble material (15 min at 15,000 x g), the supernatant fraction was divided in 3 aliquots. One aliquot (total) was kept at 4°C during the complete procedure and then subjected to trichloroacetic acid precipitation. The other 2 aliquots received 25 μl (bed volume) of protein A-Sepharose® beads (Sigma, P3391) that were preincubated with either 3 μl of an anti-EGFP serum88 or a control serum, and incubated for 2 h at 4°C with gentle shaking. After removing the supernatant fraction, the beads were washed 3 times with immunoprecipitation buffer and once with 1X phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.1 mM KH2PO4). Immunoprecipitated proteins were eluted with 45 µl of Laemmli sample buffer and the corresponding immunodepleted supernatant fractions were subjected to trichloroacetic acid precipitation.
Statistical analysis
Stastistics were performed on the VassarStats statistical computation website (http://vassarstats.net/). A 2-sample t-Test for independent samples was used to analyze the results. The significance levels were set at p < 0.05 (denoted by*) and p < 0.01 (denoted by**).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. P. Agostinis (KU Leuven, Belgium) for the SV40 large T antigen-transformed Atg5+/+, atg5−/−, and Perk+/+ MEFs, Dr. T. Yanagawa (Niigata University, Japan) for the spontaneously transformed Sqstm1+/+ MEFs, Dr. G. Dodt (University of Tübingen, Germany) for the PEX5−/− human fibroblasts, and Dr. S. Ghandour (University of Strasbourg, France) for the murine oligodendrocyte cell line. We are also grateful to Dr. M. Baes (KU Leuven, Belgium), Dr. T. Yoshimori (National Institute of Genetics, Japan), Dr. T. Voets (KU Leuven, Belgium), and Dr. S. M. Di Pietro (Colorado State University, CO, USA) for the plasmids encoding myc-tagged MmPEX5(L), EGFP-MAP1LC3B, LAMP1-EGFP, or LAMP2A-EGFP, respectively.
Funding
This work was supported by grants from the 'Fonds voor Wetenschappelijk Onderzoek-Vlaanderen (Onderzoeksprojecten G.0754.09 and G095315N)' (to MF and PVV), the KU Leuven (OT/09/045, OT/14/100, and DBOF/10/059) (to MF and PVV), and by FEDER funds through the Operational Competitiveness Program, ,COMPETE, and by national funds through FCT,Fundação para a Ciência e a Tecnologia, under the projects FCOMP-01–0124-FEDER-019731 (PTDC/BIA-BCM/118577/2010) and FCOMP-01–0124-FEDER-022718 (PEst-C/SAU/LA0002/2011) (to JEA). MN was supported by a FLOF fellowship from the Department of Cellular and Molecular Medicine (KU Leuven). TF was supported by Fundação para a Ciência e a Tecnologia, Programa Operacional Potencial Humano do QREN, and Fundo Social Europeu.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147: 728-41; PMID:22078875; http://dx.doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 2.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 2012; 22: 407-17; PMID:22748206; http://dx.doi.org/ 10.1016/j.tcb.2012.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15: 713-20; PMID:23817233; http://dx.doi.org/ 10.1038/ncb2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27: 107-32; PMID:21801009; http://dx.doi.org/ 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- 5.Araki Y, Ku WC, Akioka M, May AI, Hayashi Y, Arisaka F, Ishihama Y, Ohsumi Y. Atg38 is required for autophagy-specific phosphatidylinositol 3-kinase complex integrity. J Cell Biol 2013; 203: 299-313; PMID:24165940; http://dx.doi.org/ 10.1083/jcb.201304123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014; 157: 65-75; PMID:24679527; http://dx.doi.org/ 10.1016/j.cell.2014.02.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 2014; 53: 167-78; PMID:24462201; http://dx.doi.org/ 10.1016/j.molcel.2013.12.014 [DOI] [PubMed] [Google Scholar]
