

Abstract
Several neurodegenerative diseases involve loss of catecholamine neurons—Parkinson disease is a prototypical example. Catecholamine neurons are rare in the nervous system, and why they are vulnerable in PD and related disorders has been mysterious. Accumulating evidence supports the concept of “autotoxicity”—inherent cytotoxicity of catecholamines and their metabolites in the cells in which they are produced. According to the “catecholaldehyde hypothesis” for the pathogenesis of Parkinson disease, long-term increased build-up of 3,4-dihydroxyphenylacetaldehyde (DOPAL), the catecholaldehyde metabolite of dopamine, causes or contributes to the eventual death of dopaminergic neurons. Lewy bodies, a neuropathologic hallmark of PD, contain precipitated alpha-synuclein. Bases for the tendency of alpha-synuclein to precipitate in the cytoplasm of catecholaminergic neurons have also been mysterious. Since DOPAL potently oligomerizes and aggregates alpha-synuclein, the catecholaldehyde hypothesis provides a link between alpha-synucleinopathy and catecholamine neuron loss in Lewy body diseases. The concept developed here is that DOPAL and alpha-synuclein are nodes in a complex nexus of interacting homeostatic systems. Dysfunctions of several processes, including decreased vesicular sequestration of cytoplasmic catecholamines, decreased aldehyde dehydrogenase activity, and oligomerization of alpha-synuclein, lead to conversion from the stability afforded by negative feedback regulation to the instability, degeneration, and system failure caused by induction of positive feedback loops. These dysfunctions result from diverse combinations of genetic predispositions, environmental exposures, stress, and time. The notion of catecholamine autotoxicity has several implications for treatment, disease modification, and prevention. Conversely, disease modification clinical trials would provide key tests of the catecholaldehyde hypothesis.
Keywords: Alpha-synuclein, Autotoxicity, Catecholamine, DOPAL, Parkinson disease
1. Introduction
The burden of diseases of senescence is increasing as the population ages. Neurodegenerative diseases pose major challenges both to public health and medical science. In general, symptoms of these diseases are treatable, but the treatments do not reverse the neurodegeneration. Theoretically, disease progression might be retarded if the pathogenetic process were detected early and effective disease-modifying treatment instituted in a pre-symptomatic phase.
Parkinson disease (PD) was the first neurodegenerative disease for which the underlying neurochemical abnormality was identified—severe depletion of the catecholamine dopamine (DA) in the striatum (Ehringer & Hornykiewicz, 1960). Alleviation of the deficiency by levodopa treatment was revolutionary in the history of medical neuroscience (Cotzias, 1971). All current approved treatments of PD work directly or indirectly by countering effects of striatal DA depletion. While often effective in alleviating symptoms, no PD treatment has been proven to slow the loss of nigrostriatal neurons.
Almost a century ago, in his thesis published in 1919, Constantin Tretiakoff described for the first time two of what are now considered to be characteristic neuropathologic features of PD—a loss of pigmentation in the substantia nigra in the midbrain and nigral “corps de Lewy” (Lewy bodies). The latter designation was in recognition of the description 6 years previously, by Friedrich Lewy, of intra-neuronal hyaline inclusions in patients with paralysis agitans.
Substantia nigra depigmentation likely has a neurochemical basis—loss of neurons that contain DA since DA auto-oxidizes spontaneously to form melanin (from the Greek word for black). Tretiakoffs discoveries about nigral depigmentation and Lewy bodies in substantia nigra neurons in PD, and subsequent findings showing that putamen DA is severely depleted in PD (Kish et al., 1988; Wilson et al., 1996; Hornykiewicz, 1998) and that Lewy bodies contain abundant precipitated alpha-synuclein (Spillantini et al., 1997; Mezey et al., 1998) lead to two sets of questions, which to a major extent inspired this review.
First, catecholamine neurons are rare in the nervous system. Why are they lost in PD? What makes them different from neurons of other transmitter types? What renders catecholamine neurons, including nigral dopaminergic neurons and striatal dopaminergic terminals, susceptible?
Second, Lewy bodies contain abundant aggregated alpha-synuclein, and at least in rare forms of familial PD abnormalities of the alpha-synuclein gene are etiologic (Polymeropoulos et al., 1997; Singleton et al., 2003). Why does alpha-synuclein tend to precipitate in catecholaminergic neurons in PD?
The title of this review is a proposed answer to the first set of questions. The thesis developed here is that the unusual vulnerability of catecholamine neurons is related to inherent cytotoxicity of catecholamines and their metabolites in the cells in which they are produced—“catecholamine autotoxicity.” Catecholamines spontaneously oxidize to form quinones, chromes, polydopamine, condensation products (e.g., salsolinol), melanin, and neuromelanin. Catecholamines are also subject to enzymatic oxidation mediated by monoamine oxidase (MAO), with the immediate products being hydrogen peroxide and aldehydes. As discussed in detail in this review, there are numerous potential pathogenetic links between the aldehydes and alpha-synuclein. One of them, aldehyde-induced oligomerization of alpha-synuclein, may help explain alpha-synuclein precipitation in Lewy bodies within monoaminergic neurons in PD.
2. Overview of the autotoxicity concept
In this review much attention will be given to the “catecholaldehyde hypothesis” (Panneton et al., 2010; Goldstein, Holmes et al., 2011; Goldstein, Sullivan et al., 2011; Goldstein, Sullivan et al., 2012; 2013). Briefly, the preponderance of intra-neuronal metabolism of endogenous DA occurs via formation of the catecholaldehyde, 3,4-dihydroxyphenylacetaldehyde (DOPAL), which is toxic. In general, the toxicity occurs by two routes—peroxidation of lipid membranes due to generation of reactive oxygen species and inactivation of enzymes and transporters due to protein cross-linking (Fig. 1).
Fig. 1.
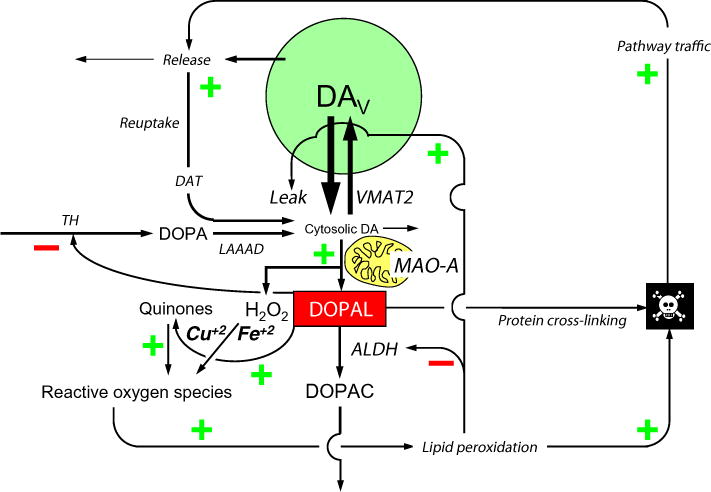
Overview of putative pathways of catecholamine autotoxicity in a dopaminergic neuron. Dopamine (DA) is formed in the cytosol from the action of L-aromatic-amino-acid decar-boxylase (LAAAD) on DOPA, which is the immediate product of the rate-limiting enzymatic step in DA synthesis, hydroxylation of tyrosine mediated by tyrosine hydroxylase (TH). Cytosolic DA can auto-oxidize spontaneously or undergo enzymatic oxidation catalyzed by monoamine oxidase-A (MAO-A) in the outer mitochondrial membrane; however, the main fate of cytosolic DA is vesicular uptake via the type 2 vesicular monoamine transporter (VMAT2). Vesicular DA (DAV) can leak from the vesicles into the cytosol or undergo exocytotic release. Most of released DA is taken back up into the cytosol via the cell membrane DA transporter (DAT). The action of MAO-A on cytosolic DA yields hydrogen peroxide (H2O2) and 3,4-dihydroxyphenylacetaldehyde (DOPAL). H2O2 reacts with metal ions to produce reactive oxygen species (in this case hydroxyl ions), resulting in oxidative injury including peroxidation of lipid membranes. DOPAL cross-links with amino acids in proteins; this can inactivate enzymes (e.g., TH) and transporters. DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH) to form 3,4-dihydroxyphenylacetic acid (DOPAC). A minor pathway of metabolism (not shown) is enzyme-catalyzed reduction to form 3,4-dihydroxyphenylethanol. Stimulatory relationships are indicated by (+) and inhibitory relationships by (−). De-stabilizing positive feedback loops are indicated when all signs in a loop are +.
As explained below in detail, we do not mean to imply here that DOPAL is the cause of PD or of any other neurodegenerative disease. Rather, according to the catecholaldehyde hypothesis, DOPAL is a node in a complex network of interacting homeostatic systems. Dysfunctions of several processes—including decreased vesicular sequestration of cytoplasmic DA and decreased DOPAL metabolism by aldehyde dehydrogenase (ALDH)—result from diverse combinations of genetic predispositions, environmental exposures, stress, and time. These abnormalities lead to conversion from the stability afforded by negative feedback regulation to the instability, degeneration, and system failure caused by induction of positive feedback loops (Goldstein, 2013).
Alpha-synuclein may be another node in this pathogenetic nexus. A key connection in this network (Fig. 2) is that DOPAL potently oligomerizes alpha-synuclein (Burke et al., 2008; Jinsmaa et al., 2014). Moreover, by a variety of means (Lotharius et al., 2002; Volles & Lansbury, 2002; Mosharov et al., 2006; Chu et al., 2012), alpha-synucleinopathy seems to interfere with vesicular sequestration of cytoplasmic catecholamines, biasing toward increased DOPAL production (Fig. 3).
Fig. 2.
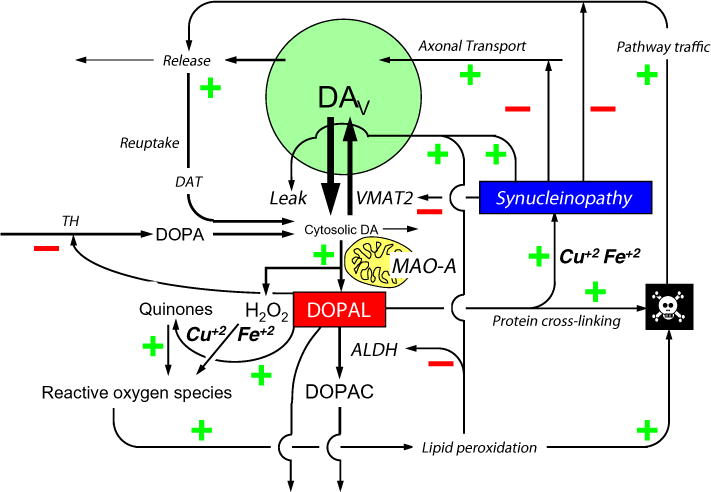
Putative toxic interactions between DOPAL and alpha-synucleinopathy in a dopaminergic neuron. According to the autotoxicity concept, DOPAL and alpha-synuclein are nodes in a complex pathogenetic nexus. DOPAL oligomerizes alpha-synuclein, rendering the protein toxic. Divalent metal cations augment DOPAL-induced oligomerization of alpha-synuclein.
Fig.3.
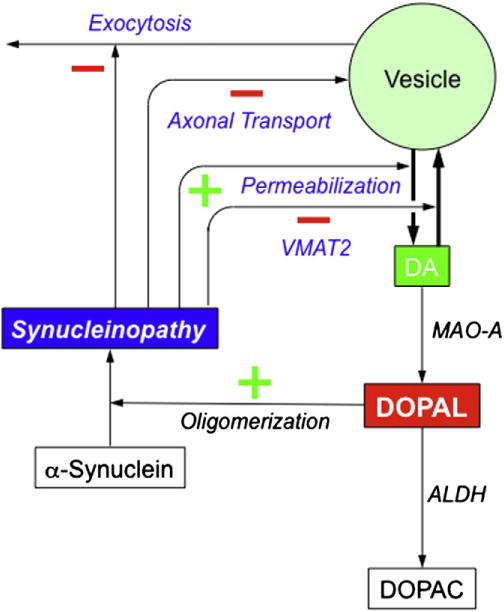
Potential mechanisms of synucleinopathy-induced augmentation of DOPAL production. Synucleinopathy might increase DOPAL formation by inhibiting VMAT2 expression or functions, permeabilizing vesicles, interfering with axonal transport (thereby decreasing vesicle numbers in striatal dopaminergic terminals), and inhibiting exocytosis.
While the diagram of the autotoxicity concept in Fig. 2 is complex, it is likely that there are other factors and inter-relationships that are missing. The neurotoxicity symbolized by the “skull and crossbones” icon can come about by several destabilizing positive feedback loops (indicated by all the signs in a given cycle being positive) or loss of negative feedback regulation (the loss indicated by an even number of negative signs in a cycle). For introductory purposes it suffices to emphasize that according to the catecholaldehyde hypothesis DOPAL occupies a central position in this network, and there are several ways that the combination of DOPAL with alpha-synucleinopathy might be cytotoxic.
3. Spontaneous oxidation of catecholamines
Catecholamines oxidize when they are exposed to even a weak oxidizing potential. This is a basis for liquid chromatography with electrochemical detection for assaying catecholamines (Goldstein et al., 1981). The effluent from the liquid chromatographic column is subjected to oxidation at a low oxidizing potential. Whereas other compounds do not oxidize at such a low potential, catecholamines do (Mosharov et al., 2003).
3.1. Quinones
Since catecholamines contain a catechol residue, they oxidize to semi-quinones or orthoquinones. Quinones of catecholamines are in turn oxidizing agents and electrophiles. Upon exposure to a reducing potential they regenerate the parent catecholamines. Detection of the reducing current is a basis of the use of a series oxidizing and then reducing electrodes in liquid chromatography with electrochemical detection. This improves sensitivity and specificity by detecting only the reversibly oxidized species (Eisenhofer et al., 1986).
Quinones covalently modify and damage cellular macromolecules (e.g., by bonding to the sulfhydryl residue of cysteine in proteins) and generate reactive oxygen species (Creveling, 2000). Oxidized DA can react with DNA to form depurinating adducts at guanine and adenine (Cavalieri et al., 2002) and inhibit NADH oxidase and mitochondrial complex I (Gautam & Zeevalk, 2011). Mitochondrial toxicity exerted by quinones impairs bio-energetic functions and evokes apoptosis (Hastings, 2009; Bisaglia et al., 2010; Jana et al., 2011). DA quinone can also interfere with proteasomal functions (Zhou & Lim, 2009). Consistent with the notion of DA quinone toxicity, VMAT or MAO-A overexpression protects against toxicity exerted by intracellular DA in response to L-DOPA exposure, while inhibition of these pathways potentiates L-DOPA toxicity in catecholaminergic PC12 cells (Weingarten & Zhou, 2001). Resveratrol, quercetin, and (−) epigallocatechin gallate, all of which are anti-oxidants, effectively preserve neuronal cell viability upon exposure to L-DOPA (Peritore et al., 2012). On the other hand, since macrophage migration inhibitory factor and glutathione S-transferase protect PC12 cells against intracellular DA cytotoxicity (Weingarten & Zhou, 2001), the toxicity may also occur via means other than oxidative injury.
3.2. Aminochrome
DA quinone can also cyclize to form DA-chrome (aminochrome), which in turn can generate the catechol, 5,6-dihydroxyindole, followed by oxidation to indole-5,6-quinone and eventually to formation of melanin (Fig. 2). Aminochrome has been shown to be toxic to murine mesencephalic MN9D cells at concentrations as low as 50 μM (Linsenbardt et al., 2009). In the mesencephalic cell line, MN9, aminochrome induces caspase-independent apoptosis (Linsenbardt et al., 2012). Aminochrome can also be formed from auto-oxidation of DA in the setting of hydrogen peroxide and a peroxidase, yielding a superoxide anion (Hastings, 1995), and activity of the peroxidase is increased in post-mortem midbrain tissue from PD patients (De Iuliis et al., 2002).
