

Abstract
Autophagy is a potentially inimical pathway and together with apoptosis, may be activated by similar stress stimuli that can lead to cell death. The molecular cues that dictate the cell fate choice between autophagy and apoptosis remain largely unknown. Here we report that the proapoptotic protein BBC3/PUMA (BCL2 binding component 3) is a bona fide substrate of chaperone-mediated autophagy (CMA). BBC3 associates with HSPA8/HSC70 (heat shock 70kDa protein 8), leading to its lysosome translocation and uptake. Inhibition of CMA results in stabilization of BBC3, which in turn sensitizes tumor cells to undergo apoptosis. We further demonstrate that upon TNF (tumor necrosis factor) treatment, IKBKB/IKKβ (inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase β)-mediated BBC3 Ser10 phosphorylation is crucial for BBC3 stabilization via blocking its degradation by CMA. Mechanistically, Ser10 phosphorylation facilitates BBC3 translocation from the cytosol to mitochondria. BBC3 stabilization resulting from either Ser10 phosphorylation or CMA inhibition potentiates TNF-induced apoptotic cell death. Our findings thus reveal that the selective degradation of BBC3 underlies the prosurvival role of CMA and define a previously unappreciated proapoptotic role of IKBKB that acts through phosphorylation-mediated stabilization of BBC3, thereby promoting TNF-triggered apoptosis.
Keywords: apoptosis, BBC3, chaperone-mediated autophagy, IKBKB, IKKβ, PUMA, TNF
Abbreviations
- BBC3/PUMA
BCL2 binding component 3
- BCL2L11/BIM
BCL2-like 11
- CA
constitutively active
- CDKN1A/p21
cyclin-dependent kinase inhibitor 1A (p21, Cip1)
- CHUK/IKKα
conserved helix-loop-helix ubiquitous kinase
- CMA
chaperone-mediated autophagy
- CHX
cycloheximide
- DOX
doxorubicin
- IKBKB/IKKβ
inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase β
- HSPA8/HSC70
heat shock 70kDa protein 8
- LAMP2A
lysosomal-associated membrane protein type 2A
- NFKB1/NF-κB
nuclear factor of kappa light polypeptide gene enhancer in B-cells 1
- NFKBIA/IκBα
nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, α
- PNS
postnuclear supernatant
- shRNA
short hairpin RNA
- TNF
tumor necrosis factor
- TP53/p53
tumor protein 53.
Introduction
Chaperone-mediated autophagy (CMA) plays an essential role in a diverse range of pathophysiological processes, including aerobic glycolysis, neurodegeneration, and killing of dormant cancer cells.1-8 A variety of cellular stress stimuli, such as nutrient deprivation, exposure to toxic compounds, and mild oxidative stress can potentiate CMA activity.9,10 In CMA, cytosolic substrates with a KFERQ-like motif are recognized by a chaperone-cochaperone complex that transports them to the lysosomal surface.11 After interacting with a lysosomal receptor, LAMP2A (lysosomal-associated membrane protein 2A),12 substrate proteins undergo unfolding and translocation across the membrane into the lysosomal lumen under the assistance of a luminal chaperone (lyso-HSPA8), where they get degraded. Interestingly, change in CMA activity under stress conditions correlates very well with lysosomal membrane LAMP2A levels.13 Notably, depletion of LAMP2A triggered cell death in lung cancer cells,6 indicating that CMA activity is crucial for cell survival.
BH3-only protein BBC3 is a proapoptotic BCL−2 family member that drives the apoptotic response to a wide range of cellular insults. Genetic studies using bbc3 knockout mice revealed a crucial role of BBC3 in the induction of apoptosis triggered by distinct apoptotic signals, including genotoxic damage, cytokine deprivation, dexamethasone, staurosporine, and PMA.14 Distinct transcriptional programs have been reported to regulate BBC3. Genotoxic insults including γ-irradiation and chemotherapeutic drugs induce BBC3 by TP53-dependent transactivation.15,16 In addition to DNA damage signals, a variety of stress stimuli can induce BBC3 in a TP53-independent manner. Transcription factors including FOXO members, NFKB1/NF-κB (nuclear factor of kappa light polypeptide gene enhancer in B-cells 1), and SMAD4 proteins can induce BBC3 in response to growth factor deprivation, TNF or TGFB treatment, respectively.17-19 In addition to transcriptional control, BBC3 has been found to undergo post-translational modification and is subject to proteasome-mediated degradation,20,21 or caspase-dependent degradation.22
In the present study, we found BBC3 is subject to CMA-dependent degradation. Our data demonstrate that the cytoprotective role of CMA under basal conditions or upon exposure to stress signals is largely mediated by BBC3. Therefore, the interaction between BBC3 and CMA identifies a potentially important point of convergence of the apoptotic and autophagic machinery. Furthermore, our results revealed that TNF-mediated BBC3 induction is fine-tuned via both post-translational and transcriptional mechanisms.
Results
Inhibition of CMA leads to BBC3 induction
BBC3 is critical for apoptosis induced by a wide range of stress signals that act through a variety of transcriptional factors. However, regulation of BBC3 at post-translational levels remains elusive. To investigate the signaling pathway that regulates BBC3 stabilization, we treated a variety of tumor cell lines with proteasomal inhibitor or lysosomal inhibitors. Upregulation of BBC3 was only observed in cells exposed to lysosomal inhibitors, but not proteasome inhibitor MG132, which induced CDKN1A/P21 (cyclin-dependent kinase inhibitor 1A [p21, Cip1]) stabilization (Fig. S1A and B). By contrast, the expression levels of BCL2L11/BIM (BCL2-like 11), another BH3-only family member, remain unchanged in response to lysosomal inhibitors (Fig. S1B). There are 3 different types of autophagy: macroautophagy, microautophagy, and CMA.23-25 To determine which autophagic pathway regulates BBC3 protein abundance, we first depleted key CMA molecules in human tumor cell lines bearing either wild-type or mutant/inactivated TP53. Ablation of LAMP2A or HSPA8, genes encoding 2 major CMA components, resulted in upregulation of BBC3 in all cancer cell lines tested (Fig. 1A) without affecting its mRNA expression levels (Fig. S1C). This result further indicates that TP53 is dispensable for CMA inhibition-induced BBC3 accumulation. Moreover, CMA ablation in a noncancerous cell line, human embryonic kidney 293 (HEK293) cells, induced BBC3 accumulation as well (Fig. S1D). To test if CMA depletion could increase the steady state levels of BBC3 protein, we measured the half-life of endogenous BBC3 in CMA-deficient cells. Silencing HSPA8 or LAMP2A significantly extended the half-life of BBC3 (Fig. 1B and C), suggesting CMA regulates BBC3 protein stability.
Figure 1.

Inhibition of CMA leads to BBC3 induction. (A) Representative western blots and densitometric data (n = 3 or 4) showing that CMA ablation leads to BBC3 upregulation. The indicated tumor cells were infected with vectors containing control, LAMP2A, or HSPA8 shRNA and then lysed. (B) Representative immunoblots (of n ≥ 3) showing that loss of CMA stabilizes BBC3. H1299 cells expressing the indicated shRNAs were treated with 40 μg/ml CHX and then harvested at the indicated times. (C) Relative BBC3 protein level shown in (B) was quantified. (D) Representative Western blots (n = 3) showing that LAMP2A depletion further promotes BBC3 induction upon serum withdrawal. H1299 cells were infected with control or LAMP2A shRNA lentiviruses for 48 h followed by serum deprivation for 48 h. Cell lysates were harvested for immunoblotting analysis. (E) Western blotting analysis of BBC3, TP53, and CDKN1A levels in HCT116 cells. Cells first infected with vectors containing shCon or BBC3 shRNAs, then with shCon or LAMP2A shRNAs were treated with 0.5 μM DOX for 6 h and then harvested. (F) FACS analysis of cell death by PI staining in HCT116 cells stably expressing shRNA constructs as in (E) showing that CMA inhibition induces cell death in a BBC3-dependent manner. (G) Percentage of early apoptotic cells after 24 h of DOX exposure in cells stably expressing shRNA constructs as in (E) was determined by ANXA5 and PI staining, which shows CMA inhibition induces cell death upon DNA damage in a BBC3-dependent manner. Data were represented as mean ±SEM; P < 0.01** and 0.001***, n = 4, t test. Quantification of BBC3 protein levels was done relative to loading control.
