

Abstract
We report on the molecular characterization of a microdeletion of approximately 2.5Mb at 2p11.2 in a female baby with left congenital aural atresia, microtia, and ipsilateral internal carotid artery agenesis. The deletion was characterized by fluorescence in situ hybridization, array comparative genomic hybridization, and whole genome re-sequencing. Among the genes present in the deleted region, we focused our attention on the FOXI3 gene. Foxi3 is a member of the Foxi class of Forkhead transcription factors. In mouse, chicken and zebrafish Foxi3 homologues are expressed in the ectoderm and endoderm giving rise to elements of the jaw as well as external, middle and inner ear. Homozygous Foxi3−/− mice have recently been generated and show a complete absence of the inner, middle, and external ears as well as severe defects in the jaw and palate. Recently, a 7-bp duplication within exon 1 of FOXI3 that produces a frameshift and a premature stop codon was found in hairless dogs. Mild malformations of the outer auditory canal (closed ear canal) and ear lobe have also been noted in a fraction of FOXI3 heterozygote Peruvian hairless dogs. Based on the phenotypes of Foxi3 mutant animals, we propose that FOXI3 may be responsible for the phenotypic features of our patient. Further characterization of the genomic region and the analysis of similar patients may help to demonstrate this point.
Keywords: congenital aural atresia, agenesis of internal carotid artery, FOXI3, deletion, array-CGH
INTRODUCTION
Congenital Aural Atresia (CAA) is a rare malformation of the ear in humans. It presents unilaterally more often than bilaterally. Its characteristics can vary from a narrow external auditory canal and hypoplasia of the tympanic membrane and middle ear cleft to a complete absence of middle-ear structures and anotia. CAA might be present as an isolated malformation but is also seen as a feature in syndromes and chromosomal anomalies, like in deletions of the long arm of chromosome 18 [Altmann, 1955; Cremers et al., 1988; Schinzel, 2002]. To our knowledge, TSHZ1 (OMIM614427: teashirt zinc finger homeobox 1) mapped to 18q22.3 may be the only reported gene whose hemizygosity leads to congenital aural atresia as a result of haploinsufficiency [Feenstra et al., 2007].
Craniofacial abnormalities often arise during development of the pharyngeal arches. The complexity of arch derivatives is reflected in their development, which requires an intricate orchestration of interactions between the ectoderm, endoderm and mesoderm, together with neural crest cells that populate each arch [Chai and Maxson, 2006; Szabo-Rogers et al., 2010]. In the last 15 years, much progress has been made in identifying the molecular signals that coordinate the early development of pharyngeal arches [Minoux and Rijli, 2010]. For example, secreted signals such as Sonic Hedgehog and Fibroblast Growth Factor 8 are crucial for correct craniofacial development and exert their influence through regulation of a number of transcription factor networks present in the developing pharyngeal arches [Ahlgren andBronner-Fraser, 1999; Trumpp et al., 1999;David and Rosa, 2001; Yamagishi et al., 2003; Brito et al., 2006; Haworth et al., 2007]. Elucidating the function of transcription factors during craniofacial development has provided much insight into the etiology of craniofacial abnormalities [Cox, 2004].
The Foxi class of Forkhead transcription factors has been shown to play important roles in early craniofacial development. Foxi genes (foxi1 in zebrafish and Foxi3 in amniotes) are expressed in early non-neural ectoderm, later becoming restricted to the preplacodal region that will give rise to all craniofacial sensory organs [Solomon et al., 2003; Ohyama and Groves, 2004a; Streit, 2007; Khatri and Groves, 2013], followed by expression in the ectoderm and endoderm of the pharyngeal arches [Ohyama and Groves, 2004b; Nissen et al., 2003; Solomon et al., 2003; Khatri and Groves, 2013; Edlund et al., 2014]. Homozygous zebrafish mutants of foxi1 and mouse Foxi3 mutants fail to form an inner ear and lack many derivatives of the pharyngeal arches including the jaw [Solomon et al., 2003; Nissen et al., 2003; Edlund et al., 2014].
