

Abstract
To increase the efficacy of currently used anti-cancer genotoxins, one of the current efforts is to find agents that can sensitize cancer cells to genotoxins so that the efficacious doses of genotoxins can be lowered to reduce deleterious side-effects. In this study, we reported that a synthetic RasGAP-derived peptide GAP159 could enhance the effect of chemotherapeutic agent cisplatin (CDDP) in human colon carcinoma HCT116 cells. Our results showed that GAP159 significantly increased the CDDP-induced cytotoxicity and apoptosis in HCT116 cells. This synergistic effect was associated with the inhibitions of phospho-AKT, phospho-ERK and NF-κB. In mouse colon tumor CT26 animal models, GAP159 combined with CDDP significantly suppressed CT26 tumor growth, and GAP159 alone showed slight inhibitory effect. Our data suggests that co-treatment of GAP159 and chemotherapeutics will become a potential therapeutic strategy for colon cancers.
KEY WORDS: GAP159, CDDP, Chemosensitization, Colon cancer, Apoptosis
Graphical abstract
GAP159, a synthetic RasGAP-derived peptide, targets and down-regulates G3BP, enhances cisplatin induced cytotoxicity and apoptosis in HCT116 cells in vitro and mouse colon tumor CT26 animal models in vivo.
1. Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide and has a high mortality rate. Despite recent advances in diagnostic modalities for CRC, it also act as a major cause of cancer death in affluent countries with over one million cases worldwide and a disease-specific mortality of approximately 33% in the developed world1. Chemotherapy is one of the commonly used strategies in CRC treatment. Conventional chemotherapeutic drugs such as cisplatin (CDDP), adriamycin and 5-fluorouracil (5-FU) often have severe side effects that limit their efficacy2. Therefore, there is a critical need to develop more effective strategies for the cancer therapy. Combination therapy with multiple drugs or modalities is a common practice in the treatment of cancers, which can achieve therapeutic effects greater than those of provided by a single drug or modality, and can reduce the side effects and resistance to drugs.
RasGAP is a regulator of Ras and Rho GTP-binding proteins. It can induce both anti- and pro-apoptotic signals depending on the extent of its cleavage by caspase3,4. RasGAP is a substrate of caspase; when cleaved at position 455, N-terminal fragment of the production protects cells from apoptosis in a Ras/PI3K/Akt-dependent manner5. As increase of caspase activity, the N-terminal fragment can further cleave into two smaller fragments at position 157 which can induce apoptosis4 and markedly reduce Akt activity. A synthetic peptide TAT-RasGAP317–326 was found to efficiently increase the extent of apoptosis induced by a variety of genotoxins and other anti-cancer treatments in several tumor cell lines both in vitro and in vivo3,6–8, which depends on functional p53 and PUMA (p53-upregulated modulator of apoptosis)9. Cui et al.10 reported that two novel peptides derived from RasGAP SH3 domain with cytotoxicity on tumor cells showed an obvious sensitizing effect on CDDP. However, they had no significant effects on normal cells, but the mechanism was not clear. Previously, we have reported two peptides showed promising anti-tumor activity in vitro and in vivo through down-regulation of G3BPs11,12. G3BPs proteins are dramatically over-expressed in human cancers, especially in breast, head and neck, colon, and lung13–15. G3BPs are involved in a variety of growth-related signaling pathways that are involved in carcinogenesis and metastasis16–18, including NF-κB, Ras signaling and the ubiquitin proteasome system13,19–21.
In this study, we demonstrated a new synthetic peptide GAP159 derived from RasGAP SH3 domain enhanced the cytotoxicity and apoptotic potential of CDDP in human colon cancer HCT116 cells. The combination of GAP159 and CDDP showed a significant synergy in HCT116 cells. We found GAP159 could down-regulate G3BPs, and several G3BP-related molecules of transduction signaling pathways were detected including ERK, NF-κB and Akt pathways. The antitumor effects of GAP159 alone or combined with CDDP were also evaluated in mouse colon tumor CT26 model.