Serum starvation is known to activate CMA, thereby degrading CMA substrates. However, serum deprivation can also induce BBC3 via transcriptional machinery.17,26 We assessed if CMA ablation in serum-starved cells would further stabilize BBC3. Transcriptional induction of BBC3 was observed following serum deprivation for 48 h (Fig. S1E). Strikingly, depletion of LAMP2A, the rate-limiting factor in CMA process, resulted in further accumulation of BBC3 (Fig. S1F), indicating BBC3 is subject to both transcriptional and post-translational regulation under serum-deprivation conditions.
BH3-only proteins are essential initiators of apoptosis. The increased BBC3 protein levels in CMA-depleted cells prompted us to test if silencing CMA components can sensitize tumor cells to genotoxic insults. In the absence of DNA damage, knockdown LAMP2A in wild-type HCT116 cells resulted in profound cell death, which can be completely blocked by BBC3 depletion (Fig. 1E and F). Doxorubicin (DOX) is an anthracycline antibiotic that is used widely in treatment of cancers. Upon DOX treatment, BBC3 is subject to TP53-dependent transcriptional upregulation, which in turn leads to apoptotic cell death. When cells were exposed to DOX treatment, silencing LAMP2A led to significant induction of early apoptosis, which again can be abolished by codepletion of BBC3 (Fig. 1E and G). Consistent with previous findings, we observed stabilization and activation of TP53 in CMA-depleted cells,6 (Fig. 1E). This raised the possibility that BBC3-dependent cell death in these cells may require functional TP53 signaling. To address this, we monitored cell viability in HCT116 TP53−/− cells depleted of LAMP2A. Knockdown of LAMP2A failed to trigger cell death in the absence of TP53 (Fig. S1G), suggesting that an intact TP53 signaling is required for CMA blockage-induced cytotoxicity in our experimental settings. A plausible explanation for this TP53-dependency might be due to a requirement for TP53 to maintain the basal levels of BBC3. In the absence of TP53, Upregulated BBC3 levels upon CMA depletion may not be enough for activating mitochondrial death. This is further supported by evidence obtained from other TP53-null or TP53-mutant cancer cells, where LAMP2A depletion resulted in profound upregulation of BBC3 without activation of cell death (Fig. 1A; Fig. S1G). Taken together, these data suggest that BBC3 is subject to CMA-mediated degradation, and the prosurvival role of CMA is largely mediated by proteolytic degradation of BBC3.
BBC3 interacts with CMA components
To test if BBC3 is targeted to lysosome, immunofluorescent staining (IF) was carried out to detect BBC3 localization. We observed high levels of colocalization of ectopic BBC3 with the lysosome-associated membrane proteins LAMP2A (Fig. 2A) in the presence of the lysosomal inhibitor ammonium chloride (NH4Cl), suggesting a possible interaction between BBC3 and CMA components. In CMA, HSPA8 functions to recruit target proteins to the lysosome for degradation. The direct interaction between HSPA8 and BBC3 was detected by NiNTA pull-down assay (Fig. 2B). The endogenous HSPA8-BBC3 complex was further confirmed by reciprocal immunoprecipitation (Fig. 2C and D). Similarly, we found an association between endogenous LAMP2A and BBC3 (Fig. 2D). These results demonstrate that BBC3 forms protein complex with LAMP2A and HSPA8.
Figure 2.
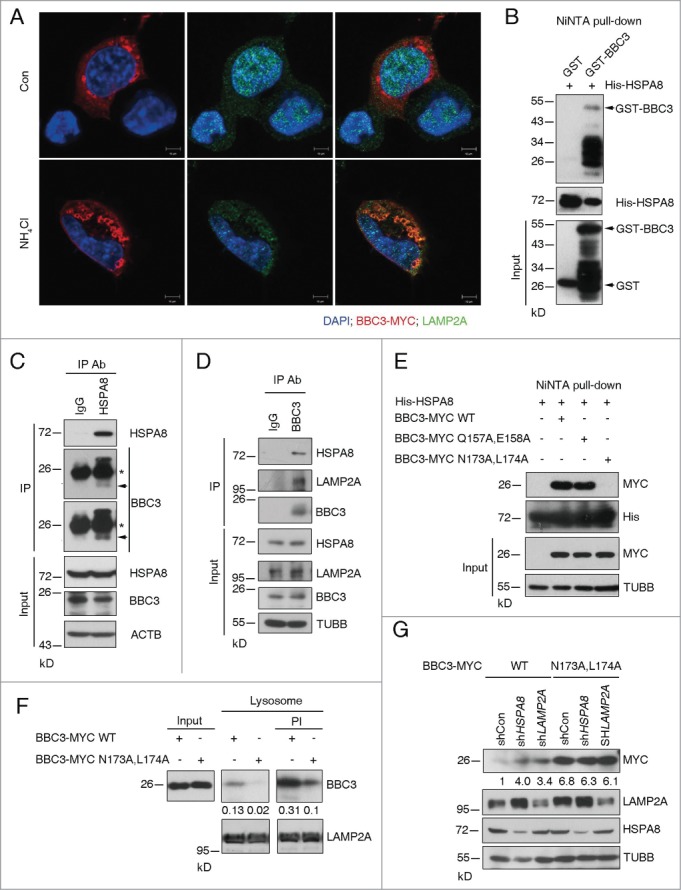
For figure legend, see page 1627.
Proteins undergoing CMA-mediated lysosomal degradation often contain a loosely defined KFERQ motif important for HSPA8 binding.27 The KFERQ motif usually contains 5 residues, including a critical glutamine (Q) residue that is preceded or followed by 4 amino acids consisting of a basic (R or K), an acidic (E or D), or a bulky hydrophobic residue (I, L, V, or F).28 Inspection of BBC3 amino acid sequence reveals 2 imperfect HSPA8 binding motifs at the C terminus of BBC3 (Fig. S2A),155RRQEE159, and 169RVLYNL174, respectively. We tested whether they may serve as HSPA8 interacting sequences by incubating purified His-HSPA8 with cellular lysates containing ectopically expressed wild-type or mutant BBC3 (Q157A and E158A or N173A and L174A). Figure 2E showed that N173A and L174A mutation almost completely abolished the interaction between BBC3 and HSPA8, suggesting 169RVLYNL174 is crucial for mediating BBC3 recognition by HSPA8. By contrast, 155RRQEE159 is not required for HSPA8-BBC3 binding.
CMA substrates usually can be directly translocated into isolated lysosomes. We next performed in vitro incubation of the immunopurified BBC3 with isolated lysosomes to determine if BBC3 can be taken up by lysosomes. Incubation of wild-type BBC3 with intact liver lysosomes untreated or pretreated with protease inhibitors (PI) showed that BBC3 bound to the lysosomal membrane and was selectively taken up by lysosomes (Fig. 2F). By contrast, the association of the N173A and L174A mutant protein with the lysosomal membrane and its translocation into the lysosomal lumen were dramatically reduced (Fig. 2F). We noticed that the protein level of ectopically expressed BBC3N173A,L174A-myc was much higher compared to that of BBC3-myc WT (Fig. 2G; Fig. S2B). A plausible explanation for this phenomenon may lie in the fact that this N173A and L174A mutant is HSPA8-binding defective; therefore, it is refractory to CMA-mediated degradation. To test if the N173A and L174A mutant may confer resistance to lysosomal degradation, we depleted HSPA8 or LAMP2A in cells expressing ectopic wild-type or N173A and L174A mutant BBC3. As expected, the N173A and L174A mutant failed to be further stabilized in response to CMA depletion (Fig. 2G), suggesting blocking HSPA8 binding is sufficient to disrupt CMA-mediated BBC3 degradation, thereby stabilizing BBC3. As expected, N173A and L174A mutant BBC3 exhibited increased intracellular cytotoxicity compared to wild-type BBC3 when ectopically expressed in HCT116 cells (Fig. S2B and C). Taken together, these data demonstrate BBC3 is a bona fide CMA substrate.
