

Abstract
We have recently shown that E1A protein of human adenovirus downregulates epidermal growth factor receptor (EGFR) expression and induces apoptosis in head and neck (HNSCC) and lung cancer cells independently of their p53 status. E1A has five isoforms of which the major ones E1A12S and E1A13S regulate transcription of cellular genes by binding to transcriptional modulators such as pRB, CtBP, p300 and p400. In this study, we have identified E1A12S isoform to have the highest effect on EGFR suppression and induction of apoptosis in HNSCC cells. Similar to Ad5, E1A12S from human adenovirus types 2, 3, 9 and 12 suppressed EGFR, whereas E1A12S of adenovirus types 4 and 40 had no effect on EGFR expression. Using deletion mutants of E1A12S we have shown that interaction of E1A with p400, but not p300 or pRB, is required for EGFR suppression and apoptosis. Inhibition of p400 by short hairpin RNA confirmed that HNSCC cells with reduced p400 expression were less sensitive to E1A-induced suppression of EGFR and apoptosis. p300 function was shown to be dispensable, as cells expressing E1A mutants that are unable to bind p300, or p300 knockout cells, remained sensitive to E1A-induced apoptosis. In summary, this study identifies p400 as an important mediator of E1A-induced downregulation of EGFR and apoptosis.
Keywords: p400, EGFR, E1A, apoptosis, head and neck cancer
Introduction
Activation of epidermal growth factor receptor (EGFR) is associated with increased tumour growth, metastasis and adverse outcome in many epithelial cancers, particularly squamous cell carcinomas of lung and head and neck (HNSCC) (Prenzel et al., 2001; Kalyankrishna and Grandis, 2006). Because EGFR is overexpressed in many cancers and its inhibition causes tumour growth inhibition, novel and effective agents that inhibit EGFR can have a major clinical and commercial benefit. So far, several strategies have been used to block EGFR activity, including monoclonal antibodies, antisense oligonucleotides, ligand–toxin and immunotoxin conjugates, as well as tyrosine kinase inhibitors (reviewed in Prenzel et al., 2001).
The E1A gene of the subgroup C of human adenovirus types 2 and 5 is essential for adenoviral replication, the primary E1A transcript produces five distinct mRNA isoforms; 13S, 12S, 11S, 10S and 9S (Frisch and Mymryk, 2002). E1A interacts with a number of cellular regulatory proteins to reprogramme gene expression and to induce cell growth (Frisch and Mymryk, 2002). In particular, E1A targets pRB family members, the p300/CBP acetyltransferases and the CtBP family of transcriptional repressors (Egan et al., 1988). CtBPs are important regulators of both gene transcription and organelle assembly. They are involved in a large number of transcriptional regulatory networks and intracellular signalling pathways, and play an essential pro-survival role in tumour-derived cells (Bergman and Blaydes, 2006). The N-terminal end of E1A binds and interferes with the function of p300 and CBP, two highly related transcriptional co-activators that regulate the expression of genes involved in growth inhibition and differentiation (reviewed in Samuelson et al., 2005). The N-terminal portion of E1A also binds two unrelated proteins, p400 and TRRAP, which interact with each other and function in large multi-subunit complexes that regulate gene transcription by affecting chromatin organization (Fuchs et al., 2001). Interaction of E1A with p300/CBP is seemingly not required for induction of ARF, p53 and apoptosis in normal fibroblasts, whereas E1A interaction with the p400-containing complex has been shown to be important (Samuelson et al., 2005). The p400 complex was shown to be part of the p21WAF1/CIP1/sid1 pathway; it inhibits transcription of p21WAF1 through p53 and the senescence pathway (Chan et al., 2005). We and others have shown that E1A12S, but not E1A13S, suppresses transcription from the p21WAF1 promoter resulting in cell-cycle arrest (Najafi et al., 2003; Flinterman et al., 2005). In addition, we have shown that induction of apoptosis in normal rat kidney and mouse embryonic fibroblast (MEF) cells requires the interaction of E1A with p300 and pRB (Mymryk et al., 1994; Samuelson and Lowe, 1997). Other studies using rodent and human cells have demonstrated that activation of apoptosis is predominantly mediated by interaction of E1A with p300 (Chiou and White, 1997; Querido et al., 1997). In some cases, the induction of apoptosis by E1A is clearly related to the stabilization of p53 (Debbas and White, 1993; Lowe and Ruley, 1993). However, E1A can clearly stimulate apoptosis independently of p53 (Flinterman et al., 2003; Samuelson et al., 2005). E1A has been shown to have antitumour and antimetastatic functions in cancer cells by suppressing overexpression of c-erbB2/HER-2, a member of the EGFR tyrosine kinase family (reviewed in Yu and Hung, 1998). We have recently shown that E1A downregulates EGFR expression, resulting in the induction of apoptosis in a panel of HNSCC cell lines (Flinterman et al., 2003). Furthermore, overexpression of EGFR under a heterologous promoter conferred resistance to E1A-induced apoptosis. This provides direct evidence that E1A-induced EGFR downregulation mediates induction of apoptosis. All cell lines used in our study lacked functional p53, suggesting that in these cells E1A-induced EGFR suppression and induction of cell death is p53-independent (Flinterman et al., 2003).
Here, we have used all five E1A isoforms as well as a panel of E1A mutant and deletion constructs to identify the E1A region/s important for EGFR suppression and induction of apoptosis. The 12S-encoded E1A was found to be the most effective suppressor of EGFR expression in HNSCC cells. Using deletion mutants of E1A12S interaction of E1A with p400, but not p300 or pRB, was found to be important for the ability of E1A to suppress EGFR. Silencing of p400 by short hairpin RNA (shRNA) confirmed that HNSCC cells with reduced p400 expression were less sensitive to E1A-induced EGFR suppression and apoptosis. However, p300 knockout in HCT116 colon cancer cells did not block E1A-induced apoptosis. In summary, this study identifies p400 as an important component involved in the regulation of EGFR and may lead to the discovery of novel control mechanisms of EGFR expression.
Results
Adenovirus type 2 E1A12S isoform induces downregulation of EGFR expression
The effect of each of the five Ad2 E1A isoforms, fused to green fluorescent protein (GFP), on EGFR expression was investigated in EGFR overexpressing HN5 cells (Figure 1a). The expression of the E1A12S protein (green) resulted in a significant suppression of EGFR (red) 48 h after transfection. Expression of E1A13S showed some reduction in EGFR levels but to a lesser extent than E1A12S. Expression of other E1A isoforms was ineffective in EGFR downregulation (Figure 1b). Both E1A12S- and E1A13S-encoded proteins contain a region between amino acids 27 and 98, which is absent in the E1A9S, E1A10S and E1A11S products. These results suggest that this region, which includes the conserved region (CR) 1 domain, is important for E1A-induced EGFR downregulation. The CR1 domain of E1A contains binding sites for both p300 and p400 proteins. Plasmids encoding the conserved regions CR1, CR2 and CR3 on their own were similarly examined in HN5 cells. No effect on EGFR expression was observed by the expression of individual CRs, possibly owing to their cytoplasmic localization, as detected by indirect immunofluorescent microscopy (data not shown).