2. Materials and methods
2.1. Peptide synthesis
GAP159, a cell-permeable peptide, was synthesized at Wuhan KatyGen Pharmaceuticals (Hubei, China). The peptides were dissolved in deionized water at a final concentration of 10 mmol/L and stored at −20 °C until further use.
2.2. Cell culture and reagents
Human colon carcinoma HCT116 cells were purchased from ATCC (Manassas, VA, USA). Cells were grown in DMEM (Gibco, Grand Island, USA) supplemented with 10% fetal bovine serum, 2 mmol/L l-glutamine, and 1% penicillin–streptomycin at 37 °C in the presence of 5% CO2. Cisplatin (Sigma-Aldrich, St Louis, MO, USA) was diluted in PBS at a final concentration of 20 mmol/L and stored at −20 °C.
2.3. MTT assays
HCT116 cells were seeded in 96-well plates at a density of 3×103 cells/well in 200 μL medium. Then the cells were treated with various concentrations of GAP159 alone or combined with CDDP. After drug exposure for indicated hours, the MTT solution (5 g/L) was added to the plates. The cells were incubated at 37 °C for another 4 h. The formazan, which is derived from MTT by living cells, was dissolved in 150 μL DMSO per well, and the absorbance was detected at 570 nm. All MTT experiments were performed in triplicate and repeated at least 3 times. The percentage of cytotoxicity was calculated as follows: cytotoxicity (%)=[(1−OD570 of experimental well)/OD570 of control well]×100%.
2.4. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay
Cells were treated with 10 μmol/L and 30 μmol/L GAP159 alone or combined with 10 μmol/L CDDP. TUNEL staining was performed in situ using the DeadEnd fluorometric TUNEL system kit (Promega, Madison, Wisconsin, USA) according to the manufacturer׳s protocol. The images were captured by an image analysis system (Eclipse TE2000-U, Nikon, Japan). The number of TUNEL positive cells was divided by the total number of cells to determine the percentage of apoptosis.
2.5. Western blot analysis
Whole cell lysates were used for immunoblotting as previously described22. Briefly, cells were washed with cold PBS, and then lysed with RIPA lysis buffer containing protease and phosphatase inhibitor. Floating cells or cell fragments were collected and lysed with the above lysates. Protein concentrations were determined by Bradford assay. Equal amounts of protein were separated by SDS-PAGE and transferred to polyvinylidene difluoride membrane (Millipore Corp., Bedford, MA, USA). Membranes were blocked in TBST containing 5% nonfat skim milk at room temperature for 2 h and probed with primary antibodies overnight at 4 °C. Then the membranes were blotted with an appropriate horseradish peroxidase-linked secondary antibody. Electochemiluminescence was performed according to the manufacturer׳s instructions with ChemiImager 5500 imaging system (Alpha Innotech Co., CA, USA). Antibodies against Akt, phospho-Akt (Ser423), ERK and phospho-ERK were from Cell Signaling Technology (Danvers, MA, USA). G3BP2 was from Abcam (Cambridge, MA, USA), and anti-β-actin was from Sigma (St Louis, MO, USA). Other anti-G3BP1, HRP-linked goat anti-rabbit and anti-mouse secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
2.6. In vivo tumor mouse model
Male BALB/c mice (Institute for Experimental Animals, Chinese Academy of Medical Sciences & Peking Union Medical College) were injected with 1.5 million mouse colon carcinoma CT26 cells subcutaneously on the right flank. The following day and thereafter daily for GAP159 and every other day for CDDP, the mice were injected intraperitoneally with PBS, 10 mg/kg, 20 mg/kg and 40 mg/kg of GAP159, 1 mg/kg CDDP, or a combination of GAP159 and CDDP. After 11 days, all mice were weighed and sacrificed, and the tumors were excised. Tumors were weighed, and the mean tumor weight was calculated. The mean treated tumor weight/mean control tumor weight ×100% is subtracted from 100% to give the tumor growth inhibition (TGI) for each group.
