

Abstract
Sarcoidosis is a multisystem granulomatous disorder invariably affecting the lungs. It is a disease with noteworthy variations in clinical manifestation and disease outcome and has been described as an “immune paradox” with peripheral anergy despite exaggerated inflammation at disease sites. Despite extensive research, sarcoidosis remains a disease with undetermined aetiology. Current evidence supports the notion that the immune response in sarcoidosis is driven by a putative antigen in a genetically susceptible individual. Unfortunately, there currently exists no reliable biomarker to delineate the disease severity and prognosis. As such, the diagnosis of sarcoidosis remains a vexing clinical challenge. In this review, we outline the immunological features of sarcoidosis, discuss the evidence for and against various candidate etiological agents (infective and noninfective), describe the exhaled breath condensate, a novel method of identifying immunological biomarkers, and suggest other possible immunological biomarkers to better characterise the immunopathogenesis of sarcoidosis.
1. Introduction
Sarcoidosis is a multisystem, inflammatory disorder of obscure aetiology. Its defining histopathology is the existence of noncaseating epithelioid granulomas with accompanying mononuclear cell infiltration and microarchitecture destruction [1, 2].
Although sarcoidosis involves the lungs in >90% of cases, it also affects the heart, skin, eye, and central nervous system [3]. This accounts for its heterogeneous clinical manifestation which ranges from having no symptoms to severe consequences, namely respiratory insufficiency, cardiac death, neurological disease, and blindness [4].
Sarcoidosis has been reported in all ethnic and racial groups with the majority of studies recording a peak incidence of 20–39 years of age for both males and females and a bimodal distribution whereby women have another peak incidence at 65–69 [4].
Disease remission occurs in as many as two-thirds of patients, usually in the first 3 years after diagnosis. Other patients have chronic unremitting sarcoidosis which may subsequently lead to lung fibrosis [1]. The erratic clinical course has impelled research into biomarkers that could delineate disease severity and outcome [5]. To date, there exist no reliable and practical biomarkers for sarcoidosis [6]. Moreover, despite earnest research efforts, the immunopathogenesis and aetiology underpinning sarcoidosis remains elusive [3].
This review outlines the current understanding of sarcoidosis, with reference to ex vivo lymphocyte stimulation in the peripheral blood and bronchoalveolar lavage fluid (BALF) of sarcoidosis patients, describes the exhaled breath condensate, an innovative method of identifying immunological markers, and proposes novel immunological markers to better characterise the immunopathogenesis of sarcoidosis.
2. Immunopathogenesis of Sarcoidosis
2.1. Key Features of the Pathological Process
2.1.1. Immune Paradox
Sarcoidosis can be described as an “immune paradox.” Peripheral anergy is observed despite exaggerated inflammation at disease sites [7]. This is demonstrated by a reduced delayed-type hypersensitivity to tuberculin and common antigens [8]. It has been postulated that underpinning this paradoxical situation is a disequilibrium between effector and regulatory lymphocytes (Treg cells), notably CD4+CD25brightFoxP3+ cells [7]. These cells accumulate in the periphery of the granuloma and peripheral blood of patients with active disease and exert anti-proliferative effects on naïve T cells. They only weakly suppress TNFα production [9], therefore allowing granuloma formation. Others argue that the intense immune response at disease sites results in activated T cells gathering at these disease sites and consequent peripheral blood lymphopenia [10]. Still others have suggested that with disease chronicity, immunosuppressive CD8+ T cells become more abundant peripherally, resulting in an anergic response [11].
2.1.2. Granuloma Formation
The noncaseating epithelioid granuloma is the histologic hallmark of sarcoidosis. Its centre is hypothesized to contain a poorly degraded antigen, surrounded by macrophages that will differentiate to epithelioid cells which subsequently fuse to form multinucleated giant cells. CD4+ T helper cells are interspersed in the granuloma while CD8+ T cells, regulatory T cells, fibroblasts, and B cells surround the periphery [4, 12]. Birefringent crystals, Hamazaki-Wesenberg, Schaumann bodies, and asteroid bodies may also be present but are nonspecific [13].
2.2. Immune Reactions in Sarcoidosis
2.2.1. Antigen Presentation
The interplay of antigen-presenting dendritic cells (DCs) and naïve CD4+ T-cells is necessary for granuloma formation [18]. DCs phagocytose the inciting sarcoid antigen. They journey to lymph nodes where they mature and prime the adaptive immune system by displaying the antigen peptide on the surface major histocompatibility complex (MHC) class II peptide groove. A specific T cell receptor (TCR) fixes its variable region to the antigen-MHC complex and is activated [18]. To optimise this activation, CD28, a costimulatory signalling molecule on T cells, interacts with CD86 on DCs [15]. The DCs also produce a battery of mediators which facilitates the sarcoid immune reaction (Figure 1) [20].
Figure 1.

The immunopathogenesis of sarcoidosis (a proposed model). The presumptive sarcoid antigen is engulfed by circulating dendritic cells. Serum amyloid A proteins can also interact with Toll-like receptor 2 and be presented to T cells via major histocompatibility complex Class II to specific T cell receptors (TCRs) along with processed antigen peptides. Ligation of costimulatory molecules CD28, CD86, and BTNL2 optimises the activation of T cells. Thereafter, a myriad of inflammatory mediators is released. Activated T cells are highly TH1 polarised. They release IL-2 which causes clonal proliferation of T cells. Furthermore, upon TCR activation, T-bet production increases. T-bet upregulates and perpetuates the production of IFNγ which facilitates granuloma formation. Antigen clearance and increased IL-10 levels facilitate disease remission. Disease chronicity results in a predominance of TH2 cytokines which leads to lung remodelling by fibrosis (adapted from: [2, 5, 12, 14–19]).
2.2.2. T-Helper 1 (TH1) Immune Response
The CD4+ T cells that trigger the granuloma formation are strongly TH1 polarised. Upon TCR activation, the expression of IFNγ and Tbx21 genes in CD4+ T cells becomes more pronounced. Interleukin-12 (IL-12) secreted by DCs is a TH1 polarising cytokine. With the aid of STAT4, IL-12 facilitates IFNγ expression. IFNγ binds to IFN receptors and stimulates STAT1 which promotes Tbx21 gene expression of T-bet. T-bet enhances IFNγ gene transcription competence and ultimately increases the production of IFNγ (Figure 1). T-bet also up regulates IL-12 β receptor (IL-12βR) expression and antagonises Gata3, a transcription factor that regulates TH2 differentiation. This amplifies the responsiveness of CD4+ T-cells to IL-12 and inhibits IL-4 and IL-13 (cytokines that facilitate the fibroproliferative response) production [16]. IL-18 upregulates IL-12βR and IFNγ expression while IL-12 increases IL-18 receptor expression on CD4+ T cells. Therefore, IL-12 and IL-18 act synergistically to promote the formation of sarcoid granulomas [20–23].
IFNγ is highly expressed in the BALF of sarcoidosis patients. IFNγ inhibits the expression of macrophage peroxisome proliferator-activated receptor γ (PPARγ), a negative regulator of inflammation. Under normal physiological conditions, macrophages constitutively express PPARγ. PPARγ promotes macrophage IL-10 production which inhibits the release of TNFα, IL-12 and matrix metalloproteinase (MMP) from DCs. In sarcoidosis, IFNγ production inhibits the expression of the immunosuppressive cytokine, IL-10 (Figure 1). This leads to an increase in the production of TNFα, IL-12, and MMP and induces chemokines CXCL-9, CXCL-10, and CXCL-11 production which, through the ligation of a T-cell receptor, CXCR3, induce T-cell chemotaxis. MMPs cause lung damage and fibrosis and the chemokines attract more Tcells and myeloid cells into the inflammatory milieu. Moreover, increased TNFα and decreased IL-10 expression liberate DCs from the inhibition by macrophages, initiating a self-amplifying inflammatory loop [19].
TNFα produced by the DCs also encourages CD4+ T-cell proliferation and survival, directly through the induction of T-cell IL-2R [24, 25] and indirectly by causing DC to mature into antigen presenting cells [26]. IL-15 is capable of promoting CD4+ T-cell survival by binding to IL-2R. These IL-15 responses are upregulated in the presence of TNFα [27].
Finally, CD4+ T-cell activation also increases IL-2 production. IL-2 is a local survival, differentiation, and growth factor of T cells. Autocrine IL-2 production results in the clonal proliferation of CD4+ T cells (Figure 1) [2, 28].
2.3. Persistent Granulomatous Inflammation
Persistent granulomatous inflammation can be attributed to the inability of the immune regulatory mechanisms to limit the duration of the inflammatory process [12].
2.3.1. Serum Amyloid A Protein
Serum amyloid A (SAA) proteins are extensively deposited in sarcoid granulomas. SAA triggers cytokine release by interacting with Toll-like receptor 2. This results in the amplification of TH1 responses to local pathogenic antigens. The inflammatory response is potentiated as SAA proteins readily accumulate and release more soluble SAA peptides into the surrounding tissue [29, 30].
