

Abstract
The remarkable metabolic differences between cancer cells and normal cells result in the potential for targeted cancer therapy. The upregulation of glutaminolysis provides energetic advantages to cancer cells. The recently described link between glutaminolysis and autophagy, mediated by MTORC1, may constitute an attractive target for therapeutic strategies. A combination of therapies targeting simultane-ously cell signaling, cancer metabolism, and autophagy can solve therapy resistance and tumor relapse problems, commonly observed in patients treated with most of the current targeted therapies. In this review we summarize the mechanistic link between glutaminolysis and autophagy, and discuss the impacts of these processes on cancer progression and the potential for therapeutic intervention.
Keywords: autophagy, cancer, glutaminolysis, MTOR, prolyl hydroxylases, ROS, α-ketoglutarate
Abbreviations
- αKG
α-ketoglutarate
- AKT1/PKB
v-akt murine thymoma viral oncogene homolog 1
- AMPK
5′-AMP-activated protein kinase
- ATG
autophagy related
- ATM
ATM serine/threonine kinase
- BCL2
B-cell CLL/lymphoma 2
- BECN1
Beclin 1, autophagy related
- BNIP3
BCL2/adenovirus E1B 19kDa interacting protein 3
- BPTES
bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulphide
- DDR
DNA damage response
- DON
6-diazo-5-oxo-L-norleucine
- EGCG
epigallocatechin gallate
- FAN1/FANCD2
FANCD2/FANCI-associated nuclease 1
- FOXO3
forkhead box O3
- GLS/GLS1/KGA
glutaminase
- GLS2/LGA
glutaminase 2 (liver mitochondrial)
- GLUD1
glutamate dehydrogenase 1
- GLUL
glutamate-ammonia ligase
- GSH
glutathione
- MAPK8/JNK1
mitogen-activated protein kinase 8
- MAP1LC3
microtubule-associated protein 1 light chain 3
- MTOR
mechanistic target of rapamycin
- MTORC1
mechanistic target of rapamycin complex 1
- MTORC2
mechanistic target of rapamycin complex 2
- NADPH
nicotinamide adenine dinucleotide phosphate
- SQSTM1/p62
sequestosome 1
- EGLN/PHD
egl-9 family hypoxia-inducible factor
- RB1CC1/FIP200
RB1-inducible coiled-coil 1
- RRAG
Ras-related GTP binding
- RHEB
RAS homolog enriched in brain
- ROS
reactive oxygen species
- SLC1A5
solute carrier family 1 (neutral amino acid transporter) member 5
- SLC7A5
solute carrier family 7 (amino acid transporter light chain, L system), member 5
- SLC3A2
solute carrier family 3 (amino acid transporter heavy chain), member 2
- TCA
tricarboxyic acid
- TFEB
transcription factor EB
- TSC
tuberous sclerosis
- ULK1/2
unc-51 like autophagy activating kinase 1/2.
Introduction
Mammalian cells control their proliferation very tightly to maintain intracellular homeostasis and tissue architecture and function. In contrast, cancer cells exhibit several features that impair this homeostasis, such as uncontrolled proliferation and metabolic reprogramming, among others.1 The high proliferation exhibited by cancer cells requires both the deregulation of proliferative control and a constant supply of nutrients. Thus, tumor cells acquire mutations in genes regulating proliferation, allowing cell growth independently of proliferative cues.2 To satisfy their high demand for nutrients, cancer cells undergo a metabolic reprogramming that stimulates anabolism through numerous metabolic pathways. Those pathways ultimately lead to a high dependency of the cancer cell on specific nutrients.3 Among those nutrients, glutamine has been described as crucial for many types of tumors.4-8 This amino acid is metabolized within the mitochondrion through an enzymatic process termed glutaminolysis, whereby glutamine is converted to α-ketoglutarate (αKG), an intermediate of the tricarboxylic acid (TCA) cycle.9 In highly proliferating cells, citrate produced in the TCA cycle is redirected into the cytosol for the production of NADPH and fatty acids. The production of αKG though glutaminolysis replenishes the TCA cycle, a process called anaplerosis.10,11 However, as we will discuss in this review, anaplerosis is not the only advantage provided by glutaminolysis to proliferating cancer cells.4,10,12
The high proliferation rate of cancer cells, along with the excessive anabolism occurring within the tumor, leads to a limited nutrient accessibility, to a hypoxic context, and to a leak of electrons within the mitochondria of tumor cells.13 Those conditions partially account for the increased levels of reactive oxygen species (ROS) exhibited by cancer cells, which, in turn, induce additional mutations in the DNA, as well as damage in macromolecules and organelles.13 Glutamine and leucine are potent inhibitors of macroautophagy (hereafter referred as autophagy), a key catabolic mechanism that cells use to degrade long-lived proteins and organelles.14 The major downstream effector mediating this inhibition is the mechanistic target of rapamycin complex 1 (MTORC1), a central regulator of cell growth, mRNA translation, autophagy, and metabolism.15-17 The mechanism underlying the detection of amino acids by MTORC1 is not completely understood.18,19 The remarkable rearrangement of the cellular metabolism during malignant transformation frequently correlates with the hyperactivation of MTORC1.20 However, autophagy is not necessarily low in all tumors that display hyperactive MTORC1, and the significance of these events for cancer remains to be fully understood. The mechanistic connection between glutaminolysis and MTORC1, and their effect on autophagy, constitute an attractive source of targets for the development of future therapies against cancer. In this review, we discuss the connection between glutaminolysis and autophagy in cancer progression, and their impact on molecular therapies targeting metabolic transformation.
Glutamine metabolism and malignant transformation
Glutamine is the most abundant free amino acid in the blood and a main physiological source of nitrogen in mammalian cells.4,5 Nevertheless, despite being a nonessential amino acid from a biochemical point of view, glutamine becomes physiologically essential in conditions of high proliferation.21 The increased consumption of glutamine has been linked to the dysregulation of oncogenes and tumor suppressors.22,23 Thus, Wise et al.22 in 2008 described the finding that MYC increases the expression of the cellular transporter of glutamine, and enhances the consumption of glutamine in cancer cells. Through glutaminolysis, glutamine is first deamidated to glutamate, in an irreversible reaction catalyzed by the enzyme GLS (glutaminase). Then, glutamate is further deaminated to αKG by the enzyme GLUD1/GLUD (glutamate dehydrogenase) (Fig. 1).9 GLS exists in 2 isoforms, GLS/GLS1/KGA (the kidney-type) and GLS2/LGA (the liver-type), encoded by the genes GLS and GLS2, respectively.24 GLS, which is the isoform that mainly accounts for the glutaminase activity of tumor cells, is inhibited by glutamate and is distributed ubiquitously.25,26 In contrast, GLS2 is mainly expressed in the liver and cannot be inhibited by glutamate.24,25,27 Additionally, whereas GLS2 is activated by ammonium, GLS is not.28,29 As glutamate levels regulate the activity of GLS, the production of αKG through glutaminolysis also requires an increase in the activity of GLUD1. Importantly, leucine, a key amino acid from a signaling standpoint, is an allosteric activator of GLUD1, and thus, upregulates the production of αKG from glutamate, preventing the inhibition of GLS.30,31 This cooperative effect between glutamine and leucine is also observed at the level of membrane transporters. Glutamine is taken up by the cell through the transporter SLC1A5. Thereafter, glutamine is extruded from the cell by a dimeric bidirectional antiporter termed SLC7A5-SLC3A2, which at the same time introduces leucine inside the cell.32 Thus, glutamine modulates the intracellular levels of leucine, which in turn activates GLUD1, enhancing the production of αKG via glutaminolysis. Mitochondrial αKG can be exported to the cytosol through SLC25A11, the mitochondrial αKG/malate carrier protein, in exchange for cytosolic malate (Fig. 1). Net release of αKG from the mitochondria toward the cytosol occurs when malate recycles in exchange for cytosolic phosphate.33,34
Figure 1.
