

Abstract
A systems biology approach was used to comprehensively examine the impact of renal disease and hemodialysis (HD) on patient response during critical illness. To achieve this we examined the metabolome, proteome, and transcriptome of 150 patients with critical illness, stratified by renal function. Quantification of plasma metabolites indicated greater change as renal function declined, with the greatest derangements in patients receiving chronic HD. Specifically, 6 uremic retention molecules, 17 other protein catabolites, 7 modified nucleosides, and 7 pentose phosphate sugars increased as renal function declined, consistent with decreased excretion or increased catabolism of amino acids and ribonucleotides. Similarly, the proteome showed increased levels of low-molecular weight proteins and acute phase reactants. The transcriptome revealed a broad-based decrease in mRNA levels among patients on HD. Systems integration revealed an unrecognized association between plasma RNASE1 and several RNA catabolites and modified nucleosides. Further, allantoin, N1-methyl-4-pyridone-3-carboxamide, and n-acetylaspartate were inversely correlated with the majority of significantly down-regulated genes. Thus, renal function broadly affected the plasma metabolome, proteome, and peripheral blood transcriptome during critical illness; changes not effectively mitigated by hemodialysis. These studies allude to several novel mechanisms whereby renal dysfunction contributes to critical illness.
Keywords: acute kidney injury, chronic kidney disease, gene expression, hemodialysis, sepsis
Introduction
Sepsis, defined as the Systemic Inflammatory Response Syndrome (SIRS) due to infection, is a major cause and consequence of acute and chronic kidney disease.1, 2 Hemodialysis (HD) can be used to mitigate renal failure although it is not a perfect substitute. Some metabolites and low-molecular weight proteins accumulate despite HD.1, 3 These analytes have not been comprehensively characterized and so their relationships to renal dysfunction, morbidity, and death are not fully known.
Recent methodological improvements in metabolome, proteome and transcriptome profiling have enabled comprehensive characterization of molecular perturbations in disease. Integrating ‘omic data can generate novel hypotheses of disease pathophysiology. Moreover, integrative metabolomic, proteomic and transcriptomic analyses can internally validate molecular findings.4 When combined with qualitative clinical phenotypes, integrative ‘omic studies can improve diagnostic, prognostic, and therapeutic strategies.4-9 However, in order to avoid false discoveries, these technologies must be applied to well-defined clinical populations with deep and accurate phenotype data with variance decomposition to control for unsuspected sources of variability.4, 6, 8, 10, 11
We utilized such an approach to analyze the Community Acquired Pneumonia and Sepsis Outcome Diagnostic (CAPSOD) study. This observational study was designed to comprehensively identify blood metabolites, proteins, and transcriptional changes differentiating early community-acquired sepsis from non-infectious SIRS and to differentiate 28-day survivors from non-survivors at the time of initial presentation.4 Interestingly, CAPSOD study analysis identified renal function as a major determinant of acute host response to sepsis and SIRS in survivors and non-survivors. We have therefore more fully investigated the associations of the peripheral blood metabolome, proteome, and transcriptome with renal function in patients at the time they present to the Emergency Department with sepsis or other SIRS-associated illnesses. The results presented herein provide a complete observational analysis of host response to critical illness in those developing acute kidney injury (AKI) or who were on chronic HD.
Results
Patient demographics and Clinical Characteristics
A previously analyzed 150-subject cohort from the CAPSOD study (Figure 1) identified renal dysfunction as the greatest contributor to variance in the plasma metabolome (explaining 44% of total variance).4 We therefore stratified these 150 subjects by renal functional category in order to explore renal-dependent systems biology. The Acute Kidney Injury Network (AKIN) criteria were used to classify renal dysfunction in those subjects who were not receiving chronic hemodialysis.12 The four categories were AKI0 (no significant increase in serum creatinine; n=65), AKI1 (serum creatinine increase of ≥ 0.3 mg/dl, or 150% to 200% above baseline; n=41), AKI2/3 (serum creatinine increase of more than 200% above baseline, or ≥ 4.0 mg/dl with an acute increase of at least 0.5mg/dl; n=20), and chronic hemodialysis (HD; n=24) (Table 1). AKIN Stage 2 and 3 were combined for this analysis due to small subject number. All HD patients were on dialysis prior to study enrollment. Acute Physiology and Chronic Health Evaluation II (APACHE II) scores, a measure of acute illness correlating with mortality, were highest in AKI2/3 (mean 22.8 ± 8.3) and lowest in AKI0 (mean 13.8 ± 7.5). Mean age was similar across AKI categories, ranging from 51.6 to 67.4 years. The majority of subjects in each group were black (range 55-92%). For those with sepsis, the frequency of infectious agents was similar among the four renal function groups with the exception of S. aureus infections, which were higher in the HD group. Mortality was highest among patients with AKI2/3 (40%) and lowest among patients receiving chronic HD (12.5%).
Figure 1.
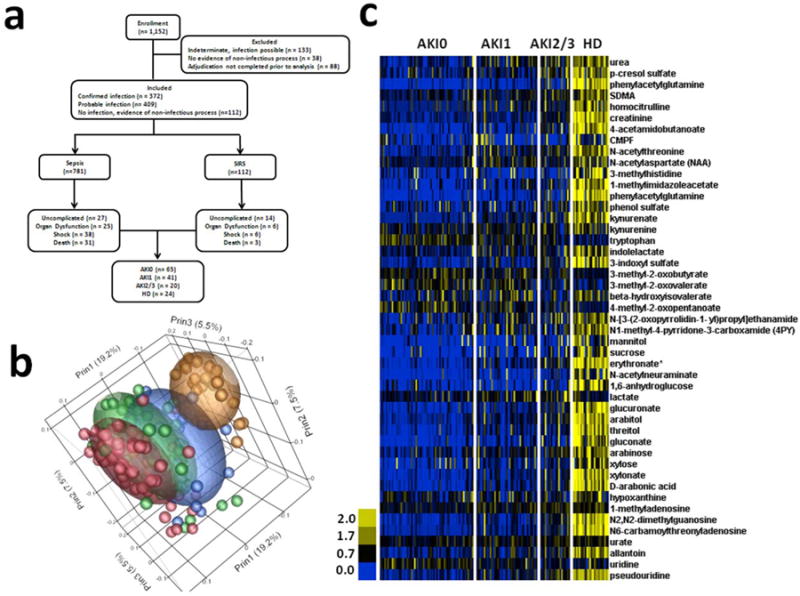
a) CONSORT diagram for the analyzed cohort. b) PCA with Pearson's product-moment correlation. AKI0 (Red, n = 65); AKI1 (Green, n = 41); AKI2/3 (Blue, n = 20); HD (gold, n = 24). c) Cell plot of representative significant metabolomic changes.
