

Abstract
Intranasal drug administration is receiving increased attention as a delivery method for bypassing the blood–brain barrier and rapidly targeting therapeutics to the CNS. However, rapid mucociliary clearance in the nasal cavity is a major hurdle. The purpose of this study was to evaluate the effect of mucoadhesive polymers in enhancing the delivery of nimodipine microemulsion to the brain via the intranasal route. The optimized mucoadhesive microemulsion was characterized, and the in vitro drug release and in vivo nasal absorption of drug from the new formulation were evaluated in rats. The optimized formulation consisted of Capmul MCM as oil, Labrasol as surfactant, and Transcutol P as co-surfactant, with a particle size of 250 nm and zeta potential value of −15 mV. In vitro and ex vivo permeation studies showed an initial burst of drug release at 30 min and sustained release up to 6 h, attributable to the presence of free drug entrapped in the mucoadhesive layer. In vivo pharmacokinetic studies in rats showed that the use of the mucoadhesive microemulsion enhanced brain and plasma concentrations of nimodipine. These results suggest that incorporation of a mucoadhesive agent in a microemulsion intranasal delivery system can increase the retention time of the formulation and enhance brain delivery of drugs.
KEY WORDS: Blood–brain barrier, Entrapment, Permeation, Pharmacokinetics, Nasal mucosa
Graphical abstract
Intranasal delivery helps in bypassing the blood–brain barrier and targeting the therapeutics directly to the CNS. However, rapid mucociliary clearance in nasal cavity is a major hurdle. To solve this problem, a novel mucoadhesive microemulsion system for nimodipine was developed which led to improved brain drug delivery via intranasal route.
1. Introduction
The delivery of therapeutics to the central nervous system (CNS) is not effective for greater than 98% of small molecules and for nearly 100% of large molecules, despite the presence of an immense network of cerebral vasculature1. The lack of effectiveness is due to the presence of the blood–brain barrier (BBB), which prevents most foreign substances, even many beneficial therapeutics, from entering the brain from the circulating blood. Although some small molecule, peptide and protein therapeutics given systemically reach the brain parenchyma by crossing the BBB, high systemic doses are generally needed to achieve therapeutic levels, resulting in adverse effects2. Therapeutics can be introduced directly into the CNS by intracerebroventricular or intraparenchymal injections; however, for multiple dosing regimens, both delivery methods are invasive, risky, and expensive techniques requiring surgical expertise. Additional limitations to the utility of these methods are inadequate CNS exposure due to slow diffusion from the injection site and rapid turnover of the cerebrospinal fluid (CSF). Intranasal delivery has come to the forefront as an alternative to invasive delivery methods to bypass the BBB and rapidly target therapeutics directly to the CNS. This delivery method utilizes pathways along olfactory and trigeminal nerves innervating the nasal passages3, 4, 5.
Nimodipine is a dihydropyridine calcium channel blocker used in the treatment of cerebrovascular spasm and senile dementia. The major problem associated with nimodipine is extensive first pass metabolism, low solubility and low oral bioavailability (5%–10%) in humans, thus accounting for low brain concentration. Nimodipine is a P-glycoprotein substrate, another characteristic contributing to its low oral bioavailability. P-glycoprotein-mediated drug efflux may also contribute to low brain drug levels. Intranasal nimodipine administration has been shown to achieve higher drug levels in brain as compared to the oral route6. The problem of low solubility of nimodipine was addressed by using 2-hydroxypropyl-β-cyclodextrin or diheptanoyl phosphatidylcholine as solubilizers to prepare water-soluble dosage forms of nimodipine. However, these approaches could not be used for intranasal delivery, since the therapeutic dose of nimodipine for intravenous (i.v.) administration is 0.5–2 mg7, 8, 9, and the effective nasal delivery volume in human is ≤400 µL (200 µL per nostril). This problem was resolved by developing a nimodipine-loaded microemulsion for intranasal delivery which achieved higher brain concentrations as compared to the oral route10. Microemulsion-based delivery systems have many characteristics which make them suitable for intranasal drug delivery. These include ease of preparation (due to spontaneous formation), thermodynamic stability, transparent and elegant appearance, increased drug loading, enhanced penetration through the biological membranes, increased bioavailability11, 12, and less inter- and intra-individual variability in drug pharmacokinetics13.
The aim of the present study was to develop an in situ gelling mucoadhesive microemulsion system for intranasal delivery of nimodipine. This formulation was designed to attain higher levels of nimodipine in the brain, overcome P-glycoprotein mediated drug efflux, reduce mucociliary drug clearance, and prolong drug release from the formulation. A thermosensitive polymer was used which converted from solution to gel upon intranasal administration. The bioadhesive property of the gel permits a prolonged adherence to the nasal mucosa, resulting in sustained drug release from the matrix. The new formulation was then characterized for various physicochemical parameters. A pharmacokinetic study of in situ gelling mucoadhesive microemulsion containing nimodipine was performed in rats to determine the concentration of nimodipine in the brain, plasma and nasal mucosa following intranasal administration. Results were compared with the pharmacokinetic profile of nimodipine achieved with suspension and microemulsion preparations.
2. Materials and methods
2.1. Materials
Nimodipine and nifedipine were obtained as a gift sample from Astron Research Ltd., Ahmedabad, India. Carbopol 934 P was obtained as a gift sample from Lubrizol Advanced Materials India Pvt. Ltd., Mumbai, India. Capmul MCM was purchased from Abitec Corporation, Janesville WI. Labrafil M 1944 CS, Peceol, Maisine 35-1, Labrasol and Transcutol P were purchased from Gattefosse, France. PEG 400, Pluronic F 68 and 127 were purchased from Sigma-Aldrich Co., MO, USA. Tween 80 was obtained from S.D. Fine Chemicals, Mumbai, India. Sodium hydroxide and potassium dihydrogen ortho-phosphate (analytical grade) were obtained from Qualigen Fine Chemicals, Mumbai, India. All the solvents and chemicals used for the study were of chromatographic grade and purchased from Qualigen Fine Chemicals, Mumbai, India. Heparin was purchased from Biological E. Ltd., Hyderabad, India. Heparinised capillaries were purchased from Himedia, Mumbai, India. Deionized water for HPLC was prepared in-house using a Milli-Q water purifier system (Millipore Elix, Germany).