Ser10 is crucial for protecting BBC3 from CMA-mediated degradation
Previous studies have revealed Ser10, as a major phosphorylation site of BBC3, can be phosphorylated upon serum or cytokine stimulation. By analyzing ectopically expressed BBC3 Ser10 mutants (S10A, S10D or S10E), it was suggested that all these mutants are more stable compared to wild-type BBC3.21 To ascertain the physiological effects of BBC3 Ser10 phosphorylation, we engineered BBC3S10A or BBC3S10D into the endogenous BBC3 locus in HCT116 cells (Fig. S3A). BBC3S10A/S10A or BBC3S10D/S10D was confirmed by sequencing genomic DNA for the presence of the homologous T > G or TC > GA BBC3 mutations (Fig. S3B). Notably, these knock-in mutations had no significant effect on BBC3 mRNA levels (Fig. S3C). Surprisingly, BBC3S10A exhibited reduced protein expression levels, whereas BBC3S10D was substantially stabilized compared to wild-type BBC3 (Fig. 3A). We next measured the half-life of BBC3 in wild-type and knock-in cells. Wild-type BBC3 had a half-life of approximately 130 min, whereas the half-life of BBC3S10A was shortened to 100 min (Fig. 3B and C). By sharp contrast, the half-life of BBC3S10D did not decline even after 8 h exposure to cycloheximide (CHX) (Fig. 3D and E). These results suggest that when driven by an endogenous promoter, the S10D mutation enhances the steady-state of BBC3, whereas S10A destabilizes BBC3.
Figure 3.

For figure legend, see page 1629.
The above observation that Ser10 mutants significantly influenced BBC3 stability prompted us to examine the potential role of Ser10 in CMA-mediated BBC3 degradation. Because BBC3S10D is relatively more stable, a plausible explanation is that this phospho-mimetic mutant may exert a protective role in CMA-mediated BBC3 degradation. To test this, we assessed BBC3 accumulation upon depletion of CMA. Both wild-type and BBC3S10A protein levels were profoundly increased in response to HSPA8 or LAMP2A silencing (Fig. S3D). However, BBC3S10D showed no further accumulation upon CMA inhibition (Fig. S3D), indicating the phospho-mimetic mutant S10D may stabilize BBC3 via blocking CMA-mediated BBC3 degradation.
To elucidate the molecular mechanism underlying Ser10-related BBC3 stabilization, we examined complex formation between endogenous BBC3 and HSPA8 in wild-type and knock-in cells. BBC3S10A displayed a much stronger interaction with HSPA8 than wild-type BBC3, whereas BBC3S10D exhibited profoundly reduced binding capacity with HSPA8 (Fig. 3F). These results indicate that the Ser10 mutation may regulate BBC3 stability by modulating its interaction with HSPA8. Consistently, the association of BBC3S10A with the lysosomal membrane and its translocation into the lysosomal lumen were dramatically increased, whereas serine to aspartate mutation rendered BBC3S10D refractory to lysosomal binding and uptake (Fig. 3G and H). Taken together, these data imply that Ser10 phosphorylation abrogates BBC3 recognition by chaperone protein, thereby preventing its subsequent lysosomal uptake and degradation.
BBC3 has been found to predominantly localize to the mitochondria and the C-terminal region of BBC3 may function as a mitochondrial localization domain.16 Cytosolic BBC3 is usually undetectable.29 Given that CMA only degrades cytosolic proteins, we speculate that Ser10 may modulate BBC3 subcellular localization. To test this, we compared the mitochondria-to-postnuclear supernatant (PNS) BBC3 ratio between wild-type BBC3 and BBC3S10A. The ratio was around 1.9:1 in wild type HCT116 cells, whereas in BBC3S10A cells, the ratio was less than 50% (Fig. 3I and J). This suggests that BBC3S10A could impair mitochondrial localization of BBC3. By sharp contrast, the cytosol-to-PNS ratio of BBC3 was 0.14:1 in wild-type cells, which was significantly lower compared to 0.45:1 in BBC3S10A cells (Fig. 3I and J). Depletion of HSPA8 led to increased cytosolic BBC3. However, we noticed the overall cytosol-to-PNS ratio of wild-type BBC3 did not show a significant difference between control cells and HSPA8-depleted cells (0.14:1 vs 0.25:2.2). In contrast, in BBC3S10A cells, silencing HSPA8 significantly increased the cytosol-to-PNS ratio of BBC3S10A (from 0.45:1 to 1.3:2) (Fig. 3I and J). These data imply that S10A mutation may facilitate the cytosolic localization of BBC3, which in turn promotes CMA-mediated degradation of BBC3.
As BBC3 protein levels are essential for activating apoptotic cell death in response to diverse stress signals, we further characterized if Ser10 is generally required for BBC3 accumulation and its proapoptotic function under stress signals. To this end, we exposed wild-type or knock-in cells with genotoxic insults. We noticed that, upon DOX exposure, BBC3S10A protein levels were less induced compared to wild-type BBC3, whereas BBC3S10D was further accumulated (Fig. S3E), albeit BBC3 mRNA levels were induced to a similar extent by DOX in these cells (Fig. S3F). Consistently, DOX-induced cell death was profoundly compromised in BBC3S10A cells, but was largely promoted in BBC3S10D cells (Fig. S3G). Thus, endogenous BBC3S10A is less proapoptotic whereas BBC3S10D is more potent in activating apoptosis in response to DNA damage agents.
IKBKB-dependent phosphorylation of BBC3 at Ser10 enables maximum BBC3 induction upon TNF exposure
We next set out to identify upstream kinases responsible for BBC3 Ser10 phosphorylation. IKK family members have been reported to phosphorylate BBC3 at Ser10 upon cytokine stimulation. We then assessed if constitutively active (CA) IKK complex is capable of phosphorylating Ser10 in wild-type and knock-in HCT116 cells. A rabbit polyclonal antibody was generated using a synthetic BBC3 phosphopeptide containing phosphorylated Ser10 as described before.21 Immunoblotting analysis revealed that the anti-phospho-Ser10 antibody specifically recognized phosphorylated wild-type BBC3 but not BBC3S10A in the presence of IKBKB (CA) (Fig. S4A). Notably, CHUK/IKKα (conserved helix-loop-helix ubiquitous kinase) (CA) only induced marginal BBC3 Ser10 phosphorylation, whereas IKBKB (CA) exhibited robust kinase activity at this site (Fig. S4B). Moreover, endogenous IKBKB and BBC3 formed a protein complex as detected by immunoprecipitation (Fig. 4A). In cells expressing ectopic IKBKB, the association between BBC3 and HSPA8 was significantly impaired (Fig. S4B), supporting the notion that Ser10 phosphorylation could block BBC3-HSPA8 interaction .
Figure 4.