2.7. Analysis of drug interaction
The coefficient of drug interaction (CDI) was used to analyze the synergistically inhibitory effect of the drug combination23. CDI was calculated as follows: CDI=AB/(A×B). AB is the ratio of the two-drug combination group to the control group, and A or B is the ratio of the single drug group to the control group. Therefore, CDI<1 indicates synergism, CDI<0.7 indicates a significantly synergistic effect, CDI=1 indicates additivity, and CDI>1 indicates antagonism.
2.8. Statistical analysis
Data were described as means±SD of the indicated number of separate experiments. A one-way analysis of variance was carried out for multiple comparisons. If there was significant variation between treatment and control groups, the mean values were compared by using student׳s t-test. P-values less than 0.05 were considered significant.
3. Results
3.1. Down-regulation of G3BP proteins by GAP159
According to the structure-base design of peptide against G3BP by Cui et al.10, we have reported two peptides showing promising anticancer activities alone or combined with CDDP, and revealed the mechanisms. Our cooperator designed a new peptide GAP159 through amino acid substitution, which, theoretically, had strong interaction with G3BP and was more stable. Previously, we found that down-regulation of G3BP by peptide or siRNA could inhibit cancer cell proliferation and induce apoptosis11. To figure out whether the new peptide GAP159 had the same effect on G3BP protein, we treated HCT116 cells with GAP159 alone or combined with CDDP. Western blot results showed that GAP159 could significantly reduce the level of G3BP1 and G3BP2 proteins (Fig. 1). CDDP alone had no effect on G3BP proteins.
Figure 1.
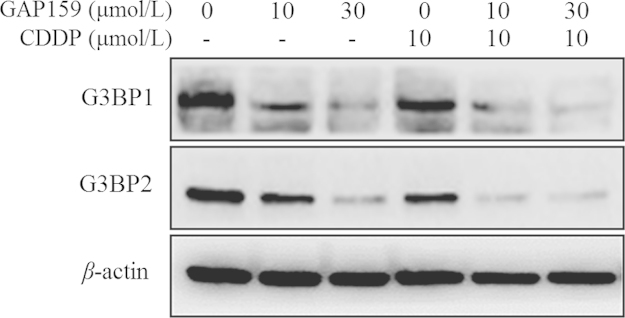
GAP159 down-regulates the level of G3BP1 and G3BP2 proteins. Cells were treated with GAP159 for 48 h; G3BP1 and G3BP2 proteins were detected by Western blot.
3.2. Inhibitory effect of GAP159 and CDDP on HCT116 cells
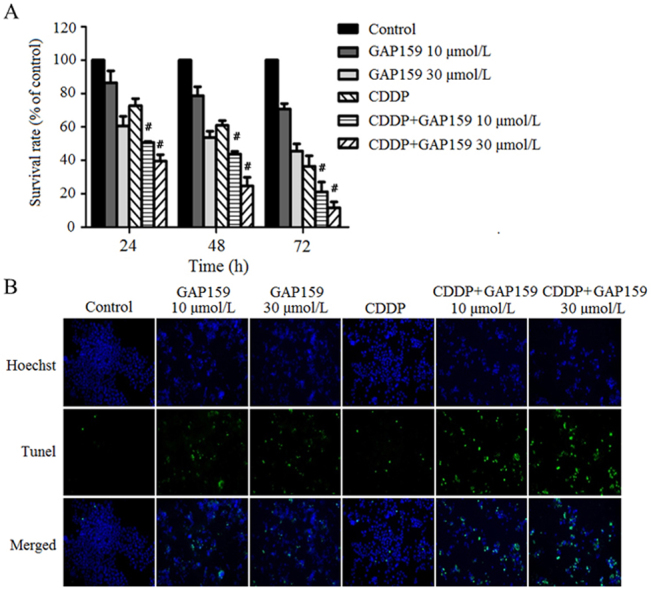
To explore whether GAP159 might be a potential therapeutic agent against HCT116 cells, we investigated the anti-proliferative activity of GAP159 with or without CDDP. After drug exposure, cell proliferation was determined by MTT assay (Fig. 2A). GAP159 alone inhibited proliferation of HCT116 cells in a dose and time-dependent manner. For 48 h treatment, the inhibition rates of 10 μmol/L and 30 μmol/L GAP159 were 21.3% and 46.34%, respectively. When treated the cells with 30 μmol/L GAP159, the inhibition rates of 24, 48 and 72 h treatment were 39.33%, 46.34%, 54.46%, respectively. The combination of GAP159 and CDDP showed significant anti-proliferative effects on HCT116 cells as compared with GAP159 or CDDP alone (P<0.05). For 72 h treatment, the inhibitory rates of 10 μmol/L and 30 μmol/L GAP159 combined with CDDP were 78.83% and 88.33%, respectively.