2.3.2. T Regulatory (Treg) Cells
Treg cells are vital for the suppression of cell-mediated immune responses. However, the Treg cells in the sarcoid granulomas (as opposed to peripheral Treg cells) have undergone extensive amplification and are therefore impaired in their ability to repress immune responses. Moreover, they secrete proinflammatory cytokines (e.g., IL-4) which encourages granuloma formation via mast cell activation and fibroblast amplification [31, 32].
2.3.3. CD1d-Restricted Natural Killer T (NKT) Cells
NKT cells have been known to moderate CD4-mediated immune responses. NKT cell numbers have been noted to be markedly reduced in sarcoid blood and BALF except in patients exhibiting Löfgren's syndrome (acute sarcoidosis characterised by uveitis, arthritis, erythema nodosum, bilateral hilar lymphadenopathy, and fever) [33]. Since Löfgren's syndrome is often associated with disease remission, reduction in the number of NKT cells can account for the persistence of sarcoidosis [34].
2.4. Remission and Progression to Fibrosis
Disease remission occurs with the suppression of macrophage and T-helper cell activity by IL-10 or when the presumptive antigen has been completely cleared (Figure 1) [4].
Persistent granulomatous inflammation can lead to fibrosis. The immunological mechanisms leading to a fibrotic outcome remain undetermined. Nonetheless, various cytokines which are able to support a fibrotic response have been found at disease sites in patients with sarcoidosis (e.g., transforming growth factor-β (TGF-β), MMP, and insulin growth factor-1 (IGF-1)) [35, 36].
It has been proposed that a switch from TH1 to TH2 cytokine predominance may occur in chronic sarcoidosis in response to persistent inflammation. TH2 cytokines such as IL-13 increase TGF-β production (Figure 1). TGF-β recruits, activates, and transforms fibroblasts into myofibroblasts which have been strongly implicated in the development of fibrosis [17, 37]. Moreover, the TH2 chemokine, CCL2, enhances fibroblast survival, augmenting the effects of TGF-β [38]. Additionally, the macrophages of patients with pulmonary fibrosis, under the influence of the TH2 cytokine milieu, express CCL18 chemokines which facilitates lung remodelling via fibrosis [39, 40].
2.5. Role of Other T Lymphocytes (TH17 and NKT Cells)
Although the majority of studies have used the TH1/TH2 model to explain the immunopathogenesis of sarcoidosis, by focusing solely on this model, there is a propensity to oversimplify the immunological process and divert research efforts away from other mechanisms.
TH17 is a novel CD4+ effector T-cell population. High levels of IL-17+/CD4+ T lymphocytes have been found in the BALF and granulomas of sarcoidosis patients, particularly in patients with active disease. They infiltrate the lungs after being recruited from the blood by the chemokine CCL20 [41]. Recently, Richmond and colleagues [42] verified the specificity of TH17 cells for mycobacterial antigens, a commonly implicated antigen for sarcoidosis. These findings suggest a possible role of TH17 in sarcoidosis disease progression.
NKT cells produce TH1 and TH2 cytokines (IFNγ and IL-4, resp.). NKT cells are mostly CD4+ and express an invariable TCR [33]. Moreover, blood NKT cells from sarcoidosis patients, when stimulated with a glycolipid stimulator, showed diminished levels of IFNγ, therefore suggesting that NKT cells exert regulatory activity which prevents disease progression [43].
3. Putative Aetiology of Sarcoidosis
The aetiology of sarcoidosis remains unclear. A myriad of observations have supported the notion that sarcoidosis can be caused by environmental and infectious agents. Moreover, based on chronic beryllium disease, an analogous granulomatous lung disease, it has been speculated that one or more antigenic stimuli may be involved in the pathogenesis of sarcoidosis. Therefore, it is highly likely that the development of a sarcoidosis reaction to an antigen depends on a combination of genetic polymorphisms, the host's immune status, and exposure to environmental agents [44].
3.1. Genetic Polymorphisms and Host Factors
3.1.1. Findings on Genome-Wide Association
Both family and genetic host studies have recognised genes that are responsible for this genetic susceptibility. Twin studies prove that monozygotic twins are more concordant for sarcoidosis than dizygotic twins. Moreover, familial aggregation of sarcoidosis can be seen worldwide. The multicentre study entitled A Case Control Etiologic Study of Sarcoidosis (ACCESS) demonstrated that sarcoidosis patients were 5 times more probable than controls to report a parent or sibling with sarcoidosis [45].
Genetic linkage studies on German families revealed a strong linkage to chromosome 6p. This led to the discovery of butyrophilin-like 2 (BTNL2), a costimulatory molecule within the MHC locus. Single nucleotide polymorphisms (rs2076530 G → A) in BTNL2 may affect T-cell regulation and activation [45].
The genome-wide association study conducted by Hofmann and colleagues [46] revealed an association for the annexin A11 gene located on chromosome 10q22.3. The annexin A11 gene regulates calcium signalling, vesicle trafficking, cell division, and apoptosis. Therefore, its dysfunction or deletion may implicate apoptotic pathways in sarcoidosis [46].
3.1.2. Human Leukocyte Antigen (HLA) Genes
HLA class II are cell surface proteins that prime the adaptive immune system to antigens. Sarcoidosis is associated with the DR subtypes of class II antigens. HLA-DRB1∗01 and HLA-DRB1∗04, are negatively associated with sarcoidosis, whereas HLA-DRB1∗03, HLA-DRB1∗11, HLA-DRB1∗12, HLA-DRB1∗14 and HLA-DRB1∗15 have been shown to increase the risk of sarcoidosis. HLA-DRB1∗03 is associated with Löfgren's syndrome (~80% of patients with Löfgren's syndrome experience disease remission). Finally, the HLA-DRB1∗1501/DQB1∗0602 haplotype was associated with severe and chronic pulmonary sarcoidosis [6, 47].
3.1.3. Non-HLA Genes
TNFα is an essential mediator for granuloma formation. Variants of the TNF gene confer a 1.5-fold increased risk of having sarcoidosis [48]. Apart from TNF, studies investigating other candidate genes (polymorphisms in the complement receptor 1 gene, NOD, and CCR2 genes) were inconclusive and had poor reproducibility between populations [49–52].
In some populations, variations in the gene that encodes for RAGE (a transmembrane receptor) have been associated with an increased risk of sarcoidosis. However, the close proximity of this gene to the MHC region makes it difficult for one to ascertain if this association is due to linkage with neighbouring HLA genes [53].
There were no associations between polymorphisms in genes for vitamin D receptor or serum angiotensin-converting enzyme [45, 54].
3.1.4. T Cell Receptor (TCR) Genes
The T-cells at sites of inflammation in sarcoidosis exhibit a restricted repertoire of TCR γδ or αβ genes. The expression of specific Vα, Vβ, or γδ+ TCR genes in blood, lung, and at sites of Kveim-Siltzbach skin reactions implies that sarcoidosis is an antigen-driven disorder. There is a subpopulation of T cells (AV2S3+ (Vα2.3) CD4+ T cells) from BALF of HLA-DR∗0301 sarcoidosis patients which is unique to sarcoidosis. Moreover, it has been shown that the amount of AV2S3+ BALF T cells at the onset of sarcoidosis correlates positively with prognosis, suggesting that AV2S3+ T cells may offer some protective function against sarcoidosis [6].
3.2. Extrinsic Factors
Numerous pathogens have been implicated and investigated in the etiology of sarcoidosis. Moreover, spatial crowding of unrelated sarcoidosis cases suggests that sarcoidosis can also be a result of exposure to environmental agents [1, 3]. Nonetheless the evidence supporting specific infectious and environmental factors varies significantly (Table 1) [3].
Table 1.