- 8.Van Veldhoven PP. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J Lipid Res 2010; 51: 2863-95; PMID:20558530; http://dx.doi.org/ 10.1194/jlr.R005959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braverman NE, Moser AB. Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta 2012; 1822: 1442-52; PMID:22627108; http://dx.doi.org/ 10.1016/j.bbadis.2012.05.008 [DOI] [PubMed] [Google Scholar]
- 10.Islinger M, Grille S, Fahimi HD, Schrader M. The peroxisome: an update on mysteries. Histochem Cell Biol 2012; 137: 547-74; PMID:22415027; http://dx.doi.org/ 10.1007/s00418-012-0941-4 [DOI] [PubMed] [Google Scholar]
- 11.Fransen M, Nordgren M, Wang B, Apanasets O. Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim Biophys Acta 2012; 1822: 1363-73; PMID:22178243; http://dx.doi.org/ 10.1016/j.bbadis.2011.12.001 [DOI] [PubMed] [Google Scholar]
- 12.Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry 2010; 49: 835-42; PMID:20050630; http://dx.doi.org/ 10.1021/bi9020378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandalio LM, Rodríguez-Serrano M, Romero-Puertas MC, del Río LA. Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Subcell Biochem 2013; 69: 231-55 [DOI] [PubMed] [Google Scholar]
- 14.Nordgren M, Fransen M. Peroxisomal metabolism and oxidative stress. Biochimie 2014; 98: 56-62; PMID:23933092; http://dx.doi.org/ 10.1016/j.biochi.2013.07.026 [DOI] [PubMed] [Google Scholar]
- 15.Brocard C, Hartig A. Peroxisome targeting signal 1: is it really a simple tripeptide? Biochim Biophys Acta 2006; 1763: 1565-73.; PMID:17007944; http://dx.doi.org/ 10.1016/j.bbamcr.2006.08.022 [DOI] [PubMed] [Google Scholar]
- 16.Fransen M. Peroxisome dynamics: molecular players, mechanisms, and (dys)functions. ISRN Cell Biology 2012; Article ID 714192; PMID:2434022424340224 [Google Scholar]
- 17.Gatto GJ Jr, Geisbrecht BV, Gould SJ, Berg JM. Peroxisomal targeting signal-1 recognition by the TPR domains of human PEX5. Nat Struct Biol 2000; 7: 1091-5; PMID:11101887; http://dx.doi.org/ 10.1038/81930 [DOI] [PubMed] [Google Scholar]
- 18.Braverman N, Dodt G, Gould SJ, Valle D. An isoform of Pex5p, the human PTS1 receptor, is required for the import of PTS2 proteins into peroxisomes. Hum Mol Genet 1998; 7: 1195-205; PMID:9668159; http://dx.doi.org/ 10.1093/hmg/7.8.1195 [DOI] [PubMed] [Google Scholar]
- 19.Francisco T, Rodrigues TA, Freitas MO, Grou CP, Carvalho AF, Sá-Miranda C, Pinto MP, Azevedo JE. A cargo-centered perspective on the PEX5 receptor-mediated peroxisomal protein import pathway. J Biol Chem 2013; 288: 29151-9; PMID:23963456; http://dx.doi.org/ 10.1074/jbc.M113.487140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Platta HW, Hagen S, Reidick C, Erdmann R. The peroxisomal receptor dislocation pathway: to the exportomer and beyond. Biochimie 2014; 98: 16-28; PMID:24345375; http://dx.doi.org/ 10.1016/j.biochi.2013.12.009 [DOI] [PubMed] [Google Scholar]
- 21.Carvalho AF, Pinto MP, Grou CP, Alencastre IS, Fransen M, Sá-Miranda C, Azevedo JE. Ubiquitination of mammalian Pex5p, the peroxisomal import receptor. J Biol Chem 2007; 282: 31267-72; PMID:17726030; http://dx.doi.org/ 10.1074/jbc.M706325200 [DOI] [PubMed] [Google Scholar]