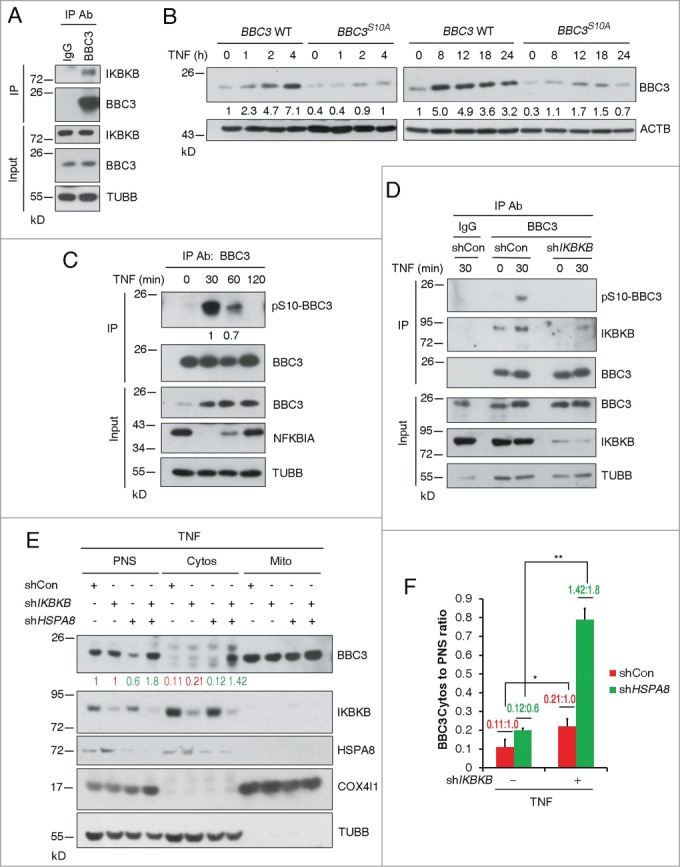
IKBKB-dependent phosphorylation of BBC3 at Ser10 enables maximum BBC3 induction upon TNF exposure. (A) Representative immunoblots of immunoprecipitation (of n = 3) using IgG or anti-BBC3 antibody showing that endogenous IKBKB associates with BBC3. (B) Representative Western blots (of n = 3) of BBC3 WT and BBC3S10A HCT116 cells incubated with 10 ng/ml TNF for the indicated times showing that serine to alanine mutation results in compromised BBC3 accumulation upon TNF exposure. The ratio of BBC3 protein normalized to ACTB relative to control (marked as 1.0) is indicated below each lane. (C) Representative immunoblots with densitometric quantification following immunoprecipitation (of n = 4) using anti-BBC3 antibody in HCT116 cells treated with TNF for 30 min following control or IKBKB shRNA lentivirus infection showing BBC3 Ser10 phosphorylation upon TNF treatment. Of note, BBC3 levels in the IP experiment do not change in this case in the TNF lane, owing to saturation of the anti-BBC3 antibody. The pSer10-BBC3 protein levels were quantified relative to BBC3 protein levels in the IP experiment. (D) Representative western blots of immunoprecipitation (of n = 3) in HCT116 cells showing that BBC3 Ser10 phosphorylation upon TNF treatment is IKBKB dependent. HCT116 cells were infected with vectors containing control or IKBKB shRNA first. At 24-h postinfection, cells were incubated with TNF for the indicated times and then harvested for IP experiment. Of note, BBC3 levels in the coIP experiment do not change in this case in the TNF and IKBKB shRNA lanes, owing to saturation of the anti-BBC3 antibody. (E) Representative Western blots of mitochondria fractionation (of n = 3) in HCT116 cells showing that depletion of IKBKB results in an increase of cytosolic BBC3 upon TNF treatment. HCT116 cells were coinfected with shIKBKB and shHSPA8 lentivirus first. At 48-h postinfection, cells were treated with TNF for 30 min and then harvested. (F) The ratio of BBC3 protein normalized to TUBB (for Cytosolic fraction) in (E) relative to control (red for shCon and green for shHSPA8) is indicated. Statistics are depicted as mean ±SEM; P < 0.05 *, and 0.01**, n = 4, t test.
Given that BBC3S10D is more stable whereas BBC3S10A is less stable, we reasoned that IKBKB-mediated Ser10 phosphorylation should stabilize BBC3. To test this, we examined BBC3 protein levels in wild-type or knock-in cells transiently transfected with the active form of IKK kinase. Interestingly, only wild-type BBC3 was stabilized upon IKBKB (CA) overexpression, whereas BBC3S10A or BBC3S10D showed minimal responses to IKBKB (CA) (Fig. S4C), albeit mRNA levels of BBC3 were all induced to a similar extent by IKBKB (CA) (Fig. S4D). This suggests that IKBKB-mediated Ser10 phosphorylation is necessary and sufficient for BBC3 stabilization.
To elucidate the physiological relevance of IKBKB-mediated BBC3 phosphorylation, we exposed cells to TNF treatment, which is known to activate IKK signaling. BBC3 is a direct target of NFKB1 and can be transcriptionally upregulated in response to TNF.18 Indeed, in cells treated with TNF for 2 h, BBC3 mRNA levels were markedly upregulated (Fig. S4E). Notably, exposure to TNF for 1 h induced detectable BBC3 protein accumulation only in wild-type cells, but not in BBC3S10A cells (Fig. 4B; Fig. S4F). Furthermore, accumulation of BBC3 protein was profoundly impaired in BBC3S10A cells following 24-h treatment with TNF (Fig. 4B; Fig. S4F), albeit transcriptional induction of BBC3 mRNA levels by TNF-induced NFKB1 activation preceded normally in wild-type and knock-in cells (Fig. S4E). In contrast, BBC3S10D protein levels remain relatively unchanged in the presence of TNF (Fig. S4G), indicating this phospho-mimetic mutation causes the maximum induction of BBC3.
To assess if TNF could stabilize BBC3 via regulating its steady-state levels, we measured BBC3 protein half-life in wild-type HCT116 cells in the presence or absence of TNF. The half-life of BBC3 was significantly extended upon TNF treatment (Fig. S5A), indicating that BBC3 is subject to both transcriptional and posttranslational regulation in response to TNF stimulation. Clearly, in order to address the precise role of IKBKB-mediated Ser10 phosphorylation in BBC3 stabilization under TNF treatment, NFKB1-mediated transactivation of BBC3 must be teased apart. To this end, we depleted RELA, the major transactivating subunit of NFKB1 and then exposed cells to TNF treatment. Depleting RELA resulted in compromised BBC3 mRNA induction in response to TNF (Fig. S5B and C). Notably, wild-type BBC3 protein was significantly accumulated whereas induction of BBC3S10A was completely abrogated upon TNF exposure (Fig. S5D). Taken together, we conclude that Ser10 phosphorylation is not only required for transient induction of BBC3 upon short exposure to TNF, but also crucial for maximum BBC3 accumulation after prolonged TNF treatment.
To assess if TNF could induce BBC3 phosphorylation at the Ser10 site, we measured the Ser10 phosphorylation status of BBC3 in cells treated with TNF. Total BBC3 protein levels were substantially increased 30 min after TNF application, and the induction was sustained over time (Fig. 4C). By contrast, TNF induced transient Ser10 phosphorylation with a peak at 30 min, which paralleled the degradation of NFKBIA/IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, α) at this time point (Fig. 4C). Notably, TNF treatment caused a transiently compromised complex formation between BBC3 and HSPA8 (Fig. S5E), further supporting the notion that Ser10 phosphorylation-mediated BBC3 stabilization is attributed to impaired CMA-dependent degradation.
TNF stimulation can activate multiple signaling pathways. We next asked if IKBKB is the major kinase responsible for TNF-mediated BBC3 phosphorylation and subsequent stabilization. To test this, BBC3 protein levels were measured following IKBKB depletion in cells treated with TNF. Silencing IKBKB abrogated TNF-induced BBC3 stabilization (Fig. S6A) and Ser10 phosphorylation (Fig. 4D). Furthermore, the turnover rate of BBC3 in cells exposed to TNF was significantly shortened upon IKBKB knockdown (Fig. S6B and C), suggesting TNF-mediated IKBKB activation is crucial for Ser10 phosphorylation and subsequent stabilization of BBC3. Paradoxically, we noticed IKBKB knockdown in fact upregulated basal BBC3 protein levels as well as mRNA transcript levels (Fig. S6A and D), despite the fact that the half-life of BBC3 was significantly shortened upon IKBKB depletion (Fig. S6E and F). We tested if shutting down transcriptional machinery could abrogate this induction of BBC3. Indeed, IKBKB depletion-induced BBC3 accumulation was completely blocked in response to RNA synthesis inhibitor actinomycin D treatment (Fig. S6G and H), suggesting that IKBKB also regulates the basal expression of BBC3 at a transcriptional level. Collectively, these data suggest that IKBKB could regulate BBC3 via 2 distinct mechanisms. Under basal conditions, IKBKB negatively regulates BBC3 transactivation. Upon TNF exposure, IKBKB could phosphorylate BBC3 at Ser10, which in turn leads to the stabilization of BBC3 by blocking CMA-mediated degradation.