List of infectious and noninfectious agents and the evidence for and against them.
| Nature | Causative agent | Evidence for | Evidence against |
|---|---|---|---|
| Infective | Mycobacterium | (i) Immunohistochemical studies showed possible remnants of cell wall deficient mycobacteria [61]. (ii) A mycobacterial cell wall component, tuberculostearic acid, was found in sarcoid specimens [62]. (iii) Techniques such as enzyme-linked immunospot assay (ELISpot) and polymerase chain reactions (PCR) have shown increasing evidence for mycobacteria in the mediastinal lymph nodes and peripheral lung tissues of sarcoidosis patients [63]. (iv) Mycobacterium tuberculosis DNA: mycobacterium tuberculosis catalase-peroxidase protein (mKatG) and circulating IgG for mKatG have been identified in sarcoidosis patients [64]. Additionally, compared to healthy controls, sarcoidosis patients have amplified T-cell responses in the peripheral blood and lungs to mKatG and mycobacteria antigens [65, 66]. (v) Mycobacterial heat-shock proteins (Mtb-hsp)70, 65, and 16 were found in the lymph nodes and sera of sarcoidosis patients [67]. (vi) Dubaniewicz and colleagues (2013) suggested that, in genetically different individuals, Mtb-hsp 16 can induce an autoimmune response in sarcoidosis. |
(i) Acid-fast stains and cultures of sarcoid specimens do not routinely demonstrate the presence of mycobacterium species. (ii) The mere presence of the mycobacterial antigens in sarcoid specimens is not proof of a causal relationship. (iii) Mycobacterial nucleic acid and antigens are not detected in many sarcoid specimens; therefore, mycobacteria may not be the sole cause of sarcoidosis [3]. |
| Propionibacterium | (i) It has been shown to be able to induce a granulomatous reaction [68]. (ii) P. acnes has been found in up to 78% of sarcoidosis sample cultures [69]. (iii) An antibody response to P. acnes proteins has been observed in 40% of BALF samples (compared to 5% in healthy controls) [70]. |
(i) Cultures from healthy controls also yield this commensal organism [71]. | |
| Viruses and other infectious pathogens | (i) Serum antibodies to herpes-like viruses (human herpes virus-8, herpes simplex virus, and Epstein-Barr virus) were elevated in patients with sarcoidosis [72]. | (i) Significant proportions of the general population have also been previously exposed to herpes-like viruses. (ii) A nonspecific polyclonal hypergammaglobulinemia, common in sarcoidosis, may explain the increased antibody titre for these viruses [73]. (iii) Viruses do not cause the epithelioid granulomas of sarcoidosis [74]. (iv) The mechanism for granuloma formation by molecular mimicry after viral exposure remains undetermined [74]. (v) Granulomatous reactions resulting from spirochetes, fungi, Tropheryma whipplei, and Borrelia species infection can be difficult to discriminate from sarcoidosis [75]. |
|
| (vi) The notion that cell-wall deficient organisms like mycobacteria, rickettsia, and chlamydia species cause sarcoidosis is founded on limited data. There is a shortage of conformation from well-controlled epidemiological and laboratory studies [75]. | |||
| Transplants | (i) Immune dysregulation following allogeneic hematopoietic cell transplantation has been shown to promote sarcoidosis in patients with susceptible HLA subtypes [76]. (ii) Individuals have developed granulomatous inflammation post lung and heart transplant from patients with sarcoidosis [77–79]. (iii) An increased incidence of sarcoidosis in closed populations may suggest an infection-related disease. |
||
|
| |||
| Noninfectious | Environmental | (i) Wood stoves and fireplaces have been associated with an increased risk of sarcoidosis [80]. (ii) Fire rescue workers, military personnel, and healthcare workers who were exposed to the dust from the destruction of the World Trade Centre in New York were found to have a higher risk of developing sarcoidosis [81, 82]. (iii) Findings from the ACCESS study showed the modest positive odds ratios (~1.5) that workplace exposure to organic solvents, dusts, pesticides, insecticides, and musty odours can increase one's risk of sarcoidosis. Reduced risk was associated with exposures to animal dander and other allergic (TH2) responses [83]. (iv) Nanoparticles of common minerals and metals can elicit a dysregulated immune response [84]. |
(i) The ACCESS study failed to identify risk factors that accrue a greater than two-fold risk (odds ratio). Moreover, it had inadequate power to ascertain the sarcoidosis risk among fire rescue workers, military personnel, and healthcare workers. Lastly, it failed to prove an association between previously hypothesised exposures (e.g., wood dust, metals, and silica) and sarcoidosis [1, 3]. |
| Autoantigens | (i) Sarcoidosis patients express low titre levels of autoantibodies. (ii) The BALF of HLA-DRB1∗0301-positive sarcoidosis patients with Löfgren's syndrome had antigenic peptides that were bound to HLA-DR molecules of lung cells that have the TCR AV2S3+ gene segment. These antigenic peptides include vimentin, ATP synthase, and lysyl tRNA synthetase, thought to be autoantigens in various conditions [85]. (iii) IFNγ enzyme-linked immunospot assays revealed a strong T-cell response to the cytoskeletal peptides of vimentin from the peripheral blood of patients with HLA-DRB1∗0301. The same was observed for ATP synthase and lysyl tRNA synthetase from BALF. Thus, this suggests a possible autoimmune response in patients with HLA-DRB1∗0301, contributing to sarcoid granulomatous inflammation [86]. |
(i) The pathological significance of autoantibodies in sarcoidosis remains unclear. The disease-specific autoantibody profile has not been described. Therefore, it has been postulated that these autoantibodies are most likely the product of general B-cell stimulation in the progression of T-cell stimulation by antigens [1]. | |
4. Ex Vivo Stimulation of BALF and Peripheral Blood Lymphocytes
Preliminary ex vivo studies that employed flow cytometry to investigate peripheral blood lymphocytes in sarcoidosis patients demonstrated a greater activation of nonstimulated CD4+ and CD8+ BALF T cells compared to peripheral blood lymphocytes. This showed that the sarcoid immune response is largely compartmentalised to disease sites [55].
The TH1/TH2 model is under scrutiny as it oversimplifies the immunopathogenesis of sarcoidosis [56]. For instance, some studies report that after lymphocyte stimulation, the proportion of CD4+ T cells expressing IL-4 and IFNγ obtained from the peripheral blood of sarcoidosis patients did not differ significantly from that of healthy controls [57, 58]. Other studies showed higher TH1 and TH2 levels of cytokine positive CD4+ T cells compared to healthy controls [59, 60], emphasising the systemic nature of the disease. The following segment clarifies this debate.
Under unstimulated conditions, the difference in the percentages of IL-4 and IFNγ secreting CD4+ lymphocytes in BALF and peripheral blood of sarcoidosis patients is insignificant [57, 58]. After BALF CD4+ lymphocytes were stimulated with ionomycin and phorbol 12-myristate acetate, there was an appreciable increase in secreted IFNγ but a decrease in IL-4 expression in sarcoidosis patients compared to controls [87]. Moreover, increased cytokine profiles have been verified by increased BALF IFNγ +/IL-4+ CD4+ T cell ratios in sarcoidosis patients. Lower ratios were demonstrated in scleroderma and in patients with idiopathic pulmonary fibrosis. It has also been shown that upon stimulation, compared with controls, there are increased numbers of CD4+IFNγ + cells in both BALF and induced sputum of patients with sarcoidosis [88, 89].
After stimulation, more T cells express TH1 than TH2 cytokines in both the BALF and peripheral blood of sarcoidosis patients and more CD4+ T cells in BALF express TH1 receptors (CXCR3, CCR5, IL-12R and IL-18R) than CD4+ T cells in the peripheral blood. [90]. Although CD4+ T cells largely express TH1 cytokines, interestingly, following stimulation, only 80% and 40% of CD4+ IL-4+ cells concurrently produce IFNγ and IL-2 respectively, thus demonstrating that activated BALF lymphocytes of sarcoidosis patients are capable of a complex, concurrent production of TH1 and TH2 cytokines [91].
To further explore this dichotomy of blood TH1/TH2 equilibrium, Nureki and colleagues [59] showed that under unstimulated conditions, TH1 and TH2 chemokines (interferon-inducible protein-10 (IP-10) and thymus and activation-regulated chemokine (TARC)) were both increased in the serum of sarcoidosis patients. This was in agreement with previous findings that demonstrated elevated BALF and peripheral blood IL-13 (a TH2 cytokine) mRNA levels [92]. Therefore, these findings reflect the systemic nature of sarcoidosis. Nonetheless, it has been suggested that TH2 cell preponderance occurs in the peripheral blood of sarcoidosis patients and that this, together with the generalised intensification of TH1 activity, gives the appearance of an increase in both TH1 and TH2 circulating cytokine expression in sarcoidosis patients compared to healthy controls [59].
TH17 cells have also been implicated in the induction of granuloma formation [93]. Flow cytometry data indicate that after stimulation, there is an increase in TH17 related cytokine levels in both BALF and peripheral blood [41]. Another study also indicated that, after stimulation, there are lower levels of IL-17A gene expression in CD4+ T cells in patients with Löfgren's syndrome compared to healthy controls [94]. These data, together with data showing heightened TH1 cytokine expression at disease sites, indicate that TH17 cells have a systemic role in patients with non-Löfgren's disease and is involved in sarcoidosis progression [14]. Therefore, further studies investigating the cytokine profiles in blood lymphocytes of patients versus healthy controls are required to assess the TH1/TH2 balance, the regulatory mechanisms in the peripheral blood of sarcoidosis patients, and the functional significance of TH17 cells.
5. Exhaled Breath Condensate: Detection of Immunological Markers
The diagnosis of sarcoidosis is never secure. Clinico-radiological findings alone are often insufficient to confirm the diagnosis of sarcoidosis. It needs to be supported by histological evidence showing noncaseating granulomas. This warrants a tissue biopsy which is invasive [12]. This makes diagnosing sarcoidosis a vexing clinical challenge, motivating researchers to look for other novel methods of diagnosing the disease.