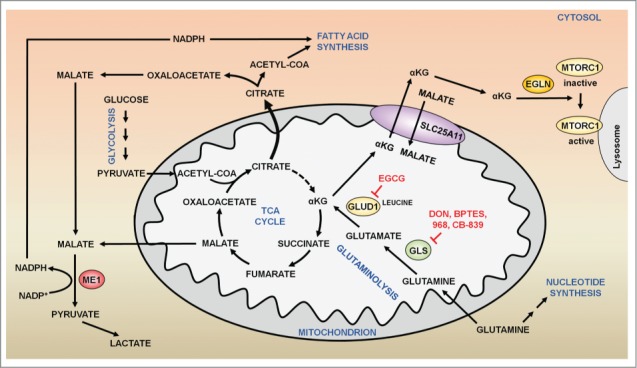
Glutamine metabolism and metabolic transformation. In highly proliferating cells, citrate is diverted away from the TCA cycle for the synthesis of lipids. In the cytoplasm, citrate is converted to acetyl-CoA and oxaloacetate by the enzyme ACLY (ATP citrate lyase). While acetyl-CoA is used for the synthesis of lipids, oxaloacetate is converted to malate. Cytosolic malate is converted to pyruvate and NADPH by ME (malic enzyme). GLS (glutaminase) deamidates glutamine into glutamate. Thereafter, glutamate is further deaminated by the enzyme GLUD1 (glutamate dehydrogenase 1) to yield αKG, which replenishes the TCA cycle. In addition, glutaminolytic αKG can be exported into the cytosol through SLC25A11, the mitochondrial αKG/malate carrier protein, where this metabolite activates EGLNs, which in turn activate MTORC1 to promote cell growth and anabolism. Several inhibitors of glutaminolysis (DON, BPTES) have shown a capacity to reduce MTORC1 activation and cell growth in cancer cells.
Proliferating cancer cells require high quantities of fatty acids and lipids to generate new membranes. To sustain the synthesis of fatty acids, citrate is diverted away from the TCA cycle to synthesize acetyl-CoA and oxaloacetate in the cytosol,35 a process catalyzed by the enzyme ACLY/ATP citrate lyase. Oxaloacetate resulting from this reaction as a co-product is then converted to malate, which subsequently is catalyzed by ME1 (malic enzyme 1, NADP[+]-dependent, cytosolic) for the production of NADPH, necessary for the synthesis of fatty acids (Fig. 1).34,36 As a consequence, the TCA cycle is disrupted, compelling cancer cells to consume alternative nutrients to re-establish the TCA cycle. Thus, as glutamine along with leucine stimulates the production of αKG, the increase in glutaminolysis is a metabolic advantage for tumor cells, reconstituting the TCA cycle (Fig. 1). Nonetheless, cancer cells consume more glutamine than the quantity required to just replenish the TCA cycle.36 Therefore, besides anaplerosis, the excessive consumption of glutamine must provide other advantages to the cancer cell. Indeed, a fraction of the malate produced from glutaminolytic αKG in the TCA cycle translocates into the cytosol, where it is converted to pyruvate by ME1.36 In addition, glutamate produced by GLS is necessary for the synthesis of glutathione (GSH), an intracellular antioxidant that contributes to mitigate the oxidative stress in proliferating cells (Figs. 2, 3).37 Glutamine is also a crucial donor of nitrogen for the synthesis of purines and pyrimidines.38 Finally, as described above, the cooperative effect of glutamine and leucine in the production of αKG converges in the activation of signaling pathways that promote cell growth. Glutaminolytic αKG enhances the GTP loading of RRAG proteins, and thus activates MTORC1 and inhibits autophagy (Figs. 2, 3).39,40 Conversely, the withdrawal of growth factors leads to the activation of FOXO3, which in turn, increases the expression of GLUL (glutamate-ammonia ligase), the enzyme that resynthesizes glutamine from glutamate.41 The increase in glutamine synthesis abrogates the production of αKG from glutaminolysis, and thus inhibits MTORC1 and enhances autophagy.39,42 Through a parallel mechanism, MTORC1 is also activated by the energetic input of glutaminolysis through the TTT-RUVBL1/2 complex, which controls the structural stability of MTORC1.43 In contrast to those observations in mammalian cells, in yeast the synthesis of glutamine rather than glutaminolysis, activates TORC1, following a Gtr1/RRAG-independent mechanism.44,45 Something similar might happen in C. elegans, where αKG addition has been reported to extend life span by inhibiting MTORC1.46 Altogether, this evidence reveals a pivotal role of glutaminolysis in cancer, linking metabolism and the MTORC1 pathway in order to promote cell growth and anabolism. This link between MTORC1 and glutaminolysis seems to operate in both directions. Indeed, MTORC1 enhances glutaminolysis by activating MYC-GLS and GLUD1, establishing a positive feedback loop that may account for the high consumption of glutamine in cancer cells.47,48 Therefore, glutaminolysis and MTORC1 mutually regulate each other in order to promote cell growth and cancer progression. In addition to that, the activation of MTORC1 by glutaminolysis also inhibits autophagy and ATM, a protein involved in the DNA damage response (DDR).40,49-51 Hence, a high rate of glutaminolysis promotes the progression of cancer at early stages, not only stimulating cell growth through the MTORC1 pathway, but also impairing the proper removal of damaged proteins and organelles through the inhibition of autophagy and through ATM-dependent DDR, which eventually would contribute to the initiation of cancer.
Figure 2.
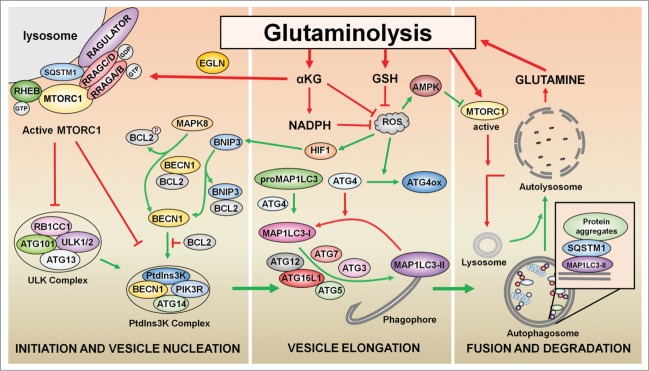
Regulation of autophagy by glutaminolysis. The generation of αKG by glutaminolysis activates EGLNs, which in turn promote MTORC1 activation. MTORC1 inhibits both the phosphatidylinositol 3-kinase (PtdIns3K) complex and ULK complex to prevent the initiation and vesicle nucleation steps of autophagy. The production of GSH, NADPH, and αKG by glutaminolysis limits the production of ROS to counteract the induction of autophagy. The accumulation of ROS induces the oxidation of ATG4, thus preventing the delipidation of the autophagy marker MAP1LC3-II, and activates MAPK8, which results in the dissociation of the BECN1-BCL2 complex. Under hypoxia, HIF1 induction activates BNIP3, which binds to BCL2 to activate autophagy by disrupting the interaction between BECN1 and BCL2. Finally, the reactivation of MTORC1 by glutaminolysis is necessary for lysosome regeneration and autophagy termination. Green arrows indicate processes that result in autophagy activation; red arrows indicate processes that result in autophagy inhibition.
Figure 3.
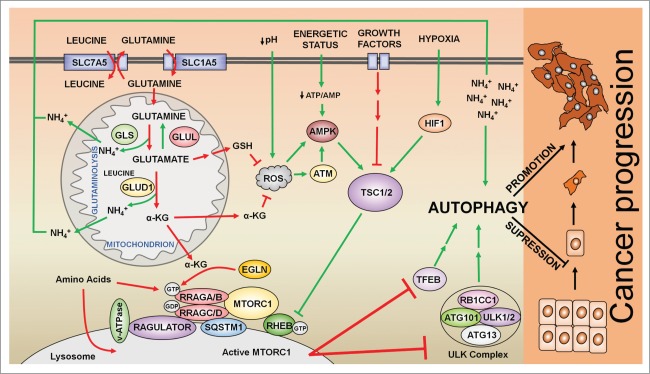
Regulation of autophagy by the MTORC1 signaling pathway in cancer cells. Glutamine is taken up by cells through the transporter SLC1A5. In addition, the antiporter SLC7A5 effluxes glutamine to introduce leucine inside of the cells. Leucine activates GLUD1 (glutamate dehydrogenase 1) allosterically, to promote the production of αKG and the activation of MTORC1 in a EGLN-dependent manner. MTORC1 integrates several inputs, most of them converging in the activation/repression of the TSC1-TSC2 complex, which inhibits RHEB and thus MTORC1. Glutaminolysis activates RRAG proteins, which promote the translocation of MTORC1 to the surface of the lysosome where MTORC1 interacts with its coactivator RHEB. Active MTORC1 induces the phosphorylation of both ULK1 and TEFB to inhibit autophagy. In addition, glutamate, a precursor of GSH (glutathione), along with αKG counteracts ROS levels to inhibit the activation of autophagy mediated by ATM, AMPK, and TSC upstream of MTORC1. Autophagy plays a dual role in cancer: while in early stages autophagy suppresses tumor progression by the removal of damaged cellular components, autophagy promotes the growth and survival of established tumors by providing nutrients and energy at later stages. Green arrows indicate processes that result in autophagy activation; red arrows indicate processes that result in autophagy inhibition.