Figure 1.

(a) Different splice variants of the E1A gene are schematically depicted. Boxes represent coding sequences. The coding sequence represented by the light grey box in E1A9S is in a different frame from the other coding sequences. The number of amino acids of the encoded E1A isoforms and the positions of the conserved regions CR1, CR2, CR3 and CR4 in the different E1A splice variants are indicated (CR positions have previously been described in Avvakumov et al., 2002, 2004). (b) Downregulation of EGFR expression by adenovirus type 2 E1A12S. HN5 cells were transiently transfected with plasmids expressing adenovirus type 2 E1A9S, E1A10S, E1A11S, E1A12S and E1A13S fused to GFP. Cells were fixed 48 h post-transfection and stained for EGFR by indirect immunofluorescent staining using anti-EGFR F4 primary antibody and Texas-Red-labelled secondary anti-mouse antibody. Cells were mounted in DAPI-containing solution. EGFR stained red and E1A-GFP was observed as green. E1A expressing cells are indicated with white arrows. Abbreviations: CR, conserved region; DAPI, 4′,6-diamino-2-phenylindole; EGFR, epidermal growth factor receptor; GFP, green fluorescent protein.
EGFR suppression by E1A of different human adenovirus types
As shown above, the adenovirus type 2 E1A12S product downregulated EGFR overexpression in HN5 cells. We then investigated the effect of the corresponding 12S encoded E1A proteins from a representative panel of five other human adenovirus subgroups. HN5 cells were transiently transfected with plasmids expressing E1A12S of human adenovirus type 3, 4, 9, 12 or 40 fused to GFP; cells were analysed 48 h after transfection. E1A12S of adenovirus types 2, 3, 9 and 12 induced EGFR downregulation, while expression of E1A12S of adenovirus types 4 and 40 had no effect on EGFR expression (Figure 2 and Table 1). Therefore, E1A12S of most, but not all human adenoviruses can suppress EGFR expression in HN5 cells.
Figure 2.
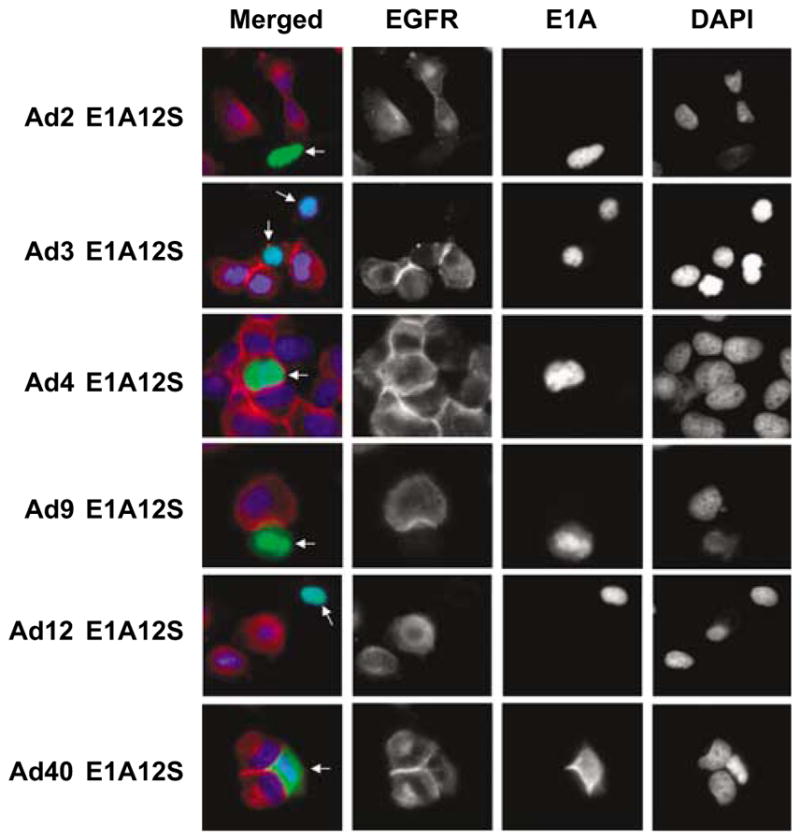
Downregulation of EGFR expression by the 12S-encoded E1A proteins of human adenovirus types 2, 3, 9 and 12, but not 4 and 40. HN5 cells were transiently transfected with GFP plasmids expressing E1A12S of different human adenovirus types. Cells were fixed 48 h post-transfection and stained for EGFR by indirect immunofluorescent staining using anti-EGFR F4 primary antibody and Texas-Red-labelled secondary anti-mouse antibody. Cells were mounted in DAPI-containing solution. EGFR stained red and E1A-GFP was observed as green. E1A expressing cells are indicated with white arrows. Abbreviations: DAPI, 4′,6-diamino-2-phenylindole; EGFR, epidermal growth factor receptor; GFP, green fluorescent protein.
Table 1.
The effect of E1A isoforms from different adenovirus subgroups on EGFR expression in HN5 cells
| Construct (all fused to GFP) | Localization | EGFR suppression | Residues from E1A12S |
|---|---|---|---|
| GFP | Cytoplasmic | No | — |
| CR1 | Cytoplasmic | No | aa 42–72 |
| CR2 | Cytoplasmic | No | aa 115–140 |
| CR3 | Cytoplasmic | No | aa 44–191 |
| C-term | Nuclear | No | aa 187–289 |
| Ad3 12S | Nuclear | Yes | — |
| Ad4 12S | Nuclear | No | — |
| Ad9 12S | Nuclear | Yes | — |
| Ad12 12S | Nuclear | Yes | — |
| Ad40 12S | Nuclear/cytoplasmic | No | — |
| Ad2 9S | Cytoplasmic | No | Contains 55 aa, bp 560–637, 1229–1315 |
| Ad2 10S | Cytoplasmic/nuclear | No | Contains 171 aa, bp 560–637, 854–974, 1229–1542 |
| Ad2 11S | Nuclear | No | Contains 217 aa, bp 560–637, 854–1112, 1229–1542 |
| Ad2 12S | Nuclear | Yes | Contains 243 aa, bp 560–974, 1229–1542 |
| Ad2 13S | Nuclear | Yes | Contains 289 aa, bp 560–1112, 1229–1542 |
Abbreviations: aa, amino acids; bp, base pair; EGFR, epidermal growth factor receptor; GFP, green fluorescent protein.