To corroborate our hypothesis that Ser10 phosphorylation may facilitate BBC3 being targeted to mitochondria, thereby blocking its degradation, we analyzed the subcellular localization of BBC3 in cells treated with TNF. We reasoned that IKBKB-deficiency should result in more cytosolic BBC3 due to reduced Ser10 phosphorylation. Indeed, HSPA8 depletion resulted in a significant increase of the cytosol-to-PNS ratio of BBC3 in cells expressing IKBKB shRNA (Fig. 4E and F), compared to the ratio of BBC3 in control cells, suggesting that IKBKB-mediated Ser10 phosphorylation could stabilize BBC3 by promoting its mitochondrial localization, which in turn abrogates its degradation by CMA.
BBC3 stabilization resulting from Ser10 phosphorylation or CMA blockage is essential for TNF-triggered apoptosis
It is widely accepted that NFKB1 activation upon TNF treatment is generally prosurvival. In addition to this, IKBKB inhibits TNF-induced apoptosis independently of NFKB1 activation via phosphorylating BAD.30 To directly validate the role of IKBKB-mediated BBC3 phosphorylation in TNF-induced apoptosis, we cotreated HCT116 cells with TNF and CHX, which is known to sensitize cells to TNF-induced apoptosis.31 Since CHX can block de novo protein synthesis, TNF-mediated transcriptional induction of BBC3 was compromised due to the termination of NFKB1 transcriptional activity in the presence of CHX (Fig. S6I). However, accumulation of BBC3 upon 1-h exposure to TNF and CHX still occurred (Fig. 5A). As expected, addition of CHX rendered HCT116 cells TNF sensitive. Approximately 30% wild-type HCT116 cells underwent apoptosis after 7-h exposure to TNF and CHX, whereas only 10% BBC3S10A cells exhibited apoptotic cell death (Fig. 5B). Most importantly, depletion of BBC3 in wild-type cells largely decreased apoptotic cell percentage upon TNF treatment (Fig. 5C and D). Collectively, these results suggest that BBC3 Ser10 phosphorylation is crucial for TNF-induced apoptosis. Given that CMA acts as a key upstream signal that regulates BBC3 proteolysis, we extended our study to determine if CMA plays a pivotal role in TNF and CHX-induced apoptosis. As shown in Figure 5E and F, CMA ablation could sensitize HCT116 cells to TNF and CHX-induced apoptosis; about 30% of the LAMP2A-depleted or the HSPA8-depleted cells underwent apoptosis after 5-h exposure to TNF and CHX, while only 22% of the control cells were apoptotic. Knockdown BBC3 significantly rescued cell death in CMA defective cells treated with TNF and CHX. These data demonstrate that inactivation of CMA can sensitize cells to TNF-triggered apoptosis via BBC3. Importantly, these results reveal a previously unrecognized proapoptotic role of IKBKB in TNF-induced apoptosis via direct, phosphorylation-mediated stabilization of BBC3.
Figure 5.
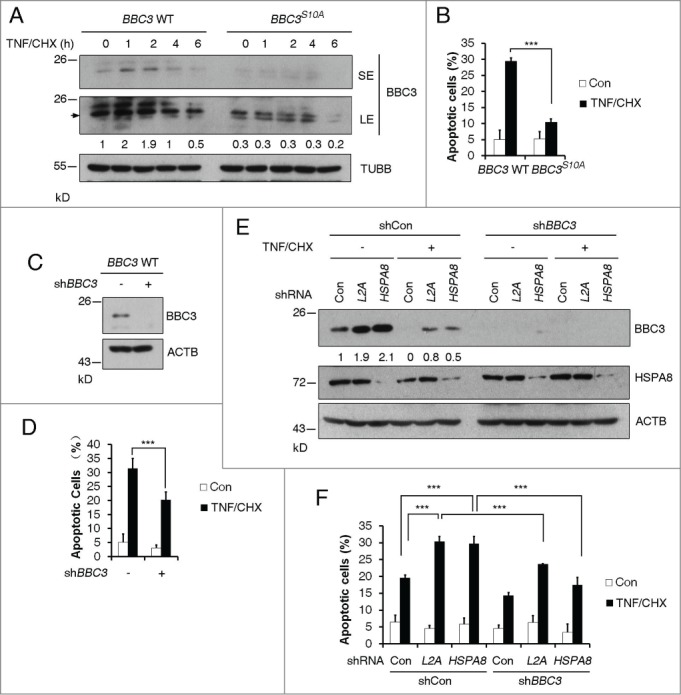
BBC3 stabilization resulting from Ser10 phosphorylation or CMA blockage is essential for TNF-triggered apoptosis. (A) Representative western blots (n=3) of BBC3 WT and BBC3S10A HCT116 cells cotreated with 10 ng/ml TNF and 2.5 μg/ml CHX for the indicated time periods. BBC3 is indicated with an arrow. SE, short exposure; LE, long exposure. Quantification of BBC3 protein levels was done relative to TUBB. (B) FACS analysis data detecting early apoptotic cells showing that BBC3S10A cells confer resistance to TNF-triggered apoptosis under TNF and CHX cotreatment for 7 h. (C) Western blots showing shRNA knockdown efficiency of BBC3 in BBC3 WT HCT116 cells. (D) FACS analysis of early apoptotic cells in (C) showing that BBC3 is required for TNF-induced cytotoxicity under TNF and CHX cotreatment. BBC3 WT cells were infected with vectors containing control or BBC3 shRNA first, and 48 h after infection, cells were cotreated with TNF and CHX for 7 h before being harvested for FACS analysis. (E) Representative immunoblots (n = 3) of BBC3 WT HCT116 cells cotreated with TNF and CHX following infection with vectors containing the indicated shRNAs. BBC3 WT HCT116 cells first infected with vectors containing shCon or BBC3 shRNAs, then with shCon, LAMP2A or HSPA8 shRNAs were cotreated with TNF and CHX for 5 h and then harvested. The BBC3 protein levels were quantified relative to ACTB. (F) FACS analysis of early apoptotic cells in response to the treatment shown in (E) showing that CMA blockage could sensitize TNF-induced apoptosis,which is protected by BBC3 depletion. Statistics are depicted as mean ± SEM; P < 0.001***, n = 4, t test.
Discussion
The fundamental function of CMA is believed to sense nutrient availability and damaged proteins. Activation of CMA under these conditions can either provide free amino acids to sustain protein synthesis or remove unwanted proteins to maintain proteostasis.32 In this report, we found BBC3 is a bona fide substrate of CMA. Depletion of CMA leads to BBC3 stabilization and reduced cell viability in the presence or absence of genotoxic insults. When cells are exposed to TNF treatment, activated IKBKB phosphorylates BBC3 at Ser10, leading to reduced cytosolic retention of BBC3, thereby blunting its degradation by CMA. We further demonstrated that activation of IKBKB upon TNF exposure exerts a proapoptotic effect in a BBC3-dependent manner (Fig. 6A and B).
Figure 6.