Figure 2.

GAP159 inhibits HCT116 cell growth. A: GAP159 synergistically enhanced the inhibitory effects of CDDP on HCT116 cells (#P<0.05 vs CDDP); B: CDI value of co-treatment of GAP159 and CDDP.
CDI was used to evaluate the nature of the interaction. As shown in Fig. 2B, the CDI values of all combination situations are under 1. For 48 h treatment, the CDI values were 0.91 and 0.75. For 72 h exposure, the CDI values were 0.81 and 0.7. These results indicate that GAP159 synergistically enhances CDDP-induced cytotoxicity in HCT116 cells.
3.3. Enhancement of CDDP-induced apoptosis by GAP159
To determine whether GAP159 potentiated CDDP-induced apoptosis, we employed TUNEL staining assay (Fig. 3A). As shown in Fig. 3B, the percentage of TUNEL positive cells is increased from 1.1% in control cells to 6.7% and 12.6% in GAP159-treated cells. GAP159 also increased CDDP-induced TUNEL positive cells from 3.8% to 22.7% and 34.7% (Fig. 3B). This result is confirmed by caspase-3/7 activity assay, which showed GAP159 markedly enhanced CDDP induced caspase-3/7 activities (Fig. 3C). Taken together, the results indicate that GAP159 enhances CDDP-induced apoptosis.
Figure 3.

GAP159 enhances CDDP induced apoptosis. A: Co-treatment of GAP159 and CDDP induced apoptosis detected by TUNEL assay; B: quantification of TUNEL positive cells (⁎P<0.05 vs Control, #P<0.05 vs CDDP); C: the changes of caspase-3/7 activity in GAP159 and/or CDDP treated cells. The results are mean±SD of three individual experiments.
3.4. Inhibition of Akt, ERK and NF-κB activations by GAP159
Previously, we found that down-regulation of G3BP by peptides and siRNAs could impair Ras activity and inhibit downstream signaling pathways11,12. Therefore, GAP159 may affect the Ras pathways including ERK and NF-κB. In addition, extraordinary activation of ERK cascades by CDDP antagonizes apoptosis24. So, we detected the effect of GAP159 on CDDP induced activations of ERK and NF-κB. Western blot results showed that GAP159 clearly inhibited CDDP induced ERK activity and had no significant effect on total ERK protein levels (Fig. 4). We also determined if the GAP159 modulated Akt activity and sensitized HCT116 cells to CDDP. Fig. 4 shows that GAP159 inhibits Akt phosphorylation, and has no effect on total Akt expression. It has been reported that NF-κB activity could be modulated by ERK and Akt25. We also found that GAP159 significantly down-regulated NF-κB (Fig. 4).
Figure 4.

Effects of GAP159 on Akt, ERK and NF-κB. A: Cells were treated with GAP159 in the absence or presence of CDDP for 48 h; B: quantification of protein levels. The protein levels were detected by Western blot.