Table 1. Patient Demographics.
| Acute Kidney Injury Stage | Stage 0 | Stage 1 | Stage 2/3 | HD |
|---|---|---|---|---|
| n | 65 | 41 | 20 | 24 |
| APACHEII | 13.8 ± 7.5 | 18.5 ± 7.9 | 22.8 ± 8.3 | 17.7 ± 5.0 |
| S. aureus | 20.0% | 9.8% | 10.0% | 33.3% |
| S. pneumoniae | 21.5% | 19.5% | 35.0% | 8.3% |
| E. coli | 12.3% | 14.6% | 5.0% | 4.2% |
| Other Etiologic Agent1 | 12.3% | 22.0% | 20.0% | 33.3% |
| Unidentified Etiologic Agent | 10.8% | 12.2% | 25.0% | 4.2% |
| No Infection | 23.1% | 22.0% | 5.0% | 16.7% |
| Death | 20.0% | 29.3% | 40.0% | 12.5% |
| Age | 59.6 ± 17.7 | 64.9 ± 16.6 | 67.4 ± 18.6 | 51.6 ± 12.1 |
| Gender (male) | 50.8% | 61.0% | 45.0% | 62.5% |
| Race (B/W/O) | 40/23/2 | 27/11/3 | 11/6/3 | 22/2/0 |
| Liver disease | 6.2% | 7.3% | 20.0% | 4.2% |
| Heart failure | 6.2% | 9.8% | 15.0% | 4.2% |
| Chronic lung disease | 30.8% | 29.3% | 30.0% | 20.8% |
| Malignancy | 13.8% | 19.5% | 5.0% | 8.3% |
Includes all identified etiologic agents other than S. aureus, S. pneumoniae, or E. coli
Data presented as mean ± standard deviation
Black, white, other (B, W, O)
Plasma metabolomic perturbations in kidney dysfunction and HD
Mass spectrometry (MS) was used to measure the levels of 370 plasma metabolites in 150 CAPSOD subjects with SIRS.4 Two hundred and forty-one of these metabolites were annotated. Clinical assays of serum creatinine, capillary lactate and serum glucose correlated well with log-transformed, normalized plasma MS values4, indicating that the MS assays were semiquantitative. Unsupervised principal components analysis (PCA) by Pearson's moment-correlation demonstrated primary segregation by renal function category (Figure 1B). Therefore, a global assessment of the plasma metabolome associated with renal function was undertaken. The group without evidence of AKI (AKI0) was defined as the reference group. Fifty-eight percent of metabolites were significantly different (−log10 p ≥ 2.0) in at least one comparison with the reference group [ANOVA with 1% false discovery rate (FDR) correction].13, 14 Most of these differences reflected elevated plasma metabolite levels in subjects with acutely impaired renal function (Figure 1C, Table S1). Furthermore, the pattern of change across groups appeared to be additive, with greater deviations in metabolite concentrations and number of affected metabolites in those with deteriorating renal function. In particular, although HD is designed to remove toxic metabolites, the vast majority of metabolites were elevated in patients with impaired renal function, and remained significantly increased in the chronic HD group (151 of 183 significantly different metabolites were increased compared to AKI0; Table S1). These results showed the profound influence of renal function on the plasma metabolome in patients with acute SIRS-associated illness, with the most pronounced effect in those who were receiving chronic hemodialysis.
We sought to characterize renal function-dependent changes in biochemical pathways among subjects with SIRS. The metabolome can be conceptually divided into four major biochemical categories: protein, carbohydrate, nucleic acid and fatty acid metabolism. As expected, significant increases in metabolites involved in protein catabolism and the urea cycle were observed in the groups with impaired renal function. Nine primary amino acids differed significantly between the AKI groups, and all but cysteine and lysine were decreased in HD patients (Table S1). The decrease in primary amino acids may have reflected increased protein catabolism which has been noted in both renal failure and sepsis.1, 15 Among patients receiving HD, the decrease may also be due to dialysis itself. In contrast, 33 of 39 (85%) significantly different amino acid derivatives (primarily catabolites) were elevated in AKI2/3 and HD patients. Urea, the principal product of protein catabolism, causes acute systemic toxicity if not cleared. Plasma urea concentrations were elevated 1.9-, 2.3-, and 3.5-fold in the AKI1, AKI2/3, and HD groups, respectively (when compared to the AKI0 group). There were also stepwise increases in the concentrations of six other known uremic retention substances with decreasing renal function. For example, there was a 19.5-fold increase in phenylacetylglutamine and a 12-fold increase in 4-acetamidobutanoate in the HD group versus the AKI0 group. Phenylacetylglutamine is involved in an alternative nitrogen elimination pathway16, whereas 4-acetamidobutanoate is a precursor of the urea cycle component ornithine. The uremic retention biochemicals P-cresol sulfate, 3-indoxyl sulfate, 3-carboxy-4-methyl-5-propyl-2-furanpropanoate (CMPF), and N1-methyl-4-pyrridone-3-carboxamide (4PY) also showed stepwise increases in concentrations as renal function declined.17 An additional 17 protein catabolites showed the same association with renal function. The increase in amino acid derivatives may be the result of increased utilization of proteins and amino acids as energy substrates, or the kidney's inability to eliminate nitrogenous waste products during renal failure, or both.
The effect of renal function on the levels of intermediates involved in energy metabolism was assessed. The concentrations of most common sugars were unchanged with respect to renal function. Notable exceptions included increased levels of sucrose and maltose. Notably, 17 nucleic acid catabolites (glucuronate, arabitol, threitol, gluconate, arabinose, xylose, xylonate, D-arabonic acid, erythronate, hypoxanthine, 1-methyladenosine, N2, N2-dimethylguanosine, N-6-carbamoylthreonyladenosine, urate, allantoin, pseudouridine, and phosphate) showed stepwise increases in plasma concentrations with decreasing renal function. These are discussed further below.
Measures of fatty acid oxidation, such as ketone bodies (3-hydroxybutyrate and acetoacetate) and free fatty acids, showed very little change with renal function, suggesting there to be no significant dysfunction in glycolysis or excess β-oxidation in relation to impaired renal function in patients with SIRS. However, there were marked increases in the metabolically inert sugar mannitol in patients with impaired renal function. Mannitol has well-known associations with elevated osmotic pressure and renal injury.18, 19 Mannitol levels were 2.8-fold higher in AKI2/3 and 11-fold higher in HD compared to AKI0. These increases were not due to exogenous administration of mannitol, and therefore appear to represent endogenous production.
Plasma proteomic perturbations in kidney dysfunction and HD
Although plasma protein homeostasis is largely regulated by the liver and other extra-renal organs and tissues, the normal kidney plays a prominent role in low molecular weight protein (LMWP) excretion and reabsorption.1 The plasma proteome in different renal function categories was therefore investigated. One-hundred-sixty-four proteins were identified, defined by the detection of two or more unique peptide sequences at <10% false-discovery rate.4 Of these, 46 (28%) showed significant differences between patients in different renal function categories (ANOVA with 5% FDR, Table S2). The majority (34/46; 74%) were increased in AKI2/3 and HD as compared to AKI0 (Figure 2 and Table S2). The changes were primarily in the complement cascade, the kallikrein-kinin pathway, acute phase reactants, and the majority of LMWPs (Table S2). Among the affected LMWPs, some have previously been implicated in renal failure such as β-2 microglobulin (B2M), cystatin C (CST3), and lipocalin 2 (LCN2).3, 20
Figure 2. Significant Differences in the Proteome.
