

Abstract
Background
Consumption of arsenic-contaminated drinking water adversely affects health. There is interindividual variation in arsenic metabolism efficiency, partially due to genetic variation in the arsenic methyltransferase (AS3MT) gene region.
Objectives
The goal of this study was to assess the overall contribution of genetic factors to variation in arsenic metabolism efficiency, as measured by the relative concentration of dimethylarsinic acid (DMA%) in urine.
Methods
Using data on genome-wide single nucleotide polymorphisms (SNPs) and urinary DMA% for 2,053 arsenic-exposed Bangladeshi individuals, we employed various SNP-based approaches for heritability estimation and polygenic modeling.
Results
Using data on all participants, the percent variance explained (PVE) for DMA% by all measured and imputed SNPs was 16% (p = 0.08), which was reduced to 5% (p = 0.34) after adjusting for AS3MT SNPs. Using information on close relatives only, the PVE was 63% (p = 0.0002), but decreased to 41% (p = 0.01) after adjusting for AS3MT SNPs. Regional heritability analysis confirmed 10q24.32 (AS3MT) as a major arsenic metabolism locus (PVE = 7%, p = 4.4 × 10–10), but revealed no additional regions. We observed a moderate association between a polygenic score reflecting elevated DMA% (composed of thousands of non-AS3MT SNPs) and reduced skin lesion risk in an independent sample (p < 0.05). We observed no associations for SNPs reported in prior candidate gene studies of arsenic metabolism.
Conclusions
Our results suggest that there are common variants outside of the AS3MT region that influence arsenic metabolism in Bangladeshi individuals, but the effects of these variants are very weak compared with variants near AS3MT. The high heritability estimates observed using family-based heritability approaches suggest substantial effects for rare variants and/or unmeasured environmental factors.
Citation
Gao J, Tong L, Argos M, Scannell Bryan M, Ahmed A, Rakibuz-Zaman M, Kibriya MG, Jasmine F, Slavkovich V, Graziano JH, Ahsan H, Pierce BL. 2015. The genetic architecture of arsenic metabolism efficiency: a SNP-based heritability study of Bangladeshi adults. Environ Health Perspect 123:985–992; http://dx.doi.org/10.1289/ehp.1408909
Introduction
Arsenic contamination of drinking water is a major public health problem in many countries, with > 137 million people in > 70 countries estimated to be exposed (International Agency for Research on Cancer 2004). Chronic exposure to arsenic has been linked to a wide array of health conditions (Rahman et al. 2009), including cancers of the lung, bladder, liver, kidney, and skin (Celik et al. 2008; Liu and Waalkes 2008; Mink et al. 2008; Yu et al. 2006; Yuan et al. 2010). Arsenic has also been associated with diabetes and cardiovascular disease, as well as neurological, reproductive, and respiratory conditions (Abhyankar et al. 2012; Golub et al. 1998; Huang et al. 2011; National Research Council 1999; Parvez et al. 2010; Vahidnia et al. 2007). Skin lesions are one of the earliest and most prevalent clinical manifestations of arsenic exposure and are considered the classical sign of arsenic toxicity (Yoshida et al. 2004).
Arsenic consumed in drinking water enters the blood stream as inorganic arsenic (iAs) [i.e., arsenite (AsIII) and arsenate (AsV)] and is metabolized primarily in the liver. According to the classical Challenger model of arsenic metabolism (Rehman and Naranmandura 2012), AsIII, the predominant form of iAs in Bangladesh, is methylated using arsenic (+3 oxidation state) methyltransferase (AS3MT) as the key enzyme and S-adenosylmethionine (SAM) as the methyl donor (Thomas et al. 2007) to produce monomethylarsonic acid (MMAV). After the reduction of MMAV to monomethylarsonous acid (MMAIII), a second methylation step produces dimethylarsinic acid (DMAV). Some DMAV can then be reduced to DMAIII (Thomas et al. 2004, 2007). The sum of urinary arsenic species (iAs, MMA, and DMA, including AsIII and AsV, MMAIII, and MMAV as well as DMAIII and DMAV) is regarded as a biomarker of recent inorganic arsenic exposure (Biggs et al. 1997), and the composition of urinary arsenic metabolites relative to total arsenic is believed to reflect arsenic methylation capacity. Higher arsenic methylation capacity is associated with lower risk for arsenical skin lesions, the classical sign of arsenic toxicity (Ahsan et al. 2007; Gao et al. 2011; Kile et al. 2011; Lindberg et al. 2007; Pierce et al. 2013; Valenzuela et al. 2005).
Familial aggregation and heritability analyses of arsenic metabolic profiles suggest that genetic factors influence interindividual variation in arsenic methylation capacity (Chung et al. 2002; Tellez-Plaza et al. 2013). Candidate gene association studies have implicated single nucleotide polymorphisms (SNPs) in the AS3MT gene region in arsenic methylation capacity (Agusa et al. 2011; Rodrigues et al. 2012; Schläwicke Engström et al. 2009), and a recent genome-wide association study (GWAS) confirmed this finding, showing two clear association signals in the AS3MT region (Pierce et al. 2012, 2013). In the GWAS, AS3MT was the only region in the genome that harbored variants showing associations of genome-wide significance. It remains unclear whether other SNPs that did not surpass the genome-wide significance threshold have weaker associations with arsenic methylation capacity.
In this study, we searched for evidence that additional genetic variants (other than the known AS3MT variants) influence arsenic methylation capacity, measured as the relative concentration of DMA in urine, using various approaches to evaluate polygenic susceptibility. We used SNP-based heritability methods to estimate the heritability in arsenic metabolism efficiency that is attributable to measured and imputed genome-wide SNPs, which we also refer to as the percent variance explained (PVE) by measured SNPs. We also used a “family-based” version of this method to estimate the full narrow-sense heritability, which reflects the additive contributions of all variants, including unmeasured rare variants (Yang et al. 2010; Zhou et al. 2013). We also conducted regional heritability analyses to estimate the heritability due to common SNPs in each segment of the genome (Nagamine et al. 2012). We used polygenic scoring (Purcell et al. 2007) to assess the polygenic contribution of arsenic metabolism variants that passed a significance threshold to skin lesion risk. In addition, we evaluated associations of 20 SNPs reported to be associated with arsenic methylation capacity in prior candidate gene studies.
Materials and Methods
Study population. The Health Effects of Arsenic Longitudinal Study (HEALS) is a large prospective cohort study of the health consequences of arsenic exposure. Details of the study design have been published previously (Ahsan et al. 2006a). A total of 11,746 healthy married adults (18–75 years of age) were enrolled in 2000–2002. At baseline, study interviewers collected information on demographic and lifestyle characteristics, conducted clinical examinations, and obtained biospecimens (blood and urine). Water samples from all 5,966 wells serving the 25-km2 study area were collected. Follow-up surveys and comprehensive physical examinations are conducted every 2 years. Approximately 1,000 of the HEALS subjects in this analysis were randomly selected to have their metabolites measured, and > 1,000 additional participants had metabolite data available due to prior ancillary studies. Only HEALS samples were used for the primary analyses described below, including chip heritability, regional heritability, and associations for candidate SNPs. For the polygenetic scoring analyses, in addition to all 2,053 HEALS samples with metabolite data, which constituted the training set, HEALS also contributed 1,285 controls and 24 skin lesion cases to the “testing” set.