3.5. GAP159 enhanced antitumor activity of CDDP in vivo
In vivo experiment was employed to confirm the synergism of GAP159 combined with CDDP. The antitumor effect evaluation was carried out using mouse colon tumor CT26 model established in BALB/c mice. The mice were intraperitoneally injected with PBS, 10 mg/kg, 20 mg/kg and 40 mg/kg GAP159 once per day, and 1 mg/kg of CDDP every other day. As evaluated on day 11 (Table 1), the inhibitory effect of 1 mg/kg CDDP was 39.2%, and 10 mg/kg, 20 mg/kg and 40 mg/kg GAP159 showed slight therapeutic efficacy with 10.6%, 18.4% and 33.5% TGI, respectively. As shown in Table 1, the combinations of CDDP with 10 mg/kg, 20 mg/kg and 40 mg/kg GAP159 were markedly displayed antitumor inhibitory effects with 51.3%, 65.5% and 69.8% TGI, respectively. These results show that GAP159 could enhance the activity of CDDP in vivo.
Table 1.
Combination treatment of GAP159 and CDDP significantly suppressed the tumor growth of BALB/c mice bearing colon CT26.
| Groups | Dosage (mg/kg) | No. of mice (begin/end) | Body weight change (g) | Tumor weight (mean±SD) | TGI (%) | CDI |
|---|---|---|---|---|---|---|
| Control | – | 10/10 | +0.44 | 1.58±0.44 | – | – |
| CDDP | 1 | 10/10 | −1.22 | 0.96±0.19a | 39.2 | – |
| GAP159 | 10 | 10/10 | −0.71 | 1.41±0.47 | 10.6 | – |
| 20 | 10/10 | −0.64 | 1.29±0.29a | 18.4 | – | |
| 40 | 10/10 | −0.89 | 1.05±0.22a | 33.5 | – | |
| GAP159 | 10+1 | 10/10 | −1.16 | 0.77±0.29a | 51.3 | 0.89 |
| +CDDP | 20+1 | 10/10 | −1.18 | 0.55±0.20a,b | 65.5 | 0.69 |
| 40+1 | 10/10 | −1.67 | 0.48±0.16a,b | 69.8 | 0.74 |
P<0.05 vs control group.
P<0.05 vs CDDP group.
4. Discussion
Chemotherapy drug CDDP is widely used for the treatment of many malignancies, including ovarian, bladder, cervical, head and neck, and colon cancer26. The cytotoxicity of CDDP is believed to be the result of the formation of platinum–DNA adducts. Despite the great efficacy in treating certain kinds of cancers, chemotherapy drugs could also damage other normal cells which cause some side effects. Specific sensitization of tumor cells to the action of chemotherapy drugs would reduce the efficacious doses of chemotherapy drugs to be used in patients, diminish the detrimental side effects on normal tissues and enhance the efficacy of chemotherapeutic treatments, which was called combination chemotherapy. In the present study, we report a synthetic peptide GAP159 strongly sensitized HCT116 cells to CDDP-induced cytotoxicity and apoptosis.
G3BP were first identified as a RasGAP SH3 domain binding protein, and the NTF2-like domain was responsible for its binding to RasGAP27. Parker et al.28 developed a monoclonal antibody directed against the NTF2-like domain of G3BP, showing a specific effect in the therapy for the G3BP-related tumors. Cui et al.10 established a G3BP NTF2-like domain and RasGAP SH3 domain interaction model, and designed two peptides showing an obvious sensitizing effect to CDDP. According to this model, our cooperators designed a new peptide GAP159 through amino acid substitution. Previously, we found that down-regulation of G3BP by peptides or siRNAs could inhibit cancer cell growth and induce apoptosis. In this study, likewise, we found GAP159 significantly down-regulated the G3BP1 and G3BP2 protein levels, and showed promising anti-proliferation effect in vitro and in vivo. Meanwhile, GAP159 alone or combined with CDDP induced HCT116 cell apoptosis, indicating a significant synergistic effect.