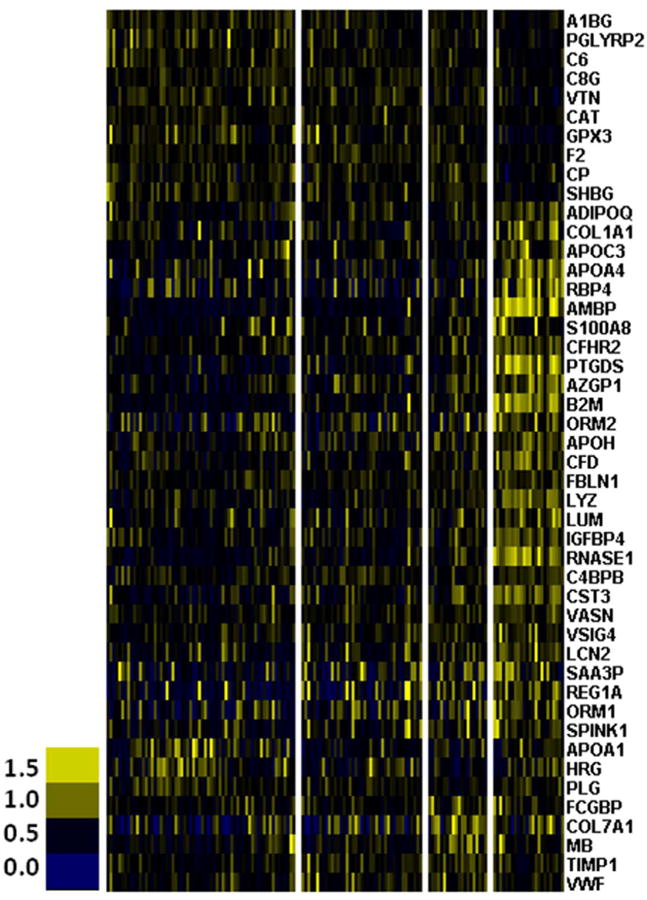
Cell plot for significant proteomic changes due to decreasing renal function. ANOVA with 5% FDR.
Blood transcriptome perturbations in kidney dysfunction and HD
The role of renal function on the blood transcriptome in patients with SIRS was investigated by mRNA sequencing and digital gene expression in 133 of the 150 subjects. Samples from 17 subjects were of insufficient quality for inclusion. Transcripts of 1,997 genes were differentially expressed across all groups (ANOVA with 1% FDR, Figure 3 and Table S3). Most of these (1,950 of 1,997 genes) differed between the HD and AKI0 groups. Moreover, 98.5% were decreased in HD. We observed similar total white blood cell counts (WBC) and differential WBC counts in the four AKI/HD categories, with the exception of AKI2/3 compared to AKI0, where both total WBC and neutrophil percent were significantly higher after Bonferroni correction (p-value = 0.0012 and 0.0164, respectively) (Table S4). This difference did not, however, account for most of the alterations in transcript abundance since only 96 genes had differential expression in AKI2/3 as compared to AKI0. Eighty (83.3%) were increased and 16 were decreased in the AKI2/3 group (Figure 3). Ingenuity Pathway Analysis of the 1,997 differentially expressed genes across AKI/HD groups showed enrichment for 55 canonical pathways (-log10 p-value ≥ 1.40; Figure 3B; Table S5). These featured dermatan sulfate biosynthesis, p38 MAPK signaling, chrondroitin sulfate biosynthesis, heparin sulfate biosynthesis, and fatty acid oxidation.
Figure 3. Significant transcriptomic differences.

Heatmap with Pearson's moment-correlations for significantly different gene expression due to decreasing renal function (ANOVAs with 5% FDR correction). 1,997 genes were significantly different from the reference group, AKI0. IPA for top 10 canonical pathways affected by AKI/HD for b) 1,997 significantly different transcripts, c) 1,058 genes correlated with allantoin, d) 949 genes correlated with 4PY and e) 916 genes correlated with NAA.
Integrative analysis of metabolome, proteome, and transcriptome
Integration of multiple, orthogonal ‘omic datasets has the potential to unveil and internally validate biological pathways of particular importance to a disease state that may go unrecognized in single datasets. We previously reported an integrated analysis of the proteome and metabolome in these patients.4 That analysis was notable for the significant positive correlations between plasma ribonuclease A family 1 (RNASE1) protein concentration and multiple RNA catabolites (seven pentose phosphate sugar metabolites and 7 modified nucleosides). RNASE1 is secreted into plasma by vascular endothelial cells and is maintained at a concentration of 300–400 μg/L.21 It is stored in the Weibel-Palade-bodies of endothelial cells, and is released upon stimulation with prothrombotic or proinflammatory agents like thrombin and vascular endothelial growth factor. Plasma RNASE1 acts to hydrolyze both extracellular single stranded and double stranded RNA released as a result of cell damage or associated with viral infection. When stratified by renal dysfunction, the strongest association between RNASE1 and these RNA catabolites was in patients with AKI2/3 and HD. Specifically, the levels of RNASE1 protein, sugar metabolites (glucuronate, arabitol, threitol, gluconate, arabinose, xylose, and xylonate), and modified nucleosides (hypoxanthine, 1-methyladenosine, N2, N2-dimethylguanosine, N-6-carbamoylthreonyladenosine, urate, allantoin, and pseudouridine) were significantly higher in patients with AKI2/3 or HD than in AKI0 controls (Table S1 and S2). This presumably reflects decreased renal clearance or increased production of RNASE1 as a consequence of SIRS, exacerbated by AKI.
An integrated analysis of the metabolome and transcriptome was also performed to define additional pathways and associations affected by renal function. We performed cross-correlation analysis of the 1,997 significantly different transcripts with the values of 215 significantly different metabolites in 133 matched patient samples. We found that 8,477 correlations were significant (−log10 p-value ≥ 4.4) and further assessed them for biologically plausible gene expression-metabolite interactions (Table S6). Eighty percent (6749/8477) of the significant correlations were accounted for by just 11 metabolites (Table 2).
Table 2. Metabolites associated with the greatest number of significantly different transcripts.
| Metabolites | # of Correlated Genes | Fraction of Total Significant Correlations (5637 total correlations) | Fraction of Significant Associations (1700 total genes) |
|---|---|---|---|
| X-04498 | 751 | 0.133 | 0.442 |
| N1-methyl-4-pyridone-3-carboxamide (4PY) | 742 | 0.132 | 0.436 |
| allantoin | 616 | 0.109 | 0.362 |
| n-acetylaspartate | 400 | 0.071 | 0.235 |
| homocitrulline | 351 | 0.062 | 0.206 |
| tryptophan | 213 | 0.038 | 0.125 |
| 3-methylhistidine | 205 | 0.036 | 0.121 |
| X-12556 | 172 | 0.031 | 0.101 |
| pseudouridine | 139 | 0.025 | 0.082 |
| urea | 131 | 0.023 | 0.077 |
| n-acetylalanine | 114 | 0.020 | 0.067 |
| X-12688 | 114 | 0.020 | 0.067 |
| 1-5-anhydroglucatol | 113 | 0.020 | 0.066 |
| 2-hydroxyglutarate | 98 | 0.017 | 0.058 |
| X-13553 | 76 | 0.013 | 0.045 |
| indolelactate | 64 | 0.011 | 0.038 |
| caproate | 61 | 0.011 | 0.036 |
| kynurenine | 61 | 0.011 | 0.036 |
| methylcysteine | 50 | 0.008 | 0.029 |
| arabitol | 42 | 0.007 | 0.025 |
Four metabolites, allantoin, N1-methyl-4-pyrridone-3-carboxamide (4PY), n-acetylaspartate (NAA), and the GC/MS unidentified metabolite X-04498 accounted for the greatest number of significant gene expression-metabolite correlations (1,058, 949, 916, and 906, respectively). Allantoin is an end-stage degradation product of purine catabolism produced via peroxidation of uric acid and protein degradation.22 4PY, a degradation product of nicotinamide-adenine dinucleotide (NAD), is a uremic retention molecule that can inhibit poly (ADP-ribose) polymerase (PARP-1) activity.23, 24 NAA is the second most abundant metabolite found in neuronal tissue and is thought to function as a neurotransmitter as well as a reservoir for acetyl-CoA and acetate.25 Reduced levels of NAA in the brain are seen in brain injury and have been linked to uremic encephalopathy.26 X-04498 is a metabolite identified by GC/MS whose spectral signature has been recurrently observed in human plasma and serum but whose structural information is unknown.