3.3. Polydopamine
At high concentrations in aqueous solutions, DA polymerizes spontaneously to form polydopamine (Lee et al., 2007). Polydopamine is an efficient and versatile industrial coating agent; whether intra-neuronal, endogenous polydopamine exists is unknown.
3.4. Salsolinols
Non-oxidative condensation of DA with acetaldehyde yields salsolinol (1-methyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline) via the Pictet–Spengler reaction. Endogenous salsolinols are found in brain areas characterized by high DA synthesis and turnover such as the ventral midbrain and striatum (DeCuypere et al., 2008). In neuroblastoma SH-SY5Y cells, salsolinol has been reported to be toxic (IC50 value of 34 μM), via inhibition of mitochondrial complex II (Storch et al., 2000). In PC12 cells, parkin knockdown elevates cellular oxidative stress, salsolinol and N-methylsalsolinol levels, which seem to be responsible for the higher cell mortality in Parkin-deficient cells upon exposure to exogenous hydrogen peroxide (Su et al., 2013). Since parkin deficiency releases MAO from inhibitory restraint (Jiang et al., 2006), it is also possible that parkin-deficient PC12 cells have a tendency to increased enzymatic metabolism of cytoplasmic DA to form DOPAL and hydrogen peroxide. N-methylsalsolinol can be neurotoxic via generation of hydroxyl radicals (Maruyama et al., 1995), and PD has been reported to be associated with elevated CSF levels of N-methylsalsolinol (Maruyama et al., 1996). In PD brain, caudate tissue levels of stereoisomers of salsolinol and N-methylsalsolinol are decreased, perhaps reflecting loss of dopaminergic terminals.
It is unclear whether or how these toxicological or post-mortem studies of spontaneous auto-oxidation products of DA relate to the pathophysiology of PD. First, in post-mortem studies of PD patients it is difficult to separate effects of chronic levodopa treatment from those of the underlying disease process. Second, studies involving exposures of cells to high concentrations of L-DOPA or DA may saturate MAO, augmenting production of DA quinone and aminochrome. Third, since DOPAL auto-oxidizes to semi- and ortho-quinones, formation of quinoproteins does not necessarily depend on DA quinone. Fourth, the action of MAO-A on DA yields hydrogen peroxide, which can combine with DA to form aminochrome. Fifth, the relative roles of spontaneous vs. enzymatic oxidation in the fate of endogenous DA have not been compared, and one may reasonably presume that enzyme-catalyzed oxidation predominates. Sixth, although many reports have suggested toxic effects of auto-oxidation products (Graham, 1978; Berman & Hastings, 1999; Stokes et al., 1999; Bisaglia et al., 2007; Hastings, 2009; Jana et al., 2011), there is little evidence that excessive spontaneous auto-oxidation of endogenous catecholamines occurs in clinical PD (Maruyama et al., 1996; DeCuypere et al., 2008).
4. Monoamine oxidase and the enzymatic oxidation of catecholamines
MAO occupies apivotal position in the intra-neuronal metabolism of catecholamines (Fig. 3). Across multiple brain areas, MAO activity is highest in the putamen, globus pallidus, substantia nigra, hypothalamus, and mammillary bodies (Riederer & Youdim, 1986).
Two isoforms of MAO exist—MAO-A and MAO-B. MAO-B is expressed to a greater extent in the striatum and brain overall (Riederer & Youdim, 1986). In the human brain, MAO-B inhibition attenuates MAO activity more potently than does MAO-A inhibition. Catecholaminergic neurons, however, express mainly MAO-A (Youdim et al., 2006). Chronic inhibition of MAO-A increases both NE release in the frontal cortex (Finberg et al., 1993) and microdialysate DA responses to locally administered levodopa in the striatum, whereas MAO-B inhibition exerts little effect (Finberg et al., 1995). DOPAL production is attenuated by MAO-A inhibition but not by MAO-B inhibition (Fornai et al., 2000). PD patients who had been treated with the MAO-B inhibitor deprenyl were reported to have increased post-mortem DA contents in the caudate nucleus, globus pallidus, putamen, and substantia nigra (Riederer & Youdim, 1986). This finding suggests that DA released from dopaminergic neurons can be taken up by local non-dopaminergic neurons and metabolized by MAO-B.
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) produces Parkinsonism in a variety of species including dogs (Johannessen et al., 1989), primates (Burns et al., 1983), and humans (Burns et al., 1985). The neurotoxicity depends on MPTP being converted by MAO-B to the potent dopaminergic neurotoxin MPP+ (Schinelli et al., 1988) in non-dopaminergic cells (Chiba et al., 1984), followed by uptake of MPP+ into dopaminergic neurons. MAO-B inhibition therefore protects against MPTP-induced neurotoxicity (Johannessen et al., 1989; Adeyemo et al., 1993). MPTP-induced neurotoxicity is relatively selective for dopaminergic neurons (Burns et al., 1985). Bases for this selectivity have not been explored. Acetaldehyde enhances MPP+ neurotoxicity and delays the elimination of MPP+ from the striatum (Zuddas et al., 1989). These findings suggest that acetaldehyde interferes with the intra-neuronal metabolism of MPP+.
4.1. Hydrogen peroxide production
Enzymatic oxidation of catecholamines produces catecholaldehydes and hydrogen peroxide. Via the Fenton reaction hydrogen peroxide in the presence of Fe+2 and a proton yields Fe+3, water, and a hydroxyl radical. Hydroxyl radicals are highly toxic and disrupt cellular and intra-cellular membranes by lipid peroxidation (Gutteridge, 1984). An alternative fate of hydrogen peroxide is reaction with the enzyme catalase to form water and oxygen. A peroxidase catalyzes formation of aminochrome from DA and hydrogen peroxide (De Iuliis et al., 2002), and activity of the peroxidase is increased in post-mortem midbrain tissue in PD.
Exposure of neuronal cells to levodopa generates hydrogen peroxide, and treatment with resveratrol or bioflavinoids attenuates levodopa-induced cytotoxicity (Peritore et al., 2012).
4.2. 3,4-Dihydroxyphenylacetaldehyde
As explained in detail below, DOPAL is produced by the enzymatic action of MAO-A on DA. Although essentially all of the intra-neuronal metabolism of DA occurs via DOPAL (Eisenhofer et al., 2004a; Marchitti et al., 2007), DOPAL levels are much lower than those of other metabolites, because of efficient conversion of DOPAL to DOPAC by ALDH and (to a lesser extent) to 3,4-dihydroxyphenylethanol (DOPET) by aldehyde/aldose reductase (AR). Until relatively recently there were no reports about endogenous DOPAL in cells or tissues. As of this writing, PubMed lists only about 60 studies about DOPAL, and of these only a few have involved measurements of endogenous DOPAL concentrations.
5. Vesicular uptake and catecholamine autotoxicity
Under resting conditions the cytoplasmic contents of catecholamines are below the limit of detection in rat pheochromocytoma PC12 cells and cultured midbrain neurons and are at most 7.5 μM in adrenomedullary chromaffin cells (Mosharov et al., 2003, 2006). Low cytoplasmic catecholamine levels reflect efficient vesicular sequestration via the type 2 vesicular monoamine transporter (VMAT2) and enzymatic oxidation catalyzed by monoamine oxidase-A (MAO-A) in the outer mitochondrial membrane. In adrenomedullary chromaffin cells the combination of reserpine to block vesicular uptake and pargyline to block MAO increases cytoplasmic catecholamine content by about 6-fold (Mosharov et al., 2003).
Vesicular catecholamines leak continuously into the cytoplasm (Eisenhofer et al., 2004b), where a portion undergoes enzymatic oxidation catalyzed by MAO-A to form catecholaldehydes (Li et al., 2001; Eisenhofer et al., 2004a; Burke et al., 2008; Rees et al., 2009; Goldstein, 2010).
5.1. Determinants of vesicular uptake and leakage
Uptake of cytoplasmic catecholamines into vesicles depends on the type-2 vesicular monoamine transporter (VMAT2). Mice with genetically determined very low VMAT2 activity have drastically decreased vesicular uptake (Miller et al., 2001; Caudle et al., 2007).
Retention of catecholamines within vesicles utilizes a proton pump. Proton pumping generates an electrochemical gradient across the vesicular membrane, via a vacuolar, magnesium-associated ATPase (Holz, 1978; Eiden et al., 2004). Catecholamines are accumulated in the vesicles at the expense of the proton gradient, at a ratio of one translocated amine per two translocated protons. Many PD patients are treated with proton pump inhibitors for gastrointestinal reflux, peptic ulcer disease, etc. Whether this treatment affects vesicular storage of catecholamines is unknown.
Catecholamine vesicles containing both norepinephrine and DA-beta-hydroxylase are delivered to terminals via axonal transport (Dahlstrom & Haggendal, 1970; Dahlstrom & Heiwall, 1975; Dahlstrom et al., 1975; Larsson et al., 1986). Therefore, factors interfering with axoplasmic transport might decrease the size of the vesicular pool at the level of the terminals and consequently decrease vesicular uptake of cytoplasmic DA.
5.2. Vesicular sequestration and neurotoxicity
Vesicular uptake is a key neuroprotective process in catecholaminergic neurons (Guillot & Miller, 2009). Interference with vesicular uptake augments the toxicity of methamphetamine (Fumagalli et al., 1999; Vergo et al., 2007; Guillot et al., 2008), MPTP (German et al., 2000; Staal & Sonsalla, 2000; Mooslehner et al., 2001), and 6-hydroxydopamine (Sun et al., 2004). Since an early effect of MPTP treatment is to decrease vesicular uptake (M.K. Chen et al., 2008), there is the potential for a cytotoxic positive feedback loop, in which toxin-induced impairment of vesicular uptake in turn promotes build-up of the toxin in the cytoplasm, killing the neuron.
Some pesticides attenuate vesicular uptake. In human neuroblastoma-derived SH-SY5Y cells, rotenone inhibits VMAT2, redistributing DA from vesicles to cytoplasm (Watabe & Nakaki, 2007, 2008). In rat pheochromocytoma PC12 cells, rotenone down-regulates VMAT2 while increasing MAO activity (Sai et al., 2008). This combination would be expected not only to build up cytoplasmic DA but also to increase DOPAL formation. DDT and its metabolites also inhibit VMAT2 (Hatcher et al., 2008). Dieldrin decreases striatal 3H-DA-derived radioactivity (Hatcher et al., 2007). This might reflect denervation and decreased vesicular uptake of cytoplasmic 3H-DA. The pesticide and complex I inhibitor, rotenone, produces several neurobehavioral and neuropathologic abnormalities resembling those in PD, and at least part of this effect may be from inhibition of vesicular uptake (Holz, 1978).
Other evidence suggesting a key role of vesicular uptake in preventing catecholamine autotoxicity comes from experiments in which cells that do not possess VMAT2 activity are exposed to increased cytoplasmic DA. CHO cells over-expressing LAAAD and exposed to levodopa have DA-induced cytotoxicity, which is attenuated by concurrent expression of VMAT2 (Weingarten & Zhou, 2001). Mice with widespread expression of the cell membrane DA transporter (DAT) have substantial striatal neurodegeneration (L. Chen et al., 2008). Because of the proximity in the striatum of dopaminergic terminals and medium spiny neurons, which do not express VMAT2, DAT expression in the neurons would be expected to cause an influx of DA and cytotoxicity mediated by oxidative injury. This study provided in vivo evidence that chronic exposure to unregulated cytosolic DA alone is sufficient to cause neurodegeneration.
Studies of mice with inherited extremely low VMAT2 activity have noted motor and non-motor neurobehavioral abnormalities resembling those in PD, including catecholamine neuron loss (Caudle et al., 2007; Taylor et al., 2009, 2014). Such mice also have extreme susceptibility to neurotoxicity from exposure to MPTP (Mooslehner et al., 2001). A recent study involving bacterial artificial chromosome transgenic mice expressing human alpha-synuclein (Janezic et al., 2013) reported age-dependent loss of nigrostriatal DA neurons, motor abnormalities resembling those in PD, and an altered distribution of vesicles in dopaminergic axons in the dorsal striatum, independently of alpha-synuclein aggregation.
5.3. Vesicular sequestration and clinical Parkinsonism
Genetic and gene expression abnormalities of VMAT2 are associated with clinical Parkinsonism. Platelet VMAT2 gene expression is decreased in PD (Sala et al., 2010). VMAT2 gene mutation produces a severe pediatric syndrome that includes Parkinsonism (Rilstone et al., 2013).
Post-mortem Western blotting and immunohistochemistry have demonstrated marked reductions in VMAT2 immunoreactivity in the putamen in PD (Miller et al., 1999). Since the decrease of VMAT2 in PD parallels that of the DA membrane transporter, the results may be explained by denervation. Based on PET imaging using 18F-AV-133, a VMAT2 ligand, VMAT2 binding is markedly decreased in PD (Okamura et al., 2010), but again this could be due to denervation.
Results of a novel clinical in vivo neuroimaging/neurochemical study provided the first evidence for abnormal vesicular storage of catecholamines in sympathetic nerve terminals in PD, after taking denervation into account (Goldstein, Holmes et al., 2011). This study asked whether sympathetic denervation is associated with decreased sequestration of catecholamines into storage vesicles within sympathetic neurons. 18F-DA was used to track myocardial uptake and retention of catecholamines. Concurrently, the fate of intraneuronal 18F-DA was followed by assessment of arterial plasma levels of the 18F-DA metabolite 18F-DOPAC. The ratio of myocardial 18F-DA to arterial 18F-DOPAC provided an index of vesicular sequestration. Tracer concentrations were measured in patients with PD with or without orthostatic hypotension (PD+ OH, PD-No-OH); PAF, a Lewy body disease without Parkinsonism; MSA, a non-Lewy body synucleinopathy; and controls. PD and PAF patients had substantially decreased vesicular 18F-DA uptake and accelerated 18F-DA loss, compared to normal values in MSA patients and controls. It was concluded that sympathetic denervation in Lewy body diseases is associated with decreased vesicular sequestration of cytoplasmic catecholamines in the residual sympathetic nerves. A previous in vivo study examining the fate of putamen 18F-DOPA-derived radioactivity in PD had found accelerated loss of radioactivity (Goldstein et al., 2008). In retrospect, the accelerated loss was consistent with decreased vesicular storage of intra-neuronal 18F-DA.
A recent post-mortem neurochemical study examined whether there is decreased vesicular storage in residual putamen dopaminergic terminals in PD (Goldstein et al., 2013). Theoretically, the DA:DOPA concentration ratio indicates vesicular sequestration for a given amount of DA synthesis. This index was validated in transgenic mice with very low VMAT2 activity. In PD putamen, vesicular sequestration is estimated to be decreased by 89% in the remaining putamen dopaminergic terminals.
6. Aldehyde detoxification and catecholamine autotoxicity
The aldehyde dehydrogenase (ALDH) gene superfamily is evolutionarily ancient (Jackson et al., 2011; Vasiliou et al., in press). Aldehydes have long posed challenges to cellular homeostasis, such as by binding covalently to proteins and thereby altering their functions.
6.1. Aldehyde Dehydrogenase genes
The human genome is known to possess 19 different ALDH genes. Aldehydes are obligate intermediates in the metabolism of endogenous monoamines. ALDH1A1 and ALDH2, expressed respectively in the cytoplasm and mitochondria, seem most relevant to catecholamine autotoxicity. Both isoforms are expressed in substantia nigra dopaminergic neurons (Wey et al., 2012).