- 22.Oliveira ME, Gouveia AM, Pinto RA, Sá-Miranda C, Azevedo JE. The energetics of Pex5p-mediated peroxisomal protein import. J Biol Chem 2003; 278: 39483-8; PMID:12885776; http://dx.doi.org/ 10.1074/jbc.M305089200 [DOI] [PubMed] [Google Scholar]
- 23.Francisco T, Rodrigues TA, Pinto MP, Carvalho AF, Azevedo JE, Grou CP. Ubiquitin in the peroxisomal protein import pathway. Biochimie 2014; 98: 29-35; PMID:23954799; http://dx.doi.org/ 10.1016/j.biochi.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 24.Platta HW, Hagen S, Erdmann R. The exportomer: the peroxisomal receptor export machinery. Cell Mol Life Sci 2013; 70:1393-411; PMID:22983384; http://dx.doi.org/ 10.1007/s00018-012-1136-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiozawa K, Maita N, Tomii K, Seto A, Goda N, Akiyama Y, Shimizu T, Shirakawa M, Hiroaki H. Structure of the N-terminal domain of PEX1 AAA-ATPase. Characterization of a putative adaptor-binding domain. J Biol Chem 2004; 279: 50060-8; PMID:15328346; http://dx.doi.org/ 10.1074/jbc.M407837200 [DOI] [PubMed] [Google Scholar]
- 26.Fujiki Y, Nashiro C, Miyata N, Tamura S, Okumoto K. New insights into dynamic and functional assembly of the AAA peroxins, Pex1p and Pex6p, and their membrane receptor Pex26p in shuttling of PTS1-receptor Pex5p during peroxisome biogenesis. Biochim Biophys Acta 2012; 1823: 145-9; PMID:22079764; http://dx.doi.org/ 10.1016/j.bbamcr.2011.10.012 [DOI] [PubMed] [Google Scholar]
- 27.Yokota S. Formation of autophagosomes during degradation of excess peroxisomes induced by administration of dioctyl phthalate. Eur J Cell Biol 1993; 61: 67-80; PMID:8223709 [PubMed] [Google Scholar]
- 28.Iwata J, Ezaki J, Komatsu M, Yokota S, Ueno T, Tanida I, Chiba T, Tanaka K, Kominami E. Excess peroxisomes are degraded by autophagic machinery in mammals. J Biol Chem 2006; 281: 4035-41; PMID:16332691; http://dx.doi.org/ 10.1074/jbc.M512283200 [DOI] [PubMed] [Google Scholar]
- 29.Till A, Lakhani R, Burnett SF, Subramani S. Pexophagy: the selective degradation of peroxisomes. Int J Cell Biol 2012; Article ID 512721; PMID:22536249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, Lamark T, Jauregui M, Law K, Lippincott-Schwartz J, et al.. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci 2013; 126: 939-952; PMID:23239026; http://dx.doi.org/ 10.1242/jcs.114819 [DOI] [PubMed] [Google Scholar]
- 31.Nordgren M, Wang B, Apanasets O, Fransen M. Peroxisome degradation in mammals: mechanisms of action, recent advances, and perspectives. Front Physiol 2013; Article ID 145; PMID:23785334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamashita SI, Abe K, Tatemichi Y, Fujiki Y. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy 2014; 10: 1549-64; PMID:25007327; http://dx.doi.org/ 10.4161/auto.29329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim I, Lemasters JJ. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal 2011; 14: 1919-28; PMID:21126216; http://dx.doi.org/ 10.1089/ars.2010.3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubio N, Verrax J, Dewaele M, Verfaillie T, Johansen T, Piette J, Agostinis P. p38(MAPK)-regulated induction of p62 and NBR1 after photodynamic therapy promotes autophagic clearance of ubiquitin aggregates and reduces reactive oxygen species levels by supporting Nrf2-antioxidant signaling. Free Radic Biol Med 2014; 67: 292-303; PMID:24269898; http://dx.doi.org/ 10.1016/j.freeradbiomed.2013.11.010 [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012; 8: 1462-76; PMID:22889933; http://dx.doi.org/ 10.4161/auto.21211 [DOI] [PubMed] [Google Scholar]