Ingenuity Pathway Analysis for the gene expression changes associated with allantoin (1,058 transcripts), 4PY (949 transcripts) and NAA (916 transcripts) was performed. Allantoin was associated with 68 enriched canonical pathways (-log10 p-value ≥ 1.40; Figure 3C; Table S7) related to p38 MAPK signaling, P2Y purigenic receptor signaling, and inflammation. Expression of RNA, autophagy, and n-glycosylation of protein were the top disease and function categories (-log10 p-value = 3.5×1010, 2.4 ×105, 7.7×105, respectively; Table S8). For 4PY, 47 canonical pathways were significantly enriched (-log10 p-value ≥ 1.40; Figure 3D; Table S9). Most of the pathways related to inflammatory signaling, hypoxia, and p38 MAPK signaling. The disease and function pathways enriched among 4PY-associated gene expression differences were similar to those observed for allantoin Table S10). Likewise, NAA-associated gene expression differences demonstrated similar changes to the canonical (Figure 3E; Table S11) and disease and function pathways (Table S12) observed with allantoin and 4PY.
Discussion
Renal dysfunction is associated with high morbidity, mortality, and healthcare costs.1, 2 Numerous studies have described metabolic, protein, or gene transcription changes in renal disease although, to our knowledge, this is the first comprehensive and integrated ‘omic analysis performed concomitantly in a single cohort of patients.
In patients with AKI, many derangements in the alternative nitrogen processing pathways and urea metabolism were noted that provide a comprehensive picture of the molecular biology attendant to renal dysfunction during critical illness. In these patients, plasma levels of seven primary amino acids were decreased, whereas those of 33 amino-acid catabolites were increased. Two of the latter were related to alternative nitrogen processing pathways, including phenylacetylglutamine, which was elevated 19.5-fold16; and 4-acetamidobutanoate, which was elevated 12-fold. Four urea retention molecules (P-cresol sulfate, 3-indoxyl sulfate, CMPF, and 4PY) were also increased.17 Interestingly, most of the amino-acid catabolites were also elevated in the HD group. Thus, both the urea cycle and alternative nitrogen-waste pathways appear to be perturbed in AKI during SIRS-associated illness. Moreover, HD fails to correct these perturbations. This observation may be clinically significant since amino-acid derivatives and protein degradation products have been implicated in the generation of uremic symptoms and can be ameliorated by low-protein diets.1 Additional studies are warranted to explore the utility of amino acid catabolites as markers of hemodialysis efficacy and subsequent optimization during acute concurrent illness.
Several of the amino-acid catabolites that differed in renal dysfunction have also been implicated in endothelial dysfunction. Of the 33 amino-acid derivatives that were increased in association with worsening renal function, two were phenyl compounds associated with endothelial dysfunction (phenylacetylglutamine and P-cresol sulfate).27-29 In sepsis, endothelial dysfunction promotes thrombus formation in the microvasculature, which contributes to the development of AKI.30 Similarly, endothelial impairment also occurs during CKD progression.31 Phenylacetylglutamine and P-cresol sulfate elevations have been identified in several different animal models of chronic renal dysfunction. These substances are toxic to endothelial cells and have been associated with increased risk of cardiac complications in the setting of renal dysfunction.27-29 Although increased levels of these metabolites are thought to be the result of worsening renal function, recent studies suggest they increase early in disease and may instead increase the risk of renal disease progression.32 Thus they may serve both as novel biomarkers for early renal dysfunction during acute illness and therapeutic targets in the prevention of renal disease progression.
Overproduction of reactive oxygen species (ROS) is an established mediator of renal damage both in sepsis30 and several types of CKD, mediated by endothelial injury as well as glomerular and tubular cell damage.33, 34 Several urea retention compounds and alternative nitrogen pathway metabolites (including 3-indoxyl sulfate, CMPF, and P-cresol) were increased in patients with renal dysfunction and have been shown to increase ROS production.35-37 For example, 3-indoxyl sulfate is an antioxidant at physiologic levels, but at high concentrations, it enhances intercellular ROS production in endothelial cells.38 The generation of ROS appears to promote acute vascular damage through inflammation, and proliferation of smooth muscle cells.39 Thus, 3-indoxyl sulfate and related compounds may mediate some of the vascular injury and increased cardiovascular morbidity and mortality associated with renal disease. They represent potential therapeutic targets to improve survival in patients with sepsis-associated renal dysfunction.
Endothelial dysfunction is not the sole cause of renal disease progression. Progressive tubular dysfunction and injury is a hallmark of many types of AKI including sepsis-associated AKI40 and also plays an important role in worsening CKD.33, 34 Mannitol, which was significantly increased in AKI2/3 (2.8-fold) and HD groups (11.8-fold), has been implicated in tubular toxicity. In renal ischemia, mannitol can improve renal blood flow.41 However, ischemia does not contribute significantly to sepsis-mediated AKI. Moreover, mannitol leads to tubular injury at high osmotic pressure.18, 19, 42-44 Mannitol regulates renal transcription indirectly via changes in osmolality that affect the osmosensitive transcription factor, nuclear factor of activated T cells 5 (NFAT5).45 Thus, mannitol elevation in AKI and HD may represent an allostatic response exacerbated by SIRS-associated illnesses, such as septic shock, and may indirectly contribute both to endothelial and tubular injury. The failure of HD to remove mannitol suggests that additional studies are warranted to explore its tubular toxicity, and its potential as a therapeutic target in AKI.