ALDH1A1 gene expression is a criterion in the identification of dopaminergic neurons derived from induced pluripotent stem cells (Cai et al., 2010; Xi et al., 2012). Interestingly, ALDH1A1 isozymes are also markers of human melanoma stem cells (Luo et al., 2012), and melanoma is associated statistically with PD (Gao et al., 2009; Inzelberg et al., 2011; Liu et al., 2011). One may reasonably speculate that the link between melanoma and PD may be from decreased ALDH activity promoting neurotoxicity from DOPAL while also promoting survival of cancer cells that otherwise would be killed by endogenous aldehydes as a protective process.
East Asian people are well known to have a high frequency of alcohol sensitivity related to dominant negative mutation of the ALDH2 gene. Decreased ALDH2 activity in this population is also associated with increased risks for various types of cancer, myocardial infarction, and alcoholic liver disease (Song et al., 2011). The ALDH2 gene has been implicated in dependence on addictive drugs (Wang et al., 2012). ALDH2 inhibition decreases cocaine seeking, via generation of tetrahydropapaveroline (Yao et al., 2010), which is discussed below in a condensation of DOPAL with DA.
6.2. Non-genetic influences on ALDH activity
Two types of non-genetic factors affect ALDH activity—lipid peroxidation products and pesticides.
6.2.1. Lipid peroxidation products
4-Hydroxy-nonenal (4HNE) and malondialdehyde (MDA) are common aldehyde products of lipid peroxidation. Protein adducts of 4HNE are found at increased frequency in nigral neurons from PD patients (Yoritaka et al., 1996). Importantly, both 4HNE and MDA inhibit ALDH, and they therefore inhibit the metabolic breakdown of DOPAL (Florang et al., 2007; Rees et al., 2007; Jinsmaa et al., 2009, 2011). The same lipid peroxidation products promote formation of alpha-synuclein oligomers (Nasstrom et al., 2011). Since oxidative injury from a variety of sources yields lipid peroxidation products, ALDH inhibition by 4HNE and MDA may help explain relatively selective loss of dopaminergic neurons in response to generalized oxidative stress.
6.2.2. Pesticides
Epidemiological studies have implicated exposure to farming chemicals such as insecticides, fungicides, and herbicides as a risk factor for PD (Tanner et al., 2011); however, mechanisms for this association have been obscure (Bronstein et al., 2009). The fungicide/pesticide, benomyl, has been shown to inhibit ALDH in vitro (Fitzmaurice et al., 2013), an effect that investigators have recognized in terms of potential relevance to PD pathogenesis.
In PC12 cells, rotenone increases cellular levels of DOPAL and DOPET while decreasing levels of DOPAC (Lamensdorf et al., 2000a), a combination that indicates decreased ALDH activity. Increased DOPAL formation in this setting contributes to rotenone-induced cytotoxicity (Lamensdorf et al., 2000b). Knockdown of MAO-A mitigates rotenone-induced apoptosis (Fitzgerald et al., 2014), consistent with DOPAL, hydrogen peroxide, or both contributing to the toxicity of this pesticide.
Dieldrin exposure induces oxidative damage to nigrostriatal dopaminergic terminals of mice, accompanied by decreased striatal DOPAC and homovanillic acid levels but without decreased striatal DA content (Hatcher et al., 2007). This neurochemical pattern would be consistent with ALDH inhibition, although theoretically decreased MAO-A activity could also explain these findings.
6.3. Aldehyde Dehydrogenase activity and Parkinsonism
Experiments and observations related to ALDH activity provide a means to distinguish auto-oxidation from enzymatic-catalyzed oxidation as bases for catecholamine autotoxicity. If cytoplasmic catecholamines were toxic due to auto-oxidation, then the status of ALDH activity would be irrelevant to the toxicity, whereas if they were toxic due to enzymatic-catalyzed oxidation to form DOPAL, then ALDH inhibition would be associated with increased toxicity. Diethyldithiocarbamate, which is thought to be the active metabolite of the classic ALDH inhibitor disulfiram (Antabuse) enhances the neurotoxic effects of MPTP in mice (Corsini et al., 1985). This finding indirectly suggests that at least part of the neuronal damage exerted by MPTP is mediated by DOPAL.
If ALDH inhibition played a pathogenic role in PD, then one would expect there to be decreased ALDH activity in PD, and congenital ALDH inhibition would be expected to be associated with development of Parkinsonism in animal models. Since ALDH acts on DOPAL to form DOPAC, the ratio of DOPAC:DOPAL concentrations provides an index ALDH activity. The finding of low striatal DOPAC:DOPAL ratios in mice with double knockout of the genes encoding ALDH1A1 and ALDH2 (ALDH1A1,2 knockouts) validates this index (Goldstein et al., 2013). Putamen DOPAC:DOPAL ratios are decreased in patients with sporadic PD (Goldstein et al., 2013), indicating that decreased ALDH activity occurs in PD.
Aldehyde/aldose reductase (AR) genes are in a super-family of aldo-keto reductases encoding at least 114 different proteins (Hyndman et al., 2003). AR constitutes a minor alternative pathway in the metabolism of DOPAL, whereas AR plays a major role in the metabolism of DOPEGAL, the catecholaldehyde of NE. This difference explains why the main end-product of the intra-neuronal metabolism of DA is the acid, DOPAC, whereas the main end-product of the intra-neuronal metabolism of NE is the glycol, DHPG (Goldstein et al., 1988; Eisenhofer et al., 2004a). The specific AR genes determining the reduction of catecholaldehydes have not yet been identified.
7. The catecholaldehyde hypothesis
Catecholaldehydes are obligate intermediates in the intra-neuronal metabolism of endogenous catecholamines (Eisenhofer et al., 2004a). Although as early as 1952 it had been suggested that products of the enzymatic oxidation of catecholamines, catecholaldehydes, are potentially toxic (Blaschko, 1952), the first report about measurement of DOPAL in human brain was published in 1993 (Mattammal et al., 1993).
Early evidence that DOPAL can be toxic to catecholaminergic cells (rat neostriatal synaptosomes, PC12 cells, and cultured fetal rat dissociated mesencephalic tissue) was reported two years later (Mattammal et al., 1995). In rat nigral dopaminergic neurons, exogenously administered DOPAL in amounts as low as 0.5 nmol was found to be toxic, whereas DA and its other metabolites showed no evidence of neurotoxicity at 5 times higher doses (Burke et al., 2003).
Although exogenously administered DOPAL is well known to be cytotoxic, it was not known whether DOPAL produced intracellularly is toxic until a 2012 report about effects of vesicular uptake blockade on levels of DOPAL in rat pheochromocytoma PC12 cells (Goldstein, Sullivan et al., 2012). Catechols were assayed in PC12 cells after reserpine to block vesicular uptake, with or without inhibition of enzymes metabolizing DOPAL—daidzein for ALDH and AL1576 for aldehyde reductase. Vesicular uptake was quantified by methods based on 6F- or 13C-DA incubation; DOPAL cytotoxicity by apoptosis responses to exogenous DA, with or without daidzein + AL1576; and DOPAL-induced synuclein oligomerization by synuclein dimer production during DOPA incubation, with or without inhibition of L-aromatic-amino-acid decarboxylase or MAO. Reserpine inhibited vesicular uptake by 95–97% and increased DOPAL levels in cells and medium. Reserpine and inhibition of DOPAL metabolism exerted additive effects on cell DOPAL content. DOPAL contributed to DA-evoked apoptosis and DOPA-evoked synuclein dimerization. The findings fit with decreased vesicular sequestration of cytosolic catecholamines and impaired catecholaldehyde detoxification contributing to the catecholaminergic denervation that characterizes PD.
A report applying a liquid chromatographic–electrochemical assay method for measuring endogenous DOPAL levels (Burke et al., 1999) yielded questionable results, because the representative chromatograph of urinary DOPAL, catecholamines, and related metabolites was of poor quality, and the reported values for urinary concentrations of catechols did not agree with those from other groups (Kagedal & Goldstein, 1988). Failure of levodopa given with an ALDH inhibitor to produce striatal DA deficiency (Legros et al., 2004) put a damper on research related to the catecholaldehyde hypothesis.
The recent development of a sensitive, specific assay method to detect and quantify DOPAL in post-mortem brain tissue was a major advance that enabled further exploration of the catecholaldehyde hypothesis for the death of catecholamine neurons in PD. A study of postmortem putamen concentrations of catechols in PD (Goldstein, Holmes et al., 2011; Goldstein, Sullivan et al., 2011) provided the first evidence for DOPAL build-up relative to DA in the putamen in PD. In the same post-mortem neurochemical study, the first evidence suggesting decreased activity of ALDH was reported.
7.1. Mechanisms of 3,4-dihydroxyphenylacetaldehyde autotoxicity
There are multiple potential mechanisms of DOPAL cytotoxicity. First and foremost, intracellular aldehydes can bind covalently with proteins, altering functions of proteins such as enzymes and transporters. DOPAL contains two reactive functional groups—the aldehyde and the catechol. Protein modification by DOPAL involves both a thiol-reactive quinone because of oxidation of the catechol and formation of adducts with amine nucleophiles of proteins and potential cross-linking because of the aldehyde (Rees et al., 2009). An example of protein modification by DOPAL is covalent modification and inhibition of tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine biosynthesis (Mexas et al., 2011). The inhibition of TH is somewhat reversible, since removal of DOPAL results in time- and concentration-dependent recovery of enzyme activity.
Second, DOPAL auto-oxidizes to form a semi-quinone or orthoquinone (Anderson et al., 2011). As described in detail above for DA quinone, DOPAL quinones are oxidizing agents that would be expected to promote generation of reactive oxygen species, resulting in lipid peroxidation and thereby disruption of cell, vesicular, and mitochondrial membranes. In aqueous media, DOPAL quinone exists exclusively in the carbonyl-hydrated form. Judging from the peculiar appearance of the DOPAL chromatographic peak by liquid chromatography with electrochemical detection, it seems likely that intracellular DOPAL can exist in multiple molecular forms that are in equilibrium with each other.
Third, DOPAL condenses with DA to form the isoquinoline, tetrahydropapaveroline (THP) (Walsh et al., 1970; Weiner, 1978). Increased THP formation has long been suspected of mediating cytotoxic effects of exogenously administered levodopa, and PD patients receiving levodopa treatment have detectable THP in urine (Cashaw, 1993). In human embryonic kidney HEK-293 and mouse neuroblastomaneuro-2A cells, exogenously administered THP is cytotoxic, with potency about that of salsolinol (Storch et al., 2002). THP selectively inhibits phosphorylated (activated) TH (Kim et al., 2005; Yao et al., 2010) and dose- and time-dependently aggregates neurofilament-L, with glutamate, proline, and lysine residues of this structural protein being particularly sensitive. THP also damages DNA, apparently via hydroxyl radical formation (Kobayashi et al., 2006).
Fourth, alpha-synuclein oligomers are widely suspected of being pathogenic in PD (Winner et al., 2011), and DOPAL oligomerizes alpha-synuclein (Burke et al., 2008). DOPAL is far more potent in this regard than is either DA or DOPAC. DOPAL concentration-dependently increases formation of alpha-synuclein dimers, whereas hydrogen peroxide does not, and in PC12 cells over-expressing alpha-synuclein, MAO inhibition by pargyline prevents levodopa-induced synuclein dimerization (Goldstein, Sullivan et al., 2012). In human neuroblastoma SH-SY5Y cells over-expressing alpha-synuclein, DA exposure augments synuclein oligomerization and cytotoxicity (Yamakawa et al., 2010); this study did not take into account the possibility of intracellular conversion of DA to DOPAL by MAO.
Recent studies have indicated that oligomerized or fibrillar alpha-synuclein is transmissible cell to cell (Lee et al., 2010; Luk et al., 2012). Since DOPAL is a neutral compound, it should readily cross cell membranes and might also be transmissible cell to cell. Thus, in response to locally administered levodopa, DOPAL is detected in vivo in striatal microdialysate (Colzi et al., 1996), and in PC12 cells, vesicular uptake blockade results in increased DOPAL concentrations in the medium (Goldstein, Sullivan et al., 2012).
7.2. Putamen 3,4-dihydroxyphenylacetaldehyde in Parkinson disease
A pivotal post-mortem neurochemical study examined contributions of decreased vesicular storage and decreased ALDH activity to DOPAL build-up in the putamen in PD (Goldstein et al., 2013). Theoretically, the DA:DOPA concentration ratio indicates vesicular uptake, and the DOPAC:DOPAL ratio indicates ALDH activity. These indices were validated in transgenic mice with very low vesicular uptake (VMAT2-Lo) or with knockouts of the genes encoding ALDH1A1 and ALDH2 (ALDH1A1,2 KO). The indices were then applied in PD putamen, and percentage decreases in vesicular uptake and ALDH activity estimated in PD. In PD putamen, vesicular uptake was estimated to be decreased by 89% and ALDH activity decreased by 70%. Therefore, elevated DOPAL levels in PD putamen reflect a combination of decreased vesicular sequestration of cytosolic DA and decreased DOPAL detoxification by ALDH.
In PC12 cells, inhibition of MAO-A attenuates the increase in intra-cellular reactive oxygen species evoked by exposure to exogenous DA. This effect of MAO-A inhibition suggests that the oxidative stress evoked by DA depends on either DOPAL, on hydrogen peroxide produced by the deamination, or both (Jana et al., 2011). MAO-A inhibition exerts only mild attenuation of the protein adduct formation, mitochondrial dysfunction, cell death, and apoptosis induced by exposure to exogenous DA (Jana et al., 2011; Goldstein, Sullivan et al., 2012). MAO inhibition by pargyline blocks rotenone-induced augmentation of NMDA currents in rat substantia nigra slices. This suggests that DOPAL or hydrogen peroxide mediates the toxic effect of rotenone (Wu & Johnson, 2011).
In support of the catecholaldehyde hypothesis, a recent study of locus ceruleus and myocardial sympathetic neurons documented noradrenergic neuronal loss in VMAT2-Lo mice (Taylor et al., 2014). Since NE is synthesized within vesicles, and VMAT2-Lo mice have neurochemical evidence of severely decreased NE formation, central and peripheral noradrenergic denervation in VMAT2-Lo mice seems to reflect cytotoxicity exerted by cytoplasmic DA via DOPAL rather than by cytoplasmic NE via DOPEGAL.
7.3. Genetic abnormalities and the catecholaldehyde hypothesis
Support for the catecholaldehyde hypothesis must include evidence that etiologic genetic abnormalities lead to processes involving catecholamine autotoxicity. Specifically, if decreased vesicular sequestration, decreased ALDH activity, and DOPAL build-up were part of the death process in dopaminergic neurons, then one would expect that genotypic abnormalities leading to familial Parkinsonism would have in common the ability to evoke this pattern. The following discussion, while highly speculative, presents a rationale for such commonality (Fig. 4).
Fig. 4.
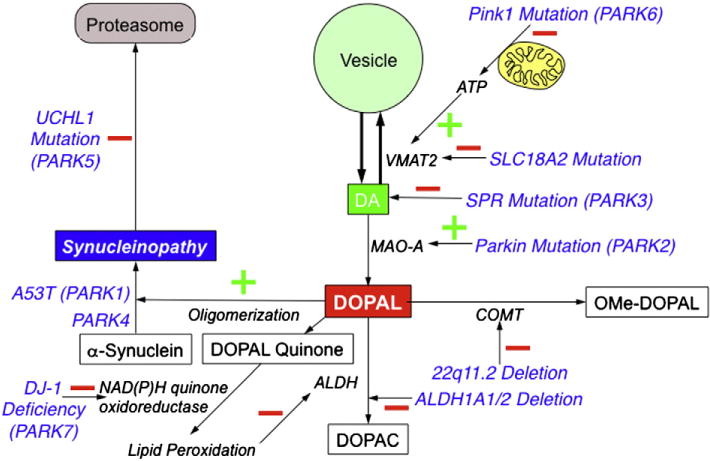
Potential sites of interaction between genotypic abnormalities and catecholamine autotoxicity. See text for meanings of abbreviations.