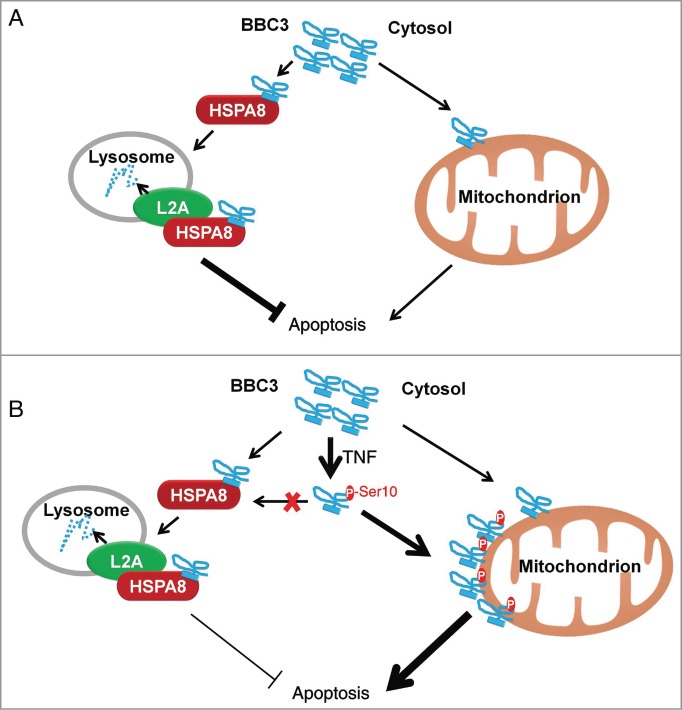
Proposed model of CMA-dependent degradation of BBC3 and phosphorylation-mediated stabilization of BBC3 in response to TNF stimulation. Schematic models under basal conditions (A) and upon TNF treatment (B) are shown.
Regulation of the steady-state of BBC3 appears to be complex, as different groups have reported different modes of regulation. These discrepancies may have resulted from different experimental designs. Two prior reports have found that BBC3 is subject to proteasomal degradation. In both studies, ectopically expressed BBC3 instead of endogenous BBC3 is assessed for its degradation rate in the presence of a proteasome inhibitor.20,21 It is therefore unclear if the same result can be obtained in a physiological setting. These studies raise the possibility that different regulatory mechanisms may exist for the basal and stress-induced BBC3 accumulation.
TNF elicits different cellular responses, ranging from cell survival, proliferation, and paradoxically, apoptosis. NFKB1 activation was shown to direct BBC3 transactivation in the presence of TNF. Our data suggest that the process whereby BBC3 induction is regulated during sustained TNF signaling occurs in 2 stages. The first stage is direct and rapid, activation of IKBKB upon TNF treatment for 30 min leads to BBC3 Ser10 phosphorylation and subsequent inhibition of its degradation by CMA, thereby inducing a transient accumulation of BBC3. The second stage involves the transactivation of BBC3 by NFKB1 after 2-h exposure to TNF, leading to a further upregulation of BBC3 protein. Importantly, the transient phosphorylation of BBC3 at Ser10 is crucial for sustained BBC3 induction after 24-h exposure to TNF. This is supported by the observation that BBC3S10A exhibited compromised induction upon prolonged TNF stimulation. Therefore, our findings establish a causal relationship between BBC3 phosphorylation and protein stabilization. Furthermore, our results uncovered that BBC3 induction is fine-tuned via both post-translational and transcriptional mechanisms in response to TNF signaling.
It is generally accepted that CMA is only induced when needed, but otherwise maintained at a basal level. However, emerging data suggest that even basal levels of CMA are important for maintaining normal cellular homeostasis. Unlike macroautophagy, which upon loss-of-function, can both promote and block cell death in a stress type-dependent manner, activation of CMA is in general prosurvival. Inhibition of CMA reduces cell proliferation and increases cell death in cancer cells.6,7 Consistent with these observations, we found that CMA ablation under basal conditions drastically induced cell death in cancer cells with intact TP53. Upon stress treatment, inhibition of CMA sensitized cells to apoptosis. We provide substantial evidence to support that BBC3 is the crucial downstream effector mediating the proapoptotic process caused by CMA inhibition. These findings suggest that CMA exerts its cytoprotective effect through restraining proapoptotic factors like BBC3. We have previously demonstrated that BBC3 is required for cytokine withdrawal-induced apoptosis.17 When cells are deprived of growth factors or nutrients, CMA is activated to allow adaptation to low nutrient conditions, therefore promoting cell survival. This provides the most rational explanation for CMA-dependent degradation of BBC3, given that BBC3 functions as a crucial proapoptotic factor when nutrients are scarce. Additionally, the proapoptotic activity of BBC3 is essential for initiating apoptosis in response to a wide range of stress stimuli, including hypoxia,33 endoplasmic reticulum (ER) stress,34 and mitochondrial perturbation,35 therefore, it remains to be elucidated if CMA-mediated BBC3 degradation plays a role in a broad spectrum of pathophysiological situations.
Materials and Methods
Cell culture
Human cancer cell lines (H1299, HCT116, HT29, HeLa), HEK293, and HEK293T cells were purchased from American Type Culture Collection (CRL-5803™, CCL−247™, HB-8247™, CCL−2™, CRL-1573™, CRL-3216™). Cells were cultured in McCoy 5A (Sigma, M4892) or DMEM (GIBCO, 12800-017) medium supplemented with 10% fetal bovine serum (Gemini, 900-108). To deprive cells of serum, plates were extensively washed with D-PBS (Hyclone, SH30256.01B) and fresh medium without serum was added.
Antibodies and reagents
The antibodies and reagents used were as follows: Polyclonal rabbit antibody against phospho-Ser10 of BBC3 (pS10) were raised by immunizing rabbits with the peptide ARARQEGS(pS)PEPVEGLC (residues 2 to 18 of human BBC3-α, bcl−2-binding component 3 isoform 4), BBC3 (Santa Cruz Biotechnology, sc-19187, sc-374223, sc-28226), TP53 (DO-1; Santa Cruz Biotechnology, sc-126), NFKBIA (Santa Cruz Biotechnology, sc-371), RELA (Santa Cruz Biotechnology, sc-372), HA (F7; Santa Cruz Biotechnology, sc-7392), HA (3F10; Roche, 11867423001), LAMP2A (Abcam, ab18528), HSPA8 (Abcam, ab19136), MYC (9B11; Cell Signaling Technology, 2276), CHUK (Cell Signaling Technology, 2682), IKBKB (Cell Signaling Technology, 2678), and GST (Cell Signaling Technology, 2622), CDKN1A (BD Biosciences, 550827), COX4I1/COXIV (Molecular Probes, A21347), BBC3 (ProScience, 3043), ACTB/β-ACTIN (Sigma, A1978), TUBB/β-TUBULIN (Sigma, T4026), MG132 (Calbiochem, 474790), cycloheximide (Calbiochem, 239763), TNF (Peprotech, 300-01B), chloroquine (Sigma, C6628), ammonium chloride (Sigma, A9434), doxorubicin (Sigma, D1515), actinomycin D (Sigma, A9415), Ni-Sepharose (GE Healthcare, 17-5268-02), and protein G sepharose (GE Healthcare, 17-0618-02). Note: different anti-BBC3 antibodies may give different background signals when applied for western blotting.
Lentivirus-mediated shRNA
The shRNA hairpin (See Table 1 for shRNA sequences) was inserted into pLV-H1-EF1α-Puromycin lentiviral vectors and lentiviral particles were generated as described.36
Table 1.