The Bangladesh Vitamin E and Selenium Trial (BEST) is a 2 × 2 factorial randomized chemoprevention trial evaluating the effects of vitamin E and selenium supplementation on nonmelanoma skin cancer risk (Argos et al. 2013). A total of 7,000 individuals have been randomized to one of four treatment arms: vitamin E only (100 IU/day), L-selenomethionine only (200 μg/day), both vitamin E and selenium, and placebo. All participants were required to have existing arsenic-related skin lesions to be eligible. BEST participants are residents of roughly the same geographic area as HEALS, and the studies have very similar protocols, questionnaires, and biospecimen collection procedures. Biological samples, including blood and urine, were collected at baseline, along with clinical and covariate data. In this study, 1,990 BEST participants living in the Araihazar area were randomly selected for genotyping. These 1,990 skin lesions cases were included in the polygenic scoring analyses only, as a part of the “testing set.”
SNP genotyping. A sample of 5,499 individuals was selected from HEALS (n = 3,454) and BEST (n = 2,045) for genome-wide SNP genotyping using Illumina’s Cyto12 SNP array (~ 300,000 SNPs). For HEALS, DNA was extracted from clotted blood using Flexigene DNA kits (catalog no. 51204; from QIAGEN. For BEST, DNA was extracted from whole blood using the QIAamp 96 DNA Blood Kit (catalog no. 51161; QIAGEN). Genotyping methods and quality control have been described previously (Pierce et al. 2012, 2013). Genotyping was conducted in two batches. A total of 5,354 participants and 257,747 SNPs passed our quality control (QC) filters. QC included sample-level filters (excluding samples with call rate < 0.97, outlying heterozygosity values, and sex mismatches) and marker-level filters (excluding SNPs with call rates < 0.95 and Hardy–Weinberg p < 10–10, and minor allele frequency < 0.01) as described previously (Pierce et al. 2012, 2013). The total genotyping rate among eligible samples was 99.8%. Genotype imputation was conducted using MaCH (Markov Chain Haplotyping algorithm) software and the HapMap 3 GIH reference panel (Gujarati Indians in Houston), yielding genotypes for 1,211,988 SNPs after QC, restricting to SNPs with an imputation accuracy of r2 > 0.3 (Li et al. 2010).
Measurements of arsenic in water and urine. Urinary arsenic was measured at the Trace Metals Core Laboratory at Columbia University, which is a member of the QC program run by the Institute de Sante Publique du Quebec and uses their QC samples to standardize the measurements of urinary arsenic. The laboratory has consistently measured urinary arsenic concentration with correlation > 0.97 for blinded quality control samples. Urinary creatinine was measured by a colorimetric diagnostics kit (Sigma). The sum of urinary arsenic concentration was divided by creatinine to obtain creatinine-adjusted total arsenic concentration (micrograms per gram creatinine) (Basu et al. 2005). Of the 3,364 genotyped HEALS participants who passed QC, 2,053 had existing data on arsenic metabolites, as described previously (Ahsan et al. 2007). High-performance liquid chromatography (HPLC) was used to separate arsenobetaine, arsenocholine, iAsV, iAsIII, MMA, and DMA (Reuter et al. 2003), and their concentrations were measured using inductively coupled plasma-mass spectrometry with dynamic reaction cell. Because AsIII can oxidize to AsV during sample transport, storage, and preparation, we express total iAs as AsIII + AsV. iAs%, MMA%, and DMA% were calculated as percentages of the sum of urinary arsenic, after subtracting arsenobetaine and arsenocholine (forms of nontoxic organic arsenic from dietary sources) from total arsenic. Drinking water arsenic concentrations were analyzed by graphite furnace atomic absorption or, when concentrations were < 5 μg/L, by inductively coupled plasma-mass spectrometry (Cheng et al. 2004; van Geen et al. 2003).
Ascertainment of skin lesions. At baseline and at each follow-up interview of HEALS, skin lesions were ascertained using a structured protocol by trained study physicians. Through the whole-body examination, the study physician recorded the presence or absence of melanosis, leukomelanosis, and keratosis as well as their location, size, and shape. For the purposes of this analysis, skin lesion cases were defined as participants diagnosed with any type of skin lesion. In BEST, skin lesions were evaluated using protocols similar to those used in HEALS. All BEST participants had existing arsenic-related skin lesions at baseline.
Estimation of variance in arsenic metabolism efficiency explained by SNPs (i.e., heritability). Our analysis sample was composed of 2,053 HEALS participants with data on genome-wide SNPs and arsenic metabolites. Because HEALS participants are selected from a relatively small geographic region, a subset of our participants are genetically related to another participant, as described previously (Pierce et al. 2012). We used the DMA% variable to represent arsenic metabolism efficiency because it is strongly and inversely correlated with both iAs% and MMA% and because DMA% showed the strongest association with 10q24.32 variants in our prior GWAS (Pierce et al. 2012).
To estimate the PVE in DMA% by genetic factors (i.e., the “heritability”), we used a linear mixed model (LMM) approach originally proposed by Yang et al. (2010). This method is often referred to as genomic restricted maximum likelihood estimation (GREML). The general purpose of the GREML method is to estimate the proportion of variation in a phenotype that is due to all measured SNPs. This is fundamentally different from the traditional GWAS approach because our goal is to estimate variance explained by all SNPs, as opposed to testing individual SNPs for association with a phenotype. The GREML method is well established, has been described in detail, and exploits the fact that genotypic similarity (i.e., “relatedness,” measured using SNPs) will be correlated with phenotypic similarity for phenotypes that are influenced by genetic variation. The GREML method can utilize data on very distantly related individuals, individuals that are typically considered “unrelated” in traditional GWAS. A LMM is used to estimate the PVE by measured SNPs for a phenotype, as implemented in the Genome-wide Complex Trait Analysis (GCTA) software package (Yang et al. 2011). For a detailed description of the analytic method, see Supplemental Material, “LMM Analysis.”
To quantify genetic similarity between individuals, we constructed an n-by-n genetic relationship matrix (GRM), where n is the sample size (n = 2,053) and each element represents the degree to which a pair of individuals are related. Each element of the GRM is the genome-wide proportion of alleles shared IBS (identical by state) between two participants, as described by Yang et al. (2011), referred to here as “KIBS.” Under circumstances where the individuals are closely related, KIBS is a good estimate of allele sharing IBD, KIBD (identical by descent, where the shared alleles are inherited from the same ancestor), because KIBS will capture information on all variants in the genome. However, KIBS is not an ideal estimate of KIBD for distantly related individuals because it will primarily capture only information on measured SNPs (Zaitlen et al. 2013). Thus, SNP-based heritability estimates obtained from very distantly related individuals will tend to be lower than the true narrow-sense heritability.
Using the GREML method, we obtained three different types of PVE/heritability estimates. First we estimated PVE using all participants (using the full IBS-based GRM). Next, we estimated PVE using a modified GRM in which distant relatives were assumed to be unrelated (i.e., KIBS values < 0.05 were set to zero), producing an estimate of the IBD-based GRM (Zaitlen et al. 2013). This provides an estimate of the full narrow-sense heritability (h2), which includes the additive effects of all genetic variation, including nongenotyped variants, but it is prone to bias due to shared environment. This h2 estimate is comparable to those generated in family-based heritability studies. We also estimated the PVE after excluding individuals from close-relative pairs to produce a data set of only distantly related individuals (all KIBS < 0.05). This method provides an estimate of the heritability due to measured SNPs (hg2). The PVE estimate based on the full GRM (the first one described above) is essentially a mix of h2 and hg2. Covariates included in the LMM were age (continuous), sex (men vs. women), batch (batch 1 vs. 2, binary), water arsenic quartiles (categorical), smoking status (nonsmoker, former smoker, and current smoker, categorical), and body mass index (BMI; ≥ 10.2, 18.5–25.0, and ≥ 25.0 kg/m2, categorical). Twenty principal components (PCs; continuous) were included to minimize potential biases caused by population structure; PCs were generated using EIGENSTRAT (Patterson et al. 2006). PVE analyses were first run using only genotyped SNPs to construct the GRM, and then run again using both genotyped and imputed SNPs to construct the GRM.