We have reported evidence for decreased vesicular sequestration in the PARK1 and PARK4 forms of familial PD due to alpha-synucleinopathy (Goldstein, Holmes et al., 2011). In PARK2, which is from parkin gene mutation, the gene product inhibits MAO-A (Jiang et al., 2006, 2012). Hypofunctional parkin mutation would be expected to release MAO-A from inhibitory restraint and thereby promote DOPAL production. Parkin knockout mice have augmented levodopa-induced neurotoxicity (Casarejos et al., 2005). PARK3 involves mutation of the gene encoding sepiapterin reductase (SPR), which catalyzes the final step in the biosynthesis of tetrahydrobiopterin, a key co-factor in tyrosine hydroxylation and thereby DA production. This could lead to Parkinsonism directly.
PARK5 is from mutation of the gene encoding ubiquitin carboxyl-terminal esterase L1 (UCHL1), which plays a role in proteasomal clearance of degraded cytoplasmic proteins (Snyder & Wolozin, 2004). Since UCHL1 inhibition increases cytoplasmic alpha-synuclein levels (Cartier et al., 2012), UCHL1 mutation might interfere with vesicular catecholamine storage in a manner similar to that in PARK1 and PARK4.
PARK6 is from mutation of the pink1 gene, the product of which is thought to modulate mitochondrial function during periods of cellular oxidative stress (Cookson, 2012). Since vesicular uptake is an ATP-dependent process, mitochondrial dysfunction would be expected to impair vesicular uptake of cytoplasmic DA. Thus, Complex 1 inhibition increases cytoplasmic DOPAL levels (Lamensdorf et al., 2000a), and in this setting DOPAL contributes to cell death (Lamensdorf et al., 2000b).
PARK7 is related to mutations of the gene encoding DJ-1. DJ-1 deficiency decreases activity of NAD(P)H quinone oxidoreductase (Clements et al., 2006). Since both DA and DOPAL auto-oxidize spontaneously to form potentially harmful quinones (Anderson et al., 2011), hypofunctional DJ-1 mutation might promote autotoxicity by decreasing the ability to detoxify the quinones.
With regard to lrrk2 mutation, which causes PARK8, the intra-neuronal functions of leucine-rich repeat kinase and consequences of mutations of the gene remain unknown (Rideout & Stefanis, 2014). Analysis of 18F-DOPA PET data from our reported patient with PD from LRRK2 mutation (Goldstein et al., 2007) has revealed markedly accelerated loss of putamen 18F-DOPA-derived radioactivity, which would be consistent with decreased vesicular sequestration of cytoplasmic 18F-DA generated from 18F-DOPA.
Variations in the gene or gene expression ALDH1A1 or ALDH2 have been reported to be associated with PD (Galter et al., 2003; Mandel et al., 2005; Galvin et al., 2009; Grunblatt et al., 2010; Fitzmaurice et al., 2014). Disabling these genes in mice produces congenital DOPAL build-up and aging-related neurobehavioral and neuropathologic abnormalities resembling those in PD (Wey et al., 2012).
Deletion at the 22q11.2 chromosomal locus is associated with markedly increased risk of early-onset PD (Butcher et al., 2013). Since this locus contains the gene encoding catechol-O-methyltransferase, 22q11.2 deletion would be expected to decrease metabolism of DOPAL (Rees et al., 2009).
Mutations in the gene encoding glucocerebrosidase underlie Gaucher disease. Patient with Gaucher disease or heterozygous carriers of the mutated Gaucher disease gene have an increased risk of developing PD (Goker-Alpan et al., 2006; Sidransky et al., 2009). Bases for the link between glucocerebrosidase gene mutations and PD remain unknown. The finding of reduced cardiac 123I-metaiodobenzylguanidine-derived radioactivity in Gaucher/PD (Lebouvier et al., 2014) fits with decreased cardiac sympathetic innervation, inactivation of the cell membrane NE transporter, or decreased vesicular sequestration.
Perhaps the most telling evidence for human genetic pathways intersecting with catecholamine autotoxicity comes from a recent report of a neurological syndrome including Parkinsonism in a family with inherited mutation of the gene encoding the type 2 vesicular monoamine transporter (VMAT2) (Rilstone et al., 2013). This would be expected to attenuate vesicular sequestration directly.
8. Network aspects of autotoxicity: interactions with alpha-synuclein
In PD, diverse pathogenetic routes from different etiologic genotypic abnormalities, environmental exposures, stress, and time might converge eventually in a common death process that involves catecholamine autotoxicity. Although researchers might agree on this proposal, most of the literature on these factors has involved studies exploring them in isolation. A more integrative approach seems required to understand chronic clinical disorders involving catecholamine systems—especially PD (Goldstein, 2013).
DOPAL may interact in many ways with alpha-synuclein (Fig. 2). As noted above, DOPAL potently oligomerizes alpha-synuclein (Burke et al., 2008). This section discusses more indirect interactions between the catecholaldehyde and the protein.
Nigrostriatal overabundance of alpha-synuclein leads to decreased vesicle density (Gaugler et al., 2012). This may decrease vesicular uptake of cytoplasmic DA and thereby augment DOPAL production for a given amount of DA synthesis.
In primary neuronal cultures of Drosophila, alpha-synuclein exposure causes time-dependent, selective dopaminergic neurodegeneration (Park et al., 2007). Blockade of TH by alpha-methyl-para-tyrosine prevents and TH gene overexpression augments the early neurodegeneration, indicating dependence of the lesion on endogenous DA. Importantly, overexpression of a Drosophila VMAT also prevented synuclein-mediated neurodegeneration. This study did not explore mechanisms by which cytoplasmic DA build-up mediates the neurodegeneration.
The lipid peroxidation products 4-oxo-2-nonenal and 4HNE promote formation of alpha-synuclein oligomers (Nasstrom et al., 2011). Since 4HNE also inhibits ALDH (see above), and DOPAL oligomerizes alpha-synuclein, interactions among DOPAL, lipid peroxidation products, and alpha-synuclein may involve multiple deleterious positive feedback loops.
Alpha-synuclein has been reported to inhibit DA uptake into rat striatal synaptosomes (Adamczyk et al., 2006) and to inhibit VMAT2 activity in alpha-synuclein stably transfected SH-SY5Y cells (Guo et al., 2008). Proto-fibrillar alpha-synuclein permeabilizes vesicles by a porelike mechanism (Volles & Lansbury, 2002). This would be expected to enhance leakage of endogenous DA and augment DOPAL production. Therefore, alpha-synucleinopathy may decrease vesicular sequestration of cytoplasmic DA both by decreasing vesicular uptake and increasing vesicular leakage.
A new human mesencephalic cell line, MESC2, produces DA and contains wild-type human alpha-synuclein. A53T mutation of the gene encoding alpha-synuclein was the first genotypic abnormality shown to cause familial PD (Polymeropoulos et al., 1997). When the A53T form of alpha-synuclein was over-expressed in differentiated MESC2 cells, this caused VMAT2 down-regulation and increased enhanced cytoplasmic DA immunofluorescence and intracellular levels of superoxide. The results suggest that A53T alpha-synuclein expression impedes vesicular DA storage, with consequent DA build-up in the cytoplasm (Lotharius et al., 2002).
Recent studies have examined dopaminergic neurotransmission in adult rats that had undergone unilateral nigral injection of an adeno-associated virus-alpha-synuclein vector, causing overexpression of alpha-synuclein in the nigral DA neurons (Decressac et al., 2012; Lundblad et al., 2012). The earliest change seen in the striatum is marked reduction in DA reuptake as measured by in vivo amperometry, consistent with early dysfunction of the cell membrane DA transporter. This is followed by impaired DA release and axonal damage. Only later is there a decrease in overall striatal innervation density or intra-axonal alpha-synuclein aggregation. These results suggest that early pre-degenerative changes in the handling of DA may drive a neurodegenerative process that is initiated at the level of the terminals and axons. The mechanism of the early apparent decrease in DA reuptake is unclear, since there is no decrease in expression of the cell membrane DA transporter. Moreover, in vivo amperometry does not quantify DA specifically compared to other readily oxidized catechols.
Metal ions are thought to contribute to the pathogenesis of PD (Barbeau, 1984; Sofic et al., 1988; Riederer et al., 1989; Gorell et al., 1999; Double et al., 2000). One possible mechanism for such a role is by promoting alpha-synuclein oligomerization. Alpha-synuclein contains binding sites for copper, iron, and manganese ions, and under certain conditions, metal ions oligomerize alpha-synuclein (Paik et al., 1999; Wang et al., 2010). it has been proposed that a unique copper-induced oligomer mediates synuclein toxicity (Brown, 2009; Wright et al., 2009); however, induction of alpha-synuclein oligomerization by exposure to metal ions requires supra-physiologic concentrations of metal ions, long reaction times, and coupling agents (Paik et al., 2000).
A recent study showed that DOPAL interacts with divalent metalcations (Cu2+, Fe2+, Mn2+) in oligomerizing alpha-synuclein (Jinsmaa et al., 2014), with potencies of Cu2+ > Fe2+ > Mn2+. Other DA metabolites, DA itself, Cu1+, Fe3+, and metal ions alone or in combination with DA had no effect. The findings show that divalent metal cations augment the already potent oligomerization of alpha-synuclein exerted by DOPAL alone.
9. Therapeutic implications of catecholamine autotoxicity
Examination of the network diagrammed in Fig. 2 brings to mind several types of treatment or prevention strategies that might be efficacious by ameliorating catecholamine autotoxicity. Conversely, clinical experimental trials would provide key tests of the catecholaldehyde hypothesis, just as levodopa treatment tested Hornykiewicz’s notion of a nigrostriatal dopaminergic lesion underlying Parkinsonism (Cotzias, 1971; Hornykiewicz, 2001, 2008).
At this point it is unclear how these approaches could be developed in a manner that takes into account the blood–brain barrier and targets central catecholaminergic neurons. Techniques based on optogenetics (Kravitz et al., 2010; Tonnesen, 2013), or targeted gene therapy (Kells et al., 2012; Mittermeyer et al., 2012; Navarro-Yepes et al., 2014) have potential here.
9.1. Monoamine oxidase inhibition
If products of MAO acting on cytoplasmic DA (DOPAL and hydrogen peroxide) mediated catecholamine autotoxicity, then MAO inhibition would be expected to be beneficial. Almost all research attention on MAO inhibitors in PD treatment or disease modification has focused on MAO-B. This is because the dopaminergic neurotoxicity of MPTP depends on conversion of MPTP to MPP+ by MAO-B. Clinical trials of the MAO-B inhibitors selegiline (L-deprenyl) and rasagiline have yielded generally positive but highly controversial results about whether MAO-B inhibition slows the neurodegenerative process in PD (Maki-Ikola & Heinonen, 1996; Shoulson, 1998; Olanow, 2006; Olanow et al., 2009; de la Fuente-Fernandez et al., 2010; Holford & Nutt, 2011).
Since dopaminergic neurons do not express MAO-B, even if MAO-B inhibitors were shown to be efficacious the mechanism of benefit would be unclear (Nagatsu & Sawada, 2006). MAO-B inhibitors exert several other effects besides inhibiting MAO-B, and inhibition of MAO-B in glial cells might decrease formation of neurotoxins in a manner analogous to MPP+ from MPTP. Moreover, in humans L-deprenyl treatment decreases plasma levels of DHPG and DOPAC, which are the main respective intra-neuronal metabolites of NE and DA (Eisenhofer et al., 1986). This brings up the possibility that administration of a selective MAO-B inhibitor can decrease MAO-A activity in humans. A recent report noted that PD patients on an MAO-B inhibitor had a 70% decrease in MAO-A activity during long-term daily treatment (Bartl et al., 2014).
MAO-A inhibitors and non-selective MAO inhibitors are well known to have very limited clinical usefulness, due to the “cheese effect” (Elsworth et al., 1978). MAO-A is a major route of deamination of dietary tyramine found in hard cheese, red wine, and many other foodstuffs. MAO-A inhibition in the gut and liver can enable dietary tyramine to reach the systemic circulation, displace endogenous NE from vesicular stores, and produce paroxysmal hypertension. One may predict that a non-selective MAO inhibitor delivered via a skin patch would be less likely to be associated with the cheese effect than the same drug delivered via oral ingestion.
L-Threo-dihydroxyphenylserine (L-DOPS, droxidopa) is a NE prodrug that recently received approval by the US FDA for treatment of symptomatic orthostatic hypotension. L-DOPS has been reported to inhibit MAO-A and MAO-B (Naoi & Nagatsu, 1986), by mechanisms that remain obscure but do not depend on NE production.
Considering the possibility that levodopa-related cytotoxicity may be mediated by products of enzymatic oxidation, deuterated levodopa might offer a theoretically less toxic PD treatment (Malmlof et al., 2008). This is because of the deuterium isotope effect. Deuterium substitution at the alpha-carbon of levodopa stabilizes the carbon-nitrogen bond of the amine residue. As a result, deuterated DA derived from deuterated levodopa would be expected to be less susceptible to conversion to DOPAL.
9.2. Anti-oxidants, metal ion chelators, and aldehyde scavengers
The catecholaldehyde hypothesis predicts that anti-oxidants, metal ion chelators, and aldehyde scavengers might be beneficial in neuroprotection of dopaminergic neurons. Anti-oxidant pre-treatment of animals or cells can attenuate or prevent neurotoxic effects of MPTP (Blanchet et al., 2008), MPP+ (Wu et al., 1994), levodopa (Lai & Yu, 1997; Peritore et al., 2012), DA (Lai & Yu, 1997; Wu & Johnson, 2011), and aminochrome (Linsenbardt et al., 2009). Metal ions, especially ions of iron, have long been suspected to participate in the pathogenetic process (Sofic et al., 1988; Ben-Shachar & Youdim, 1993; Youdim et al., 1993; Berg et al., 2001; Gotz et al., 2004; Jomova et al., 2010). This association has rationalized testing of divalent metal ion chelation (Ben-Shachar et al., 1992), an effort that continues (Zheng et al., 2012; Weinreb et al., 2013).
Treatment with aldehyde scavengers such as hydralazine is a possibility being explored (Kaminskas et al., 2004; Burcham et al., 2012). Major challenges in applying these approaches are developing drugs that cross the blood-brain barrier and relatively selectively affect the target cells. Carnosine, a dipeptide consisting of beta-alanine and L-histidine, is an endogenous brain constituent. Carnosine is highly reactive with aldehydes (Xie et al., 2013), forming covalent adducts that are metabolized by aldose reductase (Baba et al., 2013). Carnosine might be neuroprotective (Bellia et al., 2011).
9.3. Combination drugs
Based on the network in Fig. 2, metal cations interact complexly with hydrogen peroxide, quinones, DOPAL, and alpha-synuclein in mediating catecholamine autotoxicity. It has been proposed that a combined approach involving MAO inhibition, anti-oxidant, and iron chelating properties would be superior to an approach targeting any single step in the pathogenetic network (Weinreb et al., 2011; Zheng et al., 2012; Weinreb et al., 2013; Youdim, 2013; Youdim et al., 2014). Considering that antioxidant treatment with ascorbic acid and divalent cation chelation with EDTA attenuate the augmentation by Cu2+ of DOPAL-induced alpha-synuclein oligomerization (Jinsmaa et al., 2014), drugs that interfere with the three-way interaction of DOPAL, alpha-synuclein, and divalent metal cations might constitute a novel approach for future treatment or prevention approaches.