Exhaled breath condensate (EBC) has been subjected to intensive research as it provides a noninvasive alternative for sampling the airway and alveolar space a promising source of biomarkers for a variety of lung conditions [95–97]. During exhalation, water evaporation droplets and volatile molecules (e.g., nitric oxide, carbon monoxide and hydrocarbons) diffuse as gases from the alveoli and bronchi to the mouth. They are joined by nonvolatile molecules (e.g., leukotrienes, prostanoids, urea, and cytokines) from the airway lining fluid and condense via a refrigeration device to give EBC (Figure 2) [95, 98].
Figure 2.
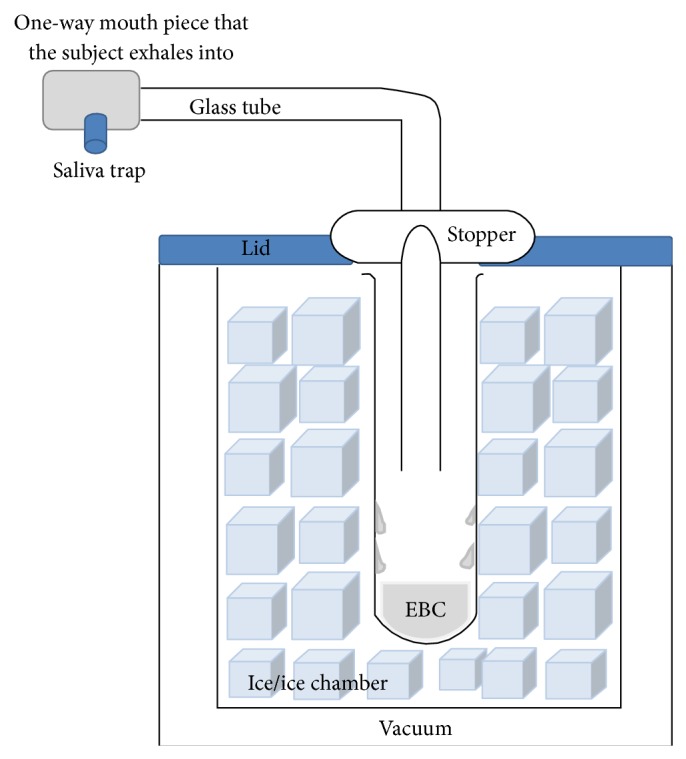
Schematic diagram of the EBC collecting apparatus. The subject blows into the mouth piece which is a one-way valve. The exhaled breath is channelled into a refrigerated collecting container.
A number of immunological biomarkers have been recognised in EBC. However, there exists no sufficiently sensitive and specific marker for diagnosing and predicting the prognosis of sarcoidosis [99].
TNFα, PAI-1, and IGF-1 levels in EBC were closely positively correlated with BALF samples from sarcoidosis patients. Conversely, IL-6 levels were negatively correlated with that which is in BALF. The propensity of IL-6 to form complex molecular forms of higher molecular weight could account for this discrepancy [100]. Another study detected TGF-β 1, PAI-1, TNFα, IL-8 and vascular endothelial growth factor in sarcoid EBC. However, the small sample size and the failure to make comparisons with healthy controls limited the usefulness of this study [101].
Exhaled eicosanoids (e.g., 8-isoprostane (8-IP)) and cysteinyl leukotrienes were also found to be elevated in the BALF and EBC of sarcoidosis patients [102]. In a later study, high initial levels of 8-IP were shown to correlate with disease persistence; therefore, it could serve as a prognostic marker [103].
Cellular and molecular biomarkers previously discovered in BALF and serum of sarcoidosis patients could also serve as biomarkers. These include eosinophils, neutrophils, serum angiotensin converting enzyme (ACE), neopterin, chitotriosidase, TGF-β, and the chemokine ligand (CCL18) [28, 40, 57, 104–107]. Other more novel markers include lysozyme, Kerbs von Lungren 6 antigen, and soluble IL-2 receptor [108, 109]. Serum levels of these biomarkers were said to reflect increased parenchymal infiltration and lymphocytic alveolitis in sarcoidosis and can thus serve as potential EBC biomarkers. Nonetheless, only a few have been shown to be sufficiently sensitive and specific [110].
Amongst the above-mentioned biomarkers, ACE is the most contentious as it has been shown to have poor sensitivity and specificity [111] and its activity is subject to the effects of gene polymorphisms. Nevertheless, it is elevated and measurable in the BALF of sarcoidosis patients and could therefore serve as a sarcoid biomarker [112].
Given the multifactorial nature of sarcoidosis, no ideal markers for detecting and monitoring the clinical course of sarcoidosis exist. It is very likely that a combination of markers will be required.
Although EBC has advantages over BAL (it is noninvasive, requires little instrumentation, does not introduce foreign substances into the lung or cause inflammatory changes, and can be repeatedly performed in sick patients) [113], the lack of reliable markers and the inability of EBC to sample specific compartments of the lungs undermine these benefits. To date, BALF remains the most relevant biological material.
6. Interferon Modulators: Novel Immunological Markers
IFNγ plays a pivotal role in the immunopathogenesis of sarcoidosis. MicroRNA-29 (miR-29) and T-bet have been shown to modulate its production [114, 115].
MicroRNAs are noncoding RNA that can inhibit the production of mRNA. The miR-29 family is made up of four members. Amongst these four, miR-29a and miR-29b were found to be downregulated in IFNγ-secreting T cells. This reduction skews the immunological response towards a TH1 lineage by initiating a positive feedback loop which enhances IFNγ production. This also suggests that the up-regulation of miR-29a and miR-29b can mitigate IFNγ expression [115, 116]. Abnormal levels of microRNA have been associated with the pathogenesis of cancers and fibrotic and obstructive lung diseases [117–119]. It has also been implicated in the fibrotic progression of sarcoidosis [120].
As previously mentioned (see immune reactions in sarcoidosis), T-bet is a transcription factor necessary for IFNγ production. It binds to a number of enhancers and to the promoter region of the IFNγ gene to promote IFNγ transcription [121]. T-bet expression has been shown to correlate with IFNγ expression [16, 122, 123] in patients with multiple sclerosis [114], coeliac disease [124], Crohn's disease [125], and Behçet's disease [126, 127]. Moreover, T-bet mRNA has been shown to be elevated in the BALF lymphocytes of patients with pulmonary sarcoidosis [128]. However, due to posttranscriptional regulation and disparities in protein and mRNA turnover rates, mRNA levels are poor proxies for protein levels [129, 130]. Unfortunately, the literature is currently deficient of studies that measure T-bet protein levels in sarcoidosis patients and studies that juxtapose miR-29 and T-bet protein levels at sarcoidosis disease sites and in the peripheral blood. Research on these fronts can offer novel insights into the “immune paradox” associated with sarcoidosis and can pave the path for novel therapeutic strategies for the disease.
7. Conclusion
Despite nearly 140 years of extensive research, the aetiology and pathogenesis of sarcoidosis and the mechanisms that regulate the immune reactions in the peripheral blood remain undetermined. Moreover, given its variable clinical manifestation and the lack of a reliable diagnostic test with uniformed reference values and measurements, diagnosing sarcoidosis remains a clinical conundrum for many physicians. Given that the majority of sarcoidosis patients have pulmonary involvement, EBC could be used as a non-invasive method to diagnose sarcoidosis. Besides being a potential immunological biomarker of sarcoidosis, interferon modulator levels in EBC and the peripheral blood can be compared to elucidate the regulatory mechanisms in the peripheral blood. Results from such studies may also explain the pathology underpinning the peripheral anergy seen in sarcoidosis.
Conflict of Interests
The authors do not have any financial conflict of interest related to this paper.