Regional heritability analysis. We also conducted genome-wide regional heritability analysis using Regional Genomic Relationship Mapping (REACTA) software (Nagamine et al. 2012). This method quantifies the contribution of a specific genomic region to the heritability of a phenotype using a mixed model that includes random effects for a specific region and a residual whole-genome effect. The whole-genome additive effect was estimated by using all SNPs to construct the GRM, whereas the regional effect was estimated using only SNPs from a specific region to estimate a local GRM. We estimated the regional heritability across all 22 autosomes among all the non-close relatives (KIBS < 0.05, n = 1,338). We analyzed 4,924 100-SNP windows for the genotyped SNPs (with an overlap of 50 SNPs between neighboring windows) and 4,787 300-SNP windows for the imputed SNPs (with an overlap of 50 SNPs between neighboring windows). p-Values for the heritability estimates were assessed using a Bonferroni-corrected p threshold (0.05/4,924 or 4,787 = 1.0 × 10–5).
Polygenic scoring. Because AS3MT variants that influence arsenic metabolism also influence arsenical skin lesion risk (Ahsan et al. 2006b; Pierce et al. 2013), we assessed the potential polygenic contribution of arsenic metabolism–related SNPs to skin lesion risk. We generated a polygenic model for DMA% using data from all 2,053 HEALS participants with arsenic metabolite data. Using this model, we generated SNP-based polygenic scores in an independent data set of 2,014 skin lesion cases (1,990 BEST samples and 24 HEALS samples) and 1,285 controls from HEALS, and we tested the score for association with case–control status. To ensure that our polygenic scoring analysis was not influenced by the contributions of highly correlated SNPs, we pruned out 170,512 SNPs to produce a data set of genotyped SNPs with no pairwise r2 values > 0.2 using the --indep-pairwise command in PLINK (http://pngu.mgh.harvard.edu/~purcell/plink/). To ensure we were evaluating associations for non-AS3MT SNPs only, we further excluded 36 SNPs within ± 1 Mb of the AS3MT transcribed region. We also removed 9,852 SNPs with low minor allele frequencies (MAF < 0.05), resulting in 77,347 SNPs that were included in the polygenic score analysis.
The polygenic analysis was conducted as follows. Among the 2,053 participants with DMA% data (the “training set”), we estimated a beta coefficient for the association between the minor allele of each SNP and DMA%, adjusting for age (continuous), sex, concentration of water arsenic (continuous), and genotyping batch (binary). For each individual in the case–control sample (the “testing set”), a polygenic score was calculated as follows: Using the results from the analysis of the training set, we first set a p-value threshold to select SNPs for inclusion in the polygenic model. Several p-value thresholds were used: 10–4, 10–3, 0.01, 0.1, 0.3, and 0.5. For each SNP with a p-value below the threshold, the number of minor alleles carried by each individual in the testing set (0, 1, or 2) was multiplied by the SNP’s beta coefficient derived from the training set. For each individual, these weighted allele counts were then summed over all SNPs passing the threshold and divided by the total number of summed SNPs to produce the polygenic score (as implemented in the PLINK “score” command) (Purcell et al. 2007). These scores were then tested for association with skin lesion status using mixed linear regression models adjusting for sex, age, and genotyping batch implemented in genome-wide efficient mixed model association (GEMMA) (Zhou and Stephens 2012). To approximate the corresponding odds ratio (OR), the beta coefficient was first divided by [x(1 – x)], where x is the proportion of cases in our sample, in order to estimate the beta from a logistic model. This quantity was exponentiated to obtain an OR.
Analysis of candidate variants identified in prior studies. We identified 20 variants in 15 genes with previously reported associations with arsenic metabolism phenotypes (Agusa et al. 2012; Breton et al. 2007; Chen et al. 2012; Chiou et al. 1997; Engström et al. 2010, 2011; Paiva et al. 2010; Porter et al. 2010; Rodrigues et al. 2012; Schläwicke Engström et al. 2009; Steinmaus et al. 2007). We examined their associations with arsenic metabolism phenotypes in our GWAS data using mixed linear regression models adjusted by sex, age, and genotyping batch. For those candidate SNPs that were not genotyped in our study, we identified proxy SNPs with r2 > 0.8 that were genotyped in our study based on HapMap2 CHB (Han Chinese in Beijing, China) and JPT (Japanese in Tokyo, Japan) data.
Standard protocol approvals, registrations, and patient consent. The study protocol was approved by the institutional review boards of The University of Chicago, Columbia University, and the Bangladesh Medical Research Council, and all study participants provided informed consent.
Results
Characteristics of HEALS participants and their associations with DMA% are shown in Table 1. In a multivariate model, older age (> 50), female sex, and lower arsenic in either water or urine were associated with higher arsenic metabolism efficiency (higher DMA%). Compared with participants with BMI between 18.5 and 25.0, people of both higher and lower BMI had elevated DMA%. No association was observed for smoking status. BEST participants do not have DMA% data and were only involved in the polygenic scoring analyses; thus, these participants are not included in Table 1.
Table 1.
Characteristics of HEALS participants and their associations with arsenic metabolism efficiency, that is, DMA% (n = 2,053).a
| Characteristic | No. (%)b | DMA% | ||
|---|---|---|---|---|
| β | SE | p-Value | ||
| Sex | ||||
| Women | 1,015 (49.4) | Referent | ||
| Men | 1,038 (50.6) | –2.98 | 0.41 | < 0.0001 |
| Age | ||||
| 17–29 | 438 (21.3) | Referent | ||
| 30–39 | 589 (28.7) | –0.06 | 0.44 | 0.90 |
| 40–49 | 557 (27.1) | 0.16 | 0.46 | 0.74 |
| 50–70 | 469 (22.8) | 1.20 | 0.51 | 0.02 |
| Water arsenic (μg/L) | ||||
| Quartile 1 (0–8) | 514 (25.3) | Referent | ||
| Quartile 2 (9–49) | 503 (24.8) | –1.04 | 0.43 | 0.02 |
| Quartile 3 (50–127) | 507 (25.0) | –1.68 | 0.43 | < 0.0001 |
| Quartile 4 (128–864) | 507 (25.0) | –2.57 | 0.43 | < 0.0001 |
| Smoking status | ||||
| Never | 1,161 (56.6) | Referent | ||
| Ever | 892 (43.5) | –0.15 | 0.44 | 0.73 |
| BMI (kg/m2) | ||||
| 10.2–18.4 | 864 (42.1) | Referent | ||
| 18.5–24.9 | 1,059 (51.6) | 0.89 | 0.32 | 0.005 |
| 25.0–51.8 | 130 (6.3) | 2.22 | 0.65 | 0.0006 |
| Urinary arsenic adjusted for creatinine (μg/g) | ||||
| Quartile 1 (11–89) | 426 (20.9) | Referent | ||
| Quartile 2 (90–176) | 556 (27.2) | –0.19 | 0.44 | 0.66 |
| Quartile 3 (177–343) | 595 (29.2) | –1.25 | 0.43 | 0.004 |
| Quartile 4 (344–8,556) | 464 (22.7) | –2.74 | 0.46 | < 0.0001 |
| Prevalent skin lesion | ||||
| No | 1,974 (96.7) | Referent | ||
| Yes | 67 (3.3) | –0.59 | 0.87 | 0.49 |
| aβ, SE, and p-values were obtained from mixed linear regression models, adjusting for age, sex, genotyping batch, smoking, BMI, and arsenic concentrations in drinking water. bCategorical variables are presented as counts and percentages. | ||||
Two types of PVE estimates for DMA% are presented in Table 2, those based on genotyped SNPs only, and those based on genotyped and imputed SNP. Below we discuss the results obtained using genotyped and imputed SNPs. The PVE estimate for DMA% was 16% (p = 0.08) when using a GRM calculated from all 2,053 participants. After adjusting for sex, age, concentration of water arsenic (quartiles), genotyping batch, BMI, and smoking status, the estimate decreased to 12% (p = 0.16). After adjustment for the top 20 principal components, the estimate changed to 15% (p = 0.10). The PVE estimate decreased to 5% after adjusting for two SNPs in the AS3MT region identified in our prior GWAS (rs9527 and rs11191527) (Pierce et al. 2012, 2013).