To better understand the proteome during AKI in SIRS, we performed concomitant mass spectrometry of plasma proteins4 and categorized those changes based on degree of renal dysfunction. Twenty-eight percent of identified plasma proteins were significantly increased in patients with declining renal function. The changes were primarily noted in the complement cascade, the kallikrien-kinin pathway, acute phase reactants, and LMWPs. These pathways have all been consistently implicated in various studies of renal disease progression.46-49 These perturbations correlate with renal disease severity and align with published studies associating these proteins/pathways with renal dysfunction.1
Concomitant analysis of the blood transcriptome showed that nearly all of the 1,997 differentially expressed genes were down-regulated in HD compared to AKI0. Although this broad-based decrease could be interpreted as a non-specific transcriptional repression, there were a number of biologically relevant pathways manifest within this broad decrease. These down-regulated genes were principally involved in p38 MAPK signaling, ERK, cell cycle regulation, immune function, and metabolism pathways1, 50, but not apoptosis.51 Rather, we hypothesize the circulating leukocyte population is unable to respond to pro-inflammatory signals despite the systemic inflammatory response. This is supported by studies demonstrating T-cell dysregulation in patients with ESRD.52, 53
Combinations of transcriptome, proteome and metabolome data may establish multi-dimensional molecular models of complex diseases that can provide insights into network responses to perturbation.4 One notable example, particularly in the AKI0 and HD groups, focused on protein. This protein is stored in vascular endothelial cells and secreted upon stimulation with prothrombotic or proinflammatory agents like thrombin or vascular endothelial growth factor. Cross correlating plasma RNASE1 concentrations with the metabolome revealed strong associations with metabolites known to cause endothelial cell injury: phenylacetylglutamine, P-cresol sulfate, and mannitol.27-29 We speculate the accumulation of such metabolites leads to endothelial cell injury and a subsequent release of RNASE1 into the circulation. Moreover, RNASE1 mitigates vascular injury by hydrolyzing extracellular RNA (eRNA) released as a result of cell damage or in association with viral infection.54 For example, RNASE1 was cardioprotective in remote ischemic preconditioning by reducing the levels of eRNA and TNF-α in arterial blood.55 This relationship is also supported in our data whereby plasma RNASE1 levels are highly correlated with RNA degradation products. In summary, SIRS, particularly in patients on chronic HD, leads to increased concentrations of endothelial-toxic metabolites (Figure 4). This stimulates RNASE1 release, which degrades eRNA resulting in increased RNA degradation products and an overall compensatory decrease in inflammation. This decreased inflammation may in part be responsible for the decreased gene expression observed in chronic HD patients.
Figure 4. Integrative model of declining renal function.
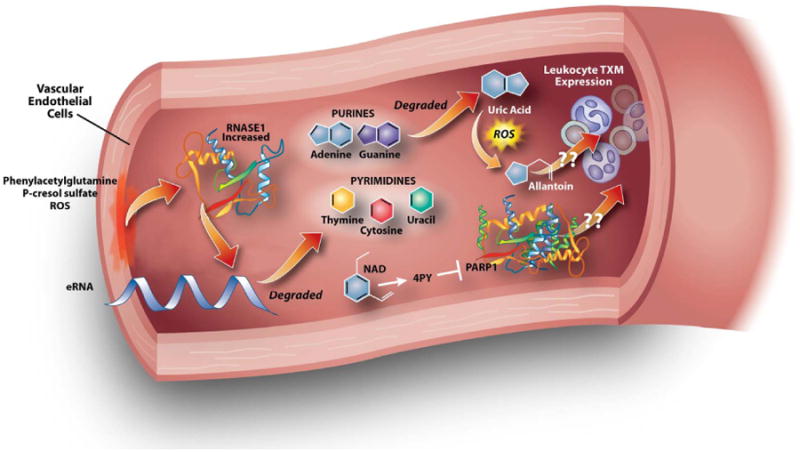
In patients with ESRD receiving HD, we hypothesized phenyacetylglutamine, p-cresol sulfate, and ROS may increase vascular endothelial injury and subsequent release of RNASE1, which then degreades eRNA. Purines are degraded into uric acid. Since humans lack functional uricase, a free-radical, non-enzymatic oxidation converts uric acid to allantoin. Increased 4PY, a degradation product of NAD, inhibits PARP1, which along with allantoin may decrease leukocyte gene transcription. These interactions between 4PY, allantoin, renal function, and gene expression are hypothetical.
Although almost all differentially expressed genes were found to be down-regulated in the HD group compared to controls, standard pathway analyses failed to identify discrete potential mechanisms responsible for this down-regulation. An appealing hypothesis was, however, that the increased metabolites observed in declining renal function states could have either direct or indirect effects on leukocyte metabolic response. They could also be leukocyte-toxic leading to global transcriptional suppression. To test this hypothesis, we performed cross-correlation of the significant metabolites and transcripts which revealed moderate correlations (Pearson's product-moment correlation coefficient -0.35 to -0.53). Within these, a few metabolites accounted for a majority of the correlations. Specifically, allantoin, 4PY, and NAA correlated with the greatest number of significantly different transcripts. 4PY is an end-stage NAD degradation product and a uremic retention molecule.23, 24 4PY is also the substrate for 4-pyridone-3-carboximide-1-β-d-ribonucleoside triphosphate (4PYTP), a novel NAD nucleotide, which has been identified as a biomarker of NAD-induced lymphocyte toxicity in children with renal failure.56 Another possible inhibitory role of 4PY in PARP1 activity has been described.24 PARP1 regulates transcription through epigenetic histone modification and transcription factor regulation.57, 58 Accordingly, we hypothesize that 4PY inhibits PARP1 activity, resulting in decreased gene expression (Figure 4). Ingenuity Pathway Analysis of genes altered by 4PY predicted that a network composed of carbohydrate metabolism, cellular function and maintenance, and RNA post-transcriptional modification was a top affected network with 30 of 35 genes within the network significantly downregulated in HD as compared to AKI0 (Figure S1). Interestingly, PARP1 was also identified as a key transcriptional regulatory target within this network. While the hypothesis is speculative, it suggests interesting metabolite/gene interactions that may alter transcription within leukocytes during renal failure. Another metabolite highly associated with decreased gene expression was allantoin, a uric acid catabolite generated by free radicals.59 Allantoin elevation in ESRD and patients receiving HD has been reported.60 Although allantoin has no known transcriptional regulatory effects in humans, we observed a strong correlation with decreased gene expression in peripheral blood. Ingenuity Pathway Analysis of genes affected by allantoin suggested that P2Y Purigenic Receptor signaling was one of the top altered canonical pathways. Dysregulated purine catabolism could lead to decreased response to inflammatory signals and globally decreased transcription. This speculation is supported by the observation that uric acid, the precursor of allantoin, plays a role in the body's inflammatory response including T-cell stimulation.61, 62 We also noted a strong association between NAA and many of the downregulated genes. NAA has been previously identified as a marker of uremic encephalopathy.26 However, NAA's relationship to transcriptome dysregulation remains unclear. This analysis also revealed that gene expression differences between HD and AKI0 were not explained entirely by cross-correlation analysis, presenting an opportunity for future research into other underlying mechanisms.
There were several limitations of this study. The effects of renal function on the plasma metabolome, proteome, and blood transcriptome were limited to patients with sepsis and other acute SIRS-associated illnesses. A component of the host response to SIRS and sepsis is increased protein catabolism and nucleotide degradation. For example, muscle wasting of 25% can occur in the first week of sepsis-related ICU care.15 Despite the relatively large and heterogeneous sample size, the patients included in this cohort are not necessarily representative of all patients with AKI, especially those without an acute concomitant illness leading to SIRS. It is also unclear how chronic kidney disease would affect a patient's systems biology. Therefore, the generalizability of our findings to renal function in other acute and chronic illnesses will require testing.
Another limitation was that phlebotomy was performed at the time patients presented at the Emergency Department with acute illness, and was not synchronized or normalized to the time since most recent dialysis session in patients receiving HD. It is assumed the plasma metabolome, proteome, and blood transcriptome differ before and after HD. However, we did not observe significant variability within the HD population that would suggest a dominant effect of time-to-HD on plasma metabolite levels. Furthermore, phlebotomy occurred before any subjects were initiated on continuous veno-venous HD (CVVHD). Therefore, we cannot comment on the effects of this form of dialysis. Among patients receiving HD, it was only those who were chronically HD-dependent that were included in this study. Patients who developed a need for HD during their hospitalization were not included as a function of study design, since samples were only collected in the first 24 hours of hospitalization. Therefore, we cannot extrapolate the impact of acute vs. chronic HD on these systems biology changes. Finally, as with any association study, we cannot assume causality. However, known biological pathways and published reports do inform likely mechanisms of interaction. Moreover, better understanding of the pathophysiology will help improve these interpretations and guide targeted experiments to interrogate implicated pathways.