Identification of E1A domains involved in EGFR suppression
Having identified E1A12S as the most effective isoform in EGFR suppression, we sought to identify the E1A12S region/s responsible for EGFR downregulation. HN5 cells were transfected with a panel of deletion/mutant E1A-GFP constructs listed in Table 2 and Figure 3a. We have previously shown that these cells express mutant p53 (Sartor et al., 1999) and wild-type pRB (Paterson et al., 1995). The C-terminal end of E1A12S (C-term; amino acids 187–289), which binds the CtBP transcriptional repressor, was used as a negative control (Figure 3a). Expression of the E1A12S mutants dl1101, dl1102, dl1103 (unable to bind p400) and Y47H/C124G (unable to bind pRB) (Wang et al., 1993) had no effect on the level of EGFR (Figure 3b). Expression of the E1A12S mutants dl1104, RG2 (unable to bind p300), dl1107 and Y47H (unable to bind pRB in some cell types such as HeLa, but able to bind pRB in other cell types such as BRK cells) (Wang et al., 1993) downregulated EGFR as efficiently as the wild-type E1A12S (Figure 3b). These results show that binding to p300 is not required for E1A-mediated EGFR suppression. In contrast, dl1101, dl1102, dl1103 mutants that are unable to bind p400 have lost the ability to suppress EGFR expression (Figure 3b). These results indicate that p400, but not p300, is required for EGFR suppression by E1A (summarized in Table 2). The role of pRB was less clear as the Y47H mutant with a mutation in the putative pRB-binding site suppressed EGFR expression, suggesting that pRB may not be required for EGFR downregulation by E1A. However, the Y47H/C124G double mutant, with mutations in both the putative and the known pRB-binding sites was inactive in EGFR downregulation.
Table 2.
The effect of E1A12S deletion mutants on EGFR expression in HN5 cells
| Constructa | Localization | EGFR suppression | Residues from E1A12S |
|---|---|---|---|
| dl1101-GFP (Δ569–634) | Nuclear | No | Deletion aa 4–25, does not bind p300 or p400 |
| dl1102-GFP (Δ635–664) | Nuclear | No | Deletion aa 25–35, does not bind p400 but still binds p300 |
| dl1103-GFP (Δ647–706) | Nuclear | No | Deletion aa 30–49, does not bind p400 or p300 |
| dl1104-GFP (Δ701–739) | Nuclear | Yes | Deletion aa 48–60, does not bind p300 |
| dl1107-GFP (Δ890–928) | Nuclear | Yes | Deletion aa 111–123, does not bind pRB |
| dl1102/06 | Partially | Deletion aa 26–35 and 90–105, does not bind p400 but still binds p300 | |
| dl1102/08 | Partially | Deletion aa 26–35 and 124–127, does not bind p400 or pRB, but still binds p300 | |
| dl1105/07 | Yes | Deletion aa 70–81 and 111–123, does not bind pRB | |
| 1–158-GFP | Cytoplasmic/nuclear | Yes | Deletion aa 159–243, binds pRB, p300, p400 |
| RG2-GFP | Nuclear | Yes | Point mutation at 2 from R to G, does not bind p300, reduced binding to pRB |
| Y47H-GFP | Nuclear | Yes | Point mutation at 47 from Y to H, does not bind pRB or p130 in HeLa cells, but does bind pRB in BRK cells (Wang et al., 1993) |
| Y47H/C124G-GFP | Nuclear | No | Point mutations at 47 from Y to H and at 124 C to G, double mutant, does not bind pRB and p107 in HeLa or in BRK cells (Wang et al., 1993). |
Abbreviations: EGFR, epidermal growth factor receptor; GFP, green fluorescent protein.
The position of base-pair deletions in the E1A12S is shown in bracket.
Figure 3.


Downregulation of EGFR expression by adenovirus type 2 E1A is p400-dependent. (a) Map showing adenovirus type 2 E1A12S deletion mutants and the regions required for binding to p300, p400 and pRB. Binding of E1A and its mutants to the indicated proteins was graded as complete (+), reduced (+/−) or none (−) (Wang et al., 1993). (b) HN5 cells were transiently transfected with plasmids expressing GFP fused to E1A12S or deletion mutants that are unable to bind p300 (dl1101, dl1103 and dl1104, RG2), p400 (dl1101 and dl1102) or pRB (dl1107, Y47H and Y47H,C124G), as well as the C-terminal region of E1A, (C-term). Cells were fixed 48 h post-transfection and stained for EGFR by indirect immunofluorescent staining using anti-EGFR F4 primary antibody and Texas-Red-labelled secondary anti-mouse antibody. Cells were mounted in DAPI-containing solution. EGFR stained red and E1A-GFP was observed as green. E1A expressing cells are indicated with white arrows. (c) H357 cells were infected with adenovirus expressing E1A12S and deletion mutants that are unable to bind p300 (dl1101), p400 (dl1101, dl1102/06 and dl1102/08) or pRB (dl1105/07) as well as control expressing only the E1A9S product. Cells were lysed 48 h post-infection and equal amounts of total protein from each sample was separated by SDS–PAGE, transferred to nitrocellulose and hybridized using different antibodies as described in the Materials and methods section. The intensity of tubulin and EGFR bands from the western blot was measured using Version 2.0 of Aida 2D Densitometry software. The amount of EGFR protein was normalized with tubulin expression level to correct for loading differences. The percentage of EGFR expression of H357 cells infected with Ad-E1A12S and its mutants was calculated with respect to the EGFR expression of control Ad-E1A9S-infected H357 cells which was set at 100%. Abbreviations: EGFR, epidermal growth factor receptor; GFP, green fluorescent protein; SDS–PAGE, lauryl sulphate–polyacrylamide gel electrophoresis.
The importance of these regions in E1A-mediated EGFR suppression was further investigated by western blot analysis. EGFR overexpressing H357 cells with mutant p53 (Sartor et al., 1999) and a functional pRB pathway (based on TGF-β-mediated accumulation of underphosphorylated pRB (Paterson et al., 1995)) were infected with adenovirus vectors encoding Ad5 E1A12S, dl1101, dl1102/06, dl1102/08, dl1105/07 or the control E1A9S, which was shown not to suppress EGFR (Figure 3a). At 48 h post-infection, cell lysates were analysed for EGFR expression. An almost complete EGFR suppression was observed in cells expressing E1A12S and significant EGFR downregulation in cells expressing dl1105/07, which contains a deletion in the pRB-binding site (Figure 3c). EGFR expression was quantified using Aida 2D Densitometry software. Normalization against the control Ad-E1A9S-infected cells showed EGFR expression was reduced to 7 and 10% by E1A12S and dl1105/07 (unable to bind pRB), respectively (Figure 3c). In contrast, control, non-infected or dl1101 (unable to bind p300 or p400)-infected cells did not show EGFR suppression. Cells expressing dl1102/06 or dl1102/08 (unable to bind p400 but still able to bind p300) showed reduced EGFR suppression (EGFR expression was 30 and 32%, respectively, as compared to control cells expressing E1A9S) but no apoptosis, as no poly (ADP ribose) polymerase (PARP) cleavage was detected (Figure 3c). Differences in the levels of E1A protein between different samples, is believed to be due to higher affinity of the M73 anti-E1A antibody, which binds to the C-terminus of exon 2 of E1A, for complete E1A12S. High-level expression of mutant E1A was shown using anti-E1A antibody M58 (data not shown). EGFR suppression by E1A12S and dl1105/07 was shown to induce apoptosis as it corresponded with the detection of PARP p85 fragment (Figure 3c). These results further confirm the importance of p400 but not p300 and pRB in the E1A-induced EGFR suppression in HNSCC cell lines HN5 and H357.