References
- 1.Chen E. S., Moller D. R. Sarcoidosis—scientific progress and clinical challenges. Nature Reviews Rheumatology. 2011;7(8):457–467. doi: 10.1038/nrrheum.2011.93. [DOI] [PubMed] [Google Scholar]
- 2.Iannuzzi M. C., Fontana J. R. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. Journal of the American Medical Association. 2011;305(4):391–399. doi: 10.1001/jama.2011.10. [DOI] [PubMed] [Google Scholar]
- 3.Saidha S., Sotirchos E. S., Eckstein C. Etiology of sarcoidosis: does infection play a role? Yale Journal of Biology and Medicine. 2012;85(1):133–141. [PMC free article] [PubMed] [Google Scholar]
- 4.Iannuzzi M. C., Rybicki B. A., Teirstein A. S. Sarcoidosis. New England Journal of Medicine. 2007;357(21):2153–2165. doi: 10.1056/NEJMra071714. [DOI] [PubMed] [Google Scholar]
- 5.Bargagli E., Mazzi A., Rottoli P. Markers of inflammation in sarcoidosis: blood, urine, BAL, sputum, and exhaled gas. Clinics in Chest Medicine. 2008;29(3):445–458. doi: 10.1016/j.ccm.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Morgenthau A. S., Iannuzzi M. C. Recent advances in sarcoidosis. Chest. 2011;139(1):174–182. doi: 10.1378/chest.10-0188. [DOI] [PubMed] [Google Scholar]
- 7.Miyara M., Amoura Z., Parizot C., et al. The immune paradox of sarcoidosis and regulatory T cells. Journal of Experimental Medicine. 2006;203(2):359–370. doi: 10.1084/jem.20050648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mathew S., Bauer K. L., Fischoeder A., Bhardwaj N., Oliver S. J. The anergic state in sarcoidosis is associated with diminished dendritic cell function. Journal of Immunology. 2008;181(1):746–755. doi: 10.4049/jimmunol.181.1.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehrenstein M. R., Evans J. G., Singh A., et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFα therapy. Journal of Experimental Medicine. 2004;200(3):277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunninghake G. W., Fulmer J. D., Young R. C., Jr. Localization of the immune response in sarcoidosis. American Review of Respiratory Disease. 1979;120(1):49–57. doi: 10.1164/arrd.1979.120.1.49. [DOI] [PubMed] [Google Scholar]
- 11.Planck A., Katchar K., Eklund A., Gripenbäck S., Grunewald J. T-lymphocyte activity in HLA-DR17 positive patients with active and clinically recovered sarcoidosis. Sarcoidosis Vasculitis and Diffuse Lung Diseases. 2003;20(2):110–117. [PubMed] [Google Scholar]
- 12.Baughman R. P., Culver D. A., Judson M. A. A concise review of pulmonary sarcoidosis. American Journal of Respiratory and Critical Care Medicine. 2011;183(5):573–581. doi: 10.1164/rccm.201006-0865CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosen Y. Pathology of sarcoidosis. Seminars in Respiratory and Critical Care Medicine. 2007;28(1):36–52. doi: 10.1055/s-2007-970332. [DOI] [PubMed] [Google Scholar]
- 14.Ahmadzai H., Wakefield D., Thomas P. S. The potential of the immunological markers of sarcoidosis in exhaled breath and peripheral blood as future diagnostic and monitoring techniques. Inflammopharmacology. 2011;19(2):55–68. doi: 10.1007/s10787-011-0079-3. [DOI] [PubMed] [Google Scholar]
- 15.Ahmadzai H., Cameron B., Chui J. J. Y., Lloyd A., Wakefield D., Thomas P. S. Peripheral blood responses to specific antigens and CD28 in sarcoidosis. Respiratory Medicine. 2012;106(5):701–709. doi: 10.1016/j.rmed.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 16.Amsen D., Spilianakis C. G., Flavell R. A. How are TH1 and TH2 effector cells made? Current Opinion in Immunology. 2009;21(2):153–160. doi: 10.1016/j.coi.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gharaee-Kermani M., Hu B., Phan S. H., Gyetko M. R. Recent advances in molecular targets and treatment of Idiopathic Pulmonary Fibrosis: focus on TGFβ signaling and the myofibroblast. Current Medicinal Chemistry. 2009;16(11):1400–1417. doi: 10.2174/092986709787846497. [DOI] [PubMed] [Google Scholar]
- 18.Noor A., Knox K. S. Immunopathogenesis of sarcoidosis. Clinics in Dermatology. 2007;25(3):250–258. doi: 10.1016/j.clindermatol.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Zaba L. C., Smith G. P., Sanchez M., Prystowsky S. D. Dendritic cells in the pathogenesis of sarcoidosis. American Journal of Respiratory Cell and Molecular Biology. 2010;42(1):32–39. doi: 10.1165/rcmb.2009-0033TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mroz R. M., Korniluk M., Stasiak-Barmuta A., Chyczewska E. Increased levels of interleukin-12 and interleukin-18 in bronchoalveolar lavage fluid of patients with pulmonary sarcoidosis. Journal of Physiology and Pharmacology. 2008;59(6):507–513. [PubMed] [Google Scholar]
- 21.Agostini C., Meneghin A., Semenzato G. T-lymphocytes and cytokines in sarcoidosis. Current Opinion in Pulmonary Medicine. 2002;8(5):435–440. doi: 10.1097/00063198-200209000-00016. [DOI] [PubMed] [Google Scholar]
- 22.Shigehara K., Shijubo N., Ohmichi M., et al. IL-12 and IL-18 are increased and stimulate IFN-γ production in sarcoid lungs. Journal of Immunology. 2001;166(1):642–649. doi: 10.4049/jimmunol.166.1.642. [DOI] [PubMed] [Google Scholar]
- 23.Szabo S. J., Dighe A. S., Gubler U., Murphy K. M. Regulation of the interleukin (IL)-12R β2 subunit expression in developing T helper 1 (Th1) and Th2 cells. Journal of Experimental Medicine. 1997;185(5):817–824. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plaetinck G., Combe M.-C., Corthesy P., et al. Control of IL-2 receptor-α expression by IL-1, tumor necrosis factor, and IL-2. Complex regulation via elements in the 5' flanking region. Journal of Immunology. 1990;145(10):3340–3347. [PubMed] [Google Scholar]
- 25.Tartaglia L. A., Goeddel D. V., Reynolds C., et al. Stimulation of human T-cell proliferation by specific activation of the 75-kDa tumor necrosis factor receptor. Journal of Immunology. 1993;151(9):4637–4641. [PubMed] [Google Scholar]
- 26.Romani N., Reider D., Heuer M., et al. Generation of mature dendritic cells from human blood An improved method with special regard to clinical applicability. Journal of Immunological Methods. 1996;196(2):137–151. doi: 10.1016/0022-1759(96)00078-6. [DOI] [PubMed] [Google Scholar]
- 27.Agostini C., Trentin L., Facco M., et al. Role of IL-15, IL-2, and their receptors in the development of T cell alveolitis in pulmonary sarcoidosis. Journal of Immunology. 1996;157(2):910–918. [PubMed] [Google Scholar]
- 28.Müller-Quernheim J., Prasse A., Zissel G. Pathogenesis of sarcoidosis. Presse Medicale. 2012;41(6):e275–e287. doi: 10.1016/j.lpm.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 29.Bargagli E., Magi B., Olivieri C., Bianchi N., Landi C., Rottoli P. Analysis of serum amyloid A in sarcoidosis patients. Respiratory Medicine. 2011;105(5):775–780. doi: 10.1016/j.rmed.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 30.Chen E. S., Song Z., Willett M. H., et al. Serum amyloid a regulates granulomatous inflammation in sarcoidosis through toll-like receptor-2. American Journal of Respiratory and Critical Care Medicine. 2010;181(4):360–373. doi: 10.1164/rccm.200905-0696OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rappl G., Pabst S., Riemann D., et al. Regulatory T cells with reduced repressor capacities are extensively amplified in pulmonary sarcoid lesions and sustain granuloma formation. Clinical Immunology. 2011;140(1):71–83. doi: 10.1016/j.clim.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 32.Rappl G., Schmidt A., Mauch C., Hombach A. A., Abken H. Extensive amplification of human regulatory T cells alters their functional capacities and targets them to the periphery. Rejuvenation Research. 2008;11(5):915–933. doi: 10.1089/rej.2008.0723. [DOI] [PubMed] [Google Scholar]
- 33.Ho L.-P., Urban B. C., Thickett D. R., Davies R. J. O., McMichael A. J. Deficiency of a subset of T cells with immunoregulatory properties in sarcoidosis. Lancet. 2005;365(9464):1062–1072. doi: 10.1016/S0140-6736(05)71143-0. [DOI] [PubMed] [Google Scholar]
- 34.Grunewald J., Eklund A. Löfgren's syndrome: human leukocyte antigen strongly influences the disease course. American Journal of Respiratory and Critical Care Medicine. 2009;179(4):307–312. doi: 10.1164/rccm.200807-1082OC. [DOI] [PubMed] [Google Scholar]