Table 2.
Estimates of the percent variance explained (PVE) by genetic factors for DMA% obtained from linear mixed regression models.
| HEALS participants | Covariate adjustment | All genotyped SNPs (n = 257,747) | All genotyped and imputed SNPs (n = 1,211,988) | ||||
|---|---|---|---|---|---|---|---|
| PVE (%) | SE | p‑Value | PVE (%) | SE | p‑Value | ||
| All participantsa (n = 2,053) | No adjustment | 13 | 10 | 0.09 | 16 | 12 | 0.08 |
| Adjusted for covariatesb | 10 | 10 | 0.15 | 12 | 12 | 0.16 | |
| Further adjusted for PCsc | 11 | 11 | 0.16 | 15 | 12 | 0.10 | |
| Adjusting for two 10q24.32 SNPs | 3 | 10 | 0.36 | 5 | 12 | 0.34 | |
| All participants, defining distant relationships as “unrelated”d (n = 2,053) | No adjustment | 48 | 13 | 0.0004 | 63 | 16 | 0.0002 |
| Adjusted for covariatesb | 42 | 14 | 0.002 | 54 | 17 | 0.001 | |
| Adjusted for two 10q24.32 SNPs | 35 | 14 | 0.007 | 41 | 17 | 0.01 | |
| PCs, principal components. aUsing the full GRM, KIBS on all individuals. The PVE is in between the full narrow-sense heritability and the heritability due to measured SNPs. bCovariates including sex, age (continuous), concentration of water arsenic (quartiles), genotyping batch, BMI, and smoking status. cTwenty principal components as additional covariates to minimize inflation in significance testing caused by population stratification. dUsing a modified GRM, with KIBS set as 0 if KIBS < 0.05 (i.e., ignoring distant relationships); this approximates the KIBD for all individuals. The PVE corresponds to the full narrow-sense heritability. After eliminating close relative pairs from the data set (KIBS > 0.05), our sample size was too small (n = 1, 338) to generate a non-zero heritability estimate using GCTA. | |||||||
The PVE estimates for DMA% based on the modified GRM in which KIBS < 0.05 were set to zero (i.e., based on all participants and defining distant relationships as unrelated) was 63% (p = 0.0002). After adjusting for covariates, the estimate decreased to 54% (p = 0.001). This estimate decreased to 41% (p = 0.01) after adjusting for the two SNPs in the AS3MT region. After eliminating close relative pairs from the data set (no KIBS > 0.05), our sample size was too small (n = 1,338) to generate a non-zero heritability estimate using GCTA (data not shown).
However, we were able to use the data set of distant relatives (no KIBS > 0.05) to conduct regional heritability analysis. The most significant regional PVE estimates were obtained for two adjacent windows in the 10q24.32 region harboring AS3MT, and these accounted for approximately 7% of the variation in DMA% (p = 4.4 × 10–10 and 8.2 × 10–8) (Figure 1, w1 and w2). The regional heritability results based on genotyped data are the same as those based on imputed data (data not shown). After Bonferroni correction, no region showed a significant PVE estimate other than 10q24.32. Regional heritability analyses using the full data set (i.e., both close and distant relatives) produced very similar results (see Supplemental Material, Figure S1).
Figure 1.
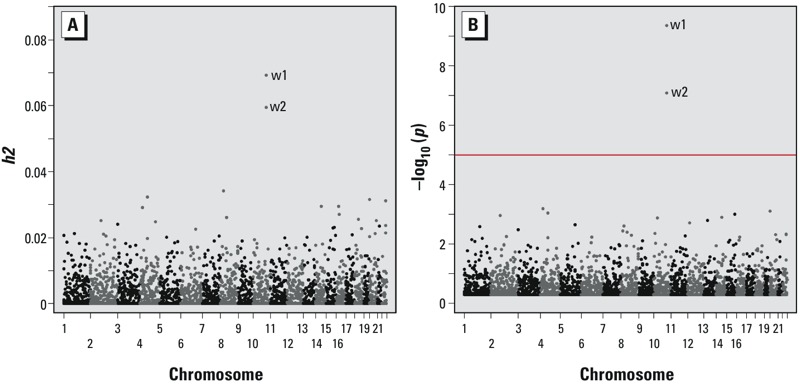
Regional heritability estimates (A) and corresponding p-values (B) for DMA%, excluding close relatives (KIBS < 0.05, n = 1,338). Estimates were obtained using measured and imputed SNPs with a window size 100 SNPs with a 50 SNP overlap between windows. A total of 4,924 tests were conducted. The red line represents the Bonferroni-corrected p-value threshold. The two adjacent/overlapping windows that surpass the p-value threshold reside in the 10q24.32 region and are labeled “w1” and “w2.”
Polygenic scores for DMA% were not significantly associated with skin lesion status when using p-value thresholds of p < 10–4, p < 10–3, and p < 0.01 (unless including AS3MT SNPs when using a threshold of < 10–4); however, polygenic scores for DMA% were associated with skin lesion status when p-value thresholds of < 0.1, < 0.3, and < 0.5 were used to construct the score (Table 3). For example, when a threshold of p < 0.5 was applied, the beta coefficient for the association polygenic scores for DMA% was –0.05 (p = 0.02), suggesting that many alleles that cause very small increases in DMA% are also inversely associated with skin lesions. The beta coefficients (and ORs) in Table 3 correspond to a one standard deviation change in the polygenic score.
Table 3.
Associations between polygenic scores for DMA% and skin lesion status.a
| p-Value threshold | non-AS3MT SNPs | AS3MT SNPs included | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of SNPs | Betab | SE | p‑Value | OR (95% CI)c | No. of SNPs | Betab | SE | p‑Value | OR (95% CI)c | |
| p < 10–4 | 11 | –0.007 | 0.007 | 0.34 | 0.97 (0.91, 1.03) | 13 | –0.02 | 0.007 | 0.01 | 0.93 (0.87, 0.98) |
| p < 10–3 | 87 | 0.001 | 0.008 | 0.89 | 1.00 (0.94, 1.07) | 89 | –0.005 | 0.008 | 0.53 | 0.98 (0.92, 1.05) |
| p < 0.01 | 801 | 0.01 | 0.01 | 0.22 | 1.06 (0.97, 1.15) | 803 | 0.01 | 0.01 | 0.35 | 1.04 (0.96, 1.14) |
| p < 0.1 | 7,810 | –0.03 | 0.02 | 0.04 | 0.87 (0.76, 0.99) | 7,812 | –0.04 | 0.02 | 0.03 | 0.86 (0.75, 0.99) |
| p < 0.3 | 23,281 | –0.04 | 0.02 | 0.04 | 0.85 (0.73, 0.99) | 23,283 | –0.04 | 0.02 | 0.03 | 0.85 (0.73, 0.98) |
| p < 0.5 | 38,644 | –0.05 | 0.02 | 0.02 | 0.82 (0.70, 0.96) | 38,646 | –0.05 | 0.02 | 0.01 | 0.82 (0.70, 0.96) |
| CI, confidence interval. aThe polygenic model was developed using all 2,053 participants with DMA% data and SNP data; the testing set was an independent set of 2,014 cases and 1,285 controls. bThe polygenic scores have been standardized, so the β coefficients from the mixed linear regression model correspond to a 1-SD change in the polygenic score, adjusted for sex, age, and genotyping batch. cOdds ratios (ORs) were calculated by dividing the beta coefficient by [x(1 – x)], where x is the proportion of cases in our sample, in order to estimate the beta from a logistic model; this quantity was exponentiated to obtain an OR. | ||||||||||
Table 4 shows associations between arsenic metabolite percentages and variants that have shown suggestive evidence of association with arsenic metabolites in prior candidate gene studies. No SNP showed significant evidence of association (p < 0.05) except for MTHFR-rs1801133 (p = 0.03 for MMA%) and DNMT1-rs2228612 (p = 0.04 for DMA% and p = 0.03 for iAs%). The directionality of association was consistent with the prior publications for MTHFR-rs1801133, but DNMT1-rs2228612 showed an association in the opposite direction to the association previously reported.