E1A induces apoptosis by downregulation of EGFR protein and mRNA
The effect of E1A on EGFR protein and mRNA levels was investigated in H357 and Saos-2 cells, using a recombinant adenovirus vector expressing wild-type E1A and a control Ad-Del vector (Yan et al., 1991). Western blot analysis showed downregulation of EGFR protein levels in Ad-E1A-infected H357 and Saos-2 cells at 24 and 48 h (Figure 4a). Complete downregulation of EGFR was observed in H357 cells at 48 h, but not in control Ad-Del-infected H357 cells. Apoptosis detected by PARP p85 antibody was shown in Ad-E1A-infected H357 and Saos-2 cells but not in non-infected or Ad-Del-infected cells (Figure 4a). 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) assay suggested that Saos-2 cells were less sensitive to E1A-induced cell death than EGFR overexpressing H357 (data not shown). At 48 h, control non-infected and Ad-Del-infected Saos-2 cells showed higher EGFR expression than at 24 h, these cells have a slow growth rate; therefore, this difference could be due to more cells being in the log growth phase at 48 h. Downregulation of EGFR was not due to reduced protein levels as actin levels were similar in all samples. We have previously shown that E1A expression results in reorganization of promyelocytic leukaemia (PML) oncogenic domains (PODs) in HN5 cells. PODs are dynamic nuclear subcompartments involved in the regulation of several cellular processes such as transcription, tumour suppression and apoptosis (reviewed in Vallian et al., 1998). In this study, western blot analysis showed induction of PML protein levels in E1A expressing Saos-2 and in H357 cells but not in the controls (Figure 4a). Semiquantitative reverse transcription polymerase chain reaction (RT–PCR) showed that similar to protein levels, at 48 h EGFR mRNA levels were downregulated in Ad-E1A-infected H357 cells compared with controls (Figure 4b).
Figure 4.
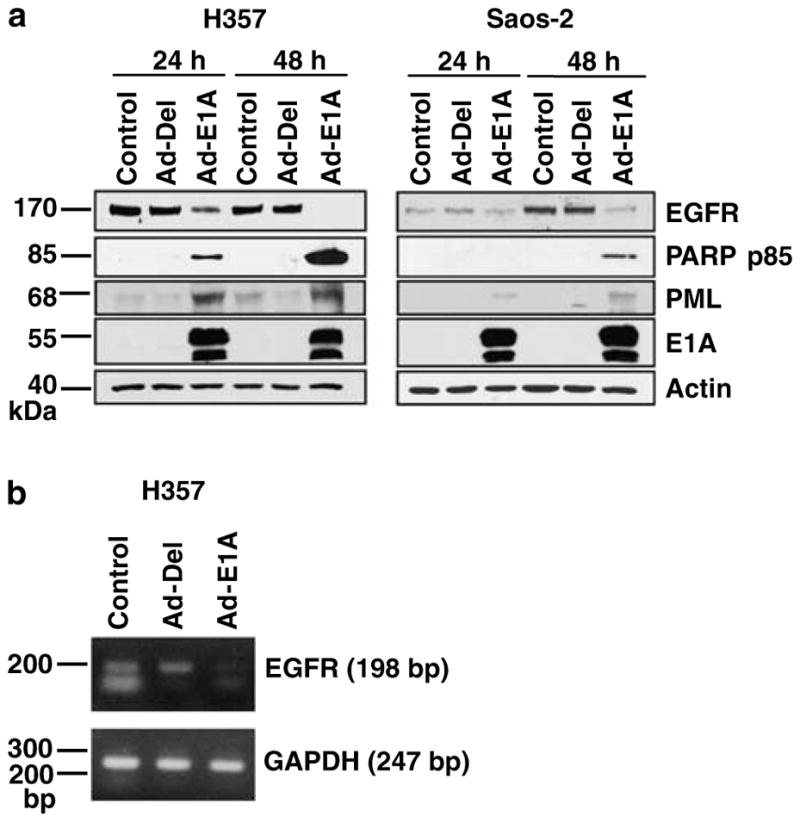
Adenovirus type 2 E1A downregulates EGFR protein and mRNA. (a) Western blot analysis showing expression of EGFR, PARP p85, E1A and actin using F4, G734A, M58 and anti-β-actin primary antibodies, respectively. H357 and Saos-2 cells were infected with Ad-E1A or control Ad-Del adenoviral vectors at a MOI of 10. Cells were lysed 24 and 48 h after infection, equal amounts of total protein from each sample was separated by SDS–PAGE, transferred to nitrocellulose and hybridized to different antibodies as described in the Materials and methods section. (b) E1A-mediated downregulation of EGFR mRNA. H357 cells were either untreated or infected with Ad-Del or Ad-E1A at a MOI of 10 and harvested 48 h after infection. PCR was performed on cDNA generated from whole mRNA. Specific EGFR was amplified using GAPDH as an internal control. The sizes of amplified fragments are indicated. Abbreviations: cDNA, complementary DNA; EGFR, epidermal growth factor receptor; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; MOI, multiplicity of infection; PARP, poly (ADP ribose) polymerase; PCR, polymerase chain reaction; SDS–PAGE, lauryl sulphate–polyacrylamide gel electrophoresis.
Role of p300 in E1A-induced cell death
Previous studies have implicated p300 as a key target for E1A-induced apoptosis (Mymryk et al., 1994; Chiou and White, 1997). Using E1A deletion mutants (Figure 3 and Table 2), p300 binding was found not to be required for E1A-induced EGFR suppression and induction of apoptosis. To further investigate the role of p300, wild-type HCT116 or syngeneic p300−/− colon cancer cells were infected with adenovirus expressing E1A12S (Figure 5). MTT cell viability assay showed efficient killing of p300−/− HCT116 cells by E1A12S (Figure 5a). Western blot analysis showed undetectable levels of p53 in untreated HCT116 cells, which are p53+/+ (Figure 5b, right panel). E1A expression induced the elevation of p53 protein levels in both HCT116 and p300−/− HCT116 cells. However, apoptosis, indicated by PARP p85, was detectable only in the E1A expressing p300−/− cells, confirming that E1A-mediated induction of cell death is through a p300- and p53-independent pathway. These data are in agreement with the study of Iyer et al. (2004), showing that HCT116 cells were more sensitive to DNA-damage-induced apoptosis in the absence of p300 (Iyer et al., 2004). The effect of E1A on EGFR expression was also examined in the HCT116 cells. However, these cells were shown to have undetectable EGFR levels and therefore the results were inconclusive (data not shown). In summary, these results suggest that p300 is not required for E1A-induced cell death. The morphological analysis (data not shown) and detection of PARP p85 suggest that p300−/− HCT116 cells are more sensitive to E1A-induced apoptosis than parental HCT116.
Figure 5.