These phenotypes, together with the expression patterns of zebrafish foxi1 and mouse and chicken Foxi3 suggest these three genes may be functional homologues. In addition, mouse Foxi3 is expressed in a number of ectodermal appendages such as whisker follicles, hair follicles, and tooth germs, [Drogemuller et al., 2008] where it is regulated by the ectodysplasin signaling pathway [Shirokova et al., 2013]. The first spontaneous mutation of FOXI3 gene was identified in Mexican and Peruvian hairless dogs and Chinese crested dogs. All three breeds have an identical 7 base pair duplication early in the coding sequence before the DNA binding domain predicted to result in a functional null allele [Drogemuller et al., 2008]. The phenotype of hairless dogs is inherited as a monogenic autosomal semi-dominant trait and is classified as canine ectodermal dysplasia (CED) because these dogs have missing or abnormally shaped teeth in addition to sparse or absent hair [O'Brien et al., 2005]. The frameshift mutation within the FOXI3 coding sequence and its embryonic expression pattern identified FOXI3 as a regulator of ectodermal appendage development [Drogemuller et al., 2008]. However, to date, no human families carrying FOXI3 mutations have been identified. Here, we provide the first report on a human case with a deletion of the entire FOXI3 gene in a female with left congenital aural atresia and ipsilateral internal carotid artery (ICA) agenesis. Our analysis of the patient, together with further analysis of the craniofacial abnormalities of FOXI3 mutant dogs presented here, suggests FOXI3 mutations may be responsible for congenital aural atresia in humans and dogs.
MATERIAL AND METHODS
Clinical Report
The proposita is the second child of nonconsanguineous parents. By history, her father had hypoacusia and tinnitus, so he underwent left ear surgery when he was 25; no further information was available about the operation. She was born at 38 weeks of gestation by caesarean. At birth, she presented with a left small dysmorphic pinna (microtia), complete atresia of the left external auditory canal (EAC), mild facial asymmetry, deviation of the mouth during crying. She came to our observation at the age of 16 days for further investigation of episodes of cyanosis during feeds. Velopharyngeal insufficiency without cleft of hard or soft palate was demonstrated. The neonate had normal hair and normal skin with normal sweating. No other malformations were evident, such as vertebral, cardiac or renal abnormalities. The phenotypic features of the patient are shown in Figure 1.
Figure 1.

Phenotypic features of the girl. A) mild facial asymmetry, B) right normal ear, C) left small dysmorphic pinna (microtia).
Temporal bone computerized tomography (CT) revealed left mixed stenosis of the entire external auditory canal including its membranous and bony portions, with a small middle ear cavity and hypoplastic mastoid complex (Fig. 2A,B,C). The left ossicular chain was dysmorphic with fusion and rotation of the malleolar – incudal articulation. Aberrant course of mastoid portion of left facial nerve was also noticed. Inner ear structures and internal auditory canals were bilaterally normal. No abnormalities of the EAC and middle ear structures were seen on the right. Interestingly, the left spinous foramen and carotid canal were not visible at the level of skull base, suggesting the persistence of the stapedial artery and agenesis of the internal carotid artery (Fig. 2D, E). The patient subsequently underwent an MR angiogram (MRA) showing the absence of flow-related signal intensity within the left ICA. The ipsilateral ophthalmic artery and middle cerebral artery originated from the posterior communicating artery while a patent anterior communicating artery supplied the left anterior cerebral artery. Brain MRI was otherwise normal. At the age of 4 months her growth and psychomotor development was normal. Left deafness was demonstrated by brainstem auditory evoked response which failed to show both peripheral and brainstem components on the left side. A normal brain stem auditory evoked response was recorded on the right side.
Figure 2.