Regulation and role of autophagy in cancer cells
During autophagy, cellular components are sequestered into vesicles called autophagosomes and then delivered to lysosomes for degradation (Fig. 2). This catabolic process generates both precursor compounds and energy supply for macromolecular synthesis and metabolic needs. Autophagy is orchestrated by several proteins known as ATG (autophagy-related) proteins through a multistep process that includes initiation and vesicle nucleation, vesicle elongation and fusion, and finally, degradation of the autophagosomal content.52-54 Upstream of ATG proteins, autophagy is coordinated by several signaling pathways including those that control tumorigenesis.55,56 Among these pathways, the MTORC1 pathway ensures the regulation of autophagy in response to metabolic and environmental stresses (e.g., nutrients and energy limitation, hypoxia, acidic pH, and oxidative stress) (Fig. 3).57,58 MTORC1 inhibition promotes autophagy by activating the ULK complex, constituted by ULK1/2, ATG13, RB1CC1/FIP200, and ATG101. The active ULK complex phosphorylates BECN1, a key component of the class III phosphatidylinositol 3-kinase complexes (one of which includes PIK3C3, PIK3R4, BECN1 and ATG14), to promote autophagy.59-61 Upstream of MTORC1, the TSC1-TSC2 complex and the AMPK protein coordinate growth factor and energy signaling cascades.62,63 In a parallel mechanism, MTORC1 also inhibits autophagy through the phosphorylation and the cytosolic retention of the transcriptional factor TFEB, which controls the expression of genes involved in the execution of autophagy.64 Although MTORC1 inhibition during nutrient restriction activates autophagy, the reactivation of MTORC1 by recycled nutrients resulting from autophagy is necessary for the restoration of lysosomes and for the termination of autophagy (Fig. 2).65
Autophagy is primarily a prosurvival adaptive response that enables cells to tolerate unfavorable conditions, including those to which cancer cells are exposed.16,55,66-70 In response to metabolic stress, caused by limited nutrient and oxygen, autophagy is induced resulting in the degradation of some cellular components and their recycling. This provides nutrients and energy necessary to sustain metabolism; in the case of cancer cells, this ensures tumor growth and survival. In contrast, by virtue of its ability to selectively degrade damaged cellular components, autophagy has been proposed to limit the accumulation of harmful components such as damaged DNA or ROS, thereby preventing tumor initiation. Thus, autophagy plays a dual role in cancer, either suppressing or supporting tumorigenesis, depending on the stage and context of the cancer.
The evidence that autophagy can prevent tumor formation comes from mouse studies in which autophagy is impaired by genetic manipulation of Atg genes. Mice with allelic loss of the essential autophagic gene Becn1, with tissue specific knockout of Atg7, or with mosaic deletion of Atg5 have increased susceptibility to development of tumors, in particular those of the liver, relative to wild-type mice.71-73 In contrast, in advanced stages of tumorigenesis, autophagy may contribute to tumor progression by providing nutrients that allow cell survival under stressful conditions (e.g., oncogene activation, changes in metabolism, hypoxia, ROS accumulation, and acidic pH).66,67,69,74 Thus, RRAS-induced transformation and tumorigenesis requires autophagy to sustain tumor metabolism and growth.75 Similarly, BRAFV600E-lung driven tumors become addicted to autophagy to sustain mitochondrial glutamine metabolism and tumor growth.76 Furthermore, the deletion of Atg5 and Atg7 causes benign liver adenomas that do not progress to hepatocellular carcinoma, suggesting that autophagy is required for tumor progression into more aggressive stages.73 Moreover, the expression of the core autophagy gene MAP1LC3 (a marker of the autophagy process) is increased in samples of aggressive tumors and correlates with the risk of metastatic disease and with a poor patient outcome.77,78 Autophagy promotes metastasis by limiting detachment-induced cell death (anoikis) during extracellular matrix detachment of cancer cells.79 Autophagy also contributes to the survival of dormant disseminated tumor cells for extremely prolonged periods.80 However, although allelic loss of BECN1 is found in some tumors,71 the complete deletion of BECN1 has not been observed, which suggests that BECN1 is necessary for tumorigenesis and for the maintenance of the malignant state.81
Mechanistic link between glutaminolysis and autophagy
Mortimore and Schworer in 1977 provided the first evidence that amino acids regulate autophagy, observing that amino acid deprivation induces the accumulation of autophagosomes in perfused rat liver.82 Thereafter, Blommaart et al.83 in 1995 showed that the effect of amino acids on autophagy is mediated by MTOR (mechanistic target of rapamycin). MTOR is an atypical serine/threonine kinase that integrates several stimuli to regulate metabolic and signaling pathways.17,84 MTOR exists as 2 structurally and functionally different complexes, termed MTORC1 and MTORC2.19,84 Whereas the activation of MTORC2 is modulated mainly by growth factors, MTORC1 integrates different input cues such as growth factors, energetic status of the cell, oxygen and nutrients. Most of the upstream inputs that signal toward MTORC1 are integrated by the TSC complex, which ultimately regulates RHEB activation upstream of MTORC1 (Fig. 3). In contrast, amino acids activate MTORC1 via another family of small GTPases known as RRAG. Amino acid addition activates RRAG and promotes the translocation of MTORC1 to the surface of the lysosome, a process in which SQSTM1/p62, a protein involved in autophagy as well as other processes, also participates.85,86 Once at the surface of the lysosome, MTORC1 is activated through its direct interaction with the coactivator RHEB (Fig. 3).19,84
Although the mechanism by which MTORC1 senses amino acids is complex and not completely understood,18,19 MTORC1 can detect the presence of glutamine and leucine through glutaminolysis.12,40,87 Thus, the production of αKG through glutaminolysis activates MTORC1 and hence, inhibits autophagy. The activation of MTORC1 exerted by αKG occurs via an increase in the GTP loading of RRAGB (a member of the RRAG family), which permits the translocation of MTORC1 to the lysosome surface, and its subsequent activation.12 The activity of EGLNs/prolyl hydroxylases is crucial for this αKG-dependent activation of MTORC1. EGLNs are the oxygen sensors of the cell, that require both oxygen and αKG to hydroxylate target proteins (such as hypoxia inducible factors).88 However, in normoxic conditions, when oxygen is not limiting, EGLN activity strictly depends on intracellular αKG levels. Therefore, at a high glutaminolytic rate, increased levels of αKG activate EGLNs, which, in turn, promotes MTORC1 activation and the subsequent inhibition of autophagy. Thus, EGLNs constitute a mechanistic link between αKG production and MTORC1 activation.40 However, the interaction between glutaminolysis and MTORC1/autophagy seems to be more complex. A recent report suggests that αKG activates MTORC1 and inhibits autophagy through a parallel mechanism involving acetyl-CoA synthesis and protein acetylation.89 Furthermore, despite the inhibitory effect of glutaminolysis on autophagy, the by-product of glutaminolysis, ammonium, has a dual role in autophagy, activating this process at low concentrations (2–4 mM), and inhibiting autophagy at higher concentrations.90 This observation, however, differs from previous observations by Seglen et al., who showed that at least in hepatocytes ammonium, known to increase the intralysosomal pH, cannot activate autophagic flux, even at low concentrations.91 Although the mechanism by which ammonium induces autophagy remains largely undescribed, it seems to be independent of MTORC1-ULK.92
Another interesting molecular connection between glutaminolysis and autophagy is related to ROS production. The accumulation of ROS activates autophagy through mechanisms that affect both the core autophagy machinery and the components of signaling pathways that regulate autophagy.93,94 Several ATG proteins are redox sensitive. One well-known example is ATG4: when oxidized by ROS, ATG4 prevents the delipidation of the autophagy marker MAP1LC3, thus leading to sustained autophagy.95 ROS indirectly regulates autophagy by promoting activation of AMPK and MAPK8/JNK1, leading to the inactivation of MTORC1 and the disruption of the BECN1-BCL2 complex, respectively.96–98 ROS may also activate autophagy through the activation of ATM, a kinase that plays a key role in DDR. In turn, active ATM inhibits MTORC1 through the AMPK-TSC-RHEB axis.99 Moreover, under hypoxia, autophagy is induced through a ROS-dependent mechanism. This form of autophagy is activated through 2 distinct mechanisms: the hypoxia-inducible, factor-mediated induction of BNIP3 and BNIP3L proteins, and the activation of AMPK.100,101 Conversely, some oxidative agents inhibit autophagy by inactivating proteins that regulate autophagy, such as the TSC complex.102 Whether the regulation exerted by ROS on autophagy is dependent on the nature or the duration of oxidative stress is still unknown. Interestingly, glutaminolysis intermediates cooperate in the production of the antioxidants GSH and NADPH.10 Furthermore, αKG reacts nonenzymatically with ROS (Figs. 2, 3).103,104 Therefore, in addition to its effects on MTORC1, glutaminolysis inactivates autophagy by counteracting the production of ROS and increasing the levels of GSH, NADPH, and αKG. The link between ROS and glutaminolysis is supported by the observation that the inhibition of either GLUD1 or GLS2 leads to an increase in ROS levels.87,105 As glutamate produced by GLS sustains the synthesis of GSH, the inhibition of GLS decreases the levels of GSH and the ability of cells to counteract ROS.105,106 Similarly, a decrease in levels of GLUD1 augments the levels of ROS, probably due to a decrease in the production of NADPH and αKG.36,87 Hence, glutaminolysis counteracts the constant oxidative stress to which cancer cells are exposed.107 This, together with the advantages provided by glutaminolysis to cancer cell growth (including MTORC1 activation), renders this enzymatic process an interesting therapeutic target.