Ras and PI3K/AKT signaling pathways play an important role in cell survival, proliferation and apoptosis, and are critical for the progression of CRC29,30. Recent studies31,32 reported that activation of the PI3K pathway contributed to the resistance to MAPK kinase (MEK) inhibitors in CRC cells, suggesting that the therapy with a combination of PI3K and MEK inhibitors may be a more effective treatment for cancers33. Our results showed that inhibition of ERK and Akt enhanced CDDP-induced anti-proliferation effect12. Interaction of RasGAP and G3BP occurs only in growing cells and both proteins are recruited to activate Ras19,27, suggesting that G3BP could participate in Ras signal pathway through RasGAP. Previously, we found that knockdown of G3BP inhibited ERK and Akt activities11. Herein, GAP159 down-regulated G3BPs, and inhibited the ERK and Akt phosphorylation. NF-κB transcription factor is an important factor implicated in regulating cell survival and apoptosis. Anti-cancer drugs induced NF-κB activation attenuated their antitumor activity34. CDDP-induced activation of ERK and NF-κB played an important role in chemoresistance35,36. In our study, GAP159 inhibited ERK and Akt activities and reduced NF-κB, which might contribute to the synergistic effect of co-treatment of GAP159 and CDDP.
In summary, GAP159 could down-regulate G3BP1 and G3BP2 proteins, and sensitize HCT116 cells to CDDP induced anti-proliferation and apoptosis. GAP159-induced the suppression of ERK and Akt, and the decrease of NF-κB contributed to this synergism. Taken together, our results show that GAP159 synergistically enhances CDDP-induced cytotoxicity, suggesting that the combination of GAP159 and chemotherapy drugs might be a promising strategy for cancer therapy.
Acknowledgments
This research was supported by grants from National Basic Research Program of China (No. 2009CB521807), National Natural Science Foundation of China (Nos. 30772583 and 81302802) and National S&T Major Special Project on Major New Drug Innovation (No. 2012ZX09301-002).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.

References
- 1.Wolpin B.M., Mayer R.J. Systemic treatment of colorectal cancer. Gastroenterology. 2008;134:1296–1310. doi: 10.1053/j.gastro.2008.02.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Michod D., Widmann C. DNA-damage sensitizers: potential new therapeutical tools to improve chemotherapy. Crit Rev Oncol Hematol. 2007;63:160–171. doi: 10.1016/j.critrevonc.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Michod D., Yang J.Y., Chen J., Bonny C., Widmann C. A RasGAP-derived cell permeable peptide potently enhances genotoxin-induced cytotoxicity in tumor cells. Oncogene. 2004;23:8971–8978. doi: 10.1038/sj.onc.1207999. [DOI] [PubMed] [Google Scholar]
- 4.Yang J.Y., Michod D., Walicki J., Murphy B.M., Kasibhatla S., Martin S.J. Partial cleavage of RasGAP by caspases is required for cell survival in mild stress conditions. Mol Cell Biol. 2004;24:10425–10436. doi: 10.1128/MCB.24.23.10425-10436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang J.Y., Widmann C. The RasGAP N-terminal fragment generated by caspase cleavage protects cells in a Ras/PI3K/Akt-dependent manner that does not rely on NFkappa B activation. J Biol Chem. 2002;277:14641–14646. doi: 10.1074/jbc.M111540200. [DOI] [PubMed] [Google Scholar]
- 6.Michod D., Annibaldi A., Schaefer S., Dapples C., Rochat B., Widmann C. Effect of RasGAP N2 fragment-derived peptide on tumor growth in mice. J Natl Cancer Inst. 2009;101:828–832. doi: 10.1093/jnci/djp100. [DOI] [PubMed] [Google Scholar]