shRNA hairpin sequences
| Target gene | shRNA hairpin sequence |
|---|---|
| Control | GCA AAG AAG GCC ACT ACT ATA |
| LAMP2A | GAC TGC AGT GCA GAT GAC G |
| HSPA8 | GCT GGT CTC AAT GTA CTT AGA |
| IKBKB | GCC TGG AGA TCC AGA TCA TGA |
| BBC3 | GCA AAT GAG CCA AAC GTG A |
| RELA | GCC CTA TCC CTT TAC GTC A |
Construction of BBC3 knock-in HCT116 cells
The approach for generating targeted cells with adeno-associated virus (AAV) was as previously described.37 Targeting vectors were constructed to introduce the BBC3S10A or BBC3S10D allele in HCT116 cells using the pSEPT rAAV shuttle vector.38 Homology arms for the targeting vector were PCR-amplified from HCT116 genomic DNA using LA Taq DNA polymerase (TAKARA, DRR002B). The S10A or S10D mutation was introduced in the targeting construct by mutagenesis (Quickchange II site-directed mutagenesis kit; Stratagene, 200523). An infectious rAAV stock bearing the targeting sequence was generated and applied to HCT116 cells as described.39 Stable HCT116 cells were obtained by 0.6 mg/ml G418 selection. The resultant resistant clones were screened with primers derived from the neomycin resistance gene (5′-CCGAGCGTGCGATATGTGTG-3′, 5′-CTGCGTGTGCTGGGGTGTG-3′) and the upstream region of the left homologous arm (5′-CGGAGAACCTGCGTGCAATC-3′, 5′-GCCCAGTCATAGCCGAATAGCC-3′), followed by further screening for homologous recombination by genomic DNA PCR with primers derived from the neomycin resistance gene (5′-TCGCCTTCTTGACGAGTTCT-3′, 5′-TCGCCTTCTATCGCCTTCTT-3′) and the downstream region of the right homologous arm (5′-AGCAAGAAGTACCTGGGGAC-3′, 5′-CATGTGACAGGCAGGGAAAC-3′). Positive PCR fragments were sequenced to confirm knock-in mutation. Next, excision of the selectable element was carried out with Cre recombinase and verified by PCR. After 2 rounds of knock-in, the resultant clones were then screened for homologous recombination of both alleles by genomic PCR with primers derived from the downstream region of the left homologous arm (5′-TGGAGAAGAGTGGAGGTGTG-3′) and the upstream region of the right homologous arm (5′-TACACACACCTGCTAGACCC-3′, 5′-TCGCTGTGTTGGGAAAGTTG-3′).
Immunofluorescence staining
H1299 cells were transfected with BBC3-myc. At 48 h after transfection, cells were untreated or treated with the indicated lysosome inhibitors for 6 h. Cells were then fixed in 4% paraformaldehyde (Sigma, P6148), permeabilized with 0.2% Triton X-100 (Sigma, T8787), blocked (5% bovine serum albumin [Amresco, 9048-46-8] in PBS), and then incubated with the indicated primary antibodies. Detection was performed with corresponding fluorescent-conjugated secondary antibodies. Confocal fluorescence images were obtained with a confocal microscope (LSM 780 NLO; Carl Zeiss, Germany). The microscope images were taken using the LSM 780 confocal laser-scanning microscope system equipped with a 63× or 100×/NA 1.40 oil immersion objective lens.
Flow cytometry analysis
For cell death analysis, cells were harvested and fixed in 95% ethanol, and then stained with 20 μg/mL propidium iodide (PI; Sigma, P4170) and 200 μg/mL RNase A (Sigma, R6513) for 30 min at 37°C. Cells were then analyzed by flow cytometry using an LSRFortessa™ cell analyzer (BD Biosciences, USA). For early cell apoptosis assay, staining of cells with ANXA5/annexin V-FITC and PI was performed following the manufacturer's recommended protocol (BD PharMingen™, 556570). ANXA5-positive and PI-negative cells were detected according to standard protocol.
Quantitative real-time PCR
Total RNA was extracted with TRIzol (Invitrogen, 15596-018). CDNA was prepared using cDNA reverse transcription kit (Invitrogen, 4368814). Quantitative RT-PCR was performed on a StepOnePlus system (Applied Biosystems, Singopore) using SYBR Universal Master Mix (Applied Biosystems, 4472919). All reactions were performed in triplicate. Primer sequences were as follows:
- BBC3:
- 5′-AGAGGGAGGAGTCTGGGAGTG-3′, 5′-GCAGCGCATATACAGTATCTTACAGG-3′;
- TBP:
- 5′-GCACAGGAGCCAAGAGTGAA-3′,
- 5′- TCACAGCTCCCCACCATATT-3′.
Isolation of lysosomes
Lysosomes were isolated by following previously described procedures.9 Male Wistar rats (200 to 250 g) were fasted for 20 h before sacrifice. Livers were removed, washed with cold PBS, and homogenized in the extraction buffer (Sigma, lysosomal isolation kit, LYSISO1-1KT). After separation by density gradient centrifugation (150,000 × g for 4 h), lysosomes were isolated from lysosomal fraction and tested for LAMP2A levels by immunoblotting. The intactness of the lysosomes was assessed using the dye Neutral Red (Sigma, N2537).
Lysosome binding and uptake assay
Both lysosome binding and uptake assays were carried out as described previously.1 For binding assay, isolated lysosomes were coincubated with purified BBC3-myc fusion proteins for 20 min at 37°C and washed 2 times with MOPS buffer (10 mM 3-[N-morpholino] propane sulfonic acid, pH 7.3, 0.3 M sucrose [Amresco, 57-50-1]). For uptake assay, isolated lysosomes were treated with a cocktail of protease inhibitors (Roche, 11873580001) for 10 min on ice and incubated with the purified BBC3-myc fusion proteins for 20 min at 37°C in MOPS buffer.
Mitochondria fractionation
Mitochondria fractionation was performed according to previously described.40 Cells were resuspended in hypotonic buffer A (250 mM sucrose, 20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1×protease inhibitors) on ice for 30 min. Cells were disrupted by passing through 26-gauge needles 30 times, and then 30-gauge needles 20 times. Cell lysates were centrifuged at 950 g for 10 min at 4°C. Part of the supernatant fraction was saved as the PNS (postnuclear supernatant) fraction. The rest of the supernatant fraction was centrifuged at 10,000 g for 20 min at 4°C. The pellet was saved as the Mito (mitochondria) fraction and lysed in RIPA buffer for western blotting. The supernatant fractions was saved as the Cytos (Cytosol) fraction.
Immunoblotting and densitometry
Whole-cell lysates were prepared with Triton lysis buffer (1% Triton X-100 [Sigma, T9284], 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 10 mM sodium fluoride, 40 mM β-glycerophosphate, 1 mM sodium orthovanadate) or modified RIPA lysis buffer (0.1% SDS [Amresco, M-107], 1% sodium deoxycholate [Amresco, 302-95-4], 1% Triton X-100, 150 mM NaCl, 2 mM EDTA, 10 mM sodium fluoride, 40 mM β-glycerophosphate, 1 mM sodium orthovanadate, 50 mM Tris-HCl, pH 8.0) supplemented with 1 mM PMSF, a Complete Protease Inhibitor tablet (Roche, 11873580001) and a phosphatase inhibitor tablet (Roche, 5650615001). Cell lysates were subjected to SDS-PAGE. Densitometric quantification of the immunoblotted membranes was performed with Quantity One software (Bio-Rad). All the immunoblots shown are representative of at least 3 independent experiments.
Statistical analysis
The statistical significance of the difference between experimental groups was determined by 2-tailed unpaired Student t test. For Fig. S4E and S4F, one-way ANOVA was used for comparison of changes in the same group; 2-way ANOVA for comparison of magnitude of changes between different groups. (***, P < 0.001; **, ##, P < 0.01; *, P < 0.05 were considered significant).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Drs. Shengcai Lin and Yong Li for critical reading of the manuscript, Dr. Bert Vogelstein for providing the pSEPT-Neo vector.