This study has several notable strengths. It presents comprehensive metabolomic, proteomic, and transcriptomic profiles related to renal function in SIRS/sepsis patients. We utilized integrative ‘omics to identify potentially causal interactions for both metabolomic dysfunctions and renal toxicity. Furthermore, since renal function greatly influences outcomes in sepsis and trauma, understanding this complicated interaction may lead to better therapeutic management with protocols tailored to a dynamic fluid-phase blood environment. For example, the results presented herein identified key molecules and pathways associated with renal dysfunction, offering opportunities for targeted intervention. Viewed in the context of known pathobiology, these molecules and pathways offer likely explanations for the high morbidity and mortality in HD patients. Moreover, these same molecules and pathways should serve as diagnostic and therapeutic targets to improve upon HD-related health and longevity.
Methods
Subject Enrollment and Cohort Selection
The Community Acquired Pneumonia and Sepsis Outcome Diagnostics (CAPSOD) study11 (ClinicalTrials.gov NCT00258869) was approved by the Institutional Review Boards of the National Center for Genome Resources (Santa Fe, NM), Duke University Medical Center (Durham, NC), Durham Veterans Affairs Medical Center (Durham, NC), and Henry Ford Health Systems (Detroit, MI). Demographic and clinical information for the 150-subject cohort were obtained as previously described.4, 11 Renal function categories were based on the Acute Kidney Injury Network system12: AKI0 (no significant increase in serum creatinine; n=64), AKI1 (serum creatinine increase of ≥ 0.3 mg/dl, or 150% to 200% from patient's baseline; n=42), AKI2/3 (serum creatinine increase of more than 200% from baseline, or ≥ 4.0 mg/dl with an acute increase of at least 0.5mg/dl; n=20), and hemodialysis (HD; n=24). As there were only two subjects meeting AKI2 criteria, they were included with AKI3 subjects to create the one category. When pre-enrollment baseline values were unavailable, we used creatinine measurements obtained upon convalescence in order to calculate AKI stage.
Metabolome, Proteome, and Transcriptome Measurements
Metabolomic and proteomic analyses was performed previously4 and reanalyzed for changes dependent on renal function. Details regarding how the metabolome and proteome were measured are provided in an online data supplement.
The transcriptome was analyzed for renal function-dependent changes. Whole blood RNA was isolated using PaxGene Blood RNA kit (Qiagen) according to manufacturer's instructions. mRNA sequencing libraries were prepared from total RNA according to Illumina's mRNA-seq Sample Prep Protocol v2.0. Briefly, Random-primed cDNA was synthesized and fragments were 3′ adenylated. Illumina DNA oligonucleotide sequencing adapters were ligated and 350-500bp fragments were selected by gel electrophoresis. cDNA sequencing libraries were amplified by 18 cycles of PCR and quality was assessed with Bioanalyzer. cDNA libraries were stored at -20°C, and were sequenced on the IlluminaGAIIx instruments (54-cycle singleton reads) as previously described.63 Base calling used the Illumina Pipeline software v1.4, except for 14 samples which used v1.3. Results were aligned to the NCBI human nuclear genome reference build GRCh37/hg19 using GSNAP and Alpheus.64 Uniquely aligned reads were enumerated on a RefSeq gene-by-gene basis and expressed as aligned reads per million. Sparse gene expression (<50% of the patients have reported read counts) were removed leaving a total of 18,618 genes expressed within the cohort. Expression data has been deposited in Gene Expression Omnibus (to be provided prior to publication).
Statistical Analyses
Overlaid kernel density estimates, univariate distribution results, correlation coefficients of pair wise sample comparisons, unsupervised principal components analysis (by Pearson product-moment correlation) and Ward hierarchal clustering of Pearson product-moment correlations were performed using log2+1 transformed data as described10 with JMP Genomics 5.0 (SAS Institute Inc., Cary, NC, USA). Four renal function categories were used for analysis: AKI0, AKI1, AKI2/3, and HD. AKI0 defined the reference standard for ANOVA, assuming a 1-5% FDR correction, as noted.13, 14 Normalized data was visualized (cell plots) with Java Treeview.65 Continuous variables are presented as mean ± standard deviation unless otherwise noted. Metabolite-by-transcript interactions were identified using global cross correlation analysis.4
Ingenuity Pathway Analysis (Ingenuity Systems, Inc., Redwood, CA) was performed per manufacturer's instructions on the 1,997 significantly different genes identified, as well as genes significantly associated with allantoin, 4PY, and NAA. Canonical pathways as well as Disease and Function pathways with −log10 p ≥ 1.4 were considered significantly enriched.
Supplementary Material
Acknowledgments
We thank the study subjects. The manuscript is dedicated in memory of Melanie Mohney. Tom Gagliano performed some of the graphic arts. The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs.
Funding Sources: This work was supported in part by grants from NIH (U01AI066569, P20RR016480, HHSN266200400064C), Pfizer Inc., and Roche Diagnostics Inc. ELT and this research were also supported by Award Number 1IK2CX000530 from the Clinical Science Research and Development Service of the VA Office of Research and Development.
Footnotes
Disclosure: All authors report no relevant relationships as it pertains to the work contained in the manuscript with the following exceptions:
RPM and JM are employees of Metabolon, Inc., a fee-for-service metabolomics research services and metabolite-based diagnostics profiling company, and, as such, have affiliations with or financial involvement with Metabolon, Inc. These authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
CBC served as a scientific consultant to BioMerieux. This relationship is unrelated to the work presented in this manuscript.
VGF has grants from the NIH, MedImmune, Forest/Cerexa, Pfizer, Merck, Advanced Liquid Logics, Theravance, Novartis, and Cubist. He served as the Chair of the Merck scientific advisory board for the V710 S. aureus vaccine. He has been a consultant for Pfizer, Novartis, Galderma, Novadigm, Durata, Achaogen, Affinium, Medicines Co., Cerexa, Trius, MedImmune, Bayer, Theravance, and Cubist. He has patents pending for work that is not presented in this manuscript. He also received royalties from UpToDate and has been paid for the development of educational presentations for Cubist, Cerexa, and Theravance. These relationships are unrelated to the work presented in this manuscript.
CWW has consulted for Becton Dickinson, bioMerieux; has received grants from Roche Molecular and Qiagen. These relationships are unrelated to the work presented in this manuscript.
Supplementary information is available at Kidney International's website.