- 36.van Zutphen T, Veenhuis M, van der Klei IJ. Damaged peroxisomes are subject to rapid autophagic degradation in the yeast Hansenula polymorpha. Autophagy 2011; 7; 863-72; PMID:21490428; http://dx.doi.org/ 10.4161/auto.7.8.15697 [DOI] [PubMed] [Google Scholar]
- 37.Shibata M, Oikawa K, Yoshimoto K, Kondo M, Mano S, Yamada K, Hayashi M, Sakamoto W, Ohsumi Y, Nishimura M. Highly oxidized peroxisomes are selectively degraded via autophagy in Arabidopsis. Plant Cell 2013; 25: 4967-83; PMID:24368788; http://dx.doi.org/ 10.1105/tpc.113.116947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA 2008; 105: 20567-74; PMID:19074260; http://dx.doi.org/ 10.1073/pnas.0810611105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown AI, Kim PK, Rutenberg AD. PEX5 and ubiquitin dynamics on mammalian peroxisome membranes. PLoS Comput Biol 2014; Article ID e1003426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okumoto K, Noda H, Fujiki Y. Distinct modes of ubiquitination of peroxisome-targeting signal type 1 (PTS1) receptor Pex5p regulate PTS1 protein import. J Biol Chem 2014; 289: 14089-108; PMID:24662292; http://dx.doi.org/ 10.1074/jbc.M113.527937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bulina ME, Lukyanov KA, Britanova OV, Onichtchouk D, Lukyanov S, Chudakov DM. Chromophore-assisted light inactivation (CALI) using the phototoxic fluorescent protein KillerRed. Nat Protoc 2006; 1: 947-53; PMID:17406328; http://dx.doi.org/ 10.1038/nprot.2006.89 [DOI] [PubMed] [Google Scholar]
- 42.Nordgren M, Wang B, Apanasets O, Brees C, Veldhoven PP, Fransen M. Potential limitations in the use of KillerRed for fluorescence microscopy. J Microsc 2012; 245: 229-35; PMID:22091555; http://dx.doi.org/ 10.1111/j.1365-2818.2011.03564.x [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Shyy JY, Chien S. Fluorescence proteins, live-cell imaging, and mechanobiology: seeing is believing. Annu Rev Biomed Eng 2008; 10: 1-38; PMID:18647110; http://dx.doi.org/ 10.1146/annurev.bioeng.010308.161731 [DOI] [PubMed] [Google Scholar]
- 44.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152: 657-68; PMID:11266458; http://dx.doi.org/ 10.1083/jcb.152.4.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8: 445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woudenberg J, Rembacz KP, Hoekstra M, Pellicoro A, van den Heuvel FA, Heegsma J, van Ijzendoorn SC, Holzinger A, Imanaka T, Moshage H, et al.. Lipid rafts are essential for peroxisome biogenesis in HepG2 cells. Hepatology 2010; 52: 623-33; PMID:20683960; http://dx.doi.org/ 10.1002/hep.23684 [DOI] [PubMed] [Google Scholar]
- 47.Huybrechts SJ, Van Veldhoven PP, Brees C, Mannaerts GP, Los GV, Fransen M. Peroxisome dynamics in cultured mammalian cells. Traffic 2009; 10: 1722-33; PMID:19719477; http://dx.doi.org/ 10.1111/j.1600-0854.2009.00970.x [DOI] [PubMed] [Google Scholar]
- 48.Grou CP, Carvalho AF, Pinto MP, Huybrechts SJ, Sá-Miranda C, Fransen M, Azevedo JE. Properties of the ubiquitin-Pex5p thiol ester conjugate. J Biol Chem 2009; 284: 10504-13; PMID:19208625; http://dx.doi.org/ 10.1074/jbc.M808978200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carvalho AF, Grou CP, Pinto MP, Alencastre IS, Costa-Rodrigues J, Fransen M, Sá-Miranda C, Azevedo JE. Functional characterization of two missense mutations in Pex5p - C11S and N526K. Biochim Biophys Acta 2007; 1773: 1141-8; PMID:17532062; http://dx.doi.org/ 10.1016/j.bbamcr.2007.04.011 [DOI] [PubMed] [Google Scholar]