2.2. Formulation development
2.2.1. Estimation of solubility of nimodipine in oils, surfactants and co-surfactants
The solubility of nimodipine was studied in various oils, surfactants and co-surfactants which were selected according to their applicability in nasal formulations. The oils screened for this study include Maisine 35-1, Labrafil 1944 CS, Capmul MCM, and Peceol. Labrasol and Tween 80 were screened as surfactants and PEG-400 and Transcutol P were screened as co-surfactants. The solubility of nimodipine in various oils, surfactants and co-surfactants was determined by adding an excess of nimodipine to a cap vial containing each of the selected vehicle (2 mL)14. The mixture was heated in a 40 °C water bath to facilitate the solubilization using a vortex mixer after sealing the vials. Mixtures were allowed to equilibrate in a shaker (Remi Equipments, India) at 25 °C for 48 h. The vials were centrifuged at 3000 rpm for 10 min after the equilibrium was achieved. The samples (500 µL of supernatant) were withdrawn and the content of nimodipine was quantified by UV–visible spectrophotometer (Systronics 2201, India) at 238 nm after dilution with methanol.
2.2.2. Preparation of phase diagrams
The pseudoternary phase diagrams of oil, surfactant, co-surfactant and water were constructed using a water titration method to obtain the concentrations of components. Surfactant was blended with co-surfactant in fixed weight ratios (1:1, 2:1 and 3:1). Aliquots of each surfactant and co-surfactant mixture (Smix) were then mixed with oil at room temperature (25 °C). For each, phase diagram, the ratio of oil to the Smix was varied as 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, 1:9 (w/w)10. Water was added drop-wise to each oil–Smix mixture under vigorous stirring and then kept aside. After equilibrium, the samples were visually checked and determined as being clear microemulsions, or emulsions, or gels. Phase diagrams were constructed using Triplot software version 4.1. Once the microemulsion region was identified, nimodipine microemulsion was prepared by dissolving nimodipine into the oil–Smix mixture, followed by addition of required amount of water with stirring. The resultant microemulsions were tightly stored at ambient temperature, and their physical stability was measured by observing periodically the occurrence of phase separation.
2.2.3. Screening of mucoadhesive and gelling excipients
Various mucoadhesive and gelling excipients were screened to ascertain thermoresponsive behavior of gelling and mucoadhesive excipients. Pluronic F 127 and Pluronic F 68 were screened as gelling agents while Carbopol 934 P, chitosan, sodium alginate and sodium CMC were screened as mucoadhesive agents. Thermoreversible gels of Pluronic F 127 and Pluronic F 68 were prepared by a cold method in which Pluronic was solubilized in distilled water and phosphate buffer (pH 6.7 and 7.4, individually) at 4 °C. The solutions were kept at 4 °C until they became clear. The temperature was increased gradually from 15 to 40 °C to assess gelling behavior, visually. The gelling nature was evaluated on a small volume of sample (500 µL). Similarly, the dispersions were prepared with varying concentrations of mucoadhesive agents15. Samples were weighed accurately, soaked in water, and kept aside at room temperature overnight. The resulting samples were then examined visually for gelling behavior at room temperature.
2.2.4. Preparation of in situ gelling mucoadhesive microemulsion
The in situ gelling mucoadhesive microemulsion was prepared by dissolving nimodipine in oil, followed by addition of surfactant–cosurfactant (Smix) mixture. Mucoadhesive and gelling excipients were dissolved in aqueous phase. The final drug–oil–Smix mixture was then titrated with the aqueous phase14 to obtain mucoadhesive microemulsion and stored at refrigerated temperature (4 °C) for further studies. Microemulsion of nimodipine was also prepared in similar manner without addition of mucoadhesive polymer.
2.3. Evaluation of formulation
2.3.1. Physicochemical evaluation
2.3.1.1. Photon correlation spectroscopy
Photon correlation spectroscopy (PCS) was employed to determine the mean particle size (PCS diameter) and size distribution (poly-dispersity index, PDI). The particle size of the optimized microemulsion was determined by light scattering based on laser diffraction using Malvern zeta-sizer ZS90 (Malvern Instruments, Inc., UK) after suitable dilution. The analysis was carried out for 2 min at room temperature at angle of detection of 90°. The apparatus consisted of a He–Ne laser (5 mW) and a sample holding cell of 5 mL capacity.
2.3.1.2. Zeta potential
The measurement of zeta potential has become inextricably connected with the characterization of colloidal dispersions, as this parameter is highly useful for the assessment of the physical stability of colloidal dispersions. Zeta potential was measured using Malvern Zetasizer ZS 90 (Malvern Instruments, UK).
2.3.1.3. pH, viscosity and gelling temperature
The pH of the developed formulation was measured using pH meter (LI-120, Eutech Instrument, Singapore). The rheological property of in situ gelling mucoadhesive microemulsion was measured by Ostwalds Viscometer (Brookefield Instrument, UK). The gelling temperature was measured by keeping the formulation on a magnetic stirrer with heater. The temperature was gradually increased and the viscosity was also measured simultaneously. The temperature at which the gel formation occurred with desired viscosity was recorded as the gelling temperature.
2.3.1.4. Percentage transmittance
The prepared formulation was diluted 10 times with continuous media and acetate buffer and the effect of dilution on globule size, percentage transmittance and phase separation was checked. The percentage transmittance of the developed formulation was measured at 650 nm using UV–visible spectrophotometer keeping distilled water as a blank10.