Despite the fact that MTORC1 activation represses autophagy, autophagy is also upregulated in MTORC1 hyperactive tumors (e.g., TSC1-TSC2 tumors).62 One mechanism proposed to explain this observation relies on the influence of the microenvironment of tumor cells. Indeed, a body of evidence suggests that autophagy is induced in cancer cells by microenvironmental stresses including growth factor withdrawal (through MTORC1 inhibition), hypoxia (through hypoxia inducible factor stabilization), oxidative stress (through ROS), and acidic pH (through AMPK activation and MTORC1 inhibition).69,108,109 The tumor cell microenvironment influences the regulation of autophagy and energy metabolism not only in cancer cells but also in stromal cells. Thus, the accumulation of ROS in the microenvironment of the tumor cell promotes autophagy in surrounding stromal cells, leading to an influx of nutrients (including glutamine) from stromal cells to cancer cells.69 Subsequently, glutamine provided by stromal cells is metabolized by the cancer cells through glutaminolysis. As explained above, ammonium produced as a by-product of glutaminolysis may diffuse into the tumor cell microenvironment and amplify autophagy in stromal cells, facilitating tumor growth by providing metabolic and energy substrates to cancer cells.90 This positive feedback between stromal cells and cancer cells is a vicious circle that enables cancer cells to survive at the expense of stromal cells.90,92 As glutaminolysis on the one hand inhibits autophagy through the activation of MTORC1, and on the other hand stimulates autophagy by producing diffusible ammonium, it remains to be determined how autophagy is switched on or off in response to glutaminolysis in cancer cells. The elucidation of these mechanisms might provide new therapeutic strategies for targeting autophagy and glutaminolysis in tumor cells.
Targeting glutaminolysis and autophagy as an anticancer therapy
Several oncogenes and tumor suppressors regulate the activity of MTORC1, thus the overactivation of this pathway is commonly detected in cancer (80% among all the different types of cancer).110 Therefore, the inhibition of MTORC1 was considered as a potential strategy to treat cancer. However, clinical trials using different analogs of rapamycin, (CCI-779, RAD001, AP23573) have shown only modest effects in patient outcome, with promising results only for a few types of tumors, particularly mantle cell lymphoma, endometrial cancer, and renal cell carcinoma.111,112 Although the unsuccessful results obtained with rapamycin analogs have been explained in part by their inability to inhibit MTORC2, the treatment of lymphoblastic leukemia with PP242 (a dual inhibitor of both MTORC complexes) displays similar effects as rapamycin treatment.113 Therefore, the lack of inhibition of MTORC2 does not seem to explain the limited effect of rapamycin analogs against cancer. Alternatively, the negative feedback loop connecting MTORC1 and IRS1 increases the activation of the AKT/PKB pathway upon MTORC1 inhibition, promoting cell survival. This negative feedback loop might explain partially the lack of effectiveness of rapamycin treatment.114-116 Also, the MKNK1/Mnk-EIF4E and MAPK/ERK-RPS6KA3/RSK2 pathways participate in the resistance to rapamycin analogs.112 Finally, as we will discuss below, MTORC1 inhibition leads to autophagy activation, which permits the survival of cancer cells.
Currently the metabolism of cancer cells has been rekindled as a therapeutic target against cancer. An increasing number of reports are describing the mechanism by which oncogenes and tumor suppressors modulate metabolic pathways which are relevant for cancer cell growth.11 The link established between glutaminolysis and MTORC1 partially accounts for glutamine addiction in cancer cells.12,42,87,117 Therefore, targeting glutaminolysis, which is a process that both activates MTORC1 and provides substrates for the anabolic machinery led by MTORC1, is a rational therapeutic approach to target simultaneously metabolism and cell signaling in tumors. A logical conclusion from this connection is that the inhibition of MTORC1 can be addressed by inhibiting either the enzymes of glutaminolysis (GLS and GLUD1), or the intermediates between glutaminolysis and MTORC1, such as the EGLNs.40 Indeed, the inhibition of glutaminolysis as a cancer therapy has been considered for many years.24 Unfortunately, this strategy has not yet led to successful results. One limitation of targeting GLS as a cancer therapy is that GLS is not only required for metabolism in cancer cells, but also for the development, growth, maintenance, and physiology of normal tissues (e.g., enterocytes, glutamatergic neurons, renal ammonium excretion). Thus, inhibiting glutaminolysis might result in serious complications in the patient. Indeed, early preclinical studies with DON (6-diazo-5-oxy-L-norleucine) and with 2 other glutamine mimetic compounds (azaserine, acivicin) showed limited antitumor effects and severe toxicity (nefrotoxicity, gastrointestinal toxicity and myelosuppression).5,118 In addition, other compounds such as BPTES (an allosteric inhibitor of GLS), and 968 (an inhibitor of Rho GTPase), have been described. Although these compounds exhibit both an increased specificity against GLS isoforms and antitumor effects on several cancer cell lines, their hydrophobic nature has hindered their application in vivo.9,24 In contrast, a recent promising inhibitor of GLS has been described, CB-839, which is currently being tested in clinical trials against several types of tumors.119 The second enzyme of glutaminolysis, GLUD1, is another interesting target for cancer therapies. EGCG, an allosteric inhibitor of GLUD1, has earned attention as a putative therapeutic agent, with significant antitumor effects in preclinical studies.120 Of note, EGCG treatment leads to glucose addiction in vitro, sensitizing the cells to glucose withdrawal. Thus, EGCG treatment might synergize with therapies that target glucose metabolism.121 However, the antitumor mechanism of EGCG has not been clearly related with its ability to inhibit GLUD1.122
Another interesting option to decrease MTORC1 activity could be the inhibition of EGLNs, which links glutaminolysis with MTORC1 activation. Interestingly, several molecules able to inhibit EGLNs are currently being evaluated in clinical trials for the treatment of anemia (FG-2216, FG-4592, GSK1278863A, and AKB-6548).123 As EGLNs are well-known activators of hypoxia inducible factor degradation, an important limitation in the use of EGLN inhibitors in the treatment of cancer would be the activation of the hypoxic response mediated by hypoxia inducible factors.88,124 However, human cells present 3 different isoforms of EGLNs (EGLN1/PHD2, EGLN2/PHD1 and EGLN3/PHD3), and only EGLN1 seems to be physiologically involved in the regulation of hypoxia inducible factor signaling.125 Of note, EGLN3 interacts with SQSTM1/p62, suggesting a direct link with MTORC1 activation.85,88,126 EGLN3 has also been related with apoptosis and tumor suppression.127 Therefore, the selective inhibition of each isoform of the EGLN family is an interesting field to explore potential anticancer therapies. Hence, EGLNs are hubs where several cellular processes converge, constituting a highly interesting target against cancer.