- 50.Wang X, Herr RA, Hansen TH. Ubiquitination of substrates by esterification. Traffic 2012; 13: 19-24; PMID:21883762; http://dx.doi.org/ 10.1111/j.1600-0854.2011.01269.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Okumoto K, Misono S, Miyata N, Matsumoto Y, Mukai S, Fujiki Y. Cysteine ubiquitination of PTS1 receptor Pex5p regulates Pex5p recycling. Traffic 2011; 12: 1067-83; PMID:21554508; http://dx.doi.org/ 10.1111/j.1600-0854.2011.01217.x [DOI] [PubMed] [Google Scholar]
- 52.Amery L, Sano H, Mannaerts GP, Snider J, Van Looy J, Fransen M, Van Veldhoven PP. Identification of PEX5-related novel peroxisome-targeting signal 1 (PTS1)-binding proteins in mammals. Biochem J 2001; 357: 635-46; PMID:11463335; http://dx.doi.org/ 10.1042/0264-6021:3570635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grou CP, Francisco T, Rodrigues TA, Freitas MO, Pinto MP, Carvalho AF, Domingues P, Wood SA, Rodríguez-Borges JE, Sá-Miranda C, et al.. Identification of ubiquitin-specific protease 9X (USP9X) as a deubiquitinase acting on ubiquitin-peroxin 5 (PEX5) thioester conjugate. J Biol Chem 2012; 287: 12815-27; PMID:22371489; http://dx.doi.org/ 10.1074/jbc.M112.340158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyata N, Okumoto K, Mukai S, Noguchi M, Fujiki Y. AWP1/ZFAND6 functions in Pex5 export by interacting with cys-monoubiquitinated Pex5 and Pex6 AAA ATPase. Traffic 2012; 13: 168-183; PMID:21980954; http://dx.doi.org/ 10.1111/j.1600-0854.2011.01298.x [DOI] [PubMed] [Google Scholar]
- 55.Ivashchenko O, Van Veldhoven PP, Brees C, Ho YS, Terlecky SR, Fransen M. Intraperoxisomal redox balance in mammalian cells: oxidative stress and interorganellar crosstalk. Mol Biol Cell 2011; 22: 1440-51; PMID:21372177; http://dx.doi.org/ 10.1091/mbc.E10-11-0919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett 2006; 580: 2623-9; PMID:16647067; http://dx.doi.org/ 10.1016/j.febslet.2006.04.008 [DOI] [PubMed] [Google Scholar]
- 57.Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 2012; 19: 1880-91; PMID:22705852; http://dx.doi.org/ 10.1038/cdd.2012.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al.. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131: 1149-63; PMID:18083104; http://dx.doi.org/ 10.1016/j.cell.2007.10.035 [DOI] [PubMed] [Google Scholar]
- 59.Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci 2013; 70: 2425-41; PMID:23052213; http://dx.doi.org/ 10.1007/s00018-012-1173-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campello S, Strappazzon F, Cecconi F. Mitochondrial dismissal in mammals, from protein degradation to mitophagy. Biochim Biophys Acta 2014; 1837: 451-60; PMID:24275087; http://dx.doi.org/ 10.1016/j.bbabio.2013.11.010 [DOI] [PubMed] [Google Scholar]
- 61.Michaeli S, Galili G. Degradation of organelles or specific organelle components via selective autophagy in plant cells. Int J Mol Sci 2014; 15: 7624-38; PMID:24802874; http://dx.doi.org/ 10.3390/ijms15057624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aksam EB, de Vries B, van der Klei IJ, Kiel JA. Preserving organelle vitality: peroxisomal quality control mechanisms in yeast. FEMS Yeast Res 2009; 9: 808-20; PMID:19538506; http://dx.doi.org/ 10.1111/j.1567-1364.2009.00534.x [DOI] [PubMed] [Google Scholar]
- 63.Oku M, Sakai Y. Peroxisomes as dynamic organelles: autophagic degradation. FEBS J 2010; 277: 3289-94; PMID:20629742; http://dx.doi.org/ 10.1111/j.1742-4658.2010.07741.x [DOI] [PubMed] [Google Scholar]