Table 4.
Association between arsenic metabolism phenotypes and candidate SNPs with associations reported in prior studies.
| Gene | Reported SNP | Function | Population | Sample size | References | p for associationa | ||
|---|---|---|---|---|---|---|---|---|
| DMA% | MMA% | iAs% | ||||||
| GSTO1-1 | rs4925 | Ala140Asp | Bangladesh | 1,800 | Rodrigues et al. 2012 | 0.46 | 0.94 | 0.60 |
| Taiwan | 247 | Chen et al. 2012 | ||||||
| GSTO2-2 | rs2297235 | UTR-5 | Bangladesh | 1,800 | Rodrigues et al. 2012 | 0.96 | 0.78 | 0.54 |
| rs156697 | Asn142Asp | Chile | 207 | Paiva et al. 2010 | 0.51 | 0.72 | 0.55 | |
| CHDH | rs9001b | Glu40Ala | Argentina | 111 | Schläwicke Engström et al. 2009 | 0.51 | 0.23 | 0.79 |
| rs7626693 | Intron | Argentina | 111 | 0.28 | 0.19 | 0.44 | ||
| MTRR | rs1801394c | Ile49Met | Argentina | 111 | ||||
| GLRX | rs3822751c | Intron | Argentina | 111 | ||||
| PRDX2 | rs10427027 | 3’-UTR | Argentina | 111 | 0.26 | 0.82 | 0.21 | |
| rs12151144b | Intron | Argentina | 111 | 0.26 | 0.82 | 0.21 | ||
| DNMT | rs16999593 | His97Arg | Argentina | 111 | 0.15 | 0.59 | 0.11 | |
| TXNRD2 | rs5746847b | Intron | Argentina | 108 | Engström et al. 2010 | 0.48 | 0.61 | 0.62 |
| Apex1 | rs1130409c | Asp148Glu | Argentina | 108 | ||||
| GSTM1 | Gene deletion | Bangladesh | 97 | Breton et al. 2007 | ||||
| Taiwan | 115 | Chiou et al. 1997 | ||||||
| Argentina | 170 | Steinmaus et al. 2007 | ||||||
| GSTT1 | Gene deletion | Taiwan | 115 | Chiou et al. 1997 | ||||
| MTHFR | rs1801133 | C677T | Argentina | 170 | Steinmaus et al. 2007 | 0.053 | 0.03 | 0.20 |
| rs1801131 | A1298C | Argentina | 170 | 0.75 | 0.14 | 0.78 | ||
| GSTP1 | rs1695 | Ile105Val | Vietnam | 190 | Agusa et al. 2012 | 0.85 | 0.52 | 0.49 |
| CBS | rs234709c | Intron | Argentina | 142 | Porter et al. 2010 | |||
| rs4920037 | Intron | Argentina | 142 | 0.25 | 0.21 | 0.50 | ||
| DNMT1 | rs2228612b | Intergenic | Bangladesh | 361 | Engström et al. 2011 | 0.04 | 0.31 | 0.03 |
| DNMT3B | rs6087990 | Intergenic | Bangladesh | 361 | 0.66 | 0.15 | 0.61 | |
| DNMT3B | rs2424913 | Intergenic | Bangladesh | 361 | 0.46 | 0.19 | 0.97 | |
| ap-Values are based on a linear mixed regression model (GEMMA) to account for relatedness; adjustments include sex, age, and genotyping batch. bUsing rs2241807 data as a proxy of rs9001 (r2 = 0.81); rs10427027, rs5748485, and rs11672909 are proxies for rs12151144, rs5746847, and rs2228612 (r2 = 1.0); r2 values are based on HapMap GIH data. cNo data on tag SNPs was available for rs1801394, rs3822751, rs1130409, and rs234709. | ||||||||
Discussion
In this study, we have assessed, for the first time, the overall contribution of genetic variation to arsenic methylation capacity, as measured by DMA%, using SNP-based heritability methods. The PVE estimates obtained using only information on close relatives were 63%, consistent with estimates obtained from a recent family-based study (59%) (Tellez-Plaza et al. 2013). When distantly related individuals were included in the analysis, PVE estimates were much lower (16%). Overall, these results suggest that the excess heritability observed in studies of close relatives is due to variants not represented on the genotyping/imputing array (e.g., rare variants) or bias due to shared environmental factors. In regional heritability analyses, the AS3MT region produced the only significant PVE estimate. These results suggest that among common variants captured on our genotyping platform, AS3MT SNPs are the major genetic determinants of arsenic methylation capacity in this population and that contributions of other common variants to methylation capacity are substantially weaker than the effects of AS3MT variants.
Prior studies have examined familial aggregation patterns for arsenic methylation phenotypes. A study of Chileans with long-term exposure to high levels of arsenic in drinking water demonstrated that urinary concentrations of iAs, MMA, and DMA, as well as their ratios, were strongly correlated among siblings (r = ~ 80), after adjustment for total urinary arsenic (Chung et al. 2002). The authors observed lower correlations for father–mother pairs (r = 0.18), suggesting that genetic factors influence arsenic metabolic profiles. A population-based study in Taiwan found that patients with Blackfoot disease, an arsenic-induced peripheral vascular disease, were three times more likely to have a family history of Blackfoot disease than community controls (Chen et al. 1988), also suggesting that genetic factors influence arsenic metabolism and/or toxicity. Our heritability estimate for DMA% based on close relatives (48% or 63%) is similar to the heritability estimated in a recent study of Native American families (59%) (Tellez-Plaza et al. 2013).
The association between variants in the 10q24.32/AS3MT region with arsenic methylation capacity is consistent across many candidate gene studies (Agusa et al. 2011; Rodrigues et al. 2012; Schläwicke Engström et al. 2009) and has recently been confirmed in a GWAS (Pierce et al. 2012, 2013). In addition to AS3MT, dozens of candidate genes have been examined for association with arsenic methylation capacity in prior studies, based on various hypotheses related to methyltransferases, one-carbon metabolism, and reduction reactions (Schläwicke Engström et al. 2009). SNPs in GSTO1, GSTO2 (Paiva et al. 2010; Rodrigues et al. 2012), MTHFR (Steinmaus et al. 2007), PNP (De Chaudhuri et al. 2008), GSTM1 (Breton et al. 2007; Chiou et al. 1997; Steinmaus et al. 2007), and several other genes have even been reported to be associated with arsenic methylation capacity (Agusa et al. 2012; Engström et al. 2010, 2011; Ghosh et al. 2008; Hernández and Marcos 2008; Porter et al. 2010; Schläwicke Engström et al. 2009). However, many of these studies were limited by small sample sizes, and the genetic variants under investigation have not shown a great deal of consistency across studies (e.g., Ahsan et al. 2007; Hernández and Marcos 2008; Xu et al. 2009). In this study, we observed evidence of replication for only one SNP with a previously reported association (MTHFR rs1801133), and this association is very weak compared with SNPs in the 10q24.32 region. However, lack of replication could potentially be due to the fact that genetic variants can have different patterns of association in different populations because of population differences in linkage disequilibrium (LD) with causal variants, differences in allele frequency, and/or differences in the prevalence of environmental exposures that interact with the variant to influence the phenotype of interest.