Axial temporal CT of the patient A) shows lumpy featureless pinna (thin white arrow), absent EAC (asterisk) and complete bony atresia (white arrow). Note the absence of left carotid canal (black arrowhead) compared to the normal controlateral carotid canal (white arrowhead). Axial temporal CT at a higher level (B) depicts rotation and fusion of the incudo-malleolar joint (black arrowhead) and partial fusion of the incus to the lateral wall of the epi-tympanum (white arrow). Coronal temporal CT (C) reveals dehiscence and abnormal course of the tympanic portion of the left facial nerve (black arrowhead) compared to the normal controlateral nerve (white arrowhead). Brain MRI (D) axial T2-weighted 3D driven equilibrium technique (DRIVE) shows absent flow void in the left ICA (black arrow). 3D time-of-flight MR angiogram (E) confirms absent flow related signal in the left ICA (open white arrow). The left middle cerebral artery is directly supplied by the posterior circulation through a posterior communicating artery, whereas the left anterior cerebral artery is supplied through a patent anterior communicating artery.
Cytogenetic and Array CGH Analyses
Standard GTG banding was performed at a resolution of 400–550 bands on metaphase chromosomes from peripheral blood lymphocytes of the patient and her parents. Molecular karyotyping was performed on the patient and her parents using the Human Genome CGH Microarray Kit G3 400 (Agilent Technologies, Palo Alto, USA) with _7.2 Kb overall median probe spacing. Labeling and hybridization were performed following the protocols provided by the manufacturers. A graphical overview was obtained using the Agilent Genomic Workbench Lite Edition Software 6.5.0.18. A fluorescent in situ hybridization (FISH) experiment was performed with probes RP11–525L16 (containing FOXI3 gene; Red, Bluegnome Ltd., Cambridge UK) and subtelomeric probe dj892G20 (Green; Cytocell Aquarius, Cambridge, UK) selected according to the UCSC Human Genome Map Feb. 2009 (GRCh37/hg19).
Whole Genome Sequencing Analysis
A fragment library with 300 bp insert size was prepared from genomic DNA. One lane of Illumina HiSeq2000 paired-end reads was collected (2_100 bp). We obtained a total of 169,419,059 paired-endreads or roughly 10x coverage. The reads were mapped to the human GRCh37 reference genome with the Burrows-Wheeler Aligner (BWA) version 0.5.9-r16 with default settings and yielded 156,510,049 uniquely mapping reads [Li and Durbin, 2009]. After sorting the mapped reads by the coordinates of the sequence with Picard tools, PCR duplicates were also labeled with Picard tools (http://sourceforge.net/projects/picard/). The Genome Analysis Tool Kit (GATK version 0591, [McKenna et al., 2010]) was used to perform local realignment and to produce a cleaned BAM file. Variants calls were then made with the unified genotype module of GATK. Variant data for each sample were obtained in variant call format(version4.0) as raw calls for all samples and sites flagged using the variant filtration module of GATK. The BAM-file was visually inspected in the critical interval chr2:87,500,001–92,000,000 using the integrative genomics viewer (IGV) [Robinson et al., 2011].
RESULTS
Cytogenetic and Array CGH Analyses
The patient was initially referred for cytogenetic analysis because of external and internal ear anomalies and cranio-facial abnormalities. On the basis of the phenotype we performed an array-CGH analysis of the patient and her parents. The array-CGH analysis showed an interstitial deletion of _2.474Mb at band p11.2 of chromosome 2 inherited from the father. The deletion spanned from probe A_16_P00422994 (87,790,841 bp) to probe A_18_P13306010 (90,265,119 bp) flanking by probe A_18_P13304801 (87,776,265 bp) and probe A_16_P00424442 (91,693,395 bp) (GRCh37/hg19) (http://genome.ucsc.edu/) (Fig. 3A). FISH analysis with probe RP11–525L16 containing FOXI3 gene confirmed the deletion (Fig. 3B). Low Copy Repeats (LCR) clusters on chromosome 2 at p11.2 flank the deleted region. The distal LCR is at chr2:87,730,959–88,320,829 and the proximal LCR, which contains the immunoglobulin kappa locus (IGK), at chr2:89,055,232–90,265,886. For these two blocks a large number of variants have been reported in the normal population (Database of Genomic Variants: http://dgv.tcag.ca/dgv). Finally, there is a gap of 1.05Mb in the human GRCh37 reference genome following the proximal LCR (chr2:90,545,104–91,595,103). Thus, the deleted region consisted of the gap in the assembly and the two LCRs flanking a central single copy region with eight protein-coding genes: KRCC1 (NM_016618.1) lysine-rich coiled-coil 1, SMYD1 (OMIM 606846) SET and MYND domain containing 1, FABP1 (OMIM 134650) fatty acid binding protein 1, THNSL2 (OMIM 611261) threonine synthase-like 2, FOXI3 (OMIM612351) forkhead box I3, TEX37 (NM_152670.2) testis expressed 37, EIF2AK3 (OMIM 226980) eukaryotic translation initiation factor 2-alpha kinase 3, and RPIA (OMIM 608611) ribose 5-phosphate isomerase A. There is also one miRNA gene, MIR4780, annotated in this interval (NCBI annotation release 104) (Table I, Fig. 1C).