- 64.Manjithaya R, Nazarko TY, Farré JC, Subramani S. Molecular mechanism and physiological role of pexophagy. FEBS Lett 2010; 584: 1367-73; PMID:20083110; http://dx.doi.org/ 10.1016/j.febslet.2010.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nuttall JM, Motley AM, Hettema EH. Deficiency of the exportomer components Pex1, Pex6, and Pex15 causes enhanced pexophagy in Saccharomyces cerevisiae. Autophagy 2014; 10: 835-45; PMID:24657987; http://dx.doi.org/ 10.4161/auto.28259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tomko RJ, Funakoshi M, Schneider K, Wang J, Hochstrasser M. Heterohexameric ring arrangement of the eukaryotic proteasomal ATPases: implications for proteasome structure and assembly. Mol Cell 2010; 38: 393-403; PMID:20471945; http://dx.doi.org/ 10.1016/j.molcel.2010.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alencastre IS, Rodrigues TA, Grou CP, Fransen M, Sá-Miranda C, Azevedo JE. Mapping the cargo protein membrane translocation step into the PEX5 cycling pathway. J Biol Chem 2009; 284: 27243-51; PMID:19632994; http://dx.doi.org/ 10.1074/jbc.M109.032565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rodrigues TA, Alencastre IS, Francisco T, Brites P, Fransen M, Grou CP, Azevedo JE. A PEX7-centered perspective on the peroxisomal targeting signal type 2-mediated protein import pathway. Mol Cell Biol 2014; 34: 2917-28; PMID:24865970; http://dx.doi.org/ 10.1128/MCB.01727-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grou CP, Carvalho AF, Pinto MP, Alencastre IS, Rodrigues TA, Freitas MO, Francisco T, Sá-Miranda C, Azevedo JE. The peroxisomal protein import machinery - a case report of transient ubiquitination with a new flavor. Cell Mol Life Sci 2009; 66: 254-62; PMID:18810320; http://dx.doi.org/ 10.1007/s00018-008-8415-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gouveia AM, Guimarães CP, Oliveira ME, Reguenga C, Sá-Miranda C, Azevedo JE. Characterization of the peroxisomal cycling receptor, Pex5p, using a cell-free in vitro import system. J Biol Chem 2003; 278: 226-32; PMID:12411433; http://dx.doi.org/ 10.1074/jbc.M209498200 [DOI] [PubMed] [Google Scholar]
- 71.Gouveia AM, Guimarães CP, Oliveira ME, Sá-Miranda C, Azevedo JE. Insertion of Pex5p into the peroxisomal membrane is cargo protein-dependent. J Biol Chem 2003; 278: 4389-92; PMID:12502712; http://dx.doi.org/ 10.1074/jbc.C200650200 [DOI] [PubMed] [Google Scholar]
- 72.Cantalupo PG, Sáenz-Robles MT, Rathi AV, Beerman RW, Patterson WH, Whitehead RH, Pipas JM. Cell-type specific regulation of gene expression by simian virus 40 T antigens. Virology 2009; 386: 183-91; PMID:19201438; http://dx.doi.org/ 10.1016/j.virol.2008.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rathi AV, Sáenz Robles MT, Cantalupo PG, Whitehead RH, Pipas JM. Simian virus 40 T-antigen-mediated gene regulation in enterocytes is controlled primarily by the Rb-E2F pathway. J Virol 2009; 83: 9521-31; PMID:19570859; http://dx.doi.org/ 10.1128/JVI.00583-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Legakis JE, Koepke JI, Jedeszko C, Barlaskar F, Terlecky LJ, Edwards HJ, Walton PA, Terlecky SR. Peroxisome senescence in human fibroblasts. Mol Biol Cell 2002; 13: 4243-55; PMID:12475949; http://dx.doi.org/ 10.1091/mbc.E02-06-0322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muller M. Cellular senescence: molecular mechanisms, in vivo significance, and redox considerations. Antioxid Redox Signal 2009; 11: 59-98; PMID:18976161; http://dx.doi.org/ 10.1089/ars.2008.2104 [DOI] [PubMed] [Google Scholar]