As stated above, autophagy is upregulated in many established cancer cell types.75 Furthermore, autophagy may be activated in tumor cells in response to a variety of anticancer therapies.128-131 Autophagy confers resistance to therapy, and the inhibition of autophagy sensitizes tumor cells to cell death induced by several anticancer treatments.129 In the presence of certain anticancer agents, however, the induction of autophagy results in synergistic antitumor effects.130,131 Thus, autophagy can be considered as a double–edged sword in tumorigenesis and cancer therapy. Given the important functions of autophagy in cancer, a number of clinical trials have investigated the effect of modulation of autophagy for the treatment of tumors.132,133 Thus, the autophagy inhibitor hydroxychloroquine is being evaluated currently in phase I-II trials in combination with anticancer therapies (https//clinicaltrials.gov/).132,133 Although the inhibition of MTORC1 abrogates anabolism and cell growth, it also promotes the activation of autophagy and, as a consequence, the survival of tumor cells. Whereas the inhibition of MTOR and MTOR-activating mechanisms (such as glutaminolysis) presents several limitations as anticancer monotherapies, a combined therapy targeting both MTORC1 and autophagy may improve treatment outcome. Indeed, preclinical and clinical experiments have shown that the combination of MTORC1 and autophagy inhibitors display synergistic effects.132,134,135 Therefore, to prevent cancer cell survival and metastatic disease, the inhibition of either glutaminolysis or EGLNs might require the concomitant inhibition of autophagy.
The inhibition of MTORC1 also sensitizes cancer cells to DNA damaging agents such as melphalan, AraC, etoposide and ionizing radiation.113,136-138 The mechanism that explains this observation is related to the ability of MTORC1 to regulate the expression of FAN1/FANCD2, a protein involved in the activation of ATM. Thus, the inhibition of MTORC1 decreases FAN1, which impairs the activation of ATM and thereby the DDR of cancer cells.113,136,139 Furthermore, AMPK is another link between ATM and MTORC1, as AMPK regulates both ATM and MTORC1.50,51,140-142 Those results highlight the potential synergy of a combined therapy targeting both MTOR and DDR as a better strategy to prevent tumor resistance to chemotherapeutic agents.
Conclusion
The high consumption of glutamine is a well-known metabolic feature that provides several advantages to proliferating cancer cells. Through glutaminolysis, glutamine modulates both the activation of MTORC1 and the inhibition of autophagy. Thus, although the inhibition of glutaminolysis leads to an inhibition of MTORC1, it would also promote the activation of autophagy, which is crucial for the survival of cancer cells. Therefore, the simultaneous inhibition of both glutaminolysis and autophagy may display a synergistic effect that could improve patient outcome with lower toxicity and side effects. This therapeutic strategy could be particularly valuable in patients with tumors that consume high quantities of glutamine. Although several inhibitors of glutaminolysis have been described, the lack of specificity has hindered their path to become antitumor agents. Hence, given the high relevance of glutaminolysis for cancer cells, the rational design of new inhibitors of glutaminolysis, along with improved autophagy inhibitors, are required to provide better therapeutic options against cancer.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Jose Luis Crespo (Seville, Spain) for critically reading the manuscript.
Funding
This work was supported by funds from the following institutions: Institut National de la Santé et de la Recherche Médicale - INSERM, Université de Bordeaux, Fondation pour la Recherche Médicale, Conseil Régional d'Aquitaine, Fondation ARC pour la Recherche sur le Cancer, SIRIC-BRIO, Institut Européen de Chimie et Biologie, Institut Bergonié, Ligue contre le Cancer - Comité de la Gironde, and Institut National du Cancer.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 2.Menon S, Manning BD. Common corruption of the mTOR signaling network in human tumors. Oncogene 2008; 27(Suppl 2):S43-51; PMID:19956179; http://dx.doi.org/ 10.1038/onc.2009.352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol 2012; 30:671-8; PMID:22781696; http://dx.doi.org/ 10.1038/nbt.2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matés JM, Segura JA, Campos-Sandoval JA, Lobo C, Alonso L, Alonso FJ, Márquez J. Glutamine homeostasis and mitochondrial dynamics. Int J Biochem Cell Biol 2009; 41:2051-61; PMID:19703661; http://dx.doi.org/ 10.1016/j.biocel.2009.03.003 [DOI] [PubMed] [Google Scholar]
- 5.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 2010; 35:427-33; PMID:20570523; http://dx.doi.org/ 10.1016/j.tibs.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newsholme EA, Crabtree B, Ardawi MS. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci Rep 1985; 5:393-400; PMID:3896338; http://dx.doi.org/ 10.1007/BF01116556 [DOI] [PubMed] [Google Scholar]
- 7.Kovacevic Z, Morris HP. The Role of Glutamine in the Oxidative Metabolism of Malignant Cells. Cancer Res 1972; 32:326-33; PMID:4400467 [PubMed] [Google Scholar]
- 8.Souba WW. Glutamine and cancer. Ann Surg 1993; 218:715-28; PMID:8257221; http://dx.doi.org/ 10.1097/00000658-199312000-00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013; 4:e532; PMID:23470539; http://dx.doi.org/ 10.1038/cddis.2013.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010; 29:313-24; PMID:19881548; http://dx.doi.org/ 10.1038/onc.2009.358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009; 324:1029-33; PMID:19460998; http://dx.doi.org/ 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durán RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, Hall MN. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 2012; 47:349-58; PMID:22749528; http://dx.doi.org/ 10.1016/j.molcel.2012.05.043 [DOI] [PubMed] [Google Scholar]
- 13.Eng CH, Abraham RT. The autophagy conundrum in cancer: influence of tumorigenic metabolic reprogramming. Oncogene 2011; 30:4687-96; PMID:21666712; http://dx.doi.org/ 10.1038/onc.2011.220 [DOI] [PubMed] [Google Scholar]
- 14.Seglen PO, Gordon PB, Poli A. Amino acid inhibition of the autophagic/lysosomal pathway of protein degradation in isolated rat hepatocytes. Biochim Biophys Acta 1980; 630:103-18; PMID:7388042; http://dx.doi.org/ 10.1016/0304-4165(80)90141-5 [DOI] [PubMed] [Google Scholar]
- 15.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344-8; PMID:21127245; http://dx.doi.org/ 10.1126/science.1193497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/ 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol 2013; 14:133-9; PMID:23361334; http://dx.doi.org/ 10.1038/nrm3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol 2014; 24:1-7; PMID:24373305; http://dx.doi.org/ 10.1016/j.tcb.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yecies JL, Manning BD. Transcriptional control of cellular metabolism by mTOR signaling. Cancer Res 2011; 71:2815-20; PMID:21487041; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medina M, Núñez de Castro I. Glutaminolysis and glycolysis interactions in proliferant cells. Int J Biochem 1990; 22:681-3; PMID:2205518; http://dx.doi.org/ 10.1016/0020-711X(90)90001-J [DOI] [PubMed] [Google Scholar]
- 22.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al.. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008; 105:18782-7; PMID:19033189; http://dx.doi.org/ 10.1073/pnas.0810199105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A 2010; 107:7455-60; PMID:20378837; http://dx.doi.org/ 10.1073/pnas.1001006107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katt WP, Cerione RA. Glutaminase regulation in cancer cells: a druggable chain of events. Drug Discov Today 2014; 19:450-7; PMID:24140288; http://dx.doi.org/ 10.1016/j.drudis.2013.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krebs HA. Metabolism of amino-acids: The synthesis of glutamine from glutamic acid and ammonia, and the enzymic hydrolysis of glutamine in animal tissues. Biochem J 1935; 29:1951-69; PMID:16745865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curthoys NP, Watford M. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr 1995; 15:133-59; PMID:8527215; http://dx.doi.org/ 10.1146/annurev.nu.15.070195.001025 [DOI] [PubMed] [Google Scholar]