Figure 3.

Results of array-CGH and FISH analyses: A) Array-CGH analysis shows an interstitial deletion ~2.474 Mb at band 2p11.2 of chromosome 2. The deleted region extends between oligomers probe A_16_P00422994 (87,790,841 bp) to probe A_18_P13306010 (90,265,119 bp) flanking by probe A_18_P13304801 (87,776,265 bp) and probe A_16_P00424442 (91,693,395 bp) (Feb. 2009 (GRCh37/hg19) Assembly). The deleted region is flanked by large complex LCR clusters on chromosome 2 at p11.2 distal LCR (chr2:87,730,959–88,320,829) and proximal LCR (1.210 Mb, chr2:89,055,232–90,265,886). B) FISH experiment was performed with probes RP11-525L16 (FOXI3) (red signal) and a subtelomeric probe dj892G20 (green signal); arrow indicates the deleted chromosome 2 and arrowhead the normal chromosome 2. C) Extract from the UCSC genome browser (http://genome.ucsc.edu/) GRCh37/hg19 shows the deleted region of our patient and its gene content, including the FOXI3 locus (circled).
Table 1.
Genes located in the deleted interval
| Gene Symbol | Gene | mRNA Accession | OMIM | Known Haploinsufficiency Phenotype |
|---|---|---|---|---|
| KRCC1 | lysine-rich coiled-coil 1 | NM_016618.1 | n.a. | Unknown |
| SMYD1 | SET and MYND domain containing 1 | NM_198274.3 | 606846 | Smyd1+/− mice reported to be normal |
| FABP1 | fatty acid binding protein 1 | NM_001443.2 | 134650 | Unknown |
| THNSL2 | threonine synthase-like 2 | NM_018271.4 | 611261 | Unknown |
| FOXI3 | forkhead box I3 | NM_001135649.1 | 612351 | FOXI3+/− dogs always ectodermal dysplasia and occasionally ear malformations; Foxi3+/− mice reported to be normal |
| TEX37 | testis expressed 37 | NM_152670.2 | n.a. | Unknown |
| EIF2AK3 | eukaryotic translation initiation factor 2-alpha kinase 3 | NM_004836.5 | 226980 | EIF2AK3+/− humans and mice reported to be normal |
| RPIA | ribose 5-phosphate isomerase A | NM_144563.2 | 608611 | RPIA+/− humans reported to be healthy |
| MIR4780 | microRNA 4780 | NR_039940.1 | n.a. | Unknown |
Genome Sequence Results
The whole genome sequence analysis confirmed the results of the cytogenetic analysis (Table S1). Due to the highly complex repeat structures in the critical region of chromosome 2, it was not possible to define the exact deletion breakpoints using short-read sequence data. However, the average sequence coverage in the central single-copy region of the critical interval was roughly 50% compared to the average coverage of the genome, which is in agreement with a heterozygous deletion. The sequence data indicated full coverage and heterozygous genotypes in the patient at chr2:87,768,690 and 91,935,028, respectively. These positions thus define the maximal size of the deleted region and are in good agreement with the aCGH data.