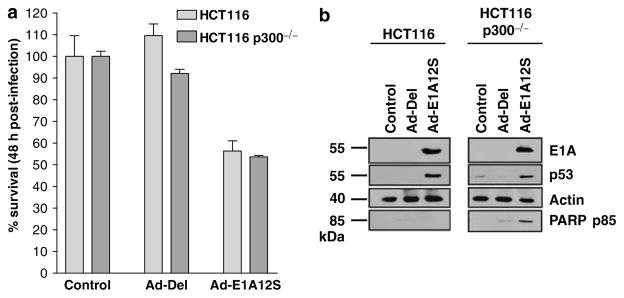
(a) MTT analysis of HCT116 and HCT116 p300−/− cells 48 h after infection with Ad-E1A12S or control Ad-Del adenoviral vectors at a MOI of 10. Error bars indicate s.d. (b) Western blot analysis of HCT116 and HCT116 p300−/− cells infected with indicated recombinant adenoviruses. Cells were lysed 24 h after infection, equal amounts of total protein from each sample was separated by SDS–PAGE, transferred to nitrocellulose and hybridized to different antibodies as described in the Materials and methods section. Abbreviations: MOI, multiplicity of infection; MTT, 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide; SDS–PAGE, lauryl sulphate–polyacrylamide gel electrophoresis.
Role of p400 in E1A-mediated downregulation of EGFR
It has recently been shown that p400 is required for E1A-induced apoptosis (Samuelson et al., 2005). We therefore investigated the role of p400 in the E1A-induced downregulation of EGFR and apoptosis in HNSCC cells. p400 protein was silenced using a p400 shRNA expressing retrovirus as described previously (Samuelson et al., 2005). Selected cell populations were established in several cancer cell lines including the HSC3 cells. These mixed cell populations were used to investigate the effect of p400 on E1A-induced apoptosis. Significant reductions in p400 protein levels were observed by western blot analysis in several p400 shRNA-selected populations derived from HSC3 cells. The HSC3-p400mp2 mixed population showing the highest inhibition of p400 protein level was used in subsequent experiments; the empty vector transduced cells (HSC3-HImp1) were used as control (Figure 6a). The p400 level was quantified using Aida 2D Densitometry software showing the reduction of p400 in HSC3-p400mp2 to 15% of the p400 expression level in HSC3-HImp1 cells (Figure 6a). MTT assay detected a significant reduction in cell death (P<0.001) at 72 h in Ad-E1A-infected HSC3-p400mp2 compared to Ad-E1A-infected HSC3-HImp1 cells (Figure 6b). PARP p85 was detected only in HSC3-HImp1 cells and not in Ad-E1A-infected HSC3-p400mp2 cells. These results indicate reduced sensitivity to apoptosis in cells with reduced p400 expression (Figure 6c). These results further confirm that p400 is an important component of E1A-induced suppression of EGFR expression and cell death.
Figure 6.

(a) Western blot analysis of HSC3 HI and p400 mixed populations. p400 was detected using anti-p400 RW144 monoclonal primary antibody and tubulin was detected using anti-tubulin monoclonal antibody (Sigma). The intensity of tubulin and p400 bands from the western blot was measured using Version 2.0 of Aida 2D Densitometry software. The amount of p400 protein was normalized with tubulin expression level to correct for loading differences. The percentage of p400 expression of HSC3-p400mp2 was calculated with respect to HSC3-HImp1, which was set at 100%. (b) MTT analysis of HSC3 HI and p400 mixed populations 48 and 72 h after infection with Ad-Del and Ad-E1A with a MOI of 10. Error bars indicate s.d. Statistical analysis was performed using Tukey’s Multiple Comparison Test, P-values are shown. (c) Western blot analysis of HSC3-HImp1 and HSC3-p400mp2 48 and 72 h after infection with indicated recombinant adenoviruses. Cells were lysed at 48 and 72 h post-infection, equal amounts of total protein from each sample was separated by SDS–PAGE, transferred to nitrocellulose and hybridized to different antibodies as described in the Materials and methods section. The percentage of EGFR expression of cells infected with Ad-E1A was calculated with respect to the EGFR expression of control Ad-Del-infected cells, which was set at 100%. Abbreviations: EGFR, epidermal growth factor receptor; MTT, 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide; SDS–PAGE, lauryl sulphate–polyacrylamide gel electrophoresis.
Expression of E1A12S of Ad2, Ad3, Ad9 and Ad12, but not Ad4 and Ad40, was shown to induce EGFR suppression (Figure 2). Therefore, we next compared the homology of the p400-binding site in these different E1A sequences. Amino acids 12–47 of Ad2 E1A have been identified as the region required for interaction with p400 (Barbeau et al., 1994; Samuelson et al., 2005). Sequence alignment of this region with the corresponding regions of the other E1A sequences used in this study (Figure 7; Avvakumov et al., 2004) did not reveal an obvious difference between the p400-binding site of E1A12S in Ad4 and Ad40 (unable to suppress EGFR) with Ad2, Ad3, Ad9 and Ad12 (able to suppress EGFR).
Figure 7.

Alignment of the homologous regions of the six different E1As used in this study (adapted from Avvakumov et al., 2004). The sequence spans amino acids 12–47 of Ad2 E1A, which corresponds to the region required for interaction to p400 (Barbeau et al., 1994; Samuelson et al., 2005). Shading is based on similarity as defined by the Blosum 45 score table.
However, protein structure prediction using the JNET neural network feature of JPRED (Cuff and Barton, 2000) suggests that amino acids 12–47 of E1A12S of Ad2, Ad3, Ad9 and Ad12 form an apparent motif family. The first half of the sequence in these four proteins forms an α-helix, with the rest of the sequence apparently having no predicted structure (Figure 1, Supplementary Data). Interestingly, Ad4 and Ad40 E1A12S seem to have a different structure in this part of the sequence. Ad4 E1A12S seems to lack the robust helical structure on the front end. Ad40 E1A12S, however, has a second helical structure in the back end of the sequence that the four EGFR suppressing sequences do not have, suggesting that the second helix alters the binding ability of the front end α-helix somehow. It can be speculated that these structural differences in the region binding p400 could provide an explanation as to why Ad4 and Ad40 E1A12S differentially regulate EGFR.
Discussion
In this study, E1A12S was shown to strongly suppress EGFR expression in HNSCC cells. E1A13S also showed some effect on EGFR suppression, but significantly less than E1A12S (Figure 1b). The other isoforms E1A9S, E1A10S and E1A11S, which lack the CR1 region, had no significant effect on EGFR expression (Figure 1b), identifying CR1 as an important region in the regulation of EGFR expression. CR1 contains binding sites for p300 and p400, as well as a putative binding site for pRB. These host proteins are found in distinct chromatin remodelling complexes that modulate gene expression either by activating or repressing transcription. The importance of these factors in the E1A-mediated regulation of EGFR was therefore investigated. Our data showed that the ability to bind p400, but not p300 or pRB, is important for E1A-induced suppression of EGFR expression and apoptosis (Figure 3). HN5 and H357 cells used in this study express wild-type pRB (Sartor et al., 1999) and H357 cells respond to TGF-β-mediated accumulation of underphosphorylated pRB protein (Paterson et al., 1995), suggesting that the pRB pathway is functional in these cells. Therefore, it is likely that E1A binds pRB to regulate transcription of some cellular proteins. However, pRB binding did not appear to be required for EGFR suppression by E1A. Specifically, E1A12S mutants lacking p400-binding sites were unable to induce apoptosis and to suppress EGFR expression in HNSCC cell lines (Figure 3). Using shRNA-mediated inhibition, we have demonstrated that HNSCC cells expressing reduced levels of p400 become resistant to E1A-induced EGFR suppression and apoptosis (Figure 6). Collectively, these results strongly suggest that p400 is an important mediator in these processes.