- 27.Szeliga M, Obara-Michlewska M. Glutamine in neoplastic cells: focus on the expression and roles of glutaminases. Neurochem Int 2009; 55:71-5; PMID:19428809; http://dx.doi.org/ 10.1016/j.neuint.2009.01.008 [DOI] [PubMed] [Google Scholar]
- 28.Verhoeven AJ, van Iwaarden JF, Joseph SK, Meijer AJ. Control of rat-liver glutaminase by ammonia and pH. Eur J Biochem 1983; 133:241-4; PMID:6852030; http://dx.doi.org/ 10.1111/j.1432-1033.1983.tb07454.x [DOI] [PubMed] [Google Scholar]
- 29.McGivan JD, Bradford NM. Characteristics of the activation of glutaminase by ammonia in sonicated rat liver mitochondria. Biochim Biophys Acta 1983; 759:296-302; PMID:6136301; http://dx.doi.org/ 10.1016/0304-4165(83)90327-6 [DOI] [PubMed] [Google Scholar]
- 30.Li M, Li C, Allen A, Stanley CA, Smith TJ. Glutamate dehydrogenase: structure, allosteric regulation, and role in insulin homeostasis. Neurochem Res 2014; 39:433-45; PMID:24122080; http://dx.doi.org/ 10.1007/s11064-013-1173-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yielding KL, Tomkins GM. An effect of L-leucine and other essential amino acids on the structure and activity of glutamic dehydrogenase. Proc Natl Acad Sci U S A 1961; 47:983-9; PMID:13787322; http://dx.doi.org/ 10.1073/pnas.47.7.983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al.. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009; 136:521-34; PMID:19203585; http://dx.doi.org/ 10.1016/j.cell.2008.11.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch 2004; 447:689-709; PMID:14598172; http://dx.doi.org/ 10.1007/s00424-003-1099-7 [DOI] [PubMed] [Google Scholar]
- 34.Meijer AJ, Van Dam K. The metabolic significance of anion transport in mitochondria. Biochim Biophys Acta 1974; 346:213-44; PMID:4613381; http://dx.doi.org/ 10.1016/0304-4173(74)90001-9 [DOI] [PubMed] [Google Scholar]
- 35.Icard P, Poulain L, Lincet H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim Biophys Acta 2012; 1825:111-6; PMID:22101401 [DOI] [PubMed] [Google Scholar]
- 36.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 2007; 104:19345-50; PMID:18032601; http://dx.doi.org/ 10.1073/pnas.0709747104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matés JM, Pérez-Gómez C, Núñez de Castro I, Asenjo M, Márquez J. Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death. Int J Biochem Cell Biol 2002; 34:439-58; PMID:11906817; http://dx.doi.org/ 10.1016/S1357-2725(01)00143-1 [DOI] [PubMed] [Google Scholar]
- 38.Salzman NP, Eagle H, Sebring ED. The utilization of glutamine, glutamic acid, and ammonia for the biosynthesis of nucleic acid bases in mammalian cell cultures. J Biol Chem 1958; 230:1001-12; PMID:13525416 [PubMed] [Google Scholar]
- 39.Durán RV, Hall MN. Glutaminolysis feeds mTORC1. Cell Cycle 2012; 11:4107-8; PMID:23095634; http://dx.doi.org/ 10.4161/cc.22632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Durán RV, MacKenzie ED, Boulahbel H, Frezza C, Heiserich L, Tardito S, Bussolati O, Rocha S, Hall MN, Gottlieb E. HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene 2013; 32:4549-56; PMID:23085753; http://dx.doi.org/ 10.1038/onc.2012.465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van der Vos KE, Eliasson P, Proikas-Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C, et al.. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat Cell Biol 2012; 14:829-37; PMID:22820375; http://dx.doi.org/ 10.1038/ncb2536 [DOI] [PubMed] [Google Scholar]
- 42.Van der Vos KE, Coffer PJ. Glutamine metabolism links growth factor signaling to the regulation of autophagy. Autophagy 2012; 8:1862-4; PMID:22996802; http://dx.doi.org/ 10.4161/auto.22152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SG, Hoffman GR, Poulogiannis G, Buel GR, Jang YJ, Lee KW, Kim BY, Erikson RL, Cantley LC, Choo AY, et al.. Metabolic Stress Controls mTORC1 Lysosomal Localization and Dimerization by Regulating the TTT-RUVBL1 / 2 Complex. Mol Cell 2013; 49:172-85; PMID:23142078; http://dx.doi.org/ 10.1016/j.molcel.2012.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stracka D, Jozefczuk S, Rudroff F, Sauer U, Hall MN. Nitrogen Source Activates TOR Complex 1 via Glutamine and Independently of Gtr/Rag. J Biol Chem 2014; 289:25010-20; PMID:25063813; http://dx.doi.org/ 10.1074/jbc.M114.574335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crespo JL, Hall MN. Elucidating TOR Signaling and Rapamycin Action: Lessons from Saccharomyces cerevisiae. Microbiol Mol Biol Rev 2002; 66:579-91; PMID:12456783; http://dx.doi.org/ 10.1128/MMBR.66.4.579-591.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, et al.. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014; 510:397-401; PMID:24828042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, et al.. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013; 153:840-54; PMID:23663782; http://dx.doi.org/ 10.1016/j.cell.2013.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Csibi A, Lee G, Yoon SO, Tong H, Ilter D, Elia I, Fendt SM, Roberts TM, Blenis J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol 2014; 24:2274-80; PMID:25220053; http://dx.doi.org/ 10.1016/j.cub.2014.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Shen C, Houghton PJ. The mTOR pathway negatively controls ATM by up-regulating miRNAs. Proc Natl Acad Sci U S A 2013; 110:11869-74; PMID:23818585; http://dx.doi.org/ 10.1073/pnas.1220898110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blagosklonny M V. Molecular damage in cancer: An argument for mtor-driven aging. Aging (Albany NY) 2011; 3:1130-41; PMID:22246147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther 2013; 15:156-69; PMID:24100703; http://dx.doi.org/ 10.4161/cbt.26726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Z, Klionsky DJ. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124-31; PMID:20034776; http://dx.doi.org/ 10.1016/j.ceb.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mizushima N, Yoshimori T, Ohsumi Y. The Role of Atg Proteins in Autophagosome Formation. Annu Rev Cell Dev Biol 2011; 27:107-32; PMID:21801009; http://dx.doi.org/ 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- 54.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15:713-20; PMID:23817233; http://dx.doi.org/ 10.1038/ncb2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lorin S, Hamaï A, Mehrpour M, Codogno P. Autophagy regulation and its role in cancer. Semin Cancer Biol 2013; 23:361-79; PMID:23811268; http://dx.doi.org/ 10.1016/j.semcancer.2013.06.007 [DOI] [PubMed] [Google Scholar]
- 56.Maiuri MC, Kroemer G. Autophagy in stress and disease. Cell Death Differ 2015; 22:365-6; PMID:25661524; http://dx.doi.org/ 10.1038/cdd.2014.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meijer AJ, Codogno P. Nutrient sensing: TOR's Ragtime. Nat Cell Biol 2008; 10:881-3; PMID:18670446; http://dx.doi.org/ 10.1038/ncb0808-881 [DOI] [PubMed] [Google Scholar]
- 58.Dunlop EA, Tee AR. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol 2014; 36:121-9; PMID:25158238; http://dx.doi.org/ 10.1016/j.semcdb.2014.08.006 [DOI] [PubMed] [Google Scholar]
- 59.Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2-11; PMID:22025673; http://dx.doi.org/ 10.1128/MCB.06159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res 2014; 24:42-57; PMID:24343578; http://dx.doi.org/ 10.1038/cr.2013.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim KH, Lee MS. Autophagy-a key player in cellular and body metabolism. Nat Rev Endocrinol 2014; 10:322-37; PMID:24663220; http://dx.doi.org/ 10.1038/nrendo.2014.35 [DOI] [PubMed] [Google Scholar]
- 62.Medvetz D, Priolo C, Henske EP. Therapeutic Targeting of Cellular Metabolism in Cells with Hyperactive mTORC1: A Paradigm Shift. Mol Cancer Res 2015; 13:3-8; PMID:25298408; http://dx.doi.org/ 10.1158/1541-7786.MCR-14-0343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem 2006; 281:34870-9; PMID:16990266; http://dx.doi.org/ 10.1074/jbc.M605488200 [DOI] [PubMed] [Google Scholar]