9.4. Compensatory activation and the timing of treatment initiation
As striatal dopaminergic terminals are lost, a variety of compensatory adjustments take place, such as increased DA release from remaining terminals (Snyder et al., 1990; Zigmond et al., 1990; Calne & Zigmond, 1991), decreased reuptake of released DA (Sossi et al., 2007), increased DA synthesis via TH (Loeffler et al., 1995; Bezard et al., 2000), upregulation of post-synaptic DA receptors (Perlmutter et al., 1987; Kaasinen et al., 2000), and increased excitability of the target striatal medium spiny neurons (Azdad et al., 2009). These adaptive changes, which exemplify compensatory activation of alternative effectors in homeostatic negative feedback loops (Goldstein, 2013), maintain dopaminergic functions until the loss of the terminals is advanced. Because of compensatory activation in PD, DA delivery to the extracellular fluid can be maintained despite decreased intra-neuronal stores. It can be shown mathematically that compensatory activation prolongs the time before the disease process manifests clinically.
A potential neurochemical correlate of compensatory activation in PD is the concentration ratio of DA:DOPAC in cerebrospinal fluid (CSF). This is because of likely differential dependence of CSF DA and CSF DOPAC on release and reuptake of DA vs. leakage of vesicular stores into the cytoplasm. Consistent with this view, across individual PD patients, CSF DOPAC is less than expected for CSF DA (Goldstein, Holmes et al., 2012); however, since decreased ALDH activity would be expected to produce the same abnormal pattern, whether CSF DA:DOPAC provides a valid index of compensatory activation in PD remains unproven. Developing means to detect compensatory activation could provide a biomarker by which to decide on appropriate timing for initiation of a neuroprotective strategy.
In the new era of individualized medicine, it may be possible to identify genetic predispositions, where “life counseling” might prevent the death of catecholamine neurons and thereby prevent PD from developing during the person’s lifetime. For instance, ALDH1A1 gene expression is decreased in the blood of patients with PD (Molochnikov et al., 2012), mice with combined ALDH1A1,2 knockout have congenitally increased striatal DOPAL levels and aging-related neurobehavioral and neuropathologic findings mimicking PD (Wey et al., 2012), PD patients have decreased putamen ALDH activity (Goldstein, Sullivan et al., 2011; 2013), and ALDH inhibition augments DOPAL responses of vesicular uptake blockade (Goldstein, Sullivan et al., 2012). From these findings one may predict that in the future people with congenitally low ALDH activity detected by neurochemical testing after early post-natal genetic profiling may be counseled to avoid careers involving high rates of exposure to agents such as pesticides that inhibit ALDH or vesicular uptake. Meanwhile, indirect epidemiological evidence suggests that exercise training might increase resilience against development of PD (Xu et al., 2010; Ahlskog, 2011), although one cannot exclude the possibility that less participation in physical activity is an early manifestation of the disease process. By tracking compensatory activation in presymptomatic individuals, one may be able to best time initiation of neuroprotective treatment, such as with drugs that inhibit MAO, detoxify catecholaldehydes, attenuate generation of reactive oxygen species due to DOPAL auto-oxidation, or interfere with DOPAL-induced oligomerization of alpha-synuclein.
10. Conclusions
Studies over the past several years have yielded evidence that PD is associated with a particular abnormal catecholamine metabolic pattern—decreased vesicular sequestration of cytoplasmic catecholamines and decreased aldehyde dehydrogenase (ALDH) activity which together build up putamen DOPAL. DOPAL generated intracellularly is cytotoxic and oligomerizes alpha-synuclein in catecholaminergic cells, and mice with congenital striatal DOPAL build-up due to ALDH1A1,2 gene knockout have aging-related neurobehavioral and neuropathologic changes mimicking those in PD. These findings are consistent with the catecholaldehyde hypothesis for the pathogenesis of PD and related disorders. Nevertheless, the body of knowledge to date is insufficient to conclude that increased catecholaldehyde production is an essential component of the pathogenic processes. The catecholaldehyde hypothesis therefore remains a hypothesis. Clinical trials based on predictions from the catecholaldehyde hypothesis would help test its value as an explanatory scientific idea.
In conclusion, autotoxicity maybe a common pathogenetic pathway in the death of catecholamine neurons. Pursuit of the findings summarized here may foster development of innovative treatment and disease modification strategies for catecholamine-related diseases such as PD.
Abbreviations
- ALDH
aldehyde dehydrogenase
- AR
aldehyde reductase
- BRA
baroreflex area
- CSF
cerebrospinal fluid
- DA
dopamine
- DHPG
3,4-dihydroxyphenylglycol
- DOPAC
3,4-dihydroxyphenylacetic acid
- DOPAL
3,4-dihydroxyphenylacetaldehyde
- 3, 4-DOPEGAL
dihydroxyphenylglycolaldehyde
- EPI
epinephrine
- 4-HNE
4-hydroxynonenal
- MAO
monoamine oxidase
- LBD
Lewy body dementia
- MSA
multiple system atrophy
- NE
norepinephrine
- OH
orthostatic hypotension
- PAF
pure autonomic failure
- PD
Parkinson disease
- PET
positron emission tomography
- REM
rapid eye movement
- SAS
sympathetic adrenergic system
- SNS
sympathetic noradrenergic system
- TH
tyrosine hydroxylase
- VMAT
vesicular monoamine transporter
Footnotes
Financial support: Writing this review was supported by the Division of Intramural Research, NINDS.
Conflict of interest
The authors affirm that they all have no actual or potential conflict of interest.
References
- Adamczyk A, Kazmierczak A, Strosznajder JB. Alpha-synuclein and its neurotoxic fragment inhibit dopamine uptake into rat striatal synaptosomes. Relationship to nitric oxide. Neurochem Int. 2006;49:407–412. doi: 10.1016/j.neuint.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Adeyemo OM, Youdim MB, Markey SP, Markey CJ, Pollard HB. L-Deprenyl confers specific protection against MPTP-induced Parkinson’s disease-like movement disorder in the goldfish. Eur J Pharmacol. 1993;240:185–193. doi: 10.1016/0014-2999(93)90897-q. [DOI] [PubMed] [Google Scholar]
- Ahlskog JE. Does vigorous exercise have a neuroprotective effect in Parkinson disease? Neurology. 2011;77:288–294. doi: 10.1212/WNL.0b013e318225ab66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DG, Mariappan SV, Buettner GR, Doorn JA. Oxidation of 3,4-dihydroxyphenylacetaldehyde, a toxic dopaminergic metabolite, to a semiquinone radical and an ortho-quinone. J Biol Chem. 2011;286:26978–26986. doi: 10.1074/jbc.M111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azdad K, Chavez M, Don Bischop P, Wetzelaer P, Marescau B, De Deyn PP, et al. Homeostatic plasticity of striatal neurons intrinsic excitability following dopamine depletion. PLoS One. 2009;4:e6908. doi: 10.1371/journal.pone.0006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba SP, Hoetker JD, Merchant M, Klein JB, Cai J, Barski OA, et al. Role of aldose reductase in the metabolism and detoxification of carnosine–acrolein conjugates. J Biol Chem. 2013;288:28163–28179. doi: 10.1074/jbc.M113.504753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeau A. Manganese and extrapyramidal disorders (a critical review and tribute to Dr. George C. Cotzias) Neurotoxicology. 1984;5:13–35. [PubMed] [Google Scholar]
- Bartl J, Muller T, Grunblatt E, Gerlach M, Riederer P. Chronic monoamine oxidase-B inhibitor treatment blocks monoamine oxidase-A enzyme activity. J Neural Transm. 2014;121:379–383. doi: 10.1007/s00702-013-1120-z. [DOI] [PubMed] [Google Scholar]
- Bellia F, Vecchio G, Cuzzocrea S, Calabrese V, Rizzarelli E. Neuroprotective features of carnosine in oxidative driven diseases. Mol Aspects Med. 2011;32:258–266. doi: 10.1016/j.mam.2011.10.009. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D, Eshel G, Riederer P, Youdim MB. Role of iron and iron chelation in dopaminergic-induced neurodegeneration: implication for Parkinson’s disease. Ann Neurol. 1992;(32 Suppl):S105–S110. doi: 10.1002/ana.410320718. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D, Youdim MB. Iron, melanin and dopamine interaction: relevance to Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 1993;17:139–150. doi: 10.1016/0278-5846(93)90038-t. [DOI] [PubMed] [Google Scholar]
- Berg D, Gerlach M, Youdim MB, Double KL, Zecca L, Riederer P, et al. Brain iron pathways and their relevance to Parkinson’s disease. J Neurochem. 2001;79:225–236. doi: 10.1046/j.1471-4159.2001.00608.x. [DOI] [PubMed] [Google Scholar]
- Berman SB, Hastings TG. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. J Neurochem. 1999;73:1127–1137. doi: 10.1046/j.1471-4159.1999.0731127.x. [DOI] [PubMed] [Google Scholar]
- Bezard E, Jaber M, Gonon F, Boireau A, Bloch B, Gross CE. Adaptive changes in the nigrostriatal pathway in response to increased 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurodegeneration in the mouse. Eur J Neurosci. 2000;12:2892–2900. doi: 10.1046/j.1460-9568.2000.00180.x. [DOI] [PubMed] [Google Scholar]
- Bisaglia M, Mammi S, Bubacco L. Kinetic and structural analysis of the early oxidation products of dopamine: analysis of the interactions with alpha-synuclein. J Biol Chem. 2007;282:15597–15605. doi: 10.1074/jbc.M610893200. [DOI] [PubMed] [Google Scholar]
- Bisaglia M, Soriano ME, Arduini I, Mammi S, Bubacco L. Molecular characterization of dopamine-derived quinones reactivity toward NADH and glutathione: implications for mitochondrial dysfunction in Parkinson disease. Biochim Biophys Acta. 2010;1802:699–706. doi: 10.1016/j.bbadis.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Blanchet J, Longpre F, Bureau G, Morissette M, DiPaolo T, Bronchti G, et al. Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1243–1250. doi: 10.1016/j.pnpbp.2008.03.024. [DOI] [PubMed] [Google Scholar]
- Blaschko H. Amine oxidase and amine metabolism. Pharmacol Rev. 1952;4:415–458. [PubMed] [Google Scholar]
- Bronstein J, Carvey P, Chen H, Cory-Slechta D, DiMonte D, Duda J, et al. Meeting report: consensus statement—Parkinson’s disease and the environment: collaborative on health and the environment and Parkinson’s Action Network (CHE PAN) conference 26–28 June 2007. Environ Health Perspect. 2009;117:117–121. doi: 10.1289/ehp.11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR. Metal binding to alpha-synuclein peptides and its contribution to toxicity. Biochem Biophys Res Commun. 2009;380:377–381. doi: 10.1016/j.bbrc.2009.01.103. [DOI] [PubMed] [Google Scholar]
- Burcham PC, Raso A, Kaminskas LM. Chaperone heat shock protein 90 mobilization and hydralazine cytoprotection against acrolein-induced carbonyl stress. Mol Pharmacol. 2012;82:876–886. doi: 10.1124/mol.112.078956. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Chung HD, Li SW. Quantitation of 3,4-dihydroxyphenylacetaldehyde and 3, 4-dihydroxyphenylglycolaldehyde, the monoamine oxidase metabolites of dopamine and noradrenaline, in human tissues by microcolumn high-performance liquid chromatography. Anal Biochem. 1999;273:111–116. doi: 10.1006/abio.1999.4196. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Kumar VB, Pandey N, Panneton WM, Gan Q, Franko MW, et al. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008;115:193–203. doi: 10.1007/s00401-007-0303-9. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Li SW, Williams EA, Nonneman R, Zahm DS. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson’s disease pathogenesis. Brain Res. 2003;989:205–213. doi: 10.1016/s0006-8993(03)03354-7. [DOI] [PubMed] [Google Scholar]
- Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci U S A. 1983;80:4546–4550. doi: 10.1073/pnas.80.14.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns RS, LeWitt PA, Ebert MH, Pakkenberg H, Kopin IJ. The clinical syndrome of striatal dopamine deficiency. Parkinsonism induced by1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) N Engl J Med. 1985;312:1418–1421. doi: 10.1056/NEJM198505303122203. [DOI] [PubMed] [Google Scholar]
- Butcher N, Kiehl T-R, Hazrati L-N, Chow E, Rogaeva D, Lang A, et al. Association between early-onset Parkinson disease and 22q11.2 deletion syndrome. Identification of a novel genetic form of Parkinson disease and its clinical implications. JAMA Neurol. 2013;70:1359–1366. doi: 10.1001/jamaneurol.2013.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Yang M, Poremsky E, Kidd S, Schneider JS, Iacovitti L. Dopaminergic neurons derived from human induced pluripotent stem cells survive and integrate into 6-OHDA-lesioned rats. Stem Cells Dev. 2010;19:1017–1023. doi: 10.1089/scd.2009.0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calne DB, Zigmond MJ. Compensatory mechanisms in degenerative neurologic diseases. Insights from parkinsonism. Arch Neurol. 1991;48:361–363. doi: 10.1001/archneur.1991.00530160025009. [DOI] [PubMed] [Google Scholar]
- Cartier AE, Ubhi K, Spencer B, Vazquez-Roque RA, Kosberg KA, Fourgeaud L, et al. Differential effects of UCHL1 modulation on alpha-synuclein in PD-like models of alpha-synucleinopathy. PLoS One. 2012;7:e34713. doi: 10.1371/journal.pone.0034713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarejos MJ, Solano RM, Menendez J, Rodriguez-Navarro JA, Correa C, Garcia de Yebenes J, et al. Differential effects of l-DOPA on monoamine metabolism, cell survival and glutathione production in midbrain neuronal-enriched cultures from parkin knockout and wild-type mice. J Neurochem. 2005;94:1005–1014. doi: 10.1111/j.1471-4159.2005.03249.x. [DOI] [PubMed] [Google Scholar]
- Cashaw JL. Determination of tetrahydropapaveroline in the urine of parkinsonian patients receiving L-dopa–carbidopa (Sinemet) therapy by high-performance liquid chromatography. J Chromatogr. 1993;613:267–273. doi: 10.1016/0378-4347(93)80141-p. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL, et al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27:8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalieri EL, Li KM, Balu N, Saeed M, Devanesan P, Higginbotham S, et al. Catechol ortho-quinones: the electrophilic compounds that form depurinating DNA adducts and could initiate cancer and other diseases. Carcinogenesis. 2002;23:1071–1077. doi: 10.1093/carcin/23.6.1071. [DOI] [PubMed] [Google Scholar]