Currently, there are no data available to indicate whether there are any physical or functional interactions between p400 and EGFR. Chromatin immunoprecipitation (ChIP) analysis has shown p400 complexes at certain c-Myc- and E2F-regulated promoters. The recruitment of p400 to these promoters was correlated with an increase in histone acetylation in the surrounding chromatin and induction of transcription (Frank et al., 2003; Taubert et al., 2004). These studies suggest that p400-containing complexes employ major chromatin-modifying functions and that they are involved in transcription regulation. Tyteca et al. (2006) have shown that it was possible to change the fate of irradiated cells from undergoing apoptosis to cell-cycle arrest by altering p400 levels. They conclude that cells with lower p400 expression become less sensitive to DNA-damage-induced apoptosis. The histone acetyl transferase Tip60 (HTATIP) is part of a multimolecular complex involved in the cellular response to DNA damage. Tip60 plays a role in cell-cycle arrest following DNA damage, by allowing p53 to activate p21WAF1 expression (reviewed in Tyteca et al., 2006). p400 is a component of the Tip60 complex and p21WAF1 gene activation by p400 knock-down is Tip60-dependent. In unstressed cells, p400 functions as a negative regulator of Tip60. Upon DNA damage Tip60 repression by p400 is abolished, resulting in the upregulation of p21WAF1 (Tyteca et al., 2006). Perhaps E1A together with p400 recruit transcription repressors such as PML, a known suppressor of EGFR transcription (Vallian et al., 1998), to regulate EGFR expression. Analysis of p400 and EGFR interactions by ChIP will help to investigate whether p400 interacts directly with the EGFR promoter. However, owing to problems with p400 antibodies, which gave high level of nonspecific binding we have so far been unable to confirm direct association between these two proteins.
Interaction of E1A with p300 was not required for the induction of cell death as the p300−/− colon cancer HCT116 cells were shown to be highly sensitive to E1A-induced apoptosis. E1A deletion studies confirmed that p300 binding is dispensable for E1A-induced apoptosis and EGFR suppression. Based on microscopic studies and the detection of PARP p85, HCT116 p300−/− cells were found to be more sensitive to E1A-induced apoptosis than wild-type cells. Interestingly, p300 exhibits an intrinsic ubiquitin ligase activity that functions in concert with MDM2 to generate poly-ubiquitinated forms of p53 that are targeted for proteasome degradation (Grossman et al., 2003). This activity of p300 is inhibited by E1A, which contributes to the stabilization of p53 and a corresponding increase in sensitivity to apoptosis inducing signals in E1A expressing cells. In this study, E1A induced p53 expression in both p300−/− and parental HCT116 cells, whereas PARP p85 was not detected in E1A expressing wild-type HCT116 cells. It is therefore not clear whether elevated levels of p53 play a role in the induction of apoptosis by E1A in the p300 knockout cells. EGFR expression in HCT116 cells was found to be very low, making the analysis of the effect of E1A expression on EGFR levels inconclusive.
EGFR is overexpressed in many types of human cancers including HNSCC. EGFR overexpression is rarely due to gene amplification and in the majority of tumours the mechanism of EGFR overexpression is unknown, but is believed to be due to altered transcriptional regulation by EGFR transactivators/suppressors. EGFR is believed to function in HNSCC as a survival factor. Many EGFR targeting strategies have been developed worldwide and candidate EGFR inhibitors including antagonistic antibodies and tyrosine kinase inhibitors are currently in phase 1–3 clinical trials (reviewed in Grandis and Sok, 2004). However, the results of these studies have so far been disappointing (Kalyankrishna and Grandis, 2006). This could be due to the complexity of EGFR signalling pathways, including its heterodimerization with other members of the HER family of tyrosine kinases such as ErbB2, ErbB3 or ErbB4. It seems that tumour cells may be sensitive to EGFR inhibition only if they are dependent on EGFR for Akt activation, cell survival and growth (Kalyankrishna and Grandis, 2006). E1A has been shown to inhibit cell proliferation of lung, ovarian and breast cancer cells overexpressing c-erbB2/HER2, a member of the EGFR family (reviewed in Yu and Hung, 1998). E1A was shown to be unable to inhibit growth of breast cancer cells with low c-erbB2/HER2 expression. Furthermore, E1A has been shown to suppress the transcription of the c-erbB2 promoter (reviewed in Yu and Hung, 1998). It would therefore be interesting to find out whether transcriptional regulators such as p400 are involved in E1A-mediated suppression of c-erbB2/HER2. Furthermore, it would be interesting to investigate whether in EGFR- or HER2-overexpressing cancer cells the function of p400 is compromised.
In summary, we have identified the E1A12S isoform as a powerful inhibitor of EGFR expression and a strong inducer of cell death in a range of solid tumour cell lines. Head and neck cancer cells in which EGFR is highly overexpressed, and presumably functions as a survival mechanism, were particularly sensitive to E1A-induced apoptosis. This function of E1A12S is shared by several, but not all, adenovirus subgroups. Additionally, p400 but not pRB and p300 function was found to be important for E1A-mediated suppression of EGFR. The exact mechanism by which E1A utilizes p400 to reduce EGFR expression, remains to be elucidated. However, our results clearly show that E1A does not simply sequester p400, as shRNA-induced reduction of p400 does not in itself reduce EGFR expression or induce apoptosis. It seems likely that E1A redirects p400 and its associated chromatin-modifying activity to novel targets, thus indirectly leading to a reduction in EGFR expression and the consequent induction of apoptosis. Comparative studies of E1A proteins from EGFR-suppressing adenovirus subgroups, with those from non-suppressing adenovirus subgroups can help in the elucidation of the details of the mechanisms involved. Given the importance of EGFR overexpression and signalling in a broad range of human cancers, the demonstrated E1A-mediated regulation of EGFR levels and the identification of p400 as an important component of this regulation may be of clinical value.
Materials and methods
Cell lines
Human HNSCC cell lines HN5, HSC3, p400 knockout HSC3, osteosarcoma cell line Saos-2, MEF PG13 retroviral packaging cell line and human embryonal kidney 293A adenoviral packaging cell line were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% FCS (fetal calf serum), 50 μg/ml streptomycin, 100 μg/ml penicillin and 1mM sodium pyruvate, all purchased from Sigma (Gillingham, UK). HNSCC cell line H357 was cultured in Nut mix (Invitrogen, Paisley, UK) supplemented with 5% FCS, 4mM L-glutamine, 25 μg/l hydrocortisone, 50 μg/ml streptomycin and 100 μg/ml penicillin. HCT116 parental and p300−/− cells were cultured in RPMI containing HEPES supplemented with 10% FCS, 50 μg/ml streptomycin and 100 μg/ml penicillin.