- 7.Pittet O., Petermann D., Michod D., Krueger T., Cheng C., Ris H.B. Effect of the TAT-RasGAP(317–326) peptide on apoptosis of human malignant mesothelioma cells and fibroblasts exposed to meso-tetra-hydroxyphenyl-chlorin and light. J Photochem Photobiol B. 2007;88:29–35. doi: 10.1016/j.jphotobiol.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 8.Rehm M., Huber H.J., Dussmann H., Prehn J.H. Systems analysis of effector caspase activation and its control by X-linked inhibitor of apoptosis protein. EMBO J. 2006;25:4338–4349. doi: 10.1038/sj.emboj.7601295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michod D., Widmann C. TAT-RasGAP317–326 requires p53 and PUMA to sensitize tumor cells to genotoxins. Mol Cancer Res. 2007;5:497–507. doi: 10.1158/1541-7786.MCR-06-0257. [DOI] [PubMed] [Google Scholar]
- 10.Cui W., Wei Z., Chen Q., Cheng Y.H., Geng L.L., Zhang J. Structure-based design of peptides against G3BP with cytotoxicity on tumor cells. J Chem Inf Model. 2010;50:380–387. doi: 10.1021/ci900404p. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H., Zhang S.H., He H.W., Zhao W.L., Chen J.H., Shao R.G. GAP161 targets and downregulates G3BP to suppress cell growth and potentiate cisplaitin-mediated cytotoxicity to colon carcinoma HCT116 cells. Cancer Sci. 2012;103:1848–1856. doi: 10.1111/j.1349-7006.2012.02361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H., Zhang S.H., He H.W., Zhao W.L., Ren K.H., Chen J.H. RasGAP-derived peptide 38GAP potentiates the cytotoxicity of cisplatin through inhibitions of Akt, ERK and NF-kappaB in colon carcinoma HCT116 cells. Cancer Lett. 2011;308:62–70. doi: 10.1016/j.canlet.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 13.Barnes C.J., Li F., Mandal M., Yang Z., Sahin A.A., Kumar R. Heregulin induces expression, ATPase activity, and nuclear localization of G3BP, a Ras signaling component, in human breast tumors. Cancer Res. 2002;62:1251–1255. [PubMed] [Google Scholar]
- 14.French J., Stirling R., Walsh M., Kennedy H.D. The expression of Ras-GTPase activating protein SH3 domain-binding proteins, G3BPs, in human breast cancers. Histochem J. 2002;34:223–231. doi: 10.1023/a:1021737413055. [DOI] [PubMed] [Google Scholar]
- 15.Guitard E., Parker F., Millon R., Abecassis J., Tocque B. G3BP is overexpressed in human tumors and promotes S phase entry. Cancer Lett. 2001;162:213–221. doi: 10.1016/s0304-3835(00)00638-8. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H., Shao R.G. G3BP: a promising target for cancer therapy. Acta Pharm Sin. 2010;45:945–951. [PubMed] [Google Scholar]
- 17.Taniuchi K., Nishimori I., Hollingsworth M.A. Intracellular CD24 inhibits cell invasion by posttranscriptional regulation of BART through interaction with G3BP. Cancer Res. 2011;71:895–905. doi: 10.1158/0008-5472.CAN-10-2743. [DOI] [PubMed] [Google Scholar]
- 18.Taniuchi K., Nishimori I., Hollingsworth M.A. The N-terminal domain of G3BP enhances cell motility and invasion by posttranscriptional regulation of BART. Mol Cancer Res. 2011;9:856–866. doi: 10.1158/1541-7786.MCR-10-0574. [DOI] [PubMed] [Google Scholar]
- 19.Gallouzi I.E., Parker F., Chebli K., Maurier F., Labourier E., Barlat I. A novel phosphorylation-dependent RNase activity of GAP-SH3 binding protein: a potential link between signal transduction and RNA stability. Mol Cell Biol. 1998;18:3956–3965. doi: 10.1128/mcb.18.7.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prigent M., Barlat I., Langen H., Dargemont C. Ikappa Balpha and IkappaBalpha/NF-kappa B complexes are retained in the cytoplasm through interaction with a novel partner, RasGAP SH3-binding protein 2. J Biol Chem. 2000;275:36441–36449. doi: 10.1074/jbc.M004751200. [DOI] [PubMed] [Google Scholar]