- Chen L, Ding Y, Cagniard B, Van Laar AD, Mortimer A, Chi W, et al. Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J Neurosci. 2008;28:425–433. doi: 10.1523/JNEUROSCI.3602-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MK, Kuwabara H, Zhou Y, Adams RJ, Brasic JR, McGlothan JL, et al. VMAT2 and dopamine neuron loss in a primate model of Parkinson’s disease. J Neurochem. 2008;105:78–90. doi: 10.1111/j.1471-4159.2007.05108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba K, Trevor A, Castagnoli N., Jr Metabolism of the neurotoxic tertiary amine, MPTP, by brain monoamine oxidase. Biochem Biophys Res Commun. 1984;120:574–578. doi: 10.1016/0006-291x(84)91293-2. [DOI] [PubMed] [Google Scholar]
- Chu Y, Morfini GA, Langhamer LB, He Y, Brady ST, Kordower JH. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain. 2012;135:2058–2073. doi: 10.1093/brain/aws133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ-1, a cancer-and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci U S A. 2006;103:15091–15096. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colzi A, Musolino A, Iuliano A, Fornai F, Bonuccelli U, Corsini GU. Identification and determination of 3,4-dihydroxyphenylacetaldehyde, the dopamine metabolite in in vivo dialysate from rat striatum. J Neurochem. 1996;66:1510–1517. doi: 10.1046/j.1471-4159.1996.66041510.x. [DOI] [PubMed] [Google Scholar]
- Cookson MR. Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harb Perspect Med. 2012;2:a009415. doi: 10.1101/cshperspect.a009415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini GU, Pintus S, Chiueh CC, Weiss JF, Kopin IJ. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxicity in mice is enhanced by pretreatment with diethyldithiocarbamate. Eur J Pharmacol. 1985;119:127–128. doi: 10.1016/0014-2999(85)90331-0. [DOI] [PubMed] [Google Scholar]
- Cotzias GC. Levodopa in the treatment of Parkinsonism. JAMA. 1971;218:1903–1908. [PubMed] [Google Scholar]
- Creveling CR. The Role of Catechol Quinone Species in Cellular Toxicity. Johnson City, TN: FP Graham Publishing Co.; 2000. [Google Scholar]
- Dahlstrom A, Haggendal J. Axonal transport of amine storage granules in sympathetic adrenergic neurons. Adv Biochem Psychopharmacol. 1970;2:65–93. [PubMed] [Google Scholar]
- Dahlstrom A, Heiwall PO. Intra-axonal transport of transmitters in mammalian neurons. J Neural Transm. 1975;(Suppl. 12):97–114. doi: 10.1007/978-3-7091-8384-7_5. [DOI] [PubMed] [Google Scholar]
- Dahlstrom A, Heiwall PO, Haggendal J, Saunders NR. Effect of antimitotic drugs on the intraaxonal transport of neurotransmitters in rat adrenergic and cholinergic nerves. Ann N Y Acad Sci. 1975;253:507–516. doi: 10.1111/j.1749-6632.1975.tb19224.x. [DOI] [PubMed] [Google Scholar]
- De Iuliis A, Burlina AP, Boschetto R, Zambenedetti P, Arslan P, Galzigna L. Increased dopamine peroxidation in postmortem Parkinsonian brain. Biochim Biophys Acta. 2002;1573:63–67. doi: 10.1016/s0304-4165(02)00331-8. [DOI] [PubMed] [Google Scholar]
- de la Fuente-Fernandez R, Schulzer M, Mak E, Sossi V. Trials of neuroprotective therapies for Parkinson’s disease: problems and limitations. Parkinsonism Relat Disord. 2010;16:365–369. doi: 10.1016/j.parkreldis.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Decressac M, Mattsson B, Lundblad M, Weikop P, Bjorklund A. Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiol Dis. 2012;45:939–953. doi: 10.1016/j.nbd.2011.12.013. [DOI] [PubMed] [Google Scholar]
- DeCuypere M, Lu Y, Miller DD, LeDoux MS. Regional distribution of tetrahydroisoquinoline derivatives in rodent, human, and Parkinson’s disease brain. J Neurochem. 2008;107:1398–1413. doi: 10.1111/j.1471-4159.2008.05709.x. [DOI] [PubMed] [Google Scholar]
- Double KL, Gerlach M, Youdim MB, Riederer P. Impaired iron homeostasis in Parkinson’s disease. J Neural Transm Suppl. 2000:37–58. doi: 10.1007/978-3-7091-6301-6_3. [DOI] [PubMed] [Google Scholar]
- Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Wien Klin Wochenschr. 1960;38:1236–1239. doi: 10.1007/BF01485901. [DOI] [PubMed] [Google Scholar]
- Eiden LE, Schafer MK, Weihe E, Schutz B. The vesicular amine transporter family (SLC18): amine/proton antiporters required for vesicular accumulation and regulated exocytotic secretion of monoamines and acetylcholine. Pflugers Arch. 2004;447:636–640. doi: 10.1007/s00424-003-1100-5. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Goldstein DS, Stull R, Keiser HR, Sunderland T, Murphy DL, et al. Simultaneous liquid-chromatographic determination of 3,4-dihydroxyphenylglycol, catecholamines, and 3,4-dihydroxyphenylalanine in plasma, and their responses to inhibition of monoamine oxidase. Clin Chem. 1986;32:2030–2033. [PubMed] [Google Scholar]
- Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004a;56:331–349. doi: 10.1124/pr.56.3.1. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Kopin IJ, Goldstein DS. Leaky catecholamine stores: undue waste or a stress response coping mechanism? Ann N Y Acad Sci. 2004b;1018:224–230. doi: 10.1196/annals.1296.027. [DOI] [PubMed] [Google Scholar]
- Elsworth JD, Glover V, Reynolds GP, Sandler M, Lees AJ, Phuapradit P, et al. Deprenyl administration in man: a selective monoamine oxidase B inhibitor without the ‘cheese effect’. Psychopharmacology (Berl) 1978;57:33–38. doi: 10.1007/BF00426954. [DOI] [PubMed] [Google Scholar]
- Finberg JP, Pacak K, Kopin IJ, Goldstein DS. Chronic inhibition of monoamine oxidase type A increases noradrenaline release in rat frontal cortex. Naunyn Schmiedebergs Arch Pharmacol. 1993;347:500–505. doi: 10.1007/BF00166742. [DOI] [PubMed] [Google Scholar]
- Finberg JP, Wang J, Goldstein DS, Kopin IJ, Bankiewicz KS. Influence of selective inhibition of monoamine oxidase A or B on striatal metabolism of l-DOPA in hemiparkinsonian rats. J Neurochem. 1995;65:1213–1220. doi: 10.1046/j.1471-4159.1995.65031213.x. [DOI] [PubMed] [Google Scholar]
- Fitzgerald JC, Ugun-Klusek A, Allen G, ADG L, Hargreaves I, Ufer C, et al. Monoamine oxidase-A knockdown in human neuroblastoma cells reveals protection against mitochondrial toxins. FASEB J. 2014;28:218–229. doi: 10.1096/fj.13-235481. [DOI] [PubMed] [Google Scholar]
- Fitzmaurice AG, Rhodes SL, Cockburn M, Ritz B, Bronstein JM. Aldehyde dehydrogenase variation enhances effect of pesticides associated with Parkinson disease. Neurology. 2014;82:419–426. doi: 10.1212/WNL.0000000000000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzmaurice AG, Rhodes SL, Lulla A, Murphy NP, Lam HA, O’Donnell KC, et al. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc Natl Acad Sci USA. 2013;110:636–641. doi: 10.1073/pnas.1220399110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florang VR, Rees JN, Brogden NK, Anderson DG, Hurley TD, Doorn JA. Inhibition of the oxidative metabolism of 3,4-dihydroxyphenylacetaldehyde, a reactive intermediate of dopamine metabolism, by 4-hydroxy-2-nonenal. Neurotoxicology. 2007;28:76–82. doi: 10.1016/j.neuro.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Fornai F, Giorgi FS, Bassi L, Ferrucci M, Alessandri MG, Corsini GU. Modulation of dihydroxyphenylacetaldehyde extracellular levels in vivo in the rat striatum after different kinds of pharmacological treatment Brain Res. 2000;861:126–134. doi: 10.1016/s0006-8993(00)02054-0. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J Neurosci. 1999;19:2424–2431. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis. 2003;14:637–647. doi: 10.1016/j.nbd.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Galvin J, Burke W, Wang J-C, Racette B, Perlmutter J, Goate A. Polymorphisms in aldehyde dehydrogenase and the risk of sporadic Parkinson disease. Parkinsonism Relat Disord. 2009;15(Suppl. 2):S155. [Google Scholar]
- Gao X, Simon KC, Han J, Schwarzschild MA, Ascherio A. Genetic determinants of hair color and Parkinson’s disease risk. Ann Neurol. 2009;65:76–82. doi: 10.1002/ana.21535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler MN, Genc O, Bobela W, Mohanna S, Ardah MT, El-Agnaf OM, et al. Nigrostriatal overabundance of alpha-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 2012;123:653–669. doi: 10.1007/s00401-012-0963-y. [DOI] [PubMed] [Google Scholar]
- Gautam AH, Zeevalk GD. Characterization of reduced and oxidized dopamine and 3,4-dihydrophenylacetic acid, on brain mitochondrial electron transport chain activities. Biochim Biophys Acta. 2011;1807:819–828. doi: 10.1016/j.bbabio.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German DC, Liang CL, Manaye KF, Lane K, Sonsalla PK. Pharmacological inactivation of the vesicular monoamine transporter can enhance 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurodegeneration of midbrain dopaminergic neurons, but not locus coeruleus noradrenergic neurons. Neuroscience. 2000;101:1063–1069. doi: 10.1016/s0306-4522(00)00385-7. [DOI] [PubMed] [Google Scholar]
- Goker-Alpan O, Giasson BI, Eblan MJ, Nguyen J, Hurtig HI, Lee VM, et al. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology. 2006;67:908–910. doi: 10.1212/01.wnl.0000230215.41296.18. [DOI] [PubMed] [Google Scholar]
- Goldstein DS. Catecholamines 101. Clin Auton Res. 2010;20:331–352. doi: 10.1007/s10286-010-0065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS. Concepts of scientific integrative medicine applied to the physiology and pathophysiology of catecholamine systems. Compr Physiol. 2013;3:1569–1610. doi: 10.1002/cphy.c130006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Eisenhofer G, Stull R, Folio CJ, Keiser HR, Kopin IJ. Plasma dihydroxyphenylglycol and the intraneuronal disposition of norepinephrine in humans. J Clin Invest. 1988;81:213–220. doi: 10.1172/JCI113298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Feuerstein G, Izzo JL, Jr, Kopin IJ, Keiser HR. Validity and reliability of liquid chromatography with electrochemical detection for measuring plasma levels of norepinephrine and epinephrine in man. Life Sci. 1981;28:467–475. doi: 10.1016/0024-3205(81)90139-9. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Bentho O, Sato T, Moak J, Sharabi Y, et al. Bio-markers to detect central dopamine deficiency and distinguish Parkinson disease from multiple system atrophy. Parkinsonism Relat Disord. 2008;14:600–607. doi: 10.1016/j.parkreldis.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Kopin IJ, Sharabi Y. Intra-neuronal vesicular uptake of catecholamines is decreased in patients with Lewy body diseases. J Clin Invest. 2011;121:3320–3330. doi: 10.1172/JCI45803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Sharabi Y. Cerebrospinal fluid biomarkers of central catecholamine deficiency in Parkinson’s disease and other synucleinopathies. Brain. 2012;135:1900–1913. doi: 10.1093/brain/aws055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Imrich R, Peckham E, Holmes C, Lopez G, Crews C, et al. Neurocirculatory and nigrostriatal abnormalities in Parkinson disease from LRRK2 mutation. Neurology. 2007;69:1580–1584. doi: 10.1212/01.wnl.0000268696.57912.64. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Sullivan P, Cooney A, Jinsmaa Y, Sullivan R, Gross DJ, et al. Vesicular uptake blockade generates the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde in PC12 Cells: relevance to the pathogenesis of Parkinson disease. J Neurochem. 2012;123:932–943. doi: 10.1111/j.1471-4159.2012.07924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Sullivan P, Holmes C, Kopin IJ, Basile MJ, Mash DC. Catechols in post-mortem brain of patients with Parkinson disease. Eur J Neurol. 2011;18:703–710. doi: 10.1111/j.1468-1331.2010.03246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Sullivan P, Holmes C, Miller GW, Alter S, Strong R, et al. Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J Neurochem. 2013;126:591–603. doi: 10.1111/jnc.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, et al. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology. 1999;20:239–247. [PubMed] [Google Scholar]
- Gotz ME, Double K, Gerlach M, Youdim MB, Riederer P. The relevance of iron in the pathogenesis of Parkinson’s disease. Ann N Y Acad Sci. 2004;1012:193–208. doi: 10.1196/annals.1306.017. [DOI] [PubMed] [Google Scholar]
- Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- Grunblatt E, Zehetmayer S, Jacob CP, Muller T, Jost WH, Riederer P. Pilot study: peripheral biomarkers for diagnosing sporadic Parkinson’s disease. J Neural Transm. 2010;117:1387–1393. doi: 10.1007/s00702-010-0509-1. [DOI] [PubMed] [Google Scholar]
- Guillot TS, Miller GW. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol Neurobiol. 2009;39:149–170. doi: 10.1007/s12035-009-8059-y. [DOI] [PubMed] [Google Scholar]
- Guillot TS, Shepherd KR, Richardson JR, Wang MZ, Li Y, Emson PC, et al. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J Neurochem. 2008;106:2205–2217. doi: 10.1111/j.1471-4159.2008.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JT, Chen AQ, Kong Q, Zhu H, Ma CM, Qin C. Inhibition of vesicular monoamine transporter-2 activity in alpha-synuclein stably transfected SH-SY5Y cells. Cell Mol Neurobiol. 2008;28:35–47. doi: 10.1007/s10571-007-9227-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutteridge JM. Lipid peroxidation initiated by superoxide-dependent hydroxyl radicals using complexed iron and hydrogen peroxide. FEBS Lett. 1984;172:245–249. doi: 10.1016/0014-5793(84)81134-5. [DOI] [PubMed] [Google Scholar]
- Hastings TG. Enzymatic oxidation of dopamine: the role of prostaglandin H synthase. J Neurochem. 1995;64:919–924. doi: 10.1046/j.1471-4159.1995.64020919.x. [DOI] [PubMed] [Google Scholar]
- Hastings TG. The role of dopamine oxidation in mitochondrial dysfunction: implications for Parkinson’s disease. J Bioenerg Biomembr. 2009;41:469–472. doi: 10.1007/s10863-009-9257-z. [DOI] [PubMed] [Google Scholar]
- Hatcher JM, Delea KC, Richardson JR, Pennell KD, Miller GW. Disruption of dopamine transport by DDT and its metabolites. Neurotoxicology. 2008;29:682–690. doi: 10.1016/j.neuro.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher JM, Richardson JR, Guillot TS, McCormack AL, Di Monte DA, Jones DP, et al. Dieldrin exposure induces oxidative damage in the mouse nigrostriatal dopamine system. Exp Neurol. 2007;204:619–630. doi: 10.1016/j.expneurol.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holford NH, Nutt JG. Interpreting the results of Parkinson’s disease clinical trials: time for a change. Mov Disord. 2011;26:569–577. doi: 10.1002/mds.23555. [DOI] [PubMed] [Google Scholar]
- Holz RW. Evidence that catecholamine transport into chromaffin vesicles is coupled to vesicle membrane potential. Proc Natl Acad Sci U S A. 1978;75:5190–5194. doi: 10.1073/pnas.75.10.5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornykiewicz O. Biochemical aspects of Parkinson’s disease. Neurology. 1998;51:S2–S9. doi: 10.1212/wnl.51.2_suppl_2.s2. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. Chemical neuroanatomy of the basal ganglia–normal and in Parkinson’s disease. J Chem Neuroanat. 2001;22:3–12. doi: 10.1016/s0891-0618(01)00100-4. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. Basic research on dopamine in Parkinson’s disease and the discovery of the nigrostriatal dopamine pathway: the view of an eyewitness. Neurodegener Dis. 2008;5:114–117. doi: 10.1159/000113678. [DOI] [PubMed] [Google Scholar]