- 64.Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012; 8:903-14; PMID:22576015; http://dx.doi.org/ 10.4161/auto.19653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu L, Mcphee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al.. Autophagy termination and lysosome reformation regulated by mTOR. Nature 2010; 465:942-6; PMID:20526321; http://dx.doi.org/ 10.1038/nature09076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kroemer G, Mariño G, Levine B. Autophagy and the Integrated Stress Response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401-10; PMID:22534666; http://dx.doi.org/ 10.1038/nrc3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang X, Overholtzer M, Thompson CB. Autophagy in cellular metabolism and cancer. J Clin Invest 2015; 125:47-54; PMID:25654550; http://dx.doi.org/ 10.1172/JCI73942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: Shaping the tumor microenvironment and therapeutic response. Trends Mol Med 2013; 19:428-46; PMID:23714574; http://dx.doi.org/ 10.1016/j.molmed.2013.04.005 [DOI] [PubMed] [Google Scholar]
- 70.Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 2014; 15:81-94; PMID:24401948; http://dx.doi.org/ 10.1038/nrm3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al.. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809-20; PMID:14638851; http://dx.doi.org/ 10.1172/JCI20039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003; 100:15077-82; PMID:14657337; http://dx.doi.org/ 10.1073/pnas.2436255100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 2011; 25:795-800; PMID:21498569; http://dx.doi.org/ 10.1101/gad.2016211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Amaravadi RK, Lippincott-schwartz J, Yin X, Weiss WA, Takebe N, Timmer W, Dipaola RS, Lotze MT. Principles and Current Strategies for Targeting Autophagy for Cancer Treatment. Clin cancer Res 2011; 17:654-66; PMID:21325294; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guo JY, Xia B, White E. Autophagy-Mediated tumor Promotion. Cell 2013; 155:1216-9; PMID:24315093; http://dx.doi.org/ 10.1016/j.cell.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, McMahon M, White E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov 2013; 3:1272-85; PMID:23965987; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res 2012; 18:370-9; PMID:22080440; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ma XH, Piao S, Wang D, Mcafee QW, Nathanson KL, Lum JJ, Li LZ, Amaravadi RK. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res 2011; 17:3478-89; PMID:21325076; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Debnath J. Detachment-induced autophagy during anoikis and lumen formation in epithelial acini. Autophagy 2008; 4:351-3; PMID:18196957; http://dx.doi.org/ 10.4161/auto.5523 [DOI] [PubMed] [Google Scholar]
- 80.Sosa MS, Bragado P, Debnath J, Aguirre-Ghiso JA. Regulation of tumor cell dormancy by tissue microenvironments and autophagy. Adv Exp Med Biol 2013; 734:73-89; PMID:23143976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gong C, Song E, Codogno P, Mehrpour M. The roles of BECN1 and autophagy in cancer are context dependent. Autophagy 2012; 8:1853-5; PMID:22960473; http://dx.doi.org/ 10.4161/auto.21996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mortimore GE, Schworer CM. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature 1977; 270:174-6; PMID:927529; http://dx.doi.org/ 10.1038/270174a0 [DOI] [PubMed] [Google Scholar]
- 83.Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 1995; 270:2320-6; PMID:7836465; http://dx.doi.org/ 10.1074/jbc.270.5.2320 [DOI] [PubMed] [Google Scholar]
- 84.Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol 2014; 15:155-62; PMID:24556838; http://dx.doi.org/ 10.1038/nrm3757 [DOI] [PubMed] [Google Scholar]
- 85.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 2011; 44:134-46; PMID:21981924; http://dx.doi.org/ 10.1016/j.molcel.2011.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008; 320:1496-501; PMID:18497260; http://dx.doi.org/ 10.1126/science.1157535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lorin S, Tol MJ, Bauvy C, Strijland A, Poüs C, Verhoeven AJ, Codogno P, Meijer AJ. Glutamate dehydrogenase contributes to leucine sensing in the regulation of autophagy. Autophagy 2013; 9:850-60; PMID:23575388; http://dx.doi.org/ 10.4161/auto.24083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boulahbel H, Durán RV, Gottlieb E. Prolyl hydroxylases as regulators of cell metabolism. Biochem Soc Trans 2009; 37:291-4; PMID:19143649; http://dx.doi.org/ 10.1042/BST0370291 [DOI] [PubMed] [Google Scholar]
- 89.Mariño G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, et al.. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell 2014; 53:710-25; PMID:24560926; http://dx.doi.org/ 10.1016/j.molcel.2014.01.016 [DOI] [PubMed] [Google Scholar]
- 90.Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal 2010; 3:ra31; PMID:20424262 [DOI] [PubMed] [Google Scholar]
- 91.Seglen PO, Grinde B, Solheim AE. Inhibition of the lysosomal pathway of protein degradation in isolated rat hepatocytes by ammonia, methylamine, chloroquine and leupeptin. Eur J Biochem 1979; 95:215-25; PMID:456353; http://dx.doi.org/ 10.1111/j.1432-1033.1979.tb12956.x [DOI] [PubMed] [Google Scholar]
- 92.Cheong H, Lindsten T, Thompson CB. Autophagy and ammonia. Autophagy 2012; 8:122-3; PMID:22170154; http://dx.doi.org/ 10.4161/auto.8.1.18078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kongara S, Karantza V. The interplay between autophagy and ROS in tumorigenesis. Front Oncol 2012; 2:171; PMID:23181220; http://dx.doi.org/ 10.3389/fonc.2012.00171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li L, Ishdorj G, Gibson SB. Reactive oxygen species regulation of autophagy in cancer: Implications for cancer treatment. Free Radic Biol Med 2012; 53:1399-410; PMID:22820461; http://dx.doi.org/ 10.1016/j.freeradbiomed.2012.07.011 [DOI] [PubMed] [Google Scholar]
- 95.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749-60; PMID:17347651; http://dx.doi.org/ 10.1038/sj.emboj.7601623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li L, Chen Y, Gibson SB. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal 2013; 25:50-65; PMID:23000343; http://dx.doi.org/ 10.1016/j.cellsig.2012.09.020 [DOI] [PubMed] [Google Scholar]
- 97.Wong CH, Iskandar KB, Yadav SK, Hirpara JL, Loh T, Pervaiz S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS One 2010; 5:e9996; PMID:20368806; http://dx.doi.org/ 10.1371/journal.pone.0009996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al.. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007; 26:2527-39; PMID:17446862; http://dx.doi.org/ 10.1038/sj.emboj.7601689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alexander A, Cai SL, Kim J, Nanez A, Sahin M, Maclean KH, Inoki K, Guan KL, Shen J, Person MD, et al.. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci 2012; 109:8352-8352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ 2008; 15:1572-81; PMID:18551130; http://dx.doi.org/ 10.1038/cdd.2008.84 [DOI] [PubMed] [Google Scholar]
- 101.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 2009; 29:2570-81; PMID:19273585; http://dx.doi.org/ 10.1128/MCB.00166-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cao J, Schulte J, Knight A, Leslie NR, Zagozdzon A, Bronson R, Manevich Y, Beeson C, Neumann CA. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J 2009; 28:1505-17; PMID:19369943; http://dx.doi.org/ 10.1038/emboj.2009.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Meijer AJ, Lorin S, Blommaart EF, Codogno P. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids 2014; PMID:24880909; http://dx.doi.org/ 10.1007/s00726-014-1765-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Andrae U, Singh J, Ziegler-Skylakakis K. Pyruvate and related alpha-ketoacids protect mammalian cells in culture against hydrogen peroxide-induced cytotoxicity. Toxicol Lett 1985; 28:93-8; PMID:4071565; http://dx.doi.org/ 10.1016/0378-4274(85)90015-3 [DOI] [PubMed] [Google Scholar]
- 105.Xiang L, Xie G, Liu C, Zhou J, Chen J, Yu S, Li J, Pang X, Shi H, Liang H. Knock-down of glutaminase 2 expression decreases glutathione, NADH, and sensitizes cervical cancer to ionizing radiation. Biochim Biophys Acta 2013; 1833:2996-3005; PMID:23954443; http://dx.doi.org/ 10.1016/j.bbamcr.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 106.Suzuki S, Tanaka T, Poyurovsky M V, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al.. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A 2010; 107:7461-6; PMID:20351271; http://dx.doi.org/ 10.1073/pnas.1002459107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 2009; 8:579-91; PMID:19478820; http://dx.doi.org/ 10.1038/nrd2803 [DOI] [PubMed] [Google Scholar]