2.3.1.5. Stability assessment
The shelf-life stability of in situ gelling mucoadhesive microemulsion system, both as a function of time and storage temperature was routinely checked by visual inspection of the samples initially on a daily and later on a weekly basis. Stable systems were identified as those free of any physical change, such as phase separation, flocculation or precipitation. Particle size of the system upon storage was also monitored to assess system stability in terms of drastic changes in the mean particle size due to coalescence or aggregation. Stability was monitored at 4 °C, ambient temperature, 37 °C and 50 °C.
2.3.2. Entrapment efficiency and drug loading
The entrapment efficiency of nimodipine loaded microemulsion was determined by a centrifugation method. Briefly, 10 µL 0.1 mol/L HCl was added into drug-loaded microemulsion, followed by centrifugation for 10 min at 20,000 rpm. The supernatant was separated and the pellets were washed thrice with distilled water. The free drug concentration was determined by UV–visible spectrophotometric analysis at 238 nm. The entrapment efficiency and drug loading of nimodipine microemulsion were calculated as per equations given below. All the measurements were performed in triplicate.
| (1) |
| (2) |
where Wa is the amount of drug added to formulation, Ws is the amount of free drug, and Wl is the weight of oil phase.
2.3.3. Estimation of mucoadhesive strength
The mucoadhesive strength was determined for the optimized formulation using Brookfields texture analyzer-QTS where one end of texture analyzer was connected to 34 mm probe, to which 907.46 mm2 goat nasal mucosa (obtained from local slaughter house) was appended16. The other end contained 40 mL of in situ gelling mucoadhesive microemulsion. Nasal mucosa was exposed to formulation for about 30 s and then allowed to detach to obtain load versus time behavior of the formulation.
2.3.4. In vitro drug release
The drug release from in situ gelling mucoadhesive microemulsion, microemulsion and suspension was estimated. In vitro drug release studies were performed using a Franz diffusion cell (12.5 mL) at 37±0.5 °C for 8 h with stirring at 400 rpm. Dialysis membrane (cellulose membrane, molecular weight cut-off between 12,000 and 14,000 Da, pore size 2.4 nm) was presoaked in simulated nasal fluid for 15 min. Presoaked membrane was mounted between the donor and receiver compartment of the diffusion cell (cross sectional area 3.14 cm2) and clamped into position. The mucoadhesive microemulsion (0.5 mL) was uniformly spread over the membrane from the donor compartment side. The reservoir fluid (simulated nasal fluid) was maintained in the receiver compartment at 37±0.5 °C at the constant volume of 12.5 mL. Samples (1 mL) were withdrawn from the receiver compartment after every hour and replaced by an equivalent amount of temperature-equilibrated fresh media. The samples were analyzed, after adequate dilutions, at 238 nm on UV–visible spectrophotometer. The same procedure was repeated for nimodipine microemulsion and suspension.
2.3.5. Ex vivo drug permeation studies
The fresh nasal mucosa was carefully excised from the nasal cavity of goat obtained from the local slaughter house. Freshly excised goat nasal mucosa was dipped immediately in phosphate buffer (pH 6.4). Cartilages were removed properly and the mucosal membrane was isolated and washed with phosphate buffer17. The mucosal preparation was mounted on the receptor compartment of a Franz diffusion cell displaying a permeation area of 3.14 cm2. Simulated nasal fluid (12.5 mL) was added to the receptor compartment. The mucosal preparation was mounted on the diffusion cell with the mucosal and serosal sides facing the donor and receiver phases, respectively. A small teflon coated magnetic bead was placed in the receptor compartment for stirring the media. The donor and receptor compartments were securely closed with a stainless steel clip. The tissue sample was pre-incubated for 20 min, maintaining temperature at 37 °C. After a pre-incubation time of 20 min, in situ gelling mucoadhesive microemulsion was added to the donor compartment on the outer surface of the nasal mucosa. The permeation study was carried out for 8 h. The aliquots were withdrawn at 15, 30 min, 1, 2, 3, 4, 5, 6 and 7 h, diluted appropriately with simulated nasal fluid, and assayed for drug concentration using HPLC at 358 nm. The chromatographic conditions for HPLC analysis of nimodipine were the same as that for plasma and brain tissue analysis (see analytical method below). The percentage drug permeation was calculated from the calibration curve of nimodipine prepared in simulated nasal fluid. The same procedure was repeated for nimodipine microemulsion and suspension.
2.3.6. Histological evaluation of nasal mucosa
Histological studies were done on excised nasal mucosa from rats (n=6) dosed with in situ gelling mucoadhesive microemulsion of nimodipine, intranasally. The animals were euthanized and mucosa was collected from all the animals 24 h post-dose. Sections (5 µm) of the nasal mucosa were collected using a rotary microtome (Model 0126, Yorco, India), followed by staining with haematoxylin and eosin. Treated and untreated specimens were compared for histological appearance with an Olympus IX51 optical microscope (Olympus Optical Co., Ltd., Tokyo, Japan). Photographs were taken with an Olympus TL4 camera.
2.3.7. Nasal absorption and brain distribution studies
2.3.7.1. Animal studies
Male Sprague-Dawley rats weighing 250–280 g were obtained from the animal house of B.V. Patel PERD Centre, Ahmedabad. Animal housing and handling was performed in accordance with Good Laboratory Practice (GLP) mentioned in CPCSEA guidelines. Animal house is registered with the Committee for the Purpose of Control and Supervision of Experiments on Animals, Ministry of Social Justice and Empowerment, Government of India, vide registration No. 1661/PO/a/12/CPCSEA, dated 21/11/2012. All experimental protocols were reviewed and accepted by the Institutional Animal Ethics Committee prior to initiation of the experiment. The animals were housed in polypropylene cages (3 animal/cage) and placed in the experimental room where they were allowed to acclimatize for a week before experiment. A 10% air exhaust conditioning unit was maintained along with a relative humidity of 60±5% and a temperature of 25±3 °C in the animal house facility. A light/dark cycle of 10 h/14 h was also regulated for the experimental animals. Amrut certified rodent diet (Maharashtra Chakan Oil Mill Ltd.) and tap water (boiling hot water cooled to room temperature) were provided ad libitum to the experimental animals.