- 21.Soncini C., Berdo I., Draetta G. Ras-GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene. 2001;20:3869–3879. doi: 10.1038/sj.onc.1204553. [DOI] [PubMed] [Google Scholar]
- 22.Ren K., Jin H., Bian C., He H., Liu X., Zhang S. MR-1 modulates proliferation and migration of human hepatoma HepG2 cells through myosin light chains-2 (MLC2)/focal adhesion kinase (FAK)/Akt signaling pathway. J Biol Chem. 2008;283:35598–35605. doi: 10.1074/jbc.M802253200. [DOI] [PubMed] [Google Scholar]
- 23.Cao S.S., Zhen Y.S. Potentiation of antimetabolite antitumor activity in vivo by dipyridamole and amphotericin B. Cancer Chemother Pharmacol. 1989;24:181–186. doi: 10.1007/BF00300240. [DOI] [PubMed] [Google Scholar]
- 24.Dent P., Grant S. Pharmacologic interruption of the mitogen-activated extracellular-regulated kinase/mitogen-activated protein kinase signal transduction pathway: potential role in promoting cytotoxic drug action. Clin Cancer Res. 2001;7:775–783. [PubMed] [Google Scholar]
- 25.Dolcet X., Llobet D., Pallares J., Matias-Guiu X. NF-κB in development and progression of human cancer. Virchows Arch. 2005;446:475–482. doi: 10.1007/s00428-005-1264-9. [DOI] [PubMed] [Google Scholar]
- 26.Rebillard A., Tekpli X., Meurette O., Sergent O., LeMoigne-Muller G., Vernhet L. Cisplatin-induced apoptosis involves membrane fluidification via inhibition of NHE1 in human colon cancer cells. Cancer Res. 2007;67:7865–7874. doi: 10.1158/0008-5472.CAN-07-0353. [DOI] [PubMed] [Google Scholar]
- 27.Parker F., Maurier F., Delumeau I., Duchesne M., Faucher D., Debussche L. A Ras-GTPase-activating protein SH3-domain-binding protein. Mol Cell Biol. 1996;16:2561–2569. doi: 10.1128/mcb.16.6.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fabienne P, Mireille K, Marc D, Isabelle B, inventors; Aventis Pharma S. A. (Antony, FR), assignee. Monoclonal antibodies directed against the G3BP protein, and uses. US patent US7001980; June 4, 2002.
- 29.Fang J.Y., Richardson B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6:322–327. doi: 10.1016/S1470-2045(05)70168-6. [DOI] [PubMed] [Google Scholar]
- 30.Repasky G.A., Chenette E.J., Der C.J. Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004;14:639–647. doi: 10.1016/j.tcb.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 31.Balmanno K., Chell S.D., Gillings A.S., Hayat S., Cook S.J. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int J Cancer. 2009;125:2332–2341. doi: 10.1002/ijc.24604. [DOI] [PubMed] [Google Scholar]
- 32.Wee S., Jagani Z., Xiang K.X., Loo A., Dorsch M., Yao Y.M. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286–4293. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 33.Engelman J.A., Chen L., Tan X., Crosby K., Guimaraes A.R., Upadhyay R. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen W., Wang X., Bai L., Liang X., Zhuang J., Lin Y. Blockage of NF-kappaB by IKKbeta- or RelA-siRNA rather than the NF-kappaB super-suppressor IkappaBalpha mutant potentiates adriamycin-induced cytotoxicity in lung cancer cells. J Cell Biochem. 2008;105:554–561. doi: 10.1002/jcb.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Persons D.L., Yazlovitskaya E.M., Cui W., Pelling J.C. Cisplatin-induced activation of mitogen-activated protein kinases in ovarian carcinoma cells: inhibition of extracellular signal-regulated kinase activity increases sensitivity to cisplatin. Clin Cancer Res. 1999;5:1007–1014. [PubMed] [Google Scholar]
- 36.Yoon H., Min J.K., Lee J.W., Kim D.G., Hong H.J. Acquisition of chemoresistance in intrahepatic cholangiocarcinoma cells by activation of AKT and extracellular signal-regulated kinase (ERK)1/2. Biochem Biophys Res Commun. 2011;405:333–337. doi: 10.1016/j.bbrc.2010.11.130. [DOI] [PubMed] [Google Scholar]