In the present study, we used four different modeling approaches to estimate heritability (i.e., PVE). First, we estimated overall heritability using the full IBS-based covariance matrix for all study participants, including closely related individuals. This estimate should fall between the full narrow-sense heritability and the heritability due to measured SNPs (hg2). Second, we estimated heritability by focusing on close relatives, using only an IBD-based kinship matrix assuming zero relatedness between pairs of individuals whose estimated relatedness was < 0.05. This is an estimate of the full narrow-sense heritability (h2), capturing contributions of rare variants, but this estimate is prone to bias due to shared environmental factors. Third, we estimated heritability due to genotyped SNPs (hg2) using the IBS-based matrix constructed after removing close relatives from the data set. This is a more conservative approach to estimating heritability, as the presense of close relatives may cause bias due to shared environmental exposures. Fourth, we conducted regional heritability analyses, obtaining many heritability estimates corresponding to many small regions of the genome. Although the low heritability observed may reflect a limited contribution of common variants to arsenic methylation capacity, we do not have ideal power to accurately estimate modest heritability values. Excluding close relatives is an impotant consideration when conducting SNP-based heritiability estimation because relatives may be more likely to share similar (unmeasured) environmental exposures that influence the phenotype, potentially inflating heritability estimates (Yang et al. 2010). We have a substantial number of related individuals in our analysis, with only 1,338 samples remaining after removing related pairs with a relationship coefficient > 0.05.
The polygenic scoring analyses suggests that there may be common SNPs with weak effects on arsenic metabolism outside of the AS3MT region. For these analyses we assumed that SNPs influencing arsenic metabolism will also influence skin lesion risk. This assumption holds for DMA%-associated variants in the AS3MT region and is supported by multiple studies reporting an inverse association between DMA% and skin lesion risk (Ahsan et al. 2007; Gao et al. 2011; Kile et al. 2011; Lindberg et al. 2007; Pierce et al. 2013; Valenzuela et al. 2005). The observation that associations are present only when less stringent p-value thresholds are used implies that there are many variants with very weak effects on arsenic metabolism that also influence skin lesion risk. In order to identify such variants with very weak effects, association studies with larger sample sizes would be needed.
Arsenic-induced skin lesions are also influenced by many nongenetic factors, and we have assessed associations for several such factors in prior studies of this population. For example, we have reported that skin lesion risk is associated with arsenic, BMI (Argos et al. 2011), dietary patterns (Pierce et al. 2011), smoking, and occupational risk factors (Melkonian et al. 2011). Although these associations are clearly important as potential determinants of arsenic toxicity, we do not consider them in our polygenic scoring analysis because they are not potential confounders of the association between a SNP (or a SNP score) and skin lesion status.
In this work, we chose to use DMA% as a measure of arsenic methylation capacity. Alternative measures of methylation capacity include iAs%, MMA%, and metabolite ratios, which are highly correlated with DMA%. We chose to present results for DMA%, in part, because in our prior GWAS (Pierce et al. 2012), DMA% showed the strongest associations with SNPs in the AS3MT region compared with iAs%, MMA%, and metabolite ratios. Furthermore, PVE estimates for MMA% or iAs% were similar to those for DMA%, but somewhat weaker in magnitude (results upon request).
Although our study is the first SNP-based heritability study of arsenic methylation capacity, it has several limitations. First, our total sample size for metabolism study was only 2,053, which is relatively small for SNP-based heritability estimation. This hindered our ability to estimate heritability with high precision and to estimate heritability using a smaller, “unrelated” subset of study participants. Larger sample size, as well as denser SNP measurements (such as genome-wide sequencing), would enhance our ability to estimate heritability and conduct polygenic scoring analysis. We were able to measure arsenic metabolites only in urine and not in other relevant specimens such as blood, although this is a limitation of most studies of arsenic metabolism.
Conclusions
In this SNP-based heritability study of arsenic metabolism efficiency, we estimated total narrow-sense heritability for DMA% to be 48–63% (using data on close relatives only), but the heritability due to measured SNPs was substantially lower (13–16%). Because the larger narrow-sense (“family-based”) estimate captures the effects of measured common variants and unmeasured rare variants (as well as shared environmental influences), and the smaller “unrelated” estimate captures the effects of measured common variants only, our results suggests that rare variants (e.g., AS3MT coding variants) and/or unknown or poorly measured environmental/lifestyle factors that cluster in families (e.g., dietary factors) make a substantial contribution of interindividual variation in arsenic methylation capacity. Moderate associations between a polygenic score for DMA% (composed of non-AS3MT SNPs) and skin lesion status were detected, suggesting the existence of additional common variants that have very weak effects on arsenic metabolism efficiency. Our regional heritability analyses did not detect additional susceptibility regions, consistent with the hypothesis that the effects of common variants outside of the 10q24.32/AS3MT region are likely to be very weak. Although these findings may not apply to other populations, our results suggest that future studies of Bangladeshi individuals with comparable exposure levels will have to have large sample sizes in order to detect associations between DMA% and common SNPs outside of the AS3MT region. Studies of rare variants may reveal genetic effects that contribute to the high heritability estimates observed in our family-based heritability analyses.
This work enhances our knowledge regarding the genetic architecture of arsenic methylation capacity in a population where the public health impact of arsenic exposure is substantial. Understanding the determinants of arsenic metabolism is critical because metabolism efficiency will likely affect the internal (or biological effective) dose, which will in turn impact risk for all arsenic-related health conditions. Understanding these determinants will improve our ability to identify high-risk subgroups and develop interventions to enhance metabolism efficiency or reduce exposure.
Supplemental Material
Footnotes
This work was supported by National Institutes of Health grants R01ES020506, P42ES010349, R01CA102484, R01CA107431, and P30CA014599.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare they have no actual or potential competing financial interests.