The animals were kept in fasting condition overnight before the experiment commenced. Animals were divided in three groups. The 1st group was dosed with nimodipine suspension, 2nd group with previously reported nimodipine microemulsion10, and 3rd with in situ gelling mucoadhesive microemulsion of nimodipine, intranasally. For the intranasal administration, 25–40 µL of the nasal formulations (concentration: 7.5 mg/mL for in situ gelling mucoadhesive microemulsion and nimodipine suspension and 6.4 mg/mL for reported nimodipine microemulsion) was administered via a polyethylene 10 tube attached to a microlitre syringe inserted 1 cm into each nostril of rat at a dose of 2 mg/kg. The blood samples of 0.5 mL were withdrawn from each rat through retro-orbital sinus at 0, 5, 15, 30 min, 1, 2, 4 and 6 h post-dose, collected into heparinized microcentrifuge tubes and centrifuged at 4000 rpm for 7 min at 4 °C. The animals were decapitated immediately after the blood collection and brain was excised at the above mentioned time points. Each brain tissue was quickly rinsed with saline and blotted up with filter paper to get rid of blood-taint and macroscopic blood vessels as much as possible. After weighing, the brain tissue samples were homogenized with 1 mL of saline at 10,000 rpm for 1 min over ice using a tissue homogenizer (Kinematica, Switzerland). The nasal mucosa was excised and homogenized in the similar process as that of brain samples. All the samples were kept frozen at −80 °C prior to HPLC analysis.
2.3.7.2. Analytical procedures
Nimodipine in brain tissue, plasma and nasal mucosa was assayed using a modified reported HPLC method10. To 200 µL brain tissue and nasal mucosa homogenates or 100 µL of plasma, 25 µL of nifedipine (20 µg/mL, internal standard) and 0.1 mL 1 mol/L NaOH were added and extracted with 1 mL extraction solvent (n-hexane/diethylether=50:50) by vortexing for 2 min. The samples were then centrifuged at 10,000 rpm for 10 min at 4 °C for tissue samples and at 2000 rpm for 10 min at 4 °C for plasma samples. This was followed by separation and transfer of the organic layer into evaporating tubes. The organic layer was evaporated under gentle stream of nitrogen till dryness. The samples were reconstituted in 100 μL mobile phase and 50 μL of it was injected onto HPLC system consisting of a PU-980 HPLC pump (Jasco, Hachioji, Tokyo, Japan) and UV-975 UV–visible detector (Jasco, Hachioji, Tokyo, Japan) set at 358 nm and a manual injection port. The data were analyzed using Borwin software version 1.3.
Chromatographic separation was achieved using a Purospher STAR column (250 mm×4.6 mm, 5 µm) maintained at room temperature. The mobile phase consisted of water:methanol:acetonitrile (25:35:40, v/v/v). The mobile phase was prepared daily and degassed before use. The flow-rate was maintained at 1.0 mL/min. Samples were quantified by determining the response (Peak areaDrug/Peak areaIS).
Calibration curves of nimodipine were prepared with plasma and respective tissues spiked with known amounts of the drug utilizing its HPLC peak area ratio to the internal standard. The linear range of nimodipine was 10–1000 ng/mL for plasma and tissue samples. The calibration curves were obtained by least square linear regression analysis using weight scheme as 1/c (c=concentration) with Borwin software version 1.3. All operations except data evaluation were carried out under thorough light shelter as nimodipine is sensitive to light-induced degradation18.
2.3.7.3. Data analysis
The maximum plasma concentration (Cmax) and the time to reach the maximum concentration (Tmax) were directly determined from the plasma concentration versus time curves. The area under the curve from 0 to t (AUC0–t) was calculated following the linear trapezoidal rule by summing the area from 0 to t h.
2.3.7.4. Statistical analysis
Statistical data analysis was performed using Graphpad Prism software where one-way analysis of variance followed by Bonferroni׳s test was applied and P≤0.05 was considered as the minimal level of significance unless indicated otherwise.
3. Results and discussion
3.1. Formulation development
3.1.1. Estimation of solubility of nimodipine in oils, surfactants and co-surfactants
Nimodipine exhibited good solubility in Capmul MCM (14.0 mg/mL) as compared to other oils. The solubility was better in Labrasol (28.20 mg/mL) and Transcutol P (47.54 mg/mL) which were chosen as surfactant and co-surfactant, respectively. The high solubility of nimodipine in Transcutol P reinforces its ability as a powerful solubilizing agent which is also seen for many other drugs. Fig. 1 shows the graphical representation of the solubility of nimodipine.
Figure 1.

Solubility of nimodipine in different oils, surfactants and cosurfactants. Data are expressed as mean±SD (n=6).
3.1.2. Preparation of phase diagrams
The selection of oils, surfactants and co-surfactants and the surfactant/co-surfactant ratios plays an important role in the formulation of microemulsion. The formulation of nimodipine microemulsion was optimized by evaluating the microemulsion regions using pseudoternary phase diagrams. A pseudoternary phase diagram of the investigated quaternary system water/Labrasol/Transcutol/Capmul MCM is presented in Fig. 2 and it shows phase diagrams with different S/CoS. The shaded areas indicate the clear o/w microemulsion system. The phase study revealed that the maximum proportion of oil was incorporated in the microemulsion system when the surfactant/co-surfactant ratio was 2:1.
Figure 2.

Pseudoternary phase diagram with varying ratios of the investigated quaternary system water/Labrasol/Transcutol/capmul MCM with (a) Smix in the ratio of 1:1 (b) Smix in the ratio of 2:1 and (c) Smix in the ratio of 3:1. The shaded areas indicate the clear o/w microemulsion system.