- 35.Henry M. T., McMahon K., Mackarel A. J., et al. Matrix metalloproteinases and tissue inhibitor of metalloproteinase-1 in sarcoidosis and IPF. European Respiratory Journal. 2002;20(5):1220–1227. doi: 10.1183/09031936.02.00022302. [DOI] [PubMed] [Google Scholar]
- 36.Zissel G., Prasse A., Müller-Quernheim J. Immunologic response of sarcoidosis. Seminars in Respiratory and Critical Care Medicine. 2010;31(4):390–403. doi: 10.1055/s-0030-1262208. [DOI] [PubMed] [Google Scholar]
- 37.Xaubet A., Marin-Arguedas A., Lario S., et al. Transforming growth factor-β1 gene polymorphisms are associated with disease progression in idiopathic pulmonary fibrosis. American Journal of Respiratory and Critical Care Medicine. 2003;168(4):431–435. doi: 10.1164/rccm.200210-1165OC. [DOI] [PubMed] [Google Scholar]
- 38.Gerke A. K., Hunninghake G. The immunology of sarcoidosis. Clinics in Chest Medicine. 2008;29(3):379–390. doi: 10.1016/j.ccm.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 39.Patterson K. C., Hogarth K., Husain A. N., Sperling A. I., Niewold T. B. The clinical and immunologic features of pulmonary fibrosis in sarcoidosis. Translational Research. 2012;160(5):321–331. doi: 10.1016/j.trsl.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prasse A., Pechkovsky D. V., Toews G. B., et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. American Journal of Respiratory and Critical Care Medicine. 2006;173(7):781–792. doi: 10.1164/rccm.200509-1518OC. [DOI] [PubMed] [Google Scholar]
- 41.Facco M., Cabrelle A., Teramo A., et al. Sarcoidosis is a Th1/Th17 multisystem disorder. Thorax. 2011;66(2):144–150. doi: 10.1136/thx.2010.140319. [DOI] [PubMed] [Google Scholar]
- 42.Richmond B. W., Ploetze K., Isom J., et al. Sarcoidosis Th17 cells are ESAT-6 antigen specific but demonstrate reduced IFN-gamma expression. Journal of Clinical Immunology. 2013;33(2):446–455. doi: 10.1007/s10875-012-9817-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi S., Kaneko Y., Seino K.-I., et al. Impaired IFN-γ production of Vα24 NKT cells in non-remitting sarcoidosis. International Immunology. 2004;16(2):215–222. doi: 10.1093/intimm/dxh020. [DOI] [PubMed] [Google Scholar]
- 44.Culver D. A. Sarcoidosis. Immunology and Allergy Clinics of North America. 2012;32(4):487–511. doi: 10.1016/j.iac.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 45.Rybicki B. A., Iannuzzi M. C., Frederick M. M., et al. Familial aggregation of sarcoidosis. A Case-Control Etiologic Study of Sarcoidosis (ACCESS) American Journal of Respiratory and Critical Care Medicine. 2001;164(11):2085–2091. doi: 10.1164/ajrccm.164.11.2106001. [DOI] [PubMed] [Google Scholar]
- 46.Hofmann S., Franke A., Fischer A., et al. Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nature Genetics. 2008;40(9):1103–1106. doi: 10.1038/ng.198. [DOI] [PubMed] [Google Scholar]
- 47.Berlin M., Fogdell-Hahn A., Olerup O., Eklund A., Grunewald J. HLA-DR predicts the prognosis in Scandinavian patients with pulmonary sarcoidosis. American Journal of Respiratory and Critical Care Medicine. 1997;156(5):1601–1605. doi: 10.1164/ajrccm.156.5.9704069. [DOI] [PubMed] [Google Scholar]
- 48.Medica I., Kastrin A., Maver A., Peterlin B. Role of genetic polymorphisms in ACE and TNF-α gene in sarcoidosis: a meta-analysis. Journal of Human Genetics. 2007;52(10):836–847. doi: 10.1007/s10038-007-0185-7. [DOI] [PubMed] [Google Scholar]
- 49.Sato H., Williams H. R. T., Spagnolo P., et al. CARD15/NOD2 polymorphisms are associated with severe pulmonary sarcoidosis. European Respiratory Journal. 2010;35(2):324–330. doi: 10.1183/09031936.00010209. [DOI] [PubMed] [Google Scholar]
- 50.Schürmann M., Valentonyte R., Hampe J., Müller-Quernheim J., Schwinger E., Schreiber S. CARD15 gene mutations in sarcoidosis. European Respiratory Journal. 2003;22(5):748–754. doi: 10.1183/09031936.03.00040602. [DOI] [PubMed] [Google Scholar]
- 51.Spagnolo P., Sato H., Grunewald J., et al. A common haplotype of the C-C chemokine receptor 2 gene and HLA-DRB1∗0301 are independent genetic risk factors for Löfgren's syndrome. Journal of Internal Medicine. 2008;264(5):433–441. doi: 10.1111/j.1365-2796.2008.01984.x. [DOI] [PubMed] [Google Scholar]
- 52.Zorzetto M., Bombieri C., Ferrarotti I., et al. Complement receptor 1 gene polymorphisms in sarcoidosis. American Journal of Respiratory Cell and Molecular Biology. 2002;27(1):17–23. doi: 10.1165/ajrcmb.27.1.4805. [DOI] [PubMed] [Google Scholar]
- 53.Campo I., Morbini P., Zorzetto M., et al. Expression of receptor for advanced glycation end products in sarcoid granulomas. American Journal of Respiratory and Critical Care Medicine. 2007;175(5):498–506. doi: 10.1164/rccm.200601-136OC. [DOI] [PubMed] [Google Scholar]
- 54.Kruit A., Ruven H. J. T., Grutters J. C., Van Den Bosch J. M. M. Angiotensin II receptor type 1 1166 A/C and angiotensin converting enzyme I/D gene polymorphisms in a Dutch sarcoidosis cohort. Sarcoidosis Vasculitis and Diffuse Lung Diseases. 2010;27(2):147–152. [PubMed] [Google Scholar]
- 55.Wahlström J., Berlin M., Sköld C. M., Wigzell H., Eklund A., Grunewald J. Phenotypic analysis of lymphocytes and monocytes/macrophages in peripheral blood and bronchoalveolar lavage fluid from patients with pulmonary sarcoidosis. Thorax. 1999;54(4):339–346. doi: 10.1136/thx.54.4.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Möllers M., Aries S. P., Drömann D., Mascher B., Braun J., Dalhoff K. Intracellular cytokine repertoire in different T cell subsets from patients with sarcoidosis. Thorax. 2001;56(6):487–493. doi: 10.1136/thorax.56.6.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Inui N., Chida K., Suda T., Nakamura H. TH1/TH2 and TC1/TC2 profiles in peripheral blood and bronchoalveolar lavage fluid celin pulmonary sarcoidosis. Journal of Allergy and Clinical Immunology. 2001;107(2):337–344. doi: 10.1067/mai.2001.112273. [DOI] [PubMed] [Google Scholar]
- 58.Prasse A., Georges C. G., Biller H., et al. Th1 cytokine pattern in sarcoidosis is expressed by bronchoalveolar CD4+ and CD8+ T cells. Clinical and Experimental Immunology. 2000;122(2):241–248. doi: 10.1046/j.1365-2249.2000.01365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nureki S.-I., Miyazaki E., Ando M., et al. Circulating levels of both Th1 and Th2 chemokines are elevated in patients with sarcoidosis. Respiratory Medicine. 2008;102(2):239–247. doi: 10.1016/j.rmed.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 60.Wahlström J., Katchar K., Wigzell H., Olerup O., Eklund A., Grunewald J. Analysis of intracellular cytokines in CD4+ and CD8+ lung and blood T cells in sarcoidosis. American Journal of Respiratory and Critical Care Medicine. 2001;163(1):115–121. doi: 10.1164/ajrccm.163.1.9906071. [DOI] [PubMed] [Google Scholar]
- 61.Alavi H. A., Moscovic E. A. Immunolocalization of cell-wall-deficient forms of Mycobacterium tuberculosis complex in sarcoidosis and in sinus histiocytosis of lymph nodes draining carcinoma. Histology and Histopathology. 1996;11(3):683–694. [PubMed] [Google Scholar]
- 62.Hanngren A., Odham G., Eklund A., Hoffner S., Stjernberg N., Westerdahl G. Tuberculostearic acid in lymph nodes from patients with sarcoidosis. Sarcoidosis. 1987;4(2):101–104. [PubMed] [Google Scholar]
- 63.Gupta D., Agarwal R., Aggarwal A. N., Jindal S. K. Molecular evidence for the role of mycobacteria in sarcoidosis: a meta-analysis. European Respiratory Journal. 2007;30(3):508–516. doi: 10.1183/09031936.00002607. [DOI] [PubMed] [Google Scholar]
