

Abstract
Maternal immune activation (MIA) is an environmental risk factor for schizophrenia, and may contribute to other developmental disorders including autism and epilepsy. Activation of pro-inflammatory cytokine systems by injection of the synthetic double-stranded RNA polyriboinosinic-polyribocytidilic acid (Poly I:C) mediates important neurochemical and behavioral corollaries of MIA, which have relevance to deficits observed in schizophrenia. We examined the consequences of MIA on forebrain expression of neuregulin-1 (NRG-1), brain-derived neurotrophic factor (BDNF) and their receptors, ErbB4 and trkB, respectively, genes associated with schizophrenia. On gestational day 14, pregnant rats were injected with Poly I:C or vehicle. Utilizing in situ hybridization, expression of NRG-1, ErbB4, BDNF, and trkB was examined in male rat offspring at postnatal day (P) 14, P30 and P60. ErbB4 mRNA expression was significantly increased at P30 in the anterior cingulate (AC Ctx), frontal, and parietal cortices, with increases in AC Ctx expression continuing through P60. ErbB4 expression was also elevated in the prefrontal cortex (PFC) at P14. In contrast, NRG-1 mRNA was decreased in the PFC at P60. Expression of BDNF mRNA was significantly upregulated in the PFC at P60 and decreased in the AC Ctx at P14. Expression of trkB was increased in two regions, the piriform cortex at P14 and the striatum at P60. These findings demonstrate developmentally and regionally selective alterations in the expression of schizophrenia-related genes as a consequence of MIA. Further study is needed to determine contributions of these effects to development of alterations of relevance to neuropsychiatric diseases.
Keywords: Poly I:C, BDNF, Neuregulin-1, trkB, ErbB4
1. Introduction
Maternal infections during pregnancy are implicated in elevated risk for several mental disorders, including schizophrenia, autism, and epilepsy (Patterson, 2002; Brown, 2006; Brown and Patterson, 2011). The strongest evidence for this linkage is in schizophrenia, a neuropsychiatric disorder affecting 1% of the population resulting from the combined influence of genetic and environmental factors. Compelling evidence demonstrates that maternal immune activation (MIA), i.e. maternal exposure to infection during pregnancy, accounts for up to one third of attributable environmental risk for schizophrenia (Brown & Derkits 2010). Evidence demonstrates that the immune response itself, rather than a specific infectious agent, is responsible for elevated risk for schizophrenia (Smith et al., 2007; Brown and Derkits, 2010).
In animal models attempting to replicate this environmental risk, prenatal exposure to infectious agents, including influenza, lipopolysaccharide and the viral mimetic polyriboinosinic-polyribocytidilic acid (Poly I:C), results in cellular, neurochemical, and behavioral alterations relevant to schizophrenia (Meyer et al., 2009; Brown and Derkits, 2010). Key alterations stem from the maternal inflammatory response rather than the virus itself, as behavioral abnormalities were induced in the absence of viral infection by injection of Poly I:C, which stimulates maternal cytokine expression (Shi et al 2003). The synthetic double-stranded Poly I:C RNA is commonly used to create a strong, acute, non-specific immune reaction via the Toll-like receptor 3, resulting in cytokine release (Fortier et al., 2004). Cytokines are polypeptides involved in the inflammatory response and whose prenatal elevation is linked to increased risk of brain damage in offspring (Yoon et al., 1997). Cytokine levels are elevated in mothers of offspring later diagnosed with schizophrenia (Brown et al., 2004). In the MIA model, both behavioral and transcriptional alterations observed in MIA offspring were also inducible through maternal exposure to the cytokine interleukin–6 (IL-6) (Smith et al., 2007). In contrast to the alterations of cytokine levels, viral RNA is not detected in the tissue of animals exposed to infections prenatally despite later adult abnormalities (Shi et al., 2005). These observations suggest stimulation of maternal pro-inflammatory cytokine systems by injection of Poly I:C mediates important neurochemical and behavioral consequences of MIA (Ozawa et al., 2006; Smith et al., 2007; Pratt et al., 2013).