The whole genome sequence data were visually inspected for protein coding variants in the central single copy region of the deletion (chr2:88,320,830–89,055,231). A total of 10 such variants were identified. At each of these variants the patient carried the variant allele in presumably hemizygous state. All these variants have been previously identified and are present in dbSNP build 138 indicating that they are most likely non-pathogenic. The sequence data confirmed intact open reading frames for the remaining copies of the eight deleted protein coding genes except for _200 nt at the extremely GC-rich 5'-end of the FOXI3 gene that were not covered by the Illumina sequence reads (Table S1). Based on these results, and on the aural phenotype of FOXI3 mutant dogs (see below), FOXI3 was considered the best candidate among the eight genes in the critical region.
Evidence for Aural Atresia in Peruvian Hairless Dogs
Recently, a 7-bp duplication within exon 1 of FOXI3 that produces a frameshift and a premature stop codon was found in hairless dogs [Drogemuller et al., 2008]. The phenotype of hairless dogs is now classified as canine ectodermal dysplasia (CED), which is inherited as a monogenic autosomal semidominant trait. Mild malformations of the external auditory canal and ear lobe are relatively occasional findings (The Society of Finnish Chinese Crested Dog, http://www.kiinanharjakoirat.fi, and personal communication with breeders of Mexican and Peruvian Hairless dogs). We performed a clinical examination, CT scan, and autopsy of one Peruvian Hairless dog that was euthanized at the age of 4 weeks due to auditory defects. The dog lacked an external auditory canal and had a small tympanic cavity on the right side, but had normal hearing and ear anatomy on the left side (Fig. 4). This observation, together with the known expression of FOXI3 in the ectoderm of the first and second pharyngeal arches [Nissen et al., 2003; Solomon et al., 2003; Ohyama and Groves, 2004; Khatri and Groves, 2013; Edlund et al., 2014; Khatri et al., 2014], suggests that FOXI3 might also have a role in the morphogenesis of external structures of the ear.
Figure 4.
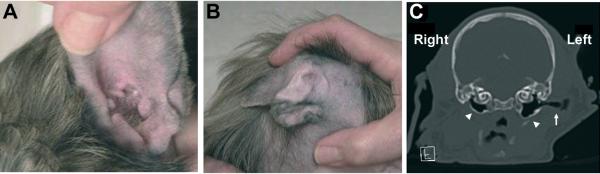
Auditory malformations in hairless dogs D) right normal ear, E) left dysmorphic pinna, F) CT scan of a 4-week-old Peruvian Hairless puppy demonstrates the absence of external auditory canal (EAC) (asterisk) and small tympanic cavity (arrowhead) on the right side compared to the normal structures, tympanic cavity (open arrowhead) and EAC (arrow) on the left side of the head. Supplementary table 1. Summary of the whole genome sequence analysis
DISCUSSION
We identified a female patient with an approximately 2.5Mb interstitial deletion of the short arm of chromosome 2 at band 2p11.2 inherited from her father. The breakpoints of the deletion fall inside two blocks of LCRs, where there are directly oriented subunits with high sequence identity that could mediate non-allelic homologous recombination (NAHR) [Shaw and Lupski, 2004]. The deleted region contains eight protein-coding genes, including FOXI3. Two patients with FOXI3 deletion are reported in the DECIPHER database (http://decipher.sanger.ac.uk/). Patient n.252352 has a 4.56Mb deletion and shows, among other abnormalities, dysmorphic large ears and widely spaced and abnormally shaped teeth, as observed in FOXI3 mutant dogs [O'Brien et al., 2005]. The other patient (DECIPHER n.253836) has a 1.86Mb deletion and intellectual disability is the sole reported phenotypic feature.
Canine FOXI3 mutants have abnormalities when present in heterozygous and homozygous state. Homozygous dogs are never born and are presumed to be embryonic lethal, similar to what is seen in zebrafish and mouse mutants [Nissen et al., 2003; Solomon et al., 2003; Edlund et al., 2014]. The FOXI3 external ear phenotype is unilateral, relatively rare, and exclusively found in heterozygous FOXI3 dogs. Dogs with the ear phenotype have been shown to be unilaterally deaf in ABR (Auditory Brainstem Response) testing. Due to the limited amount of information available from breeders, it is not clear which proportion of FOXI3 +/− dogs have abnormalities of their outer ears. This seems to be a variable phenotype due to incomplete penetrance or variable expressivity. Homozygous Foxi3 −/− mice have recently been generated and show a complete absence of the inner, middle, and external ears as well as severe defects in the jaw and palate [Edlund et al., 2014]. Homozygous mutant mice die between embryonic day 11 and birth. However, unlike dogs and humans, heterozygous Foxi3+/− mice show no apparent defects in any of these structures, on either congenic or outbred backgrounds [Edlund et al., 2014].