To establish shRNA p400 knockout, PG13 packaging cells were stably transfected with pSIN-puro p400-sh or control pSIN-puro HI-sh retroviral vectors. Supernatant from selected PG13 populations producing p400shRNA retroviral vectors was used to infect HSC3 cells, after 72 h cells were put under puromycin selection.
Plasmids
PCR products encoding the various human adenovirus type 2 E1A isoforms were cloned in-frame into the pEGFP expression vector as EcoRI/SalI fragments. E1A12S mutants with specific small in-frame deletions were similarly expressed from pEGFP, as were the 12S complementary DNAs (cDNAs) for human adenovirus types 3, 4, 9, 12 and 40. All PCR products were sequenced to ensure fidelity.
Adenovirus vectors
The adenoviruses used in this study are all derived from the Ad5 dl309 background, which lacks the E3 domain encoding genes for 14.7K, 14.5K and 10.4K, but retains the wild-type Ad-E1A region (Jones and Shenk, 1979). The following replication incompetent adenoviruses were used: Ad-E1A (dl324) containing functional E1A but with complete deletion of E1B (Yan et al., 1991), Ad-Del (dl312) as a control containing a complete deletion of E1A (Yan et al., 1991). The E1A12S virus and mutants dl1101, dl1102/06, dl1102/08 and dl1105/07 do not express the E1A13S product or the E1B proteins (Shepherd et al., 1993; Mymryk, 1998). The positions of the E1A mutations in these mutants are illustrated in Figure 3a. The E1A9S virus does not express the E1B proteins and was constructed by replacing the E1A region with the Ad2 9S E1A cDNA (Moran et al., 1986), kindly provided by Dr E Moran (Fels Institute for Cancer Research and Molecular Biology, Philadelphia, PA, USA). Adenovirus amplification and purification was essentially performed as described previously (Graham and Prevec, 1991).
MTT cell proliferation assay
Cell survival was measured by MTT assay as described previously (Flinterman et al., 2003).
Transient transfection and indirect fluorescence microscopy
Cells were seeded in Falcon eight-well culture slides (Becton Dickinson, Oxford, UK) and transfected at 50–80% confluency with 400 ng plasmid DNA preincubated with 1.4 μl Lipofectamine 2000 Reagent (Invitrogen), according to the manufacturer’s protocol. EGFR expression was detected using EGFR-specific monoclonal antibody clone F4 (gift from Professor William Gullick, Department of Biosciences, University of Kent at Canterbury, UK) followed by horse anti-mouse Texas-Red (Vector Laboratories, Peterborough, UK). Fixation and preparation of slides were carried out as described previously (Flinterman et al., 2003).
Expression analysis by RT–PCR
RT–PCR was performed as described previously (Flinterman et al., 2005). EGFR cDNA was amplified using primers EGFR-F1 (5′-CAG-CCC-ACC-TGT-GTC-AAC-AGC-3′) and EGFR-R1 (5′-AAT-AAA-TTC-ACT-GCT-TTG-TGG-3′), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA was amplified as an internal control for the RT–PCR procedure using primers GAPDH-F1 (5′-ACC-TGA-CCT-GCC-GTCTAG-AA-3′) and GAPDH-R1 (5′-TCC-ACC-ACC-CTG-TTGCTG-TA-3′). Thirty-five cycles were performed, consisting of denaturation at 94°C, annealing at 58°C and extension at 72°C (each step 30 s).
Western blot analysis
Western blotting was performed as described previously (Sartor et al., 1999). For analysis of p400 protein expression, 4–12% gradient NuPage Bis–Tris precast gels were used (Invitrogen) to resolve lysates. The antibodies used for western blot analysis were: rabbit anti-PARP p85 fragment clone G734A (Promega, Southampton, UK) in a 1:2000 dilution, mouse anti-β-actin (Sigma) in a 1:500 dilution, mouse anti-tubulin (Sigma) in a 1:4000 dilution, mouse anti-E1A clone M58 (Pharmingen, BD Biosciences, Oxford, UK) in a 1:500 dilution, mouse anti-E1A clone M73 (Oncogene Research Products) in a 1:1000 dilution, mouse anti-EGFR clone F4 in a 1:1000 dilution, rabbit anti-PML clone 3573 (gift from Dr Kun-San Chang, Department of Molecular Pathology, MD Anderson Cancer Center, USA) in a 1:10 000 dilution, mouse anti-p400 clone RW144 (gift from Dr Ho-Man Chan, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA) in a 1:2 dilution. Secondary anti-mouse and anti-rabbit antibodies linked to horseradish peroxidase (Amersham Biosciences, Piscataway, NJ, USA) were used diluted at 1:1000 and 1:2000, respectively.
Acknowledgments
We thank Professor Bill Gullick for the generous gift of EGFR antibody, Dr Kun-San Chang for PML antibody and Dr Ho-Man Chan for p400 antibody. We also thank Professor Bruce Luxon (Department of Biochemistry and Molecular Biology, University of Texas Medical Branch, Galveston, USA) for his help in using the JNET neural network feature of JPRED software for protein structure prediction and for helpful discussions. Marcella Flinterman was supported by a grant from the UK Department of Trade and Industry. We thank DTI and CRUK for supporting this study.