3.1.3. Screening of mucoadhesive and gelling excipients
Various mucoadhesive and gelling excipients were selected for their use in development of in situ gelling microemulsions. Thermoresponsive behavior was observed for Pluronics. Based on visual observations, gelling characteristics were satisfactory for both the grades of Pluronics. A blend of Pluronics showed satisfactory thermoreversible characteristics and was therefore used in the microemulsion. Gelling properties of various concentrations of Carbopol 934 P were assessed similarly. Carbopol 934 P with 0.3% (w/w) showed good gelling behavior and was selected as mucoadhesive excipient in the formulation. Table 1 shows gelling behavior of polymers at varying pH and temperatures.
Table 1.
Gelling behavior of polymers at varying pH values and temperatures.
| Excipients | Gelling at 40 °C | Gelling at room temperature | Gelling in phosphate buffer |
Gelling at small scale (500 µL) | |
|---|---|---|---|---|---|
| pH 6.7 | pH 7.4 | ||||
| Pluronic F 127 | + | NA | + | + | + |
| Pluronic F 68 | – | NA | + | + | + |
| Combination of Pluronics (F 127 20% w/w and F 68 8% w/w) | ++ | NA | ++ | ++ | + |
| Carbopol 934 P concentration (0.3% w/v) | NA | + | + | + | + |
| Carbopol 934 P concentration (0.5% w/v) | NA | + | + | + | + |
| Carbopol 934 P concentration (1.0% w/v) | NA | Excessive gelling and turbid formulation | Excessive gelling | Excessive gelling | + |
| Combination of Pluronics (F 127 20% w/w and F 68 8% w/w) and Carbopol 934 P concentration (0.3% w/v) | + | + | + | + | + |
–: Absence of gelling behavior; +: gelling at the surface; ++: satisfactory and thermoreversible behavior. NA: Not applicable.
3.1.4. Preparation of in situ gelling mucoadhesive microemulsion
The in situ gelling mucoadhesive microemulsion was optimized based on globule size and zeta potential. The optimized formulation, shown in Table 2, had the minimum particle size and maximum entrapment efficiency. The evaluated parameters are shown in Table 3. Fig. 3 shows the graph of particle size distribution. This optimized formulation was used for in vitro, ex vivo and in vivo studies.
Table 2.
Excipient composition of optimized in situ gelling mucoadhesive microemulsion.
| Role/category | Name | % w/w |
|---|---|---|
| Oil | Capmul MCM | 4 |
| Surfactant | Labrasol | 20 |
| Co-surfactant | Transcutol P | 10 |
| Aqueous phase | Water | 66 |
| Mucoadhesive | Carbopol 934P | 0.3 |
| Gelling excipient 1 | Pluronic F127 | 20 |
| Gelling excipient 2 | Pluronic F 68 | 8 |
Table 3.
Stability of microemulsion and in situ gelling mucoadhesive microemulsion after 30 days.
| Parameters | Microemulsion |
Mucoadhesive microemulsion |
||
|---|---|---|---|---|
| Initial | After 30 days | Initial | After 30 days | |
| Globule size (nm) | 150±4.5 | 170±6.5 | 250±6.7 | 280±9.7 |
| Zeta potential (mV) | −15±0.3 | −14±0.3 | −15±0.45 | −13±0.45 |
| pH | – | – | 6.7±0.2 | 6.7±0.2 |
| Viscosity (cP) | 27.5±0.3 | 26.5±0.4 | 35.5±0.7 | 36.5±0.7 |
| Gelling temperature (°C) | – | – | 28–32 | 28–32 |
| Transmittance (%) | 90±2 | 88±4 | 80±5 | 78±3 |
| Drug loading (%) | 70±2 | 69±2 | 68±2 | 65±2 |
Data are expressed as mean±SD, n=6.
Figure 3.
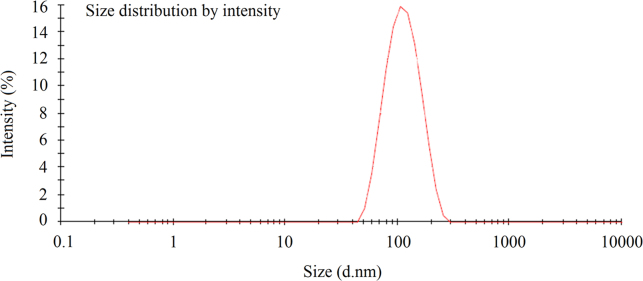
Representation of the obtained data for average particle size of in situ gelling mucoadhesive microemulsion.
3.2. Evaluation of formulation
3.2.1. Physicochemical evaluation
The physicochemical characteristics of the optimized in situ gelling mucoadhesive microemulsion are shown in Table 3. The pH was found to be 6.7, which lies within the pH range of the nasal cavity (5.5–7.5). The formulation has low viscosity and thus makes it pourable for nasal administration. The system demonstrated good transparency (>80%) with particle size in the nanometer range. The prepared formulation was physically stable for sustained periods with or without addition of nimodipine; no occurrence of phase separation or significant changes in particle size was observed. The stability of the formulation was further reinforced by the high zeta potential value. The results of the study inferred that solubility of nimodipine was enhanced in microemulsion based systems. The adhesiveness of the individual polymers and developed formulation are summarized in Table 4. It was observed that the mucoadhesive strength of the final formulation was more or less similar to that of the Carbopol 934 P. These results indicate that the formulation possessed sufficient mucoadhesiveness and to adhere to nasal mucosa upon administration.
Table 4.
Mucoadhesive strengths of individual polymers and developed formulation.
| Time (s) | Mucoadhesive strength (g) |
|||
|---|---|---|---|---|
| Carbopol 934 P | Pluronic127 | Pluronic 68 | Developed formulation | |
| 30 | 19±2.4 | 18±2.7 | 14±1.8 | 20±1.6 |
| 60 | 13±1.9 | 15±2.2 | 12±1.5 | 13±1.4 |
| 120 | 10±1.5 | 10±1.2 | 8±0.9 | 3±0.2 |
Data are expressed as mean±SD, n=6.