The phenotypic features of dog FOXI3 mutants are surprisingly similar to those present in our patient. In fact, the girl showed a left small dysmorphic pinna (microtia), complete atresia of the left external auditory canal, mild facial asymmetry, anomalies of middle ear structures. Moreover, she presented the persistence of stapedial artery, which is a remnant of second aortic arch, and the agenesis of internal carotid artery that is a derivative of the 3rd arch. Interestingly, her father reported to have had problems to left ear. Her father presented with hypoacusia and tinnitus, so he underwent left ear surgery when he was 25; no further information was available about the operation.
To our knowledge, this is the first reported case of a human FOXI3 gene deletion. Based on this observation, we propose that FOXI3 could be responsible for the phenotypic features of our patient. The other genes in the deleted region do not appear to have a causative role for the phenotype, but we cannot exclude their involvement with certainty. Further characterization of the genomic region and the analysis of other patients may definitively demonstrate this point.
Supplementary Material
ACKNOWLEDGMENTS
We thank the patient's parents for their kind participation and support. We thank the Next Generation Sequencing Platform of the University of Bern for performing the whole genome sequencing experiment. We are grateful to Marco Bertorello and Corrado Torello for their technical assistance. This work was supported by “Cinque per mille dell'IRPEF- Finanziamento della ricerca sanitaria” and “Finanziamento Ricerca Corrente, Ministero Salute (contributo per la ricerca intramurale).
REFERENCES
- Ahlgren SC, Bronner-Fraser M. Inhibition of sonic hedgehog signaling in vivo results in craniofacial neural crest cell death. Curr Biol. 1999;9:1304–1314. doi: 10.1016/s0960-9822(00)80052-4. [DOI] [PubMed] [Google Scholar]
- Altmann F. Congenital atresia of the ear in man and animals. Ann Otol Rhinol Laryngol. 1955;64:824–858. doi: 10.1177/000348945506400313. [DOI] [PubMed] [Google Scholar]
- Brito JM, Teillet MA, Le Douarin NM. An early role for sonic hedgehog from foregut endoderm in jaw development: Ensuring neural crest cell survival. Proc Natl Acad Sci USA. 2006;103:11607–11612. doi: 10.1073/pnas.0604751103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Maxson RE., Jr Recent advances in craniofacial morphogenesis. Dev Dyn. 2006;235:2353–2375. doi: 10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- Cox TC. Taking it to the max: The genetic and developmental mechanisms coordinating midfacial morphogenesis and dysmorphology. Clin Genet. 2004;65:163–176. doi: 10.1111/j.0009-9163.2004.00225.x. [DOI] [PubMed] [Google Scholar]
- Cremers CW, Teunissen E, Marres EH. Classification of congenital aural atresia and results of reconstructive surgery. Adv Otorhinolaryngol. 1988;40:9–14. doi: 10.1159/000415666. [DOI] [PubMed] [Google Scholar]
- David NB, Rosa FM. Cell autonomous commitment to an endodermal fate and behaviour by activation of Nodal signalling. Development. 2001;128:3937–3947. doi: 10.1242/dev.128.20.3937. [DOI] [PubMed] [Google Scholar]
- Drogemuller C, Karlsson EK, Hytonen MK, Perloski M, Dolf G, Sainio K, Lohi H, Lindblad-Toh K, Leeb T. A mutation in hairless dogs implicates FOXI3 in ectodermal development. Science. 2008;321:1462. doi: 10.1126/science.1162525. [DOI] [PubMed] [Google Scholar]