References
- Abhyankar LN, Jones MR, Guallar E, Navas-Acien A.2012Arsenic exposure and hypertension: a systematic review. Environ Health Perspect 120494–500.; 10.1289/ehp.1103988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agusa T, Fujihara J, Takeshita H, Iwata H. Individual variations in inorganic arsenic metabolism associated with AS3MT genetic polymorphisms. Int J Mol Sci. 2011;12:2351–2382. doi: 10.3390/ijms12042351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agusa T, Kunito T, Tue NM, Lan VT, Fujihara J, Takeshita H, et al. Individual variations in arsenic metabolism in Vietnamese: the association with arsenic exposure and GSTP1 genetic polymorphism. Metallomics. 2012;4:91–100. doi: 10.1039/c1mt00133g. [DOI] [PubMed] [Google Scholar]
- Ahsan H, Chen Y, Kibriya MG, Slavkovich V, Parvez F, Jasmine F, et al. Arsenic metabolism, genetic susceptibility, and risk of premalignant skin lesions in Bangladesh. Cancer Epidemiol Biomarkers Prev. 2007;16:1270–1278. doi: 10.1158/1055-9965.EPI-06-0676. [DOI] [PubMed] [Google Scholar]
- Ahsan H, Chen Y, Parvez F, Argos M, Hussain AI, Momotaj H, et al. Health Effects of Arsenic Longitudinal Study (HEALS): description of a multidisciplinary epidemiologic investigation. J Expo Sci Environ Epidemiol. 2006a;16:191–205. doi: 10.1038/sj.jea.7500449. [DOI] [PubMed] [Google Scholar]
- Ahsan H, Chen Y, Parvez F, Zablotska L, Argos M, Hussain I, et al. Arsenic exposure from drinking water and risk of premalignant skin lesions in Bangladesh: baseline results from the Health Effects of Arsenic Longitudinal Study. Am J Epidemiol. 2006b;163:1138–1148. doi: 10.1093/aje/kwj154. [DOI] [PubMed] [Google Scholar]
- Argos M, Kalra T, Pierce BL, Chen Y, Parvez F, Islam T, et al. A prospective study of arsenic exposure from drinking water and incidence of skin lesions in Bangladesh. Am J Epidemiol. 2011;174:185–194. doi: 10.1093/aje/kwr062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argos M, Rahman M, Parvez F, Dignam J, Islam T, Quasem I, et al. Baseline comorbidities in a skin cancer prevention trial in Bangladesh. Eur J Clin Invest. 2013;43:579–588. doi: 10.1111/eci.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Som A, Ghoshal S, Mondal L, Chaubey RC, Bhilwade HN, et al. Assessment of DNA damage in peripheral blood lymphocytes of individuals susceptible to arsenic induced toxicity in West Bengal, India. Toxicol Lett. 2005;159:100–112. doi: 10.1016/j.toxlet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Biggs ML, Kalman DA, Moore LE, Hopenhayn-Rich C, Smith MT, Smith AH. Relationship of urinary arsenic to intake estimates and a biomarker of effect, bladder cell micronuclei. Mutat Res. 1997;386:185–195. doi: 10.1016/s1383-5742(97)00012-4. [DOI] [PubMed] [Google Scholar]
- Breton CV, Kile ML, Catalano PJ, Hoffman E, Quamruzzaman Q, Rahman M, et al. 2007GSTM1 and APE1 genotypes affect arsenic-induced oxidative stress: a repeated measures study. Environ Health 639; 10.1186/1476-069X-6-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik I, Gallicchio L, Boyd K, Lam TK, Matanoski G, Tao X, et al. Arsenic in drinking water and lung cancer: a systematic review. Environ Res. 2008;108:48–55. doi: 10.1016/j.envres.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Wu MM, Lee SS, Wang JD, Cheng SH, Wu HY. Atherogenicity and carcinogenicity of high-arsenic artesian well water. Multiple risk factors and related malignant neoplasms of blackfoot disease. Arteriosclerosis. 1988;8:452–460. doi: 10.1161/01.atv.8.5.452. [DOI] [PubMed] [Google Scholar]
- Chen JW, Wang SL, Wang YH, Sun CW, Huang YL, Chen CJ, et al. Arsenic methylation, GSTO1 polymorphisms, and metabolic syndrome in an arseniasis endemic area of southwestern Taiwan. Chemosphere. 2012;88:432–438. doi: 10.1016/j.chemosphere.2012.02.059. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Zheng Y, Mortlock R, van Geen A. Rapid multi-element analysis of groundwater by high-resolution inductively coupled plasma mass spectrometry. Anal Bioanal Chem. 2004;379:512–518. doi: 10.1007/s00216-004-2618-x. [DOI] [PubMed] [Google Scholar]
- Chiou HY, Hsueh YM, Hsieh LL, Hsu LI, Hsu YH, Hsieh FI, et al. Arsenic methylation capacity, body retention, and null genotypes of glutathione S-transferase M1 and T1 among current arsenic-exposed residents in Taiwan. Mutat Res. 1997;386:197–207. doi: 10.1016/s1383-5742(97)00005-7. [DOI] [PubMed] [Google Scholar]
- Chung JS, Kalman DA, Moore LE, Kosnett MJ, Arroyo AP, Beeris M, et al. 2002Family correlations of arsenic methylation patterns in children and parents exposed to high concentrations of arsenic in drinking water. Environ Health Perspect 110729–733.; 10.1289/ehp.10581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Chaudhuri S, Ghosh P, Sarma N, Majumdar P, Sau TJ, Basu S, et al. 2008Genetic variants associated with arsenic susceptibility: study of purine nucleoside phosphorylase, arsenic (+3) methyltransferase, and glutathione S-transferase omega genes. Environ Health Perspect 116501–505.; 10.1289/ehp.10581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engström KS, Vahter M, Lindh C, Teichert F, Singh R, Concha G, et al. Low 8-oxo-7,8-dihydro-2’-deoxyguanosine levels and influence of genetic background in an Andean population exposed to high levels of arsenic. Mutat Res. 2010;683:98–105. doi: 10.1016/j.mrfmmm.2009.10.014. [DOI] [PubMed] [Google Scholar]
- Engström K, Vahter M, Mlakar SJ, Concha G, Nermell B, Raqib R, et al. 2011Polymorphisms in arsenic(+III oxidation state) methyltransferase (AS3MT) predict gene expression of AS3MT as well as arsenic metabolism. Environ Health Perspect 119182–188.; 10.1289/ehp.1002471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Yu J, Yang L. Urinary arsenic metabolites of subjects exposed to elevated arsenic present in coal in Shaanxi Province, China. Int J Environ Res Public Health. 2011;8:1991–2008. doi: 10.3390/ijerph8061991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, Banerjee M, Giri AK, Ray K. Toxicogenomics of arsenic: classical ideas and recent advances. Mutat Res. 2008;659:293–301. doi: 10.1016/j.mrrev.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Golub MS, Macintosh MS, Baumrind N. Developmental and reproductive toxicity of inorganic arsenic: animal studies and human concerns. J Toxicol Environ Health B Crit Rev. 1998;1:199–241. doi: 10.1080/10937409809524552. [DOI] [PubMed] [Google Scholar]
- Hernández A, Marcos R. Genetic variations associated with interindividual sensitivity in the response to arsenic exposure. Pharmacogenomics. 2008;9:1113–1132. doi: 10.2217/14622416.9.8.1113. [DOI] [PubMed] [Google Scholar]
- Huang CF, Chen YW, Yang CY, Tsai KS, Yang RS, Liu SH. Arsenic and diabetes: current perspectives. Kaohsiung J Med Sci. 2011;27:402–410. doi: 10.1016/j.kjms.2011.05.008. [DOI] [PubMed] [Google Scholar]
- International Agency for Research on Cancer (IARC). Arsenic in drinking-water. IARC Monogr Eval Carcinog Risk Hum. 2004;84:39–267. [PMC free article] [PubMed] [Google Scholar]