Abbreviations
- BSA
bovine serum albumin
- CR
conserved region
- DAPI
4′,6-diamino-2-phenylindole
- DMEM
Dulbecco’s modified Eagle’s medium
- EGFR
epidermal growth factor receptor
- FCS
fetal calf serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GFP
green fluorescent protein
- HA
influenza A virus haemagglutinin
- HEK 293A
human embryonal kidney 293A
- HNSCC
human head and neck squamous cell carcinoma
- MOI
multiplicity of infection
- MTT
3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- RT
reverse transcription
- SDS
lauryl sulphate
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- Avvakumov N, Kajon AE, Hoeben RC, Mymryk JS. Comprehensive sequence analysis of the E1A proteins of human and simian adenoviruses. Virology. 2004;329:477–492. doi: 10.1016/j.virol.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Avvakumov N, Wheeler R, D’Halluin JC, Mymryk JS. Comparative sequence analysis of the largest E1A proteins of human and simian adenoviruses. J Virol. 2002;76:7968–7975. doi: 10.1128/JVI.76.16.7968-7975.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeau D, Charbonneau R, Whalen SG, Bayley ST, Branton PE. Functional interactions within adenovirus E1A protein complexes. Oncogene. 1994;9:359–373. [PubMed] [Google Scholar]
- Bergman LM, Blaydes JP. C-terminal binding proteins: emerging roles in cell survival and tumorigenesis. Apoptosis. 2006;11:879–888. doi: 10.1007/s10495-006-6651-4. [DOI] [PubMed] [Google Scholar]
- Chan HM, Narita M, Lowe SW, Livingston DM. The p400 E1A-associated protein is a novel component of the p53 –> p21 senescence pathway. Genes Dev. 2005;19:196–201. doi: 10.1101/gad.1280205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou SK, White E. p300 binding by E1A cosegregates with p53 induction but is dispensable for apoptosis. J Virol. 1997;71:3515–3525. doi: 10.1128/jvi.71.5.3515-3525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuff JA, Barton GJ. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins. 2000;40:502–511. doi: 10.1002/1097-0134(20000815)40:3<502::aid-prot170>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- Egan C, Jelsma TN, Howe JA, Bayley ST, Ferguson B, Branton PE. Mapping of cellular protein-binding sites on the products of early-region 1A of human adenovirus type 5. Mol Cell Biol. 1988;8:3955–3959. doi: 10.1128/mcb.8.9.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinterman M, Gäken J, Farzaneh F, Tavassoli M. E1A-mediated suppression of EGFR expression and induction of apoptosis in head and neck squamous carcinoma cell lines. Oncogene. 2003;22:1965–1977. doi: 10.1038/sj.onc.1206190. [DOI] [PubMed] [Google Scholar]
- Flinterman M, Guelen L, Ezzati-Nik S, Killick R, Melino G, Tominaga K, et al. E1A activates transcription of p73 and Noxa to induce apoptosis. J Biol Chem. 2005;280:5945–5959. doi: 10.1074/jbc.M406661200. [DOI] [PubMed] [Google Scholar]
- Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003;4:575–580. doi: 10.1038/sj.embor.embor861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Mymryk JS. Adenovirus-5 E1A: paradox and paradigm. Nat Rev Mol Cell Biol. 2002;3:441–452. doi: 10.1038/nrm827. [DOI] [PubMed] [Google Scholar]
- Fuchs M, Gerber J, Drapkin R, Sif S, Ikura T, Ogryzko V, et al. The p400 complex is an essential E1A transformation target. Cell. 2001;106:297–307. doi: 10.1016/s0092-8674(01)00450-0. [DOI] [PubMed] [Google Scholar]
- Graham FL, Prevec L. Manipulation of adenovirus vectors. In: Murray E, editor. Methods in Molecular Biology. Vol. 7. Humana Press; Cliffton, NJ: 1991. pp. 109–128. [DOI] [PubMed] [Google Scholar]
- Grandis JR, Sok JC. Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol Ther. 2004;102:37–46. doi: 10.1016/j.pharmthera.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Grossman SR, Deato ME, Brignone C, Chan HM, Kung AL, Tagami H, et al. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science. 2003;300:342–344. doi: 10.1126/science.1080386. [DOI] [PubMed] [Google Scholar]
- Iyer NG, Chin SF, Ozdag H, Daigo Y, Hu DE, Cariati M, et al. p300 regulates p53-dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proc Natl Acad Sci USA. 2004;101:7386–7391. doi: 10.1073/pnas.0401002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones N, Shenk T. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell. 1979;17:683–689. doi: 10.1016/0092-8674(79)90275-7. [DOI] [PubMed] [Google Scholar]
- Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24:2666–2672. doi: 10.1200/JCO.2005.04.8306. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- Moran E, Grodzicker T, Roberts RJ, Mathews MB, Zerler B. Lytic and transforming functions of individual products of the adenovirus E1A gene. J Virol. 1986;57:765–775. doi: 10.1128/jvi.57.3.765-775.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mymryk JS. Database of mutations within the adenovirus 5 E1A oncogene. Nucleic Acids Res. 1998;26:292–294. doi: 10.1093/nar/26.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mymryk JS, Shire K, Bayley ST. Induction of apoptosis by adenovirus type 5 E1A in rat cells requires a proliferation block. Oncogene. 1994;9:1187–1193. [PubMed] [Google Scholar]
- Najafi SM, Li Z, Makino K, Shao R, Hung MC. The adenoviral E1A induces p21WAF1/CIP1 expression in cancer cells. Biochem Biophys Res Commun. 2003;305:1099–1104. doi: 10.1016/s0006-291x(03)00905-7. [DOI] [PubMed] [Google Scholar]
- Paterson IC, Patel V, Sandy JR, Prime SS, Yeudall WA. Effects of transforming growth factor beta-1 on growth-regulatory genes in tumour-derived human oral keratinocytes. Br J Cancer. 1995;72:922–927. doi: 10.1038/bjc.1995.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer. 2001;8:11–31. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- Querido E, Teodoro JG, Branton PE. Accumulation of p53 induced by the adenovirus E1A protein requires regions involved in the stimulation of DNA synthesis. J Virol. 1997;71:3526–3533. doi: 10.1128/jvi.71.5.3526-3533.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelson AV, Lowe SW. Selective induction of p53 and chemosensitivity in RB-deficient cells by E1A mutants unable to bind the RB-related proteins. Proc Natl Acad Sci USA. 1997;94:12094–12099. doi: 10.1073/pnas.94.22.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelson AV, Narita M, Chan HM, Jin J, de Stanchina E, McCurrach ME, et al. p400 is required for E1A to promote apoptosis. J Biol Chem. 2005;280:21915–21923. doi: 10.1074/jbc.M414564200. [DOI] [PubMed] [Google Scholar]
- Sartor M, Steingrimsdottir H, Elamin F, Gäken J, Warnakulasuriya S, Partridge M, et al. Role of p16/MTS1, cyclin D1 and RB in primary oral cancer and oral cancer cell lines. Br J Cancer. 1999;80:79–86. doi: 10.1038/sj.bjc.6690505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd SE, Howe JA, Mymryk JS, Bayley ST. Induction of the cell cycle in baby rat kidney cells by adenovirus type 5 E1A in the absence of E1B and a possible influence of p53. J Virol. 1993;67:2944–2949. doi: 10.1128/jvi.67.5.2944-2949.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubert S, Gorrini C, Frank SR, Parisi T, Fuchs M, Chan HM, et al. E2F-dependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol Cell Biol. 2004;24:4546–4556. doi: 10.1128/MCB.24.10.4546-4556.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyteca S, Vandromme M, Legube G, Chevillard-Briet M, Trouche D. Tip60 and p400 are both required for UV-induced apoptosis but play antagonistic roles in cell cycle progression. EMBO J. 2006;25:1680–1689. doi: 10.1038/sj.emboj.7601066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallian S, Chin KV, Chang KS. The promyelocytic leukemia protein interacts with Sp1 and inhibits its transactivation of the epidermal growth factor receptor promoter. Mol Cell Biol. 1998;18:7147–7156. doi: 10.1128/mcb.18.12.7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HG, Rikitake Y, Carter MC, Yaciuk P, Abraham SE, Zerler B, et al. Identification of specific adenovirus E1A N-terminal residues critical to the binding of cellular proteins and to the control of cell growth. J Virol. 1993;67:476–488. doi: 10.1128/jvi.67.1.476-488.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan DH, Chang LS, Hung MC. Repressed expression of the HER-2/c-erbB-2 proto-oncogene by the adenovirus E1a gene products. Oncogene. 1991;6:343–345. [PubMed] [Google Scholar]
- Yu D, Hung MC. The erbB2 gene as a cancer therapeutic target and the tumor- and metastasis-suppressing function of E1A. Cancer Metastasis Rev. 1998;17:195–202. doi: 10.1023/a:1006054421970. [DOI] [PubMed] [Google Scholar]