3.2.2. In vitro drug release behavior
The results of drug release studies from in situ gelling mucoadhesive microemulsion are shown in Fig. 4. The drug release behavior was studied in simulated nasal fluid. Incorporation of drug in microemulsion-based system improved drug solubilization and in vitro release, as compared to drug suspension. In situ gelling mucoadhesive microemulsion based system showed a high initial burst, due to the presence of Pluronic. Pluronic, a surfactant at low concentration, facilitates solubilization and higher drug release. The drug release at later stages was hampered due to extensive cross linking of Pluronic at higher temperature (temperature-dependent gelling). The presence of mucoadhesive agent (Carbopol 934 P) might have also restricted the drug release14. The higher initial burst release is desirable in case of immediate therapeutic intervention as in the case of patients suffering from stroke.
Figure 4.

In vitro release profile of nimodipine from different formulations. Data are expressed as mean±SD (n=6).
3.2.3. Ex vivo permeation studies
The permeation of nimodipine in situ gelling mucoadhesive microemulsion, microemulsion and suspension across the nasal mucosa is shown in Fig. 5. The drug suspension in water showed lowest permeability. Drug permeation improved in microemulsion based system, which may be attributed to the presence of surfactants and co-surfactants, which helps in solubilization. The higher permeation may be due to the transcellular uptake, high solubilization capacity as well as the potential for enhanced absorption of the microemulsion19. In situ gelling mucoadhesive microemulsion showed a high initial burst release, which may be due to the presence of Pluronic. Carbopol 934 P might have also contributed to the enhancement of drug absorption; this may be due to the opening of tight junctions, thereby facilitating transport of drugs via paracellular pathway14.
Figure 5.

Ex vivo permeation study of nimodipine from different formulations. Data are expressed as mean±SD (n=6).
3.2.4. Histological evaluation of nasal mucosa
Microscopic observations indicated that the optimized formulation had no significant effect on the histological appearance of mucosa. The epithelium layer was intact, with no alterations in basal membrane or in the superficial part of sub mucosa, and no necrosis was observed (Fig. 6). Thus the optimized formulation seemed to be safe with respect to nasal administration.
Figure 6.
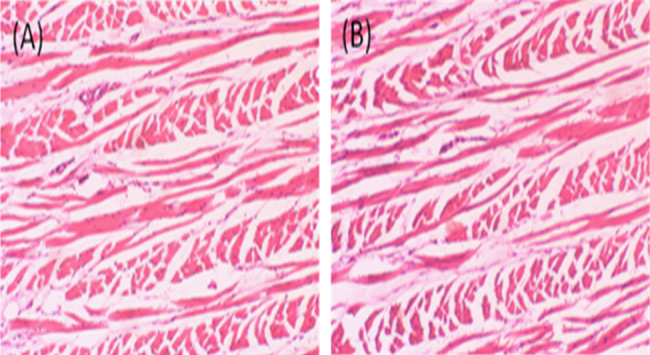
Histological photomicrographs of eosin–hematoxylin-stained nasal mucosa (10× magnification). (A) Control mucosa without application of formulation; (B) nasal mucosa after application of nimodipine in situ gelling mucoadhesive microemulsion.
3.2.5. Nasal absorption and brain distribution studies
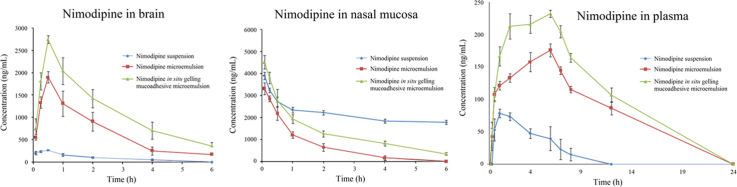
The intranasal route has been used for effective delivery of many therapeutic entities to the brain, including nimodipine as a suspension6, as well as in an o/w microemulsion system10. The microemulsion system served as an effective delivery system for nimodipine. However, an in situ gelling mucoadhesive microemulsion system was developed for nimodipine with an approach to achieve better residence time, reduced mucociliary clearance and better bioavailability as compared to the reported microemulsion system. The concentration of nimodipine in brain, nasal mucosa and plasma was determined after intranasal administration of nimodipine suspension, nimodipine microemulsion and nimodipine in situ gelling mucoadhesive microemulsion. Fig. 7a–c represents the mean brain tissue, nasal mucosa and plasma concentration–time profiles of nimodipine after intranasal administration of different formulations to rats at a dose of 2 mg/kg. Pharmacokinetic parameters of the different animal groups are shown in Table 5. Following intranasal administration, the profiles of nimodipine in brain displayed an initial absorption phase, with maximal concentrations within 30 min, followed by a rapid decline phase for all the three formulations. However, Cmax and AUC values attained with in situ gelling mucoadhesive microemulsion were significantly higher than those from the nimodipine microemulsion and nimodipine suspension treatments. The sampling time points for brain, nasal mucosa and plasma were initially up to 6 h. However, the plasma concentration was found to increase up to the last time point (6th hour). Based upon these results, another study was performed to develop an extended plasma profile of nimodipine upon intranasal administration of the three formulations. The blood sampling points for the second study were 0, 5, 15, 30 min, 1, 2, 4, 6, 7, 8, 12 and 24 h post-dose.
Figure 7.

Concentrations of nimodipine in (a) brain, (b) nasal mucosa and (c) plasma after intranasal administration of different formulations in rats. Data are expressed as mean±SD (n=6).
Table 5.