- Edlund RK, Ohyama T, Kantarci H, Riley BB, Groves AK. Foxi transcription factors promote pharyngeal arch development by regulating formation of FGF signaling centers. Dev Biol. 2014;391:158–169. doi: 10.1016/j.ydbio.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feenstra I, Vissers LE, Orsel M, van Kessel AG, Brunner HG, Veltman JA, van Ravenswaaij-Arts CM. Genotype-phenotype mapping of chromosome 18q deletions by high-resolution array CGH: an update of the phenotypic map. Am J Med Genet A. 2007;143A:1858–1867. doi: 10.1002/ajmg.a.31850. [DOI] [PubMed] [Google Scholar]
- Haworth KE, Wilson JM, Grevellec A, Cobourne MT, Healy C, Helms JA, Sharpe PT, Tucker AS. Sonic hedgehog in the pharyngeal endoderm controls arch pattern via regulation of Fgf8 in head ectoderm. Dev Biol. 2007;303:244–258. doi: 10.1016/j.ydbio.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Khatri SB, Groves AK. Expression of the Foxi2 and Foxi3 transcription factors during development of chicken sensory placodes and pharyngeal arches. Gene Expr Patterns. 2013;13:38–42. doi: 10.1016/j.gep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri SB, Edlund RK, Groves AK. Foxi3 is necessary for the induction of the chick otic placode in response to FGF signaling. Dev Biol. 2014;391:158–169. doi: 10.1016/j.ydbio.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows- Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoux M, Rijli FM. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development. 2010;137:2605–2621. doi: 10.1242/dev.040048. [DOI] [PubMed] [Google Scholar]
- Nissen RM, Yan J, Amsterdam A, Hopkins N, Burgess SM. Zebrafish foxi one modulates cellular responses to Fgf signaling required for the integrity of ear and jaw patterning. Development. 2003;130:2543–2554. doi: 10.1242/dev.00455. [DOI] [PubMed] [Google Scholar]
- O'Brien DP, Johnson GS, Schnabel RD, Khan S, Coates JR, Johnson GC, Taylor JF. Genetic mapping of canine multiple system degeneration and ectodermal dysplasia loci. J Hered. 2005;96:727–734. doi: 10.1093/jhered/esi086. [DOI] [PubMed] [Google Scholar]
- Ohyama T, Groves AK. Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis. 2004a;38:195–199. doi: 10.1002/gene.20017. [DOI] [PubMed] [Google Scholar]
- Ohyama T, Groves AK. Expression of mouse Foxi class genes in early craniofacial development. Dev Dyn. 2004b;231:640–646. doi: 10.1002/dvdy.20160. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man, 2nd Edition Chromosome Research. 2002;10:1–4. [Google Scholar]
- Shaw CJ, Lupski JR. Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet 13 Spec No. 2004;1:R57–R64. doi: 10.1093/hmg/ddh073. [DOI] [PubMed] [Google Scholar]
- Shirokova V, Jussila M, Hytonen MK, Perala N, Drogemuller C, Leeb T, Lohi H, Sainio K, Thesleff I, Mikkola ML. Expression of Foxi3 is regulated by ectodysplasin in skin appendage placodes. Dev Dyn. 2013;242:593–603. doi: 10.1002/dvdy.23952. [DOI] [PubMed] [Google Scholar]
- Solomon KS, Kudoh T, Dawid IB, Fritz A. Zebrafish foxi1 mediates otic placode formation and jaw development. Development. 2003;130:929–940. doi: 10.1242/dev.00308. [DOI] [PubMed] [Google Scholar]
- Streit A. The preplacodal region: An ectodermal domain with multipotential progenitors that contribute to sense organs and cranial sensory ganglia. Int J Dev Biol. 2007;51:447–461. doi: 10.1387/ijdb.072327as. [DOI] [PubMed] [Google Scholar]
- Szabo-Rogers HL, Smithers LE, Yakob W, Liu KJ. New directions in craniofacial morphogenesis. Dev Biol. 2010;341:84–94. doi: 10.1016/j.ydbio.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR. Cremediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev. 1999;13:3136–3148. doi: 10.1101/gad.13.23.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi H, Maeda J, Hu T, McAnally J, Conway SJ, Meyers EN, Yamagishi C, Srivastava D. Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev. 2003;17:269–281. doi: 10.1101/gad.1048903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.