- 108.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Sotgia F, et al.. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal 2012; 16:1264-84; PMID:21883043; http://dx.doi.org/ 10.1089/ars.2011.4243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Marino ML, Pellegrini P, Di Lernia G, Djavaheri-Mergny M, Brnjic S, Zhang X, Hägg M, Linder S, Fais S, Codogno P, et al.. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem 2012; 287:30664-76; PMID:22761435; http://dx.doi.org/ 10.1074/jbc.M112.339127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Howell JJ, Ricoult SJ, Ben-Sahra I, Manning BD. A growing role for mTOR in promoting anabolic metabolism. Biochem Soc Trans 2013; 41:906-12; PMID:23863154; http://dx.doi.org/ 10.1042/BST20130041 [DOI] [PubMed] [Google Scholar]
- 111.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell 2007; 12:9-22; PMID:17613433; http://dx.doi.org/ 10.1016/j.ccr.2007.05.008 [DOI] [PubMed] [Google Scholar]
- 112.Sun SY. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett 2013; 340:1-8; PMID:23792225; http://dx.doi.org/ 10.1016/j.canlet.2013.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guo F, Li J, Zhang S, Du W, Amarachintha S, Sipple J, Phelan J, Grimes HL, Zheng Y, Pang Q. mTOR kinase inhibitor sensitizes T-cell lymphoblastic leukemia for chemotherapy-induced DNA damage via suppressing FANCD2 expression. Leukemia 2014; 28:203-6; PMID:23852546; http://dx.doi.org/ 10.1038/leu.2013.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098-101; PMID:15718470; http://dx.doi.org/ 10.1126/science.1106148 [DOI] [PubMed] [Google Scholar]
- 115.Carracedo A, Ma L, Teruya-feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, et al.. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008; 118:3065-74; PMID:18725988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol 2004; 167:399-403; PMID:15533996; http://dx.doi.org/ 10.1083/jcb.200408161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Choo AY, Kim SG, Vander Heiden MG, Mahoney SJ, Vu H, Yoon SO, Cantley LC, Blenis J. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol Cell 2010; 38:487-99; PMID:20513425; http://dx.doi.org/ 10.1016/j.molcel.2010.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Catane R, Von Hoff DD, Glaubiger DL, Muggia FM. Azaserine, DON, and azotomycin: three diazo analogs of L-glutamine with clinical antitumor activity. Cancer Treat Rep 1979; 63:1033-8; PMID:380801 [PubMed] [Google Scholar]
- 119.Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et al.. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther 2014; 13:890-901; PMID:24523301; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0870 [DOI] [PubMed] [Google Scholar]
- 120.Li M, Li C, Allen A, Stanley CA, Smith TJ. The structure and allosteric regulation of mammalian glutamate dehydrogenase. Arch Biochem Biophys 2012; 519:69-80; PMID:22079166; http://dx.doi.org/ 10.1016/j.abb.2011.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang C, Sudderth J, Dang T, Bachoo RGM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res 2009; 69:7986-93; PMID:19826036; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stine ZE, Dang CV. Stress eating and tuning out: cancer cells re-wire metabolism to counter stress. Crit Rev Biochem Mol Biol 2013; 48:609-19; PMID:24099138; http://dx.doi.org/ 10.3109/10409238.2013.844093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Denny WA. Giving Anemia a Boost with Inhibitors of Prolyl Hydroxylase. J Med Chem 2012; 55:2943-4; PMID:22409479; http://dx.doi.org/ 10.1021/jm300314a [DOI] [PubMed] [Google Scholar]
- 124.Hirsilä M, Koivunen P, Günzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem 2003; 278:30772-80; PMID:12788921; http://dx.doi.org/ 10.1074/jbc.M304982200 [DOI] [PubMed] [Google Scholar]
- 125.Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1 a in normoxia. EMBO J 2003; 22:4082-90; PMID:12912907; http://dx.doi.org/ 10.1093/emboj/cdg392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rantanen K, Pursiheimo JP, Högel H, Miikkulainen P, Sundström J, Jaakkola PM. p62/SQSTM1 regulates cellular oxygen sensing by attenuating PHD3 activity through aggregate sequestration and enhanced degradation. J Cell Sci 2013; 126:1144-54; PMID:23345396; http://dx.doi.org/ 10.1242/jcs.115667 [DOI] [PubMed] [Google Scholar]
- 127.Tennant DA, Gottlieb E. HIF prolyl hydroxylase-3 mediates alpha-ketoglutarate-induced apoptosis and tumor suppression. J Mol Med (Berl) 2010; 88:839-49; PMID:20383689; http://dx.doi.org/ 10.1007/s00109-010-0627-0 [DOI] [PubMed] [Google Scholar]
- 128.Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8:528-39; PMID:21587219; http://dx.doi.org/ 10.1038/nrclinonc.2011.71 [DOI] [PubMed] [Google Scholar]
- 129.Shen S, Kepp O, Michaud M, Martins I, Minoux H, Métivier D, Maiuri MC, Kroemer RT, Kroemer G. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene 2011; 30:4544-56; PMID:21577201; http://dx.doi.org/ 10.1038/onc.2011.168 [DOI] [PubMed] [Google Scholar]
- 130.Macintosh RL, Ryan KM. Autophagy in tumour cell death. Semin Cancer Biol 2013; 23:344-51; PMID:23774296; http://dx.doi.org/ 10.1016/j.semcancer.2013.05.006 [DOI] [PubMed] [Google Scholar]
- 131.Fulda S, Kögel D. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015; PMID:25619832; http://dx.doi.org/ 10.1038/onc.2014.458 [DOI] [PubMed] [Google Scholar]
- 132.Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, et al.. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 2013; 4:e838; PMID:24113172; http://dx.doi.org/ 10.1038/cddis.2013.350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Poklepovic A, Gewirtz DA. Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy 2014; 10:1478-80; PMID:24991829; http://dx.doi.org/ 10.4161/auto.29428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rosich L, Xargay-Torrent S, López-Guerra M, Campo E, Colomer D, Roué G. Counteracting autophagy overcomes resistance to everolimus in mantle cell lymphoma. Clin Cancer Res 2012; 18:5278-89; PMID:22879389; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-0351 [DOI] [PubMed] [Google Scholar]
- 135.Grimaldi A, Balestrieri ML, D'Onofrio N, Di Domenico G, Nocera C, Lamberti M, Tonini G, Zoccoli A, Santini D, Caraglia M, et al.. The synergistic effect of everolimus and chloroquine on endothelial cell number reduction is paralleled by increased apoptosis and reduced autophagy occurrence. PLoS One 2013; 8:e79658; PMID:24244540; http://dx.doi.org/ 10.1371/journal.pone.0079658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shen C, Oswald D, Phelps D, Cam H, Pelloski CE, Pang Q, Houghton PJ. Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double-strand breaks. Cancer Res 2013; 73:3393-401; PMID:23633493; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Storozhuk Y, Hopmans SN, Sanli T, Barron C, Tsiani E, Cutz JC, Pond G, Wright J, Singh G, Tsakiridis T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer 2013; 108:2021-32; PMID:23632475; http://dx.doi.org/ 10.1038/bjc.2013.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shortt J, Martin BP, Newbold A, Hannan KM, Devlin JR, Baker AJ, Ralli R, Cullinane C, Schmitt CA, Reimann M, et al.. Combined inhibition of PI3K-related DNA damage response kinases and mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood 2013; 121:2964-74; PMID:23403624; http://dx.doi.org/ 10.1182/blood-2012-08-446096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guo F, Li J, Du W, Zhang S, O'Connor M, Thomas G, Kozma S, Zingarelli B, Pang Q, Zheng Y. mTOR regulates DNA damage response through NF-kB-mediated FANCD2 pathway in hematopoietic cells. Leukemia 2013; 27:2040-6; PMID:23538752; http://dx.doi.org/ 10.1038/leu.2013.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hardie DG. AMPK — Sensing Energy while Talking to Other Signaling Pathways. Cell Metab 2014; 20:939-52; PMID:25448702; http://dx.doi.org/ 10.1016/j.cmet.2014.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem 2002; 277:23977-80; PMID:11997383; http://dx.doi.org/ 10.1074/jbc.C200171200 [DOI] [PubMed] [Google Scholar]
- 142.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115:577-90; PMID:14651849; http://dx.doi.org/ 10.1016/S0092-8674(03)00929-2 [DOI] [PubMed] [Google Scholar]