Pharmacokinetic parameters of nimodipine from different formulations upon intranasal administration in rats.
| Group |
Cmax (ng/mL) |
Tmax (min) |
AUC0–t (ng·min/mL) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Brain | Nasal mucosa | Plasma | Brain | Nasal mucosa | Plasma | Brain | Nasal mucosa | Plasma | |
| Nimodipine suspension | 272.24±38.37 | 3909.17±166.55 | 78.50±6.50 | 30.00±0.00 | 5.00±0.00 | 60.00±0.00 | 68,012.15±2649.03 | 754,934.10±2532.60 | 580,320.78±402.39 |
| Nimodipine microemulsion | 1901.98±125.98 | 3314.19±266.62 | 175.85±10.04 | 30.00±0.00 | 5.00±0.00 | 360.00±0.00 | 271,305.80±4093.20 | 239,447.70±3870.68 | 2943,244.60±2402.96 |
| Nimodipine in situ gelling mucoadhesive microemulsion | 2747.62±91.00 | 4514.94±307.56 | 232.32±5.63 | 30.00±0.00 | 5.00±0.00 | 360.00±0.00 | 438,745.40±5846.28 | 447,739.40±4496.12 | 3950,107.50±2543.47 |
Data are expressed as mean±SD, (n=6).
Nasal absorption of nimodipine from both the microemulsion formulations into systemic circulation exhibited sustained release, with maximal plasma concentrations achieved at 6 h. The maximum plasma concentration for nimodipine upon administration as a suspension was observed at 1 h. Nimodipine concentrations were measured in nasal mucosa with an objective to determine the amount of drug retained in nasal cavity. The results inferred that significant amount of drug was present in nasal mucosa, following administration of microemulsion formulations, indicating that a large fraction of drug remains at the site of application. This may be due to incorporation of mucoadhesive and gelling excipients. However, higher concentrations of nimodipine were present in the nasal mucosa of animals dosed with nimodipine suspension, likely to be due to the larger concentrations of the drug in suspension, not able to cross the nasal mucosa. This was also reflected in the corresponding low concentration in the brain and plasma of the animals dosed with nimodipine suspension. Statistical analysis showed a significant difference (P≤0.05) in nimodipine concentrations in the plasma, brain and nasal tissue upon intranasal administration of different formulations, inferred from AUC values. These results were in agreement with the previous reports indicating the existence of an alternative pathway for brain delivery of nimodipine by formulating as o/w microemulsion10.
Despite the potential of the nasal route for effective drug delivery, it is associated with many disadvantages, including mucociliary clearance, enzymatic activity, and the barrier nature of the epithelium combined with the mucus layer. The latter is especially problematic for the nasal absorption of high molecular-weight and hydrophilic drugs10, 20, 21, 22. Therefore, the use of absorption enhancers and/or the design of suitable formulations (e.g., microemulsions and mucoadhesive delivery systems) are necessary to enhance the nasal bioavailability of these drugs23, 24. The main finding of the present study is that the combination of the surfactant Pluronics (as an absorption enhancer) and Carbopol 934 P (as a mucoadhesive agent) can significantly increase the nose-to-brain delivery of nimodipine. While the exact mechanism of this effect has not been determined, it is known that mucoadhesive additives may improve drug absorption by reducing the mucociliary clearance rate and by increasing the residence time of the drug formulation in the nasal cavity25, 26. Moreover, the use of both Labrasol and Capmul MCM (both of which are reported to inhibit P-glycoprotein activity27, 28) might also be responsible for the observed improved bioavailability of nimodipine from the developed formulation. Furthermore, some mucoadhesive polymer-containing systems may directly change epithelial tight junctions and increase drug absorption and bioavailability23, 29.
4. Conclusions
The microemulsion system containing 4% (w/w) Capmul MCM, 30% Labrasol/Transcutol P (2:1) and water was found to be optimum for intranasal delivery of nimodipine. This preparation had a higher solubility of nimodipine (up to 7.5 mg/mL), as compared to earlier reported formulation (6.4 mg/mL10). The incorporation of 0.3% (w/w) of Carbopol 934 P was considered optimum for mucoadhesive microemulsion formulation. Sufficient gelling at physiological temperature was obtained with 20% (w/w) Pluronic F 127 combined with 8% (w/w) Pluronic F 68. The formulation was found to be physically stable for 3 months at ambient conditions. In vitro and ex vivo studies showed the drug release up to 6 h with a burst release at 30 min. The burst release was due to incorporation of Pluronics and the prolonged release up to 6 h was attributed to the mucoadhesives used in the formulation. In vivo pharmacokinetic studies in rats showed a higher brain and plasma concentrations of nimodipine from in situ gelling mucoadhesive microemulsion as compared to earlier reported formulation. It can be concluded from this study that, this formulation strategy may be tried for effective targeting of other therapeutic entities to CNS which are reported to have low bioavailability and poor brain penetration.
Acknowledgments
The authors wish to acknowledge NIPER – Ahmedabad (No. NIPERA160712) for providing all the facilities and their grant of a Junior Research Scholarship to Ms. Rudree Pathak to carry out this work; Astron Research Ltd., Ahmedabad, India and Lubrizol Advanced Materials India Pvt. Ltd., Mumbai, India for providing nimodipine, nifedipine and Carbopol 934 P as gift samples, respectively.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.