References
- 1.Meyer TW, Hostetter TH. Uremia. N Engl J Med. 2007;357:1316–1325. doi: 10.1056/NEJMra071313. [DOI] [PubMed] [Google Scholar]
- 2.Himmelfarb J, Ikizler TA. Hemodialysis. N Engl J Med. 2010;363:1833–1845. doi: 10.1056/NEJMra0902710. [DOI] [PubMed] [Google Scholar]
- 3.Vanholder R, De Smet R, Glorieux G, et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003;63:1934–1943. doi: 10.1046/j.1523-1755.2003.00924.x. [DOI] [PubMed] [Google Scholar]
- 4.Langley RJ, Tsalik EL, Velkinburgh JC, et al. An integrated clinico-metabolomic model improves prediction of death in sepsis. Science translational medicine. 2013;5:195ra195. doi: 10.1126/scitranslmed.3005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tenenbaum JD, Christian V, Cornish MA, et al. The MURDOCK Study: a long-term initiative for disease reclassification through advanced biomarker discovery and integration with electronic health records. American journal of translational research. 2012;4:291–301. [PMC free article] [PubMed] [Google Scholar]
- 6.Tsalik EL, Jaggers LB, Glickman SW, et al. Discriminative value of inflammatory biomarkers for suspected sepsis. J Emerg Med. 2012;43:97–106. doi: 10.1016/j.jemermed.2011.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn SH, Tsalik EL, Cyr DD, et al. Gene expression-based classifiers identify Staphylococcus aureus infection in mice and humans. PLoS One. 2013;8:e48979. doi: 10.1371/journal.pone.0048979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsalik EL, Jones D, Nicholson B, et al. Multiplex PCR to diagnose bloodstream infections in patients admitted from the emergency department with sepsis. J Clin Microbiol. 2010;48:26–33. doi: 10.1128/JCM.01447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaas AK, Burke T, Chen M, et al. A host-based RT-PCR gene expression signature to identify acute respiratory viral infection. Science translational medicine. 2013;5:203ra126. doi: 10.1126/scitranslmed.3006280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mudge J, Miller NA, Khrebtukova I, et al. Genomic convergence analysis of schizophrenia: mRNA sequencing reveals altered synaptic vesicular transport in post-mortem cerebellum. PLoS ONE. 2008;3:e3625. doi: 10.1371/journal.pone.0003625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glickman SW, Cairns CB, Otero RM, et al. Disease progression in hemodynamically stable patients presenting to the emergency department with sepsis. Acad Emerg Med. 2010;17:383–390. doi: 10.1111/j.1553-2712.2010.00664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mehta RL, Kellum JA, Shah SV, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Critical care. 2007;11:R31. doi: 10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storey JD. A direct approach to false discovery rates. Royal Statistical Society: Series B (Statistical Methodology) 2002;64:479–498. [Google Scholar]
- 14.Storey JD, Taylor JE, Siegmund D. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach. Journal of teh Royal Statistical Society: Series B (Statistical Methodology) 2004;66:187–205. [Google Scholar]
- 15.Puthucheary ZA, Rawal J, McPhail M, et al. Acute skeletal muscle wasting in critical illness. JAMA: the journal of the American Medical Association. 2013;310:1591–1600. doi: 10.1001/jama.2013.278481. [DOI] [PubMed] [Google Scholar]
- 16.Brusilow SW. Phenylacetylglutamine may replace urea as a vehicle for waste nitrogen excretion. Pediatr Res. 1991;29:147–150. doi: 10.1203/00006450-199102000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Lesaffer G, De Smet R, Lameire N, et al. Intradialytic removal of protein-bound uraemic toxins: role of solute characteristics and of dialyser membrane. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2000;15:50–57. doi: 10.1093/ndt/15.1.50. [DOI] [PubMed] [Google Scholar]
- 18.Dorman HR, Sondheimer JH, Cadnapaphornchai P. Mannitol-induced acute renal failure. Medicine (Baltimore) 1990;69:153–159. doi: 10.1097/00005792-199005000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Visweswaran P, Massin EK, Dubose TD., Jr Mannitol-induced acute renal failure. J Am Soc Nephrol. 1997;8:1028–1033. doi: 10.1681/ASN.V861028. [DOI] [PubMed] [Google Scholar]
- 20.Park JS, Kim GH, Kang CM, et al. Application of cystatin C reduction ratio to high-flux hemodialysis as an alternative indicator of the clearance of middle molecules. Korean J Intern Med. 2010;25:77–81. doi: 10.3904/kjim.2010.25.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gansler J, Preissner KT, Fischer S. Influence of proinflammatory stimuli on the expression of vascular ribonuclease 1 in endothelial cells. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2014;28:752–760. doi: 10.1096/fj.13-238600. [DOI] [PubMed] [Google Scholar]
- 22.Johnson RJ, Lanaspa MA, Gaucher EA. Uric acid: a danger signal from the RNA world that may have a role in the epidemic of obesity, metabolic syndrome, and cardiorenal disease: evolutionary considerations. Seminars in nephrology. 2011;31:394–399. doi: 10.1016/j.semnephrol.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rutkowski B, Slominska E, Szolkiewicz M, et al. N-methyl-2-pyridone-5-carboxamide: a novel uremic toxin? Kidney international Supplement. 2003:S19–21. doi: 10.1046/j.1523-1755.63.s84.36.x. [DOI] [PubMed] [Google Scholar]
- 24.Slominska EM, Kowalik K, Smolenski RT, et al. Accumulation of poly(ADP-ribose) polymerase inhibitors in children with chronic renal failure. Pediatric nephrology. 2006;21:800–806. doi: 10.1007/s00467-006-0072-z. [DOI] [PubMed] [Google Scholar]
- 25.Moffett JR, Arun P, Ariyannur PS, et al. N-Acetylaspartate reductions in brain injury: impact on post-injury neuroenergetics, lipid synthesis, and protein acetylation. Frontiers in neuroenergetics. 2013;5:11. doi: 10.3389/fnene.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tryc AB, Alwan G, Bokemeyer M, et al. Cerebral metabolic alterations and cognitive dysfunction in chronic kidney disease. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2011;26:2635–2641. doi: 10.1093/ndt/gfq729. [DOI] [PubMed] [Google Scholar]
- 27.Vanholder R, Van Laecke S, Glorieux G. What is new in uremic toxicity? Pediatric nephrology. 2008;23:1211–1221. doi: 10.1007/s00467-008-0762-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jourde-Chiche N, Dou L, Cerini C, et al. Vascular incompetence in dialysis patients--protein-bound uremic toxins and endothelial dysfunction. Semin Dial. 2011;24:327–337. doi: 10.1111/j.1525-139X.2011.00925.x. [DOI] [PubMed] [Google Scholar]
- 29.Scholze A, Jankowski V, Henning L, et al. Phenylacetic acid and arterial vascular properties in patients with chronic kidney disease stage 5 on hemodialysis therapy. Nephron Clinical practice. 2007;107:c1–6. doi: 10.1159/000105137. [DOI] [PubMed] [Google Scholar]
- 30.Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011;22:999–1006. doi: 10.1681/ASN.2010050484. [DOI] [PubMed] [Google Scholar]
- 31.El Nahas M. The global challenge of chronic kidney disease. Kidney Int. 2005;68:2918–2929. doi: 10.1111/j.1523-1755.2005.00774.x. [DOI] [PubMed] [Google Scholar]
- 32.Niewczas MA, Sirich TL, Mathew AV, et al. Uremic solutes and risk of end-stage renal disease in type 2 diabetes: metabolomic study. Kidney Int. 2014 doi: 10.1038/ki.2013.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Small DM, Coombes JS, Bennett N, et al. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology. 2012;17:311–321. doi: 10.1111/j.1440-1797.2012.01572.x. [DOI] [PubMed] [Google Scholar]