- Kile ML, Hoffman E, Rodrigues EG, Breton CV, Quamruzzaman Q, Rahman M, et al. A pathway-based analysis of urinary arsenic metabolites and skin lesions. Am J Epidemiol. 2011;173:778–786. doi: 10.1093/aje/kwq427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg AL, Kumar R, Goessler W, Thirumaran R, Gurzau E, Koppova K, et al. 2007Metabolism of low-dose inorganic arsenic in a central European population: influence of sex and genetic polymorphisms. Environ Health Perspect 1151081–1086.; 10.1289/ehp.10026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Waalkes MP. Liver is a target of arsenic carcinogenesis. Toxicol Sci. 2008;105:24–32. doi: 10.1093/toxsci/kfn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkonian S, Argos M, Pierce BL, Chen Y, Islam T, Ahmed A, et al. A prospective study of the synergistic effects of arsenic exposure and smoking, sun exposure, fertilizer use, and pesticide use on risk of premalignant skin lesions in Bangladeshi men. Am J Epidemiol. 2011;173:183–191. doi: 10.1093/aje/kwq357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink PJ, Alexander DD, Barraj LM, Kelsh MA, Tsuji JS. Low-level arsenic exposure in drinking water and bladder cancer: a review and meta-analysis. Regul Toxicol Pharmacol. 2008;52:299–310. doi: 10.1016/j.yrtph.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Nagamine Y, Pong-Wong R, Navarro P, Vitart V, Hayward C, Rudan I, et al. 2012Localising loci underlying complex trait variation using Regional Genomic Relationship Mapping. PloS One 7e46501; 10.1371/journal.pone.0046501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council. Arsenic in Drinking Water. Washington, DC:National Academies Press. 1999. Available: http://books.nap.edu/openbook.php?isbn=0309063337 [accessed 19 February 2015]
- Paiva L, Hernández A, Martínez V, Creus A, Quinteros D, Marcos R. Association between GSTO2 polymorphism and the urinary arsenic profile in copper industry workers. Environ Res. 2010;110:463–468. doi: 10.1016/j.envres.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Parvez F, Chen Y, Brandt-Rauf PW, Slavkovich V, Islam T, Ahmed A, et al. A prospective study of respiratory symptoms associated with chronic arsenic exposure in Bangladesh: findings from the Health Effects of Arsenic Longitudinal Study (HEALS). Thorax. 2010;65:528–533. doi: 10.1136/thx.2009.119347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D.2006Population structure and eigenanalysis. PLoS Genet 2e190; 10.1371/journal.pgen.0020190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce BL, Argos M, Chen Y, Melkonian S, Parvez F, Islam T, et al. Arsenic exposure, dietary patterns, and skin lesion risk in Bangladesh: a prospective study. Am J Epidemiol. 2011;173:345–354. doi: 10.1093/aje/kwq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce BL, Kibriya MG, Tong L, Jasmine F, Argos M, Roy S, et al. 2012Genome-wide association study identifies chromosome 10q24.32 variants associated with arsenic metabolism and toxicity phenotypes in Bangladesh. PLoS Genet 8e1002522; 10.1371/journal.pgen.1002522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce BL, Tong L, Argos M, Gao J, Farzana J, Roy S, et al. Arsenic metabolism efficiency has a causal role in arsenic toxicity: Mendelian randomization and gene-environment interaction. Int J Epidemiol. 2013;42:1862–1871. doi: 10.1093/ije/dyt182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter KE, Basu A, Hubbard AE, Bates MN, Kalman D, Rey O, et al. Association of genetic variation in cystathionine-β-synthase and arsenic metabolism. Environ Res. 2010;110:580–587. doi: 10.1016/j.envres.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MM, Ng JC, Naidu R. Chronic exposure of arsenic via drinking water and its adverse health impacts on humans. Environ Geochem Health. 2009;31(suppl 1):189–200. doi: 10.1007/s10653-008-9235-0. [DOI] [PubMed] [Google Scholar]
- Rehman K, Naranmandura H. Arsenic metabolism and thioarsenicals. Metallomics. 2012;4:881–892. doi: 10.1039/c2mt00181k. [DOI] [PubMed] [Google Scholar]
- Reuter W, Davidowski L, Neubauer K, Di Bussolo J. Speciation of Five Arsenic Compounds in Urine by HPLC/ICP-MS. 2003. Available: http://www.perkinelmer.com/PDFs/Downloads/app_speciationfivearseniccompounds.pdf [accessed 20 August 2015]
- Rodrigues EG, Kile M, Hoffman E, Quamruzzaman Q, Rahman M, Mahiuddin G, et al. GSTO and AS3MT genetic polymorphisms and differences in urinary arsenic concentrations among residents in Bangladesh. Biomarkers. 2012;17:240–247. doi: 10.3109/1354750X.2012.658863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schläwicke Engström K, Nermell B, Concha G, Strömberg U, Vahter M, Broberg K. Arsenic metabolism is influenced by polymorphisms in genes involved in one-carbon metabolism and reduction reactions. Mutat Res. 2009;667:4–14. doi: 10.1016/j.mrfmmm.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Steinmaus C, Moore LE, Shipp M, Kalman D, Rey OA, Biggs ML, et al. Genetic polymorphisms in MTHFR 677 and 1298, GSTM1 and T1, and metabolism of arsenic. J Toxicol Environ Health A. 2007;70:159–170. doi: 10.1080/15287390600755240. [DOI] [PubMed] [Google Scholar]
- Tellez-Plaza M, Gribble MO, Voruganti VS, Francesconi KA, Goessler W, Umans JG, et al. 2013Heritability and preliminary genome-wide linkage analysis of arsenic metabolites in urine. Environ Health Perspect 121345–351.; 10.1289/ehp.1205305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DJ, Li J, Waters SB, Xing W, Adair BM, Drobna Z, et al. Arsenic (+3 oxidation state) methyltransferase and the methylation of arsenicals. Exp Biol Med (Maywood) 2007;232:3–13. [PMC free article] [PubMed] [Google Scholar]
- Thomas DJ, Waters SB, Styblo M. Elucidating the pathway for arsenic methylation. Toxicol Appl Pharmacol. 2004;198:319–326. doi: 10.1016/j.taap.2003.10.020. [DOI] [PubMed] [Google Scholar]
- Vahidnia A, van der Voet GB, de Wolff FA. Arsenic neurotoxicity—a review. Hum Exp Toxicol. 2007;26:823–832. doi: 10.1177/0960327107084539. [DOI] [PubMed] [Google Scholar]
- Valenzuela OL, Borja-Aburto VH, Garcia-Vargas GG, Cruz-Gonzalez MB, Garcia-Montalvo EA, Calderon-Aranda ES, et al. 2005Urinary trivalent methylated arsenic species in a population chronically exposed to inorganic arsenic. Environ Health Perspect 113250–254.; 10.1289/ehp.7519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Geen A, Zheng Y, Versteeg R, Stute M, Horneman A, Dhar R, et al. 2003Spatial variability of arsenic in 6000 tube wells in a 25 km2 area of Bangladesh. Water Resour Res 39; 10.1029/2002WR001617 [DOI] [Google Scholar]
- Xu Y, Li X, Zheng Q, Wang H, Wang Y, Sun G. Lack of association of glutathione-S-transferase omega 1 (A140D) and omega 2 (N142D) gene polymorphisms with urinary arsenic profile and oxidative stress status in arsenic-exposed population. Mutat Res. 2009;679:44–49. doi: 10.1016/j.mrgentox.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Yamauchi H, Fan Sun G. Chronic health effects in people exposed to arsenic via the drinking water: dose–response relationships in review. Toxicol Appl Pharmacol. 2004;198:243–252. doi: 10.1016/j.taap.2003.10.022. [DOI] [PubMed] [Google Scholar]
- Yu HS, Liao WT, Chai CY. Arsenic carcinogenesis in the skin. J Biomed Sci. 2006;13:657–666. doi: 10.1007/s11373-006-9092-8. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Marshall G, Ferreccio C, Steinmaus C, Liaw J, Bates M, et al. Kidney cancer mortality: fifty-year latency patterns related to arsenic exposure. Epidemiology. 2010;21:103–108. doi: 10.1097/EDE.0b013e3181c21e46. [DOI] [PubMed] [Google Scholar]
- Zaitlen N, Kraft P, Patterson N, Pasaniuc B, Bhatia G, Pollack S, et al. 2013Using extended genealogy to estimate components of heritability for 23 quantitative and dichotomous traits. PLoS Genet 9e1003520; 10.1371/journal.pgen.1003520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Carbonetto P, Stephens M.2013Polygenic modeling with Bayesian sparse linear mixed models. PLoS Genet 9e1003264; 10.1371/journal.pgen.1003264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.