References
- 1.Pardridge W.M. The blood–brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frey W.H., II Bypassing the blood-brain barrier to delivery therapeutic agents to the brain and spinal cord. Drug Deliv Technol. 2002;2:46–49. [Google Scholar]
- 3.Thorne R.G., Pronk G.J., Padmanabhan V., Frey W.H., II Delivery of insulin like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127:481–496. doi: 10.1016/j.neuroscience.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 4.Dhanda D.S., Frey W.H., II, Leopold D., Kompella U.B. Approaches for drug deposition in the human olfactory epithelium. Drug Deliv Technol. 2005;5:64–72. [Google Scholar]
- 5.Illum L. Is nose-to-brain transport of drugs in man a reality? J Pharm Pharmacol. 2004;56:3–17. doi: 10.1211/0022357022539. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Q.Z., Jiang X.G., Wu C.H. Distribution of nimodipine in brain following intranasal administration in rats. Acta Pharmacol Sin. 2004;25:522–527. [PubMed] [Google Scholar]
- 7.Yoshida A., Yamamoto M., Iloh T., Irie T., Hirayama F., Uekama K. Utility of 2-hydroxypropyl-β-cyclodextrin in an intramuscular injectable preparation of nimodipine. Chem Pharm Bull. 1990;38:176–179. doi: 10.1248/cpb.38.176. [DOI] [PubMed] [Google Scholar]
- 8.Glertlova M., Ondrias K. Nifedipine and nimodipine solubility in the presence of diheptanoyl phosphatidylcholine. Pharmazie. 1992;47:861–863. [Google Scholar]
- 9.Mück W., Breuel H.P., Kuhlmann J. The influence of age on the pharmacokinetics of nimodipine. Int J Clin Pharmacol. 1996;34:293–298. [PubMed] [Google Scholar]
- 10.Zhang Q., Jiang X., Jiang W., Lu W., Su L., Shi Z. Preparation of nimodipine-loaded microemulsion for intranasal delivery and evaluation on the targeting efficiency to the brain. Int J Pharm. 2004;275:85–96. doi: 10.1016/j.ijpharm.2004.01.039. [DOI] [PubMed] [Google Scholar]
- 11.Tenjarla S.N. Microemulsions: an overview and pharmaceutical applications. Crit Rev Ther Drug Carrier Syst. 1999;16:461–521. [PubMed] [Google Scholar]
- 12.Lieberman H.A., Rieger M.M., Banker G.S. Marcel Decker; New York: 1996. Pharmaceutical dosage forms: disperse systems. [Google Scholar]
- 13.Kovarik J.M., Muller E.A., van Bree J.B., Tetzloff W., Kathz K. Reduced inter-and intra-individual variability in cyclosporine pharmacokinetics from a microemulsion formulation. J Pharm Sci. 1994;83:444–446. doi: 10.1002/jps.2600830336. [DOI] [PubMed] [Google Scholar]
- 14.Vyas T.K., Babbar A.K., Sharma R.K., Singh S., Misra A. Preliminary brain targeting studies on intranasal mucoadhesive microemulsions of Sumatriptan. AAPS Pharm Sci Tech. 2006;7:E1–E9. doi: 10.1208/pt070108. [DOI] [PubMed] [Google Scholar]
- 15.Ved P.M., Kim K. Poly(ethelene oxide/propelene oxide) copolymer thermoreversible gelling system for the enhancement of intranasal zidovudine to the brain. Int J Pharm Sci. 2011;411:1–9. doi: 10.1016/j.ijpharm.2011.02.040. [DOI] [PubMed] [Google Scholar]
- 16.Alexander A., Ajazuddin G.T., Sawrna S.P. Various evaluation parameters used for the evaluation of different mucoadhesive dosage forms: a review. Int J Drug Formul Res. 2011;2:1–26. [Google Scholar]
- 17.Mandal S., Mandal S.D. Design and development of carbamazepine mucoadhesive microemulsion for intranasal delivery: an ex-vivo study. Int J Pharma Sci Rev Res. 2010;3:56–60. [Google Scholar]
- 18.Muck W., Bode H. Bioanalytics of nimodipine – an overview of methods. Pharmazie. 1994;49:130–139. [PubMed] [Google Scholar]
- 19.Lawrence M.J., Rees G.D. Microemulsion based media as novel drug delivery system. Adv Drug Del Rev. 2000;45:89–121. doi: 10.1016/s0169-409x(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 20.Illum L. Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci. 2000;11:1–18. doi: 10.1016/s0928-0987(00)00087-7. [DOI] [PubMed] [Google Scholar]
- 21.Illum L. Nasal drug delivery: new developments and strategies. Drug Discov Today. 2002;7:1184–1189. doi: 10.1016/s1359-6446(02)02529-1. [DOI] [PubMed] [Google Scholar]
- 22.Illum L. Nasal drug delivery-possibilities, problems and solutions. J Control Release. 2003;87:187–198. doi: 10.1016/s0168-3659(02)00363-2. [DOI] [PubMed] [Google Scholar]
- 23.Wolburg H., Wolburg-Buchholz K., Sam H., Horvát S., Deli M.A., Mack A.F. Epithelial and endothelial barriers in the olfactory region of the nasal cavity of the rat. Histochem Cell Biol. 2008;130:127–140. doi: 10.1007/s00418-008-0410-2. [DOI] [PubMed] [Google Scholar]
- 24.Pringels E., Vervaet C., Verbeeck R., Foreman P., Remon J.P. The addition of calcium ions to starch/Carbopol(R) mixtures enhances the nasal bioavailability of insulin. Eur J Pharm Biopharm. 2008;68:201–206. doi: 10.1016/j.ejpb.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Jansson B., Hägerström H., Fransén N., Edsman K., Björk E. The influence of gellan gum on the transfer of fluorescein dextran across rat nasal epithelium in vivo. Eur J Pharm Biopharm. 2005;59:557–564. doi: 10.1016/j.ejpb.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 26.Ugwoke M.I., Agu R.U., Verbeke N., Kinget R. Nasal mucoadhesive drug delivery: background, applications, trends and future perspectives. Adv Drug Deliv Rev. 2005;57:1640–1665. doi: 10.1016/j.addr.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 27.Nornoo A.O., Zheng H., Lopes L.B., Johnson-Restrepo B., Kannan K., Reed R. Oral microemulsions of paclitaxel: in situ and pharmacokinetic studies. Eur J Pharm Biopharm. 2009;71:310–317. doi: 10.1016/j.ejpb.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 28.Cornaire G., Woodley J., Hermann P., Cloarec A., Arellano C., Houin G. Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int J Pharm. 2004;278:119–131. doi: 10.1016/j.ijpharm.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 29.Lehr C.M. Lectin-mediated drug delivery: the second generation of bioadhesives. J Control Release. 2000;65:19–29. doi: 10.1016/s0168-3659(99)00228-x. [DOI] [PubMed] [Google Scholar]