- 34.Small DM, Bennett NC, Roy S, et al. Oxidative stress and cell senescence combine to cause maximal renal tubular epithelial cell dysfunction and loss in an in vitro model of kidney disease. Nephron Experimental nephrology. 2012;122:123–130. doi: 10.1159/000350726. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe H, Miyamoto Y, Honda D, et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013;83:582–592. doi: 10.1038/ki.2012.448. [DOI] [PubMed] [Google Scholar]
- 36.Dou L, Jourde-Chiche N, Faure V, et al. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. Journal of thrombosis and haemostasis: JTH. 2007;5:1302–1308. doi: 10.1111/j.1538-7836.2007.02540.x. [DOI] [PubMed] [Google Scholar]
- 37.Niwa T. Removal of protein-bound uraemic toxins by haemodialysis. Blood purification. 2013;35 Suppl 2:20–25. doi: 10.1159/000350843. [DOI] [PubMed] [Google Scholar]
- 38.Yu M, Kim YJ, Kang DH. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clinical journal of the American Society of Nephrology: CJASN. 2011;6:30–39. doi: 10.2215/CJN.05340610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujii H, Nakai K, Fukagawa M. Role of oxidative stress and indoxyl sulfate in progression of cardiovascular disease in chronic kidney disease. Therapeutic apheresis and dialysis: official peer-reviewed journal of the International Society for Apheresis, the Japanese Society for Apheresis, the Japanese Society for Dialysis Therapy. 2011;15:125–128. doi: 10.1111/j.1744-9987.2010.00883.x. [DOI] [PubMed] [Google Scholar]
- 40.Gomez H, Ince C, De Backer D, et al. A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock. 2014;41:3–11. doi: 10.1097/SHK.0000000000000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bragadottir G, Redfors B, Ricksten SE. Mannitol increases renal blood flow and maintains filtration fraction and oxygenation in postoperative acute kidney injury: a prospective interventional study. Critical care. 2012;16:R159. doi: 10.1186/cc11480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diringer MN, Zazulia AR. Osmotic therapy: fact and fiction. Neurocrit Care. 2004;1:219–233. doi: 10.1385/NCC:1:2:219. [DOI] [PubMed] [Google Scholar]
- 43.Dickenmann M, Oettl T, Mihatsch MJ. Osmotic nephrosis: acute kidney injury with accumulation of proximal tubular lysosomes due to administration of exogenous solutes. Am J Kidney Dis. 2008;51:491–503. doi: 10.1053/j.ajkd.2007.10.044. [DOI] [PubMed] [Google Scholar]
- 44.Fang L, You H, Chen B, et al. Mannitol is an independent risk factor of acute kidney injury after cerebral trauma: a case-control study. Renal failure. 2010;32:673–679. doi: 10.3109/0886022X.2010.486492. [DOI] [PubMed] [Google Scholar]
- 45.Kuper C, Beck FX, Neuhofer W. NFAT5 contributes to osmolality-induced MCP-1 expression in mesothelial cells. Mediators of inflammation. 2012;2012:513015. doi: 10.1155/2012/513015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsu SI, Couser WG. Chronic progression of tubulointerstitial damage in proteinuric renal disease is mediated by complement activation: a therapeutic role for complement inhibitors? J Am Soc Nephrol. 2003;14:S186–191. doi: 10.1097/01.asn.0000070032.58017.20. [DOI] [PubMed] [Google Scholar]
- 47.Kakoki M, Smithies O. The kallikrein-kinin system in health and in diseases of the kidney. Kidney Int. 2009;75:1019–1030. doi: 10.1038/ki.2008.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van den Brand JA, Hofstra JM, Wetzels JF. Low-molecular-weight proteins as prognostic markers in idiopathic membranous nephropathy. Clinical journal of the American Society of Nephrology: CJASN. 2011;6:2846–2853. doi: 10.2215/CJN.04020411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balla S, Nusair MB, Alpert MA. Risk factors for atherosclerosis in patients with chronic kidney disease: recognition and management. Current opinion in pharmacology. 2013;13:192–199. doi: 10.1016/j.coph.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 50.Turner CM, Arulkumaran N, Singer M, et al. Is the inflammasome a potential therapeutic target in renal disease? BMC nephrology. 2014;15:21. doi: 10.1186/1471-2369-15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hotchkiss RS, Tinsley KW, Swanson PE, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. Journal of immunology. 2001;166:6952–6963. doi: 10.4049/jimmunol.166.11.6952. [DOI] [PubMed] [Google Scholar]
- 52.Lisowska KA, Debska-Slizien A, Jasiulewicz A, et al. Influence of hemodialysis on circulating CD4(low)CD25 (high) regulatory T cells in end-stage renal disease patients. Inflammation research: official journal of the European Histamine Research Society [et al] 2014;63:99–103. doi: 10.1007/s00011-013-0679-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Betjes MG, Langerak AW, van der Spek A, et al. Premature aging of circulating T cells in patients with end-stage renal disease. Kidney Int. 2011;80:208–217. doi: 10.1038/ki.2011.110. [DOI] [PubMed] [Google Scholar]
- 54.Barrabes S, Pages-Pons L, Radcliffe CM, et al. Glycosylation of serum ribonuclease 1 indicates a major endothelial origin and reveals an increase in core fucosylation in pancreatic cancer. Glycobiology. 2007;17:388–400. doi: 10.1093/glycob/cwm002. [DOI] [PubMed] [Google Scholar]
- 55.Cabrera-Fuentes HA, Niemann B, Grieshaber P, et al. RNase1 as a potential mediator of remote ischaemic preconditioning for cardioprotection. European journal of cardio-thoracic surgery: official journal of the European Association for Cardio-thoracic Surgery. 2015 doi: 10.1093/ejcts/ezu519. [DOI] [PubMed] [Google Scholar]
- 56.Synesiou E, Fairbanks LD, Simmonds HA, et al. 4-Pyridone-3-carboxamide-1-beta-D-ribonucleoside triphosphate (4PyTP), a novel NAD metabolite accumulating in erythrocytes of uremic children: a biomarker for a toxic NAD analogue in other tissues? Toxins. 2011;3:520–537. doi: 10.3390/toxins3060520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes & development. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 58.Krishnakumar R, Kraus WL. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Molecular cell. 2010;39:736–749. doi: 10.1016/j.molcel.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meotti FC, Jameson GN, Turner R, et al. Urate as a physiological substrate for myeloperoxidase: implications for hyperuricemia and inflammation. The Journal of biological chemistry. 2011;286:12901–12911. doi: 10.1074/jbc.M110.172460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Causse E, Ribes D, Longlune N, et al. Aminothiols and allantoin in chronic dialysis patients: effects of hemodialysis sessions. Clinical nephrology. 2010;73:51–57. doi: 10.5414/cnp73051. [DOI] [PubMed] [Google Scholar]
- 61.Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–521. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 62.Kono H, Chen CJ, Ontiveros F, et al. Uric acid promotes an acute inflammatory response to sterile cell death in mice. The Journal of clinical investigation. 2010;120:1939–1949. doi: 10.1172/JCI40124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsalik EL, Langley RJ, Dinwiddie DL, et al. An integrated transcriptome and expressed variant analysis of sepsis survival and death. Genome medicine. 2014;6:111. doi: 10.1186/s13073-014-0111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller NA, Kingsmore SF, Farmer A, et al. Management of High-Throughput DNA Sequencing Projects: Alpheus. Journal of computer science and systems biology. 2008;1:132. doi: 10.4172/jcsb.1000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.