- 76.Apanasets O, Grou CP, Van Veldhoven PP, Brees C, Wang B, Nordgren M, Dodt G, Azevedo JE, Fransen M. PEX5, the shuttling import receptor for peroxisomal matrix proteins, is a redox-sensitive protein. Traffic 2014; 15: 94-103; PMID:24118911; http://dx.doi.org/ 10.1111/tra.12129 [DOI] [PubMed] [Google Scholar]
- 77.Yokota S, Oda T, Fahimi HD. The role of 15-lipoxygenase in disruption of the peroxisomal membrane and in programmed degradation of peroxisomes in normal rat liver. J Histochem Cytochem 2001; 49: 613-22; PMID:11304799; http://dx.doi.org/ 10.1177/002215540104900508 [DOI] [PubMed] [Google Scholar]
- 78.Juenemann K, Reits EA. Alternative macroautophagic pathways. Int J Cell Biol 2012; 2012: Article ID 189794; PMID:22536246; http://dx.doi.org/ 10.1155/2012/189794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ 2013; 20: 21-30; PMID:22722335; http://dx.doi.org/ 10.1038/cdd.2012.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Léon S1, Subramani S. A conserved cysteine residue of Pichia pastoris Pex20p is essential for its recycling from the peroxisome to the cytosol. J Biol Chem 2007; 282: 7424-30; http://dx.doi.org/ 10.1074/jbc.M611627200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feutz AC, Pham-Dinh D, Allinquant B, Miehe M, Ghandour MS. An immortalized jimpy oligodendrocyte cell line: defects in cell cycle and cAMP pathway. Glia 2001; 34: 241-52; PMID:11360297; http://dx.doi.org/ 10.1002/glia.1058 [DOI] [PubMed] [Google Scholar]
- 82.Brees C, Fransen M. A cost-effective approach to microporate mammalian cells with the Neon Transfection System. Anal Biochem 2014; 466: 49-50; PMID:25172131; http://dx.doi.org/ 10.1016/j.ab.2014.08.017 [DOI] [PubMed] [Google Scholar]
- 83.Amery L, Fransen M, De Nys K, Mannaerts GP, Van Veldhoven PP. Mitochondrial and peroxisomal targeting of 2-methylacyl-CoA racemase in humans. J Lipid Res 2000; 41: 1752-9; PMID:11060344 [PubMed] [Google Scholar]
- 84.Huybrechts SJ, Van Veldhoven PP, Hoffman I, Zeevaert R, de Vos R, Demaerel P, Brams M, Jaeken J, Fransen M, Cassiman D. Identification of a novel PEX14 mutation in Zellweger syndrome. J Med Genet 2008; 45: 376-83; PMID:18285423; http://dx.doi.org/ 10.1136/jmg.2007.056697 [DOI] [PubMed] [Google Scholar]
- 85.Fransen M, Van Veldhoven PP, Subramani S. Identification of peroxisomal proteins by using M13 phage protein VI phage display: molecular evidence that mammalian peroxisomes contain a 2,4-dienoyl-CoA reductase. Biochem J 1999; 340: 561-8; PMID:10333503; http://dx.doi.org/ 10.1042/0264-6021:3400561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fransen M. HaloTag as a tool to investigate peroxisome dynamics in cultured mammalian cells. Methods Mol Biol 2014; 1174: 157-70; PMID:24947380 [DOI] [PubMed] [Google Scholar]
- 87.Wilkinson KD, Gan-Erdene T, Kolli N. Derivitization of the C-terminus of ubiquitin and ubiquitin-like proteins using intein chemistry: methods and uses. Methods Enzymol 2005; 399: 37-51; PMID:16338347 [DOI] [PubMed] [Google Scholar]
- 88.Fransen M, Vastiau I, Brees C, Brys V, Mannaerts GP, Van Veldhoven PP. Potential role for Pex19p in assembly of PTS-receptor docking complexes. J Biol Chem 2004; 279: 12615-24; PMID:14715663; http://dx.doi.org/ 10.1074/jbc.M304941200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.