- Hyndman D, Bauman DR, Heredia VV, Penning TM. The aldo-keto reductase superfamily homepage. Chem Biol Interact. 2003:143–144. 621–631. doi: 10.1016/s0009-2797(02)00193-x. [DOI] [PubMed] [Google Scholar]
- Inzelberg R, Rabey JM, Melamed E, Djaldetti R, Reches A, Badarny S, et al. High prevalence of malignant melanoma in Israeli patients with Parkinson’s disease. J Neural Transm. 2011;118:1199–1207. doi: 10.1007/s00702-011-0580-2. [DOI] [PubMed] [Google Scholar]
- Jackson B, Brocker C, Thompson DC, Black W, Vasiliou K, Nebert DW, et al. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum Genomics. 2011;5:283–303. doi: 10.1186/1479-7364-5-4-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana S, Sinha M, Chanda D, Roy T, Banerjee K, Munshi S, et al. Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: implications in dopamine cytotoxicity and pathogenesis of Parkinson’s disease. Biochim Biophys Acta. 2011;1812:663–673. doi: 10.1016/j.bbadis.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A. 2013;110:E4016–E4025. doi: 10.1073/pnas.1309143110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Jiang Q, Liu W, Feng J. Parkin suppresses the expression of monoamine oxidases. J Biol Chem. 2006;281:8591–8599. doi: 10.1074/jbc.M510926200. [DOI] [PubMed] [Google Scholar]
- Jiang H, Ren Y, Yuen EY, Zhong P, Ghaedi M, Hu Z, et al. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun. 2012;3:668. doi: 10.1038/ncomms1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinsmaa Y, Florang VR, Rees JN, Anderson DG, Strack S, Doorn JA. Products of oxidative stress inhibit aldehyde oxidation and reduction pathways in dopamine catabolism yielding elevated levels of a reactive intermediate. Chem Res Toxicol. 2009;22:835–841. doi: 10.1021/tx800405v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinsmaa Y, Florang VR, Rees JN, Mexas LM, Eckert LL, Allen EM, et al. Dopamine-derived biological reactive intermediates and protein modifications: implications for Parkinson’s disease. Chem Biol Interact. 2011;192:118–121. doi: 10.1016/j.cbi.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinsmaa Y, Sullivan P, Gross D, Cooney A, Sharabi Y, Goldstein DS. Divalent metal ions enhance DOPAL-induced oligomerization of alpha-synuclein. Neurosci Lett. 2014;569:27–32. doi: 10.1016/j.neulet.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen JN, Chiueh CC, Bacon JP, Garrick NA, Burns RS, Weise VK, et al. Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the dog: effect of pargyline pretreatment. J Neurochem. 1989;53:582–589. doi: 10.1111/j.1471-4159.1989.tb07373.x. [DOI] [PubMed] [Google Scholar]
- Jomova K, Vondrakova D, Lawson M, Valko M. Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem. 2010;345:91–104. doi: 10.1007/s11010-010-0563-x. [DOI] [PubMed] [Google Scholar]
- Kaasinen V, Ruottinen HM, Nagren K, Lehikoinen P, Oikonen V, Rinne JO. Upregulation of putaminal dopamine D2 receptors in early Parkinson’s disease: a comparative PET study with [11C] raclopride and [11C]N-methylspiperone. J Nucl Med. 2000;41:65–70. [PubMed] [Google Scholar]
- Kagedal B, Goldstein DS. Catecholamines and their metabolites. J Chromatogr. 1988;429:177–233. doi: 10.1016/s0378-4347(00)83871-2. [DOI] [PubMed] [Google Scholar]
- Kaminskas LM, Pyke SM, Burcham PC. Strong protein adduct trapping accompanies abolition of acrolein-mediated hepatotoxicity by hydralazine in mice. J Pharmacol Exp Ther. 2004;310:1003–1010. doi: 10.1124/jpet.104.067330. [DOI] [PubMed] [Google Scholar]
- Kells AP, Forsayeth J, Bankiewicz KS. Glial-derived neurotrophic factor gene transfer for Parkinson’s disease: anterograde distribution of AAV2 vectors in the primate brain. Neurobiol Dis. 2012;48:228–235. doi: 10.1016/j.nbd.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Kim MN, Lee JJ, Lee MK. Inhibition of dopamine biosynthesis by tetrahydropapaveroline. Neurosci Lett. 2005;386:1–4. doi: 10.1016/j.neulet.2005.04.105. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N Engl J Med. 1988;318:876–880. doi: 10.1056/NEJM198804073181402. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Oikawa S, Kawanishi S. Mechanism of DNA damage and apoptosis induced by tetrahydropapaveroline, a metabolite of dopamine. Neurochem Res. 2006;31:523–532. doi: 10.1007/s11064-006-9044-8. [DOI] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, et al. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CT, Yu PH. Dopamine- and L-beta-3,4-dihydroxyphenylalanine hydrochloride (L-dopa)-induced cytotoxicity towards catecholaminergic neuroblastoma SH-SY5Y cells. Effects of oxidative stress and antioxidative factors. Biochem Pharmacol. 1997;53:363–372. doi: 10.1016/s0006-2952(96)00731-9. [DOI] [PubMed] [Google Scholar]
- Lamensdorf I, Eisenhofer G, Harvey-White J, Hayakawa Y, Kirk K, Kopin IJ. Metabolic stress in PC12 cells induces the formation of the endogenous dopaminergic neurotoxin, 3,4-dihydroxyphenylacetaldehyde. J Neurosci Res. 2000;60:552–558. doi: 10.1002/(SICI)1097-4547(20000515)60:4<552::AID-JNR14>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lamensdorf I, Eisenhofer G, Harvey-White J, Nechustan A, Kirk K, Kopin IJ. 3,4-Dihydroxyphenylacetaldehyde potentiates the toxic effects of metabolic stress in PC12 cells. Brain Res. 2000;868:191–201. doi: 10.1016/s0006-8993(00)02309-x. [DOI] [PubMed] [Google Scholar]
- Larsson PA, Booj S, Lundmark K, Goldstein M, Dahlstrom A. Reserpine-induced effects in the adrenergic neuron as studied with cytofluorimetric scanning. Brain Res Bull. 1986;16:63–74. doi: 10.1016/0361-9230(86)90013-4. [DOI] [PubMed] [Google Scholar]
- Lebouvier T, Clairembault T, Devos D, Pallardy A, Coron E, Neunlist M, et al. Peripheral autonomic nervous system involvement in Gaucher-related parkinsonism. J Parkinsons Dis. 2014;4:29–32. doi: 10.3233/JPD-130333. [DOI] [PubMed] [Google Scholar]
- Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-inspired surface chemistry for multifunctional coatings. Science. 2007;318:426–430. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol. 2010;6:702–706. doi: 10.1038/nrneurol.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legros H, Janin F, Dourmap N, Bonnet JJ, Costentin J. Semi-chronic increase in striatal level of 3,4-dihydroxyphenylacetaldehyde does not result in alteration of nigrostriatal dopaminergic neurones. J Neurosci Res. 2004;75:429–435. doi: 10.1002/jnr.10880. [DOI] [PubMed] [Google Scholar]
- Li SW, Lin TS, Minteer S, Burke WJ. 3,4-Dihydroxyphenylacetaldehyde and hydrogen peroxide generate a hydroxyl radical: possible role in Parkinson’s disease pathogenesis. Brain Res Mol Brain Res. 2001;93:1–7. doi: 10.1016/s0169-328x(01)00120-6. [DOI] [PubMed] [Google Scholar]
- Linsenbardt AJ, Breckenridge JM, Wilken GH, Macarthur H. Dopaminochrome induces caspase-independent apoptosis in the mesencephalic cell line, MN9D. J Neurochem. 2012;122:175–184. doi: 10.1111/j.1471-4159.2012.07756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsenbardt AJ, Wilken GH, Westfall TC, Macarthur H. Cytotoxicity of dopaminochrome in the mesencephalic cell line, MN9D,isdependent upon oxidative stress. Neurotoxicology. 2009;30:1030–1035. doi: 10.1016/j.neuro.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Gao X, Lu Y, Chen H. Meta-analysis of the relationship between Parkinson disease and melanoma. Neurology. 2011;76:2002–2009. doi: 10.1212/WNL.0b013e31821e554e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler DA, LeWitt PA, DeMaggio AJ, Juneau PL, Milbury PE, Matson WR. Markers of dopamine depletion and compensatory response in striatum and cerebrospinal fluid. J Neural Transm Park Dis Dement Sect. 1995;9:45–53. doi: 10.1007/BF02252962. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P. Effect of mutant alpha-synucleinondopamine homeostasis in a new human mesencephalic cell line. J Biol Chem. 2002;277:38884–38894. doi: 10.1074/jbc.M205518200. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad M, Decressac M, Mattsson B, Bjorklund A. Impaired neurotrans-mission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc Natl Acad Sci U S A. 2012;109:3213–3219. doi: 10.1073/pnas.1200575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Dallaglio K, Chen Y, Robinson WA, Robinson SE, McCarter MD, et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells. 2012;30:2100–2113. doi: 10.1002/stem.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki-Ikola O, Heinonen E. Study design problems of DATATOP study analysis. Ann Neurol. 1996;40:946–948. doi: 10.1002/ana.410400624. [DOI] [PubMed] [Google Scholar]
- Malmlof T, Svensson TH, Schilstrom B. Altered behavioural and neurochemical profile of l-DOPA following deuterium substitutions in the molecule. Exp Neurol. 2008;212:538–542. doi: 10.1016/j.expneurol.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Mandel S, Grunblatt E, Riederer P, Amariglio N, Jacob-Hirsch J, Rechavi G, et al. Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin–proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann N Y Acad Sci. 2005;1053:356–375. doi: 10.1196/annals.1344.031. [DOI] [PubMed] [Google Scholar]
- Marchitti SA, Deitrich RA, Vasiliou V. Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev. 2007;59:125–150. doi: 10.1124/pr.59.2.1. [DOI] [PubMed] [Google Scholar]
- Maruyama W, Abe T, Tohgi H, Dostert P, Naoi M. A dopaminergic neurotoxin, (R)-N-methylsalsolinol, increases in Parkinsonian cerebrospinal fluid. Ann Neurol. 1996;40:119–122. doi: 10.1002/ana.410400120. [DOI] [PubMed] [Google Scholar]
- Maruyama W, Dostert P, Matsubara K, Naoi M. N-methyl(R)salsolinol produces hydroxyl radicals: involvement to neurotoxicity. Free Radic Biol Med. 1995;19:67–75. doi: 10.1016/0891-5849(95)00013-n. [DOI] [PubMed] [Google Scholar]
- Mattammal MB, Chung HD, Strong R, Hsu FF. Confirmation of a dopamine metabolite in parkinsonian brain tissue by gas chromatography–mass spectrometry. J Chromatogr. 1993;614:205–212. doi: 10.1016/0378-4347(93)80310-z. [DOI] [PubMed] [Google Scholar]
- Mattammal MB, Haring JH, Chung HD, Raghu G, Strong R. Anendogenous dopaminergic neurotoxin: implication for Parkinson’s disease. Neurodegeneration. 1995;4:271–281. doi: 10.1016/1055-8330(95)90016-0. [DOI] [PubMed] [Google Scholar]
- Mexas LM, Florang VR, Doorn JA. Inhibition and covalent modification of tyrosine hydroxylase by 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite. Neurotoxicology. 2011;32:471–477. doi: 10.1016/j.neuro.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezey E, Dehejia AM, Harta G, Suchy SF, Nussbaum RL, Brownstein MJ, et al. Alpha synuclein is present in Lewy bodies in sporadic Parkinson’s disease. Mol Psychiatry. 1998;3:493–499. doi: 10.1038/sj.mp.4000446. [DOI] [PubMed] [Google Scholar]
- Miller GW, Erickson JD, Perez JT, Penland SN, Mash DC, Rye DB, et al. Immunochemical analysis of vesicular monoamine transporter (VMAT2) protein in Parkinson’s disease. Exp Neurol. 1999;156:138–148. doi: 10.1006/exnr.1998.7008. [DOI] [PubMed] [Google Scholar]
- Miller GW, Wang YM, Gainetdinov RR, Caron MG. Dopamine transporter and vesicular monoamine transporter knockout mice: implications for Parkinson’s disease. Methods Mol Med. 2001;62:179–190. doi: 10.1385/1-59259-142-6:179. [DOI] [PubMed] [Google Scholar]
- Mittermeyer G, Christine CW, Rosenbluth KH, Baker SL, Starr P, Larson P, et al. Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson’s disease. Hum Gene Ther. 2012;23:377–381. doi: 10.1089/hum.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molochnikov L, Rabey JM, Dobronevsky E, Bonucelli U, Ceravolo R, Frosini D, et al. A molecular signature in blood identifies early Parkinson’s disease. Mol Neurodegener. 2012;7:26. doi: 10.1186/1750-1326-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooslehner KA, Chan PM, Xu W, Liu L, Smadja C, Humby T, et al. Mice with very low expression of the vesicular monoamine transporter 2 gene survive into adulthood: potential mouse model for parkinsonism. Mol Cell Biol. 2001;21:5321–5331. doi: 10.1128/MCB.21.16.5321-5331.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Gong LW, Khanna B, Sulzer D, Lindau M. Intracellular patch electrochemistry: regulation of cytosolic catecholamines in chromaffin cells. J Neurosci. 2003;23:5835–5845. doi: 10.1523/JNEUROSCI.23-13-05835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Staal RG, Bove J, Prou D, Hananiya A, Markov D, et al. Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci. 2006;26:9304–9311. doi: 10.1523/JNEUROSCI.0519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatsu T, Sawada M. Molecular mechanism of the relation of monoamine oxidase B and its inhibitors to Parkinson’s disease: possible implications of glial cells. J Neural Transm. 2006;53–65(Suppl) doi: 10.1007/978-3-211-33328-0_7. [DOI] [PubMed] [Google Scholar]
- Naoi M, Nagatsu T. Inhibition of monoamine oxidase by 3,4-dihydroxyphenylserine. J Neurochem. 1986;47:604–607. doi: 10.1111/j.1471-4159.1986.tb04542.x. [DOI] [PubMed] [Google Scholar]
- Nasstrom T, Fagerqvist T, Barbu M, Karlsson M, Nikolajeff F, Kasrayan A, et al. The lipid peroxidation products 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote the formation of alpha-synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radic Biol Med. 2011;50:428–437. doi: 10.1016/j.freeradbiomed.2010.11.027. [DOI] [PubMed] [Google Scholar]
- Navarro-Yepes J, Zavala-Flores L, Anandhan A, Wang F, Skotak M, Chandra N, et al. Antioxidant gene therapy against neuronal cell death. Pharmacol Ther. 2014;142:206–230. doi: 10.1016/j.pharmthera.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura N, Villemagne VL, Drago J, Pejoska S, Dhamija RK, Mulligan RS, et al. In vivo measurement of vesicular monoamine transporter type 2 density in Parkinson disease with (18)F-AV-133. J Nucl Med. 2010;51:223–228. doi: 10.2967/jnumed.109.070094. [DOI] [PubMed] [Google Scholar]
- Olanow CW. Rationale for considering that propargylamines might be neuro-protective in Parkinson’s disease. Neurology. 2006;66:S69–S79. doi: 10.1212/wnl.66.10_suppl_4.s69. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A, et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med. 2009;361:1268–1278. doi: 10.1056/NEJMoa0809335. [DOI] [PubMed] [Google Scholar]
- Paik SR, Shin HJ, Lee JH. Metal-catalyzed oxidation of alpha-synuclein in the presence of Copper(II) and hydrogen peroxide. Arch Biochem Biophys. 2000;378:269–277. doi: 10.1006/abbi.2000.1822. [DOI] [PubMed] [Google Scholar]