- 64.Song Z., Marzilli L., Greenlee B. M., et al. Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis. Journal of Experimental Medicine. 2005;201(5):755–767. doi: 10.1084/jem.20040429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Drake W. P., Dhason M. S., Nadaf M., et al. Cellular recognition of Mycobacterium tuberculosis ESAT-6 and KatG peptides in systemic sarcoidosis. Infection and Immunity. 2007;75(1):527–530. doi: 10.1128/IAI.00732-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oswald-Richter K. A., Beachboard D. C., Zhan X., et al. Multiple mycobacterial antigens are targets of the adaptive immune response in pulmonary sarcoidosis. Respiratory Research. 2010;11, article 161 doi: 10.1186/1465-9921-11-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dubaniewicz A., Kämpfer S., Singh M. Serum anti-mycobacterial heat shock proteins antibodies in sarcoidosis and tuberculosis. Tuberculosis. 2006;86(1):60–67. doi: 10.1016/j.tube.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 68.Minami J., Eishi Y., Ishige Y., et al. Pulmonary granulomas caused experimentally in mice by a recombinant trigger-factor protein of Propionibacterium acnes. Journal of Medical and Dental Sciences. 2003;50(4):265–274. [PubMed] [Google Scholar]
- 69.Eishi Y., Suga M., Ishige I., et al. Quantitative analysis of mycobacterial and propionibacterial DNA in lymph nodes of Japanese and European patients with sarcoidosis. Journal of Clinical Microbiology. 2002;40(1):198–204. doi: 10.1128/JCM.40.1.198-204.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hiramatsu J.-I., Kataoka M., Nakata Y., et al. Propionibacterium acnes DNA detected in bronchoalveolar lavage cells from patients with sarcoidosis. Sarcoidosis Vasculitis and Diffuse Lung Diseases. 2003;20(3):197–203. [PubMed] [Google Scholar]
- 71.Ishige I., Eishi Y., Takemura T., et al. Propionibacterium acnes is the most common bacterium commensal in peripheral lung tissue and mediastinal lymph nodes from subjects without sarcoidosis. Sarcoidosis Vasculitis and Diffuse Lung Diseases. 2005;22(1):33–42. [PubMed] [Google Scholar]
- 72.Nikoskelainen J., Hannuksela M., Palva T. Antibodies to Epstein Barr virus and some other herpesviruses in patients with sarcoidosis, pulmonary tuberculosis and erythema nodosum. Scandinavian Journal of Infectious Diseases. 1974;6(3):209–216. doi: 10.3109/inf.1974.6.issue-3.01. [DOI] [PubMed] [Google Scholar]
- 73.Mitchell D. N., McSwiggan D. A., Mikhail J. R., Heimer G. V. Antibody to herpes like virus in sarcoidosis. The American Review of Respiratory Disease. 1975;111(6):880–882. doi: 10.1164/arrd.1975.111.6.880. [DOI] [PubMed] [Google Scholar]
- 74.Chen E. S., Moller D. R. Etiology of sarcoidosis. Clinics in Chest Medicine. 2008;29(3):365–377. doi: 10.1016/j.ccm.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 75.Schilstra A., Rottoli P., Jacobs J. A., Van Suylen R. J., Galluzzi P., Drent M. Case studies to explore the pitfalls in the diagnosis of sarcoidosis. Sarcoidosis Vasculitis and Diffuse Lung Diseases. 2006;23(2):135–140. [PubMed] [Google Scholar]
- 76.Pukiat S., McCarthy P. L., Hahn T., et al. Sarcoidosis-associated MHC Ags and the development of cutaneous and nodal granulomas following allogeneic hematopoietic cell transplant. Bone Marrow Transplantation. 2011;46(7):1032–1034. doi: 10.1038/bmt.2010.235. [DOI] [PubMed] [Google Scholar]
- 77.Ionescu D. N., Hunt J. L., Lomago D., Yousem S. A. Recurrent sarcoidosis in lung transplant allografts: granulomas are of recipient origin. Diagnostic Molecular Pathology. 2005;14(3):140–145. doi: 10.1097/01.pas.0000176765.26047.6f. [DOI] [PubMed] [Google Scholar]
- 78.Padilla M. L., Schilero G. J., Teirstein A. S. Donor-acquired sarcoidosis. Sarcoidosis Vasculitis and Diffuse Lung Diseases. 2002;19(1):18–24. [PubMed] [Google Scholar]
- 79.Das B. B., Shoemaker L., Kim E., Mascio C. E., Austin E. H. Severe calcification of the aorta (porcelain aorta) associated with sarcoidosis in a pediatric heart transplant recipient. Pediatric Transplantation. 2012;16(5):E162–E166. doi: 10.1111/j.1399-3046.2011.01557.x. [DOI] [PubMed] [Google Scholar]
- 80.Kajdasz D. K., Lackland D. T., Mohr L. C., Jr., Judson M. A. A current assessment of rurally linked exposures as potential risk factors for sarcoidosis. Annals of Epidemiology. 2001;11(2):111–117. doi: 10.1016/S1047-2797(00)00179-4. [DOI] [PubMed] [Google Scholar]
- 81.Sarcoidosis among U.S. Navy Enlisted Men, 1965–1993. Morbidity and Mortality Weekly Report. 1997;46(23):539–543. [PubMed] [Google Scholar]
- 82.Prezant D. J., Dhala A., Goldstein A., et al. The incidence, prevalence, and severity of sarcoidosis in New York City firefighters. Chest. 1999;116(5):1183–1193. doi: 10.1378/chest.116.5.1183. [DOI] [PubMed] [Google Scholar]
- 83.Newman L. S., Rose C. S., Bresnitz E. A., et al. A case control etiologic study of sarcoidosis: environmental and occupational risk factors. American Journal of Respiratory and Critical Care Medicine. 2004;170(12):1324–1330. doi: 10.1164/rccm.200402-249OC. [DOI] [PubMed] [Google Scholar]
- 84.Heffner D. K. The cause of sarcoidosis: the Centurial enigma solved. Annals of Diagnostic Pathology. 2007;11(2):142–152. doi: 10.1016/j.anndiagpath.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 85.Wahlström J., Dengjel J., Persson B., et al. Identification of HLA-DR-bound peptides presented by human bronchoalveolar lavage cells in sarcoidosis. Journal of Clinical Investigation. 2007;117(11):3576–3582. doi: 10.1172/JCI32401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wahlström J., Dengjel J., Winqvist O., et al. Autoimmune T cell responses to antigenic peptides presented by bronchoalveolar lavage cell HLA-DR molecules in sarcoidosis. Clinical Immunology. 2009;133(3):353–363. doi: 10.1016/j.clim.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 87.Hill T. A., Lightman S., Pantelidis P., Abdallah A., Spagnolo P., du Bois R. M. Intracellular cytokine profiles and T cell activation in pulmonary sarcoidosis. Cytokine. 2008;42(3):289–292. doi: 10.1016/j.cyto.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 88.Rottoli P., Magi B., Perari M. G., et al. Cytokine profile and proteome analysis in bronchoalveolar lavage of patients with sarcoidosis, pulmonary fibrosis associated with systematic sclerosis and idiopathic pulmonary fibrosis. Proteomics. 2005;5(5):1423–1430. doi: 10.1002/pmic.200301007. [DOI] [PubMed] [Google Scholar]
- 89.Tsiligianni I., Antoniou K. M., Kyriakou D., et al. Th1/Th2 cytokine pattern in bronchoalveolar lavage fluid and induced sputum in pulmonary sarcoidosis. BMC Pulmonary Medicine. 2005;5, article 8 doi: 10.1186/1471-2466-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Katchar K., Eklund A., Grunewald J. Expression of Th1 markers by lung accumulated T cells in pulmonary sarcoidosis. Journal of Internal Medicine. 2003;254(6):564–571. doi: 10.1111/j.1365-2796.2003.01230.x. [DOI] [PubMed] [Google Scholar]
- 91.Kunisawa A., Kawanishi Y., Tago H., Nagate A., Kasuga I., Ohyashiki K. Direct demonstration of the productive capability of cytokines at the single cell level in lung sarcoidosis using multicolor cytometry. Respiration. 2002;69(2):155–164. doi: 10.1159/000056319. [DOI] [PubMed] [Google Scholar]
- 92.Hauber H.-P., Gholami D., Meyer A., Pforte A. Increased interleukin-13 expression in patients with sarcoidosis. Thorax. 2003;58(6):519–524. doi: 10.1136/thorax.58.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bettelli E., Korn T., Oukka M., Kuchroo V. K. Induction and effector functions of TH17 cells. Nature. 2008;453(7198):1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wikén M., Idali F., Al Hayja M. A., Grunewald J., Eklund A., Wahlström J. No evidence of altered alveolar macrophage polarization, but reduced expression of TLR2, in bronchoalveolar lavage cells in sarcoidosis. Respiratory Research. 2010;11, article 121 doi: 10.1186/1465-9921-11-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Davis M. D., Montpetit A., Hunt J. Exhaled breath condensate. An overview. Immunology and Allergy Clinics of North America. 2012;32(3):363–375. doi: 10.1016/j.iac.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kazani S., Israel E. Utility of exhaled breath condensates across respiratory diseases. American Journal of Respiratory and Critical Care Medicine. 2012;185(8):791–792. doi: 10.1164/rccm.201202-0212ED. [DOI] [PubMed] [Google Scholar]
- 97.Nakamura K., Mikuniya M., Takanashi S., et al. Is analysis of exhaled breath condensate an equivalent to bronchoalveolar lavage fluid in sarcoidosis patients? Hirosaki Medical Journal. 2012;63(1):12–20. [Google Scholar]