Funding
This work is supported by National Basic Research Program of China 973 Program Grant 2015CB553802 (to H.Y.), National Natural Science Foundation of China Grant 31300627 (to L.Z.), Fundamental Research Funds for the Central Universities Grant 20720150066 (to H.Y.), by the National Science Foundation of China for Fostering Talents in Basic Research (Grant No. J1310027), and Ministry of Education of China 111 Project B06016.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004; 305:1292-5; PMID:15333840; http://dx.doi.org/ 10.1126/science.1101738 [DOI] [PubMed] [Google Scholar]
- 2.Kabuta T, Furuta A, Aoki S, Furuta K, Wada K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem 2008; 283:23731-8; PMID:18550537; http://dx.doi.org/ 10.1074/jbc.M801918200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, et al.. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 2013; 16:394-406; PMID:23455607; http://dx.doi.org/ 10.1038/nn.3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 2009; 18:4153-70; PMID:19654187; http://dx.doi.org/ 10.1093/hmg/ddp367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, Mao Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009; 323:124-7; PMID:19119233; http://dx.doi.org/ 10.1126/science.1166088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med 2011; 3:109ra17; PMID:22089453; http://dx.doi.org/ 10.1126/scitranslmed.3003182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, et al.. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 2011; 42:719-30; PMID:21700219; http://dx.doi.org/ 10.1016/j.molcel.2011.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, Pan L, Ince TA, Kroemer G, Brugge JS, et al.. Chaperone-mediated autophagy degrades mutant p53. Genes Dev 2013; 27:1718-30; PMID:23913924; http://dx.doi.org/ 10.1101/gad.220897.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuervo AM, Knecht E, Terlecky SR, Dice JF. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol 1995; 269:C1200-8; PMID:7491910 [DOI] [PubMed] [Google Scholar]
- 10.Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 2004; 15:4829-40; PMID:15331765; http://dx.doi.org/ 10.1091/mbc.E04-06-0477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dice JF. Chaperone-mediated autophagy. Autophagy 2007; 3:295-9; PMID:17404494; http://dx.doi.org/ 10.4161/auto.4144 [DOI] [PubMed] [Google Scholar]
- 12.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996; 273:501-3; PMID:8662539; http://dx.doi.org/ 10.1126/science.273.5274.501 [DOI] [PubMed] [Google Scholar]
- 13.Cuervo AM, Dice JF. Regulation of lamp2a levels in the lysosomal membrane. Traffic 2000; 1:570-83; PMID:11208145; http://dx.doi.org/ 10.1034/j.1600-0854.2000.010707.x [DOI] [PubMed] [Google Scholar]
- 14.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins BBC3 and noxa. Science 2003; 302:1036-8; PMID:14500851; http://dx.doi.org/ 10.1126/science.1090072 [DOI] [PubMed] [Google Scholar]
- 15.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. BBC3 induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7:673-82; PMID:11463391; http://dx.doi.org/ 10.1016/S1097-2765(01)00213-1 [DOI] [PubMed] [Google Scholar]
- 16.Nakano K, Vousden KH. BBC3, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7:683-94; PMID:11463392; http://dx.doi.org/ 10.1016/S1097-2765(01)00214-3 [DOI] [PubMed] [Google Scholar]
- 17.You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A, Mak TW. FOXO3a-dependent regulation of BBC3 in response to cytokine/growth factor withdrawal. J Exp Med 2006; 203:1657-63; PMID:16801400; http://dx.doi.org/ 10.1084/jem.20060353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang P, Qiu W, Dudgeon C, Liu H, Huang C, Zambetti GP, Yu J, Zhang L. BBC3 is directly activated by NF-kappaB and contributes to TNF-α-induced apoptosis. Cell Death Differ 2009; 16:1192-202; PMID:19444283; http://dx.doi.org/ 10.1038/cdd.2009.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SH, Jung YS, Chung JY, Oh AY, Lee SJ, Choi DH, Jang SM, Jang KS, Paik SS, Ha NC, et al.. Novel tumor suppressive function of Smad4 in serum starvation-induced cell death through PAK1-BBC3 pathway. Cell Death Dis 2011; 2:e235; PMID:22130069; http://dx.doi.org/ 10.1038/cddis.2011.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fricker M, O'Prey J, Tolkovsky AM, Ryan KM. Phosphorylation of BBC3 modulates its apoptotic function by regulating protein stability. Cell Death Dis 2010; 1:e59; PMID:21364664; http://dx.doi.org/ 10.1038/cddis.2010.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandow JJ, Jabbour AM, Condina MR, Daunt CP, Stomski FC, Green BD, Riffkin CD, Hoffmann P, Guthridge MA, Silke J, et al.. Cytokine receptor signaling activates an IKK-dependent phosphorylation of BBC3 to prevent cell death. Cell Death Differ 2012; 19:633-41; PMID:21997190; http://dx.doi.org/ 10.1038/cdd.2011.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Callus BA, Moujallad DM, Silke J, Gerl R, Jabbour AM, Ekert PG, Vaux DL. Triggering of apoptosis by BBC3 is determined by the threshold set by prosurvival Bcl−2 family proteins. J Mol Biol 2008; 384:313-23; PMID:18835564; http://dx.doi.org/ 10.1016/j.jmb.2008.09.041 [DOI] [PubMed] [Google Scholar]
- 23.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/ 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol 2004; 14:70-7; PMID:15102438; http://dx.doi.org/ 10.1016/j.tcb.2003.12.002 [DOI] [PubMed] [Google Scholar]
- 25.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990-5; PMID:15528435; http://dx.doi.org/ 10.1126/science.1099993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ming L, Sakaida T, Yue W, Jha A, Zhang L, Yu J. Sp1 and p73 activate BBC3 following serum starvation. Carcinogenesis 2008; 29:1878-84; PMID:18579560; http://dx.doi.org/ 10.1093/carcin/bgn150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuervo AM. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab 2010; 21:142-50; PMID:19857975; http://dx.doi.org/ 10.1016/j.tem.2009.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dice JF, Terlecky SR, Chiang HL, Olson TS, Isenman LD, Short-Russell SR, Freundlieb S, Terlecky LJ. A selective pathway for degradation of cytosolic proteins by lysosomes. Sem Cell Biol 1990; 1:449-55; PMID:2103896 [PubMed] [Google Scholar]
- 29.Essmann F, Pohlmann S, Gillissen B, Daniel PT, Schulze-Osthoff K, Janicke RU. Irradiation-induced translocation of p53 to mitochondria in the absence of apoptosis. J Biol Chem 2005; 280:37169-77; PMID:16148012; http://dx.doi.org/ 10.1074/jbc.M502052200 [DOI] [PubMed] [Google Scholar]
- 30.Yan J, Xiang J, Lin Y, Ma J, Zhang J, Zhang H, Sun J, Danial NN, Liu J, Lin A. Inactivation of BAD by IKK inhibits TNFalpha-induced apoptosis independently of NF-kappaB activation. Cell 2013; 152:304-15; PMID:23332762; http://dx.doi.org/ 10.1016/j.cell.2012.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol 2001; 21:3964-73; PMID:11359904; http://dx.doi.org/ 10.1128/MCB.21.12.3964-3973.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 2012; 22:407-17; PMID:22748206; http://dx.doi.org/ 10.1016/j.tcb.2012.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. BBC3 mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci USA 2003; 100:1931-6; PMID:12574499; http://dx.doi.org/ 10.1073/pnas.2627984100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (BBC3) and NOXA by p53. J Biol Chem 2006; 281:7260-70; PMID:16407291; http://dx.doi.org/ 10.1074/jbc.M509868200 [DOI] [PubMed] [Google Scholar]
- 35.Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH. BBC3- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ 2009; 16:1135-45; PMID:19300452; http://dx.doi.org/ 10.1038/cdd.2009.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L, Mei Y, Fu NY, Guan L, Xie W, Liu HH, Yu CD, Yin Z, Yu VC, You H. TRIM39 regulates cell cycle progression and DNA damage responses via stabilizing p21. Proc Natl Acad Sci USA 2012; 109:20937-42; PMID:23213251; http://dx.doi.org/ 10.1073/pnas.1214156110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rago C, Vogelstein B, Bunz F. Genetic knockouts and knockins in human somatic cells. Nature Protocols 2007; 2:2734-46; PMID:18007609; http://dx.doi.org/ 10.1038/nprot.2007.408 [DOI] [PubMed] [Google Scholar]
- 38.Topaloglu O, Hurley PJ, Yildirim O, Civin CI, Bunz F. Improved methods for the generation of human gene knockout and knockin cell lines. Nucleic Acids Res 2005; 33:e158; PMID:16214806; http://dx.doi.org/ 10.1093/nar/gni160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohli M, Rago C. An efficient method for generating human somatic cell gene knockouts. Discov Med 2004; 4:37-40; PMID:20705018 [PubMed] [Google Scholar]
- 40.Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 2003; 162:59-69; PMID:12847083; http://dx.doi.org/ 10.1083/jcb.200302084 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.