- Paik SR, Shin HJ, Lee JH, Chang CS, Kim J. Copper(II)-induced self-oligomerization of alpha-synuclein. Biochem J. 1999;340(Pt 3):821–828. [PMC free article] [PubMed] [Google Scholar]
- Panneton WM, Kumar VB, Gan Q, Burke WJ, Galvin JE. The neurotoxicity of DOPAL: behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS One. 2010;5:e15251. doi: 10.1371/journal.pone.0015251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SS, Schulz EM, Lee D. Disruption of dopamine homeostasis underlies selective neurodegeneration mediated by alpha-synuclein. Eur J Neurosci. 2007;26:3104–3112. doi: 10.1111/j.1460-9568.2007.05929.x. [DOI] [PubMed] [Google Scholar]
- Peritore CS, Ho A, Yamamoto BK, Schaus SE. Resveratrol attenuates L-DOPA-induced hydrogen peroxide toxicity in neuronal cells. Neuroreport. 2012;23:989–994. doi: 10.1097/WNR.0b013e32835a4ea4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter JS, Kilbourn MR, Raichle ME, Welch MJ. MPTP-induced upregulation of in vivo dopaminergic radioligand-receptor binding in humans. Neurology. 1987;37:1575–1579. doi: 10.1212/wnl.37.10.1575. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Rees JN, Florang VR, Anderson DG, Doorn JA. Lipid peroxidation products inhibit dopamine catabolism yielding aberrant levels of a reactive intermediate. Chem Res Toxicol. 2007;20:1536–1542. doi: 10.1021/tx700248y. [DOI] [PubMed] [Google Scholar]
- Rees JN, Florang VR, Eckert LL, Doorn JA. Protein reactivity of 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite, is dependent on both the aldehyde and the catechol. Chem Res Toxicol. 2009;22:1256–1263. doi: 10.1021/tx9000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout HJ, Stefanis L. The neurobiology of LRRK2 and its role in the pathogenesis of Parkinson’s disease. Neurochem Res. 2014;39:576–592. doi: 10.1007/s11064-013-1073-5. [DOI] [PubMed] [Google Scholar]
- Riederer P, Sofic E, Rausch WD, Schmidt B, Reynolds GP, Jellinger K, et al. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem. 1989;52:515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- Riederer P, Youdim MB. Monoamine oxidase activity and monoamine metabolism in brains of parkinsonian patients treated with L-deprenyl. J Neurochem. 1986;46:1359–1365. doi: 10.1111/j.1471-4159.1986.tb01747.x. [DOI] [PubMed] [Google Scholar]
- Rilstone JJ, Alkhater RA, Minassian BA. Brain dopamine-serotonin vesicular transport disease and its treatment. N Engl J Med. 2013;368:543–550. doi: 10.1056/NEJMoa1207281. [DOI] [PubMed] [Google Scholar]
- Sai Y, Wu Q, Le W, Ye F, Li Y, Dong Z. Rotenone-induced PC12 cell toxicity is caused by oxidative stress resulting from altered dopamine metabolism. Toxicol In Vitro. 2008;22:1461–1468. doi: 10.1016/j.tiv.2008.04.019. [DOI] [PubMed] [Google Scholar]
- Sala G, Brighina L, Saracchi E, Fermi S, Riva C, Carrozza V, et al. Vesicular monoamine transporter 2 mRNA levels are reduced in platelets from patients with Parkinson’s disease. J Neural Transm. 2010;117:1093–1098. doi: 10.1007/s00702-010-0446-z. [DOI] [PubMed] [Google Scholar]
- Schinelli S, Zuddas A, Kopin IJ, Barker JL, di Porzio U. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine metabolism and 1-methyl-4-phenylpyridinium uptake in dissociated cell cultures from the embryonic mesencephalon. J Neurochem. 1988;50:1900–1907. doi: 10.1111/j.1471-4159.1988.tb02495.x. [DOI] [PubMed] [Google Scholar]
- Shoulson I. DATATOP: a decade of neuroprotective inquiry. Parkinson Study Group. Deprenyl And Tocopherol Antioxidative Therapy Of Parkinsonism. Ann Neurol. 1998;44:S160–S166. [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Keller RW, Jr, Zigmond MJ. Dopamine efflux from striatal slices after intracerebral 6-hydroxydopamine: evidence for compensatory hyperactivity of residual terminals. J Pharmacol Exp Ther. 1990;253:867–876. [PubMed] [Google Scholar]
- Snyder H, Wolozin B. Pathological proteins in Parkinson’s disease: focus on the proteasome. J Mol Neurosci. 2004;24:425–442. doi: 10.1385/JMN:24:3:425. [DOI] [PubMed] [Google Scholar]
- Sofic E, Riederer P, Heinsen H, Beckmann H, Reynolds GP, Hebenstreit G, et al. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm. 1988;74:199–205. doi: 10.1007/BF01244786. [DOI] [PubMed] [Google Scholar]
- Song BJ, Abdelmegeed MA, Yoo SH, Kim BJ, Jo SA, Jo I, et al. Post-translational modifications of mitochondrial aldehyde dehydrogenase and biomedical implications. J Proteomics. 2011;74:2691–2702. doi: 10.1016/j.jprot.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossi V, de la Fuente-Fernandez R, Schulzer M, Troiano AR, Ruth TJ, Stoessl AJ. Dopamine transporter relation to dopamine turnover in Parkinson’s disease: a positron emission tomography study. Ann Neurol. 2007;62:468–474. doi: 10.1002/ana.21204. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Staal RG, Sonsalla PK. Inhibition of brain vesicular monoamine transporter (VMAT2) enhances 1-methyl-4-phenylpyridinium neurotoxicity in vivo in rat striata. J Pharmacol Exp Ther. 2000;293:336–342. [PubMed] [Google Scholar]
- Stokes AH, Hastings TG, Vrana KE. Cytotoxic and genotoxic potential of dopamine. J Neurosci Res. 1999;55:659–665. doi: 10.1002/(SICI)1097-4547(19990315)55:6<659::AID-JNR1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Storch A, Kaftan A, Burkhardt K, Schwarz J. 1-Methyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (salsolinol) is toxic to dopaminergic neuroblastoma SH-SY5Y cells via impairment of cellular energy metabolism. Brain Res. 2000;855:67–75. doi: 10.1016/s0006-8993(99)02272-6. [DOI] [PubMed] [Google Scholar]
- Storch A, Ott S, Hwang YI, Ortmann R, Hein A, Frenzel S, et al. Selective dopaminergic neurotoxicity of isoquinoline derivatives related to Parkinson’s disease: studies using heterologous expression systems of the dopamine transporter. Biochem Pharmacol. 2002;63:909–920. doi: 10.1016/s0006-2952(01)00922-4. [DOI] [PubMed] [Google Scholar]
- Su Y, Duan J, Ying Z, Hou Y, Zhang Y, Wang R, et al. Increased vulnerability of parkin knock down PC12 cells to hydrogen peroxide toxicity: the role of salsolinol and NM-salsolinol. Neuroscience. 2013;233:72–85. doi: 10.1016/j.neuroscience.2012.12.045. [DOI] [PubMed] [Google Scholar]
- Sun M, Kong L, Wang X, Holmes C, Gao Q, Zhang GR, et al. Coexpression of tyrosine hydroxylase, GTP cyclohydrolase I, aromatic amino acid decarboxylase, and vesicular monoamine transporter 2 from a helper virus-free herpes simplex virus type 1 vector supports high-level, long-term biochemical and behavioral correction of a rat model of Parkinson’s disease. Hum Gene Ther. 2004;15:1177–1196. doi: 10.1089/hum.2004.15.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, et al. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119:866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TN, Alter SP, Wang M, Goldstein DS, Miller GW. Reduced vesicular storage of catecholamines causes progressive degeneration in the locus ceruleus. Neuropharmacology. 2014;76:A97–A105. doi: 10.1016/j.neuropharm.2013.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TN, Caudle WM, Shepherd KR, Noorian A, Jackson CR, Iuvone PM, et al. Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci. 2009;29:8103–8113. doi: 10.1523/JNEUROSCI.1495-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonnesen J. Optogenetic cell control in experimental models of neurological disorders. Behav Brain Res. 2013;255:35–43. doi: 10.1016/j.bbr.2013.07.007. [DOI] [PubMed] [Google Scholar]
- Vasiliou V, Yoshida A, Lindahl R, Holmes R, Hsu LC, Agarwal P, et al. Mammalian aldehyde dehydrogenase (ALDH) genes: recommended nomenclature based on divergent evolution, chromosomal mapping, and human polymorphisms. Pharmacogenetics. 2014 in press. [PubMed] [Google Scholar]
- Vergo S, Johansen JL, Leist M, Lotharius J. Vesicular monoamine transporter 2 regulates the sensitivity of rat dopaminergic neurons to disturbed cytosolic dopamine levels. Brain Res. 2007;1185:18–32. doi: 10.1016/j.brainres.2007.09.028. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a porelike mechanism. Biochemistry. 2002;41:4595–4602. doi: 10.1021/bi0121353. [DOI] [PubMed] [Google Scholar]
- Walsh MJ, Davis VE, Yamanaka Y. Tetrahydropapaveroline: an alkaloid metabolite of dopamine in vitro. J Pharmacol Exp Ther. 1970;174:388–400. [PubMed] [Google Scholar]
- Wang TY, Lee SY, Chen SL, Chen SH, Chu CH, Huang SY, et al. The aldehyde dehydrogenase 2 gene is associated with heroin dependence. Drug Alcohol Depend. 2012;120:220–224. doi: 10.1016/j.drugalcdep.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Wang X, Moualla D, Wright JA, Brown DR. Copper binding regulates intracellular alpha-synuclein localisation, aggregation and toxicity. J Neurochem. 2010;113:704–714. doi: 10.1111/j.1471-4159.2010.06638.x. [DOI] [PubMed] [Google Scholar]
- Watabe M, Nakaki T. Mitochondrial complex I inhibitor rotenone-elicited dopamine redistribution from vesicles to cytosol in human dopaminergic SH-SY5Y cells. J Pharmacol Exp Ther. 2007;323:499–507. doi: 10.1124/jpet.107.127597. [DOI] [PubMed] [Google Scholar]
- Watabe M, Nakaki T. Mitochondrial complex I inhibitor rotenone inhibits and redistributes vesicular monoamine transporter 2 via nitration in human dopaminergic SH-SY5Y cells. Mol Pharmacol. 2008;74:933–940. doi: 10.1124/mol.108.048546. [DOI] [PubMed] [Google Scholar]
- Weiner H. Relationship between 3,4-dihydroxyphenylacetaldehyde levels and tetrahydropapaveroline formation. Alcohol Clin Exp Res. 1978;2:127–131. doi: 10.1111/j.1530-0277.1978.tb04712.x. [DOI] [PubMed] [Google Scholar]
- Weingarten P, Zhou QY. Protection of intracellular dopamine cytotoxicity by dopamine disposition and metabolism factors. J Neurochem. 2001;77:776–785. doi: 10.1046/j.1471-4159.2001.00263.x. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Amit T, Riederer P, Youdim MB, Mandel SA. Neuroprotective profile of the multitarget drug rasagiline in Parkinson’s disease. Int Rev Neurobiol. 2011;100:127–149. doi: 10.1016/B978-0-12-386467-3.00007-8. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Mandel S, Youdim MB, Amit T. Targeting dysregulation of brain iron homeostasis in Parkinson’s disease by iron chelators. Free Radic Biol Med. 2013;62:52–64. doi: 10.1016/j.freeradbiomed.2013.01.017. [DOI] [PubMed] [Google Scholar]
- Wey M, Fernandez E, Martinez PA, Sullivan P, Goldstein DS, Strong R. Neurodegeneration and motor dysfunctioninmice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson’s disease. PLoS One. 2012;7:e31522. doi: 10.1371/journal.pone.0031522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Levey AI, Rajput A, Ang L, Guttman M, Shannak K, et al. Differential changes in neurochemical markers of striatal dopamine nerve terminals in idiopathic Parkinson’s disease. Neurology. 1996;47:718–726. doi: 10.1212/wnl.47.3.718. [DOI] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In vivo demonstration that {alpha}-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JA, Wang X, Brown DR. Unique copper-induced oligomers mediate alpha-synuclein toxicity. FASEB J. 2009;23:2384–2393. doi: 10.1096/fj.09-130039. [DOI] [PubMed] [Google Scholar]
- Wu YN, Johnson SW. Dopamine oxidation facilitates rotenone-dependent potentiation of N-methyl-D-aspartate currents in rat substantia nigra dopamine neurons. Neuroscience. 2011;195:138–144. doi: 10.1016/j.neuroscience.2011.08.041. [DOI] [PubMed] [Google Scholar]
- Wu RM, Mohanakumar KP, Murphy DL, Chiueh CC. Antioxidant mechanism and protection of nigral neurons against MPP+ toxicity by deprenyl (selegiline) Ann N Y Acad Sci. 1994;738:214–221. doi: 10.1111/j.1749-6632.1994.tb21806.x. [DOI] [PubMed] [Google Scholar]
- Xi J, Liu Y, Liu H, Chen H, Emborg ME, Zhang SC. Specification of midbrain dopamine neurons from primate pluripotent stem cells. Stem Cells. 2012;30:1655–1663. doi: 10.1002/stem.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Baba SP, Sweeney BR, Barski OA. Detoxification of aldehydes by histidine-containing dipeptides: from chemistry to clinical implications. Chem Biol Interact. 2013;202:288–297. doi: 10.1016/j.cbi.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Park Y, Huang X, Hollenbeck A, Blair A, Schatzkin A, et al. Physical activities and future risk of Parkinson disease. Neurology. 2010;75:341–348. doi: 10.1212/WNL.0b013e3181ea1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa K, Izumi Y, Takeuchi H, Yamamoto N, Kume T, Akaike A, et al. Dopamine facilitates alpha-synuclein oligomerization in human neuroblastoma SH-SY5Y cells. Biochem Biophys Res Commun. 2010;391:129–134. doi: 10.1016/j.bbrc.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Yao L, Fan P, Arolfo M, Jiang Z, Olive MF, Zablocki J, et al. Inhibition of aldehyde dehydrogenase-2 suppresses cocaine seeking by generating THP, a cocaine use-dependent inhibitor of dopamine synthesis. Nat Med. 2010;16:1024–1028. doi: 10.1038/nm.2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdim MB. Multi target neuroprotective and neurorestorative anti-Parkinson and anti-Alzheimer drugs ladostigil and m30 derived from rasagiline. Exp Neurobiol. 2013;22:1–10. doi: 10.5607/en.2013.22.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdim MB, Ben-Shachar D, Eshel G, Finberg JP, Riederer P. The neurotoxicity of iron and nitric oxide. Relevance to the etiology of Parkinson’s disease. Adv Neurol. 1993;60:259–266. [PubMed] [Google Scholar]
- Youdim MB, Edmondson D, Tipton KF. The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci. 2006;7:295–309. doi: 10.1038/nrn1883. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Kupershmidt L, Amit T, Weinreb O. Promises of novel multi-target neuroprotective and neurorestorative drugs for Parkinson’s disease. Parkinsonism Relat Disord. 2014;20(Suppl. 1):S132–S136. doi: 10.1016/S1353-8020(13)70032-4. [DOI] [PubMed] [Google Scholar]
- Zheng H, Amit T, Bar-Am O, Fridkin M, Youdim MB, Mandel SA. From anti-Parkinson’s drug rasagiline to novel multitarget iron chelators with acetylcholinesterase and monoamine oxidase inhibitory and neuroprotective properties for Alzheimer’s disease. J Alzheimers Dis. 2012;30:1–16. doi: 10.3233/JAD-2012-120013. [DOI] [PubMed] [Google Scholar]
- Zhou ZD, Lim TM. Dopamine (DA) induced irreversible proteasome inhibition via DA derived quinones. Free Radic Res. 2009;43:417–430. doi: 10.1080/10715760902801533. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci. 1990;13:290–296. doi: 10.1016/0166-2236(90)90112-n. [DOI] [PubMed] [Google Scholar]
- Zuddas A, Corsini GU, Schinelli S, Barker JL, Kopin IJ, di Porzio U. Acetaldehyde directly enhances MPP+ neurotoxicity and delays its elimination from the striatum. Brain Res. 1989;501:11–22. doi: 10.1016/0006-8993(89)91021-4. [DOI] [PubMed] [Google Scholar]