- 98.Liang Y., Yeligar S. M., Brown L. A. Exhaled breath condensate: a promising source for biomarkers of lung disease. The Scientific World Journal. 2012;2012:7. doi: 10.1100/2012/217518.217518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bons J. A., Drent M., Bouwman F. G., Mariman E. C., van Dieijen-Visser M. P., Wodzig W. K. Potential biomarkers for diagnosis of sarcoidosis using proteomics in serum. Respiratory Medicine. 2007;101(8):1687–1695. doi: 10.1016/j.rmed.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 100.Rozy A., Czerniawska J., Stȩpniewska A., et al. Inflammatory markers in the exhaled breath condensate of patients with pulmonary sacroidosis. Journal of Physiology and Pharmacology. 2006;57(4, supplement):335–340. [PubMed] [Google Scholar]
- 101.Kowalska A., Pus'cin'ska E., Czerniawska J., et al. Markers of fibrosis and inflammation in exhaled breath condensate (EBC) and bronchoalveolar lavage fluid (BALF) of patients with pulmonary sarcoidosis—a pilot study. Pneumonologia i Alergologia Polska. 2010;78(5):356–362. [PubMed] [Google Scholar]
- 102.Piotrowski W. J., Antczak A., Marczak J., Nawrocka A., Kurmanowska Z., Górski P. Eicosanoids in exhaled breath condensate and BAL fluid of patients with sarcoidosis. Chest. 2007;132(2):589–596. doi: 10.1378/chest.07-0215. [DOI] [PubMed] [Google Scholar]
- 103.Piotrowski W. J., Kurmanowska Z., Antczak A., Marczak J., Górski P. Exhaled 8-isoprostane as a prognostic marker in sarcoidosis. A short term follow-up. BMC Pulmonary Medicine. 2010;10, article 23 doi: 10.1186/1471-2466-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bargagli E., Bianchi N., Margollicci M., et al. Chitotriosidase and soluble IL-2 receptor: comparison of two markers of sarcoidosis severity. Scandinavian Journal of Clinical and Laboratory Investigation. 2008;68(6):479–483. doi: 10.1080/00365510701854975. [DOI] [PubMed] [Google Scholar]
- 105.Drent M., Jacobs J. A., De Vries J., Lamers R. J. S., Liem I. H., Wouters E. F. M. Does the cellular bronchoalveolar lavage fluid profile reflect the severity of sarcoidosis? European Respiratory Journal. 1999;13(6):1338–1344. doi: 10.1034/j.1399-3003.1999.13f18.x. [DOI] [PubMed] [Google Scholar]
- 106.Rothkrantz-Kos S., Van Dieijen-Visser M. P., Mulder P. G. H., Drent M. Potential usefulness of inflammatory markers to monitor respiratory functional impairment in sarcoidosis. Clinical Chemistry. 2003;49(9):1510–1517. doi: 10.1373/49.9.1510. [DOI] [PubMed] [Google Scholar]
- 107.Ziegenhagen M. W., Rothe M. E., Schlaak M., Müller-Quernheim J. Bronchoalveolar and serological parameters reflecting the severity of sarcoidosis. European Respiratory Journal. 2003;21(3):407–413. doi: 10.1183/09031936.03.00010403. [DOI] [PubMed] [Google Scholar]
- 108.Miyoshi S., Hamada H., Kadowaki T., et al. Comparative evaluation of serum markers in pulmonary sarcoidosis. Chest. 2010;137(6):1391–1397. doi: 10.1378/chest.09-1975. [DOI] [PubMed] [Google Scholar]
- 109.Shigemura M., Nasuhara Y., Konno S., et al. Effects of molecular structural variants on serum Krebs von den Lungen-6 levels in sarcoidosis. Journal of Translational Medicine. 2012;10, article 111 doi: 10.1186/1479-5876-10-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Paone G., Leone A., Batzella S., et al. Use of discriminant analysis in assessing pulmonary function worsening in patients with sarcoidosis by a panel of inflammatory biomarkers. Inflammation Research. 2013;62(3):325–332. doi: 10.1007/s00011-012-0585-9. [DOI] [PubMed] [Google Scholar]
- 111.Biller H., Ruprecht B., Gaede K. I., Müller-Quernheim J., Zissel G. Gene polymorphisms of ACE and the angiotensin receptor AT2R1 influence serum ACE levels in sarcoidosis. Sarcoidosis Vasculitis and Diffuse Lung Diseases. 2009;26(2):139–146. [PubMed] [Google Scholar]
- 112.Allen R. K., Pierce R. J., Barter C. E. Angiotensin-converting enzyme in bronchoalveolar lavage fluid in sarcoidosis. Sarcoidosis. 1992;9(1):54–59. [PubMed] [Google Scholar]
- 113.Jackson A. S., Sandrini A., Campbell C., Chow S., Thomas P. S., Yates D. H. Comparison of biomarkers in exhaled breath condensate and bronchoalveolar lavage. American Journal of Respiratory and Critical Care Medicine. 2007;175(3):222–227. doi: 10.1164/rccm.200601-107OC. [DOI] [PubMed] [Google Scholar]
- 114.Frisullo G., Angelucci F., Caggiula M., et al. pSTAT1, pSTAT3, and T-bet expression in peripheral blood mononuclear cells from relapsing-remitting multiple sclerosis patients correlates with disease activity. Journal of Neuroscience Research. 2006;84(5):1027–1036. doi: 10.1002/jnr.20995. [DOI] [PubMed] [Google Scholar]
- 115.Liston A., Papadopoulou A. S., Danso-Abeam D., Dooley J. MicroRNA-29 in the adaptive immune system: setting the threshold. Cellular and Molecular Life Sciences. 2012;69(21):3533–3541. doi: 10.1007/s00018-012-1124-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ma F., Xu S., Liu X., et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-γ . Nature Immunology. 2011;12(9):861–869. doi: 10.1038/ni.2073. [DOI] [PubMed] [Google Scholar]
- 117.Croce C. M. Causes and consequences of microRNA dysregulation in cancer. Nature Reviews Genetics. 2009;10(10):704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ezzie M. E., Crawford M., Cho J.-H., et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. 2012;67(2):122–131. doi: 10.1136/thoraxjnl-2011-200089. [DOI] [PubMed] [Google Scholar]
- 119.Liu G., Friggeri A., Yang Y., et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. Journal of Experimental Medicine. 2010;207(8):1589–1597. doi: 10.1084/jem.20100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Crouser E. D., Julian M. W., Crawford M., et al. Differential expression of microRNA and predicted targets in pulmonary sarcoidosis. Biochemical and Biophysical Research Communications. 2012;417(2):886–891. doi: 10.1016/j.bbrc.2011.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Djuretic I. M., Levanon D., Negreanu V., Groner Y., Rao A., Ansel K. M. Erratum: Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nature Immunology. 2007;8(2):145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 122.Ji N., Sosa R. A., Forsthuber T. G. More than just a T-box: the role of T-bet as a possible biomarker and therapeutic target in autoimmune diseases. Immunotherapy. 2011;3(3):435–441. doi: 10.2217/imt.10.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang H.-T., Ge X.-S., Xue Z.-P., Li B.-Q. Role of transcription factor T-bet and Eomes in IFN-gamma secretion of different human T cell subsets. Chinese Journal of Cellular and Molecular Immunology. 2010;26(1):31–34. [PubMed] [Google Scholar]
- 124.Holtmann M. H., Neurath M. F. T helper cell polarisation in coeliac disease: any (T-)bet? Gut. 2004;53(8):1065–1067. doi: 10.1136/gut.2003.038232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Matsuoka K., Inoue N., Sato T., et al. T-bet upregulation and subsequent interleukin 12 stimulation are essential for induction or Th1 mediated immunopathology in Crohn's disease. Gut. 2004;53(9):1303–1308. doi: 10.1136/gut.2003.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li B., Yang P., Zhou H., et al. T-bet expression is upregulated in active Behçet's disease. British Journal of Ophthalmology. 2003;87(10):1264–1267. doi: 10.1136/bjo.87.10.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rajendram R., Rao N. A. Molecular mechanisms in Behçet's disease. British Journal of Ophthalmology. 2003;87(10):1199–1200. doi: 10.1136/bjo.87.10.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kriegova E., Fillerova R., Tomankova T., et al. T-helper cell type-1 transcription factor T-bet is upregulated in pulmonary sarcoidosis. European Respiratory Journal. 2011;38(5):1136–1144. doi: 10.1183/09031936.00089910. [DOI] [PubMed] [Google Scholar]
- 129.Cox B., Kislinger T., Emili A. Integrating gene and protein expression data: pattern analysis and profile mining. Methods. 2005;35(3):303–314. doi: 10.1016/j.ymeth.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 130.Hack C. J. Integrated transcriptome and proteome data: the challenges ahead. Briefings in Functional Genomics & Proteomics. 2004;3(3):212–219. doi: 10.1093/bfgp/3.3.212. [DOI] [PubMed] [Google Scholar]