Maternal immune activation offspring display behavioral abnormalities into adulthood of relevance to schizophrenia. These behavioral abnormalities include altered locomotor responsiveness to stress, amphetamine, and novel environment, increased anxiety and depressive symptomology, deficits in prepulse inhibition of startle (PPI) and latent inhibition and increased responsiveness to stimulant drugs (Shi et al., 2005; Meyer et al., 2005; Brown and Patterson, 2011; Missault et al., 2014). Animals also display impaired working memory, place preference, and novel object recognition (Meyer et al., 2009; Richtand et al., 2012; Lukasz et al., 2013). Neuroanatomically, MIA elicits many brain alterations, including increases in γ-aminobutyric (GABA) receptor expression in the hippocampus and cerebellum, and widely reported developmental and functional abnormalities of the dopaminergic mesotelencephalic system (Bakos et al 2004, Ozawa et al 2006, Romero et al 2010, Vuillermot et al 2010). Prefrontal cortical abnormalities observed in MIA offspring include elevated basal extracellular glutamate (Roenker et al 2011) and altered synaptophysin expression (Romero et al 2010). Within the hippocampus, MIA offspring exhibit glutamate system defects including decreased N-Methyl-D-aspartate (NMDA) receptor-dependent synaptic current and plasticity (Escobar et al 2011, Lante et al 2007) and elevated basal extracellular glutamate (Ibi et al 2009). Imaging studies of this animal model consistently observe increased ventricular size in MIA offspring, with volume reductions in the hippocampus, prefrontal cortex (PFC), and striatum (Meyer et al., 2009; Piontkewitz et al., 2011b). Striatal and hippocampal volume reductions preceded the onset of behavioral abnormalities in both sexes, with structural abnormalities appearing developmentally earlier in males (Piontkewitz et al 2011a, Piontkewitz et al 2011b, Piontkewitz et al 2009). Of interest, antipsychotic medications impact some of these behavioral alterations, including the increased responsiveness to amphetamine, PPI, latent inhibition and alterations in the volume of brain regions (Meyer and Feldon, 2010; Piontkewitz et al., 2011b; Richtand et al., 2011).
Exposure to maternal infection during development may interact with expression of genes mediating schizophrenia risk. Neurotrophic factors are signaling molecules important during many stages of neurodevelopment including proliferation, differentiation, and migration and continue to support neuronal health and synaptic maintenance into adulthood (Knusel et al., 1991; Poo, 2001; Yarden and Sliwkowski, 2001; Buonanno and Fischbach, 2001; Seroogy et al., 2013). Neuregulin-1 (NRG-1) and its receptor ErbB4, as well as brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, are associated with the development of schizophrenia (Stefansson et al., 2002; Chong et al., 2008; Rybakowski, 2008; Ray et al., 2014). ErbB4 and NRG-1 are associated with familial inheritance of schizophrenia and the interaction between the two genes increases the risk of schizophrenia in some studies (Stefansson et al., 2003; Nicodemus et al., 2006; Norton et al., 2006; Silberberg et al., 2006). Dysregulation of NRG-1 signaling may also contribute to the development of positive schizophrenic symptoms (Shamir et al., 2012). Whereas studies directly linking BDNF gene mutations to schizophrenia are conflicting, multiple reports find that BDNF (both mRNA and protein), as well as its high-affinity receptor trkB, are reduced in cortical regions of schizophrenia patients (Durany et al., 2001; Weickert et al., 2003, 2005; Pandya et al., 2013). We therefore sought to examine the effects of MIA on mRNA expression of these genes throughout the postnatal period in the rat forebrain, testing the hypothesis that MIA will result in aberrant regulation of NRG-1, BDNF and their respective receptors. The regions and time points examined were chosen for their relevance to the manifestation of behavioral symptoms in the MIA model.
2. Methods
2.1 Animals
Nulliparous female Sprague Dawley rats, aged 3–5 months, were obtained from Harlan Laboratories (Indianapolis, IN). Male breeders were produced within the animal facility. All animals were housed under standard conditions with food and water available ad libitum and a 12:12 light/dark cycle. All procedures were in accordance with the Guide for Care and Use of Laboratory Animals, with approval by our Institutional Animal Care and Use Committee.
2.2 Prenatal Poly I:C treatment
The female rats were acclimated for at least two weeks, and then placed in the same cage overnight with a male Sprague Dawley rat. Males were removed the next morning, considered gestational day (G) 0 (Taylor, 1986). The female rats were singly housed throughout the pregnancy. On G14, pregnant dams were weighed (weight gain > 40 grams) and injected with Poly I:C (Sigma, St. Louis, MO) (8 mg/kg, i.p.) (Bronson et al., 2011) dissolved in saline or with saline vehicle control (1 ml/kg, i.p.). Dams were weighed again 24 hours following the Poly I:C or saline injection to identify anorexia and weight loss resulting from an inflammatory response (Fortier et al., 2004). The MIA dams without weight loss and saline dams with weight loss were removed from study as the offspring of these animals have a different phenotypic response (Bronson et al., 2011). On postnatal day (P) 1, litters were culled to eight. Pups were weaned on P21 and housed 2–3 per cage by sex and litter. No more than 2 rats per litter were used in each experimental group to avoid litter effect confounds. On P14, P30 and P60 male MIA offspring were sacrificed and brains were collected and frozen in dry ice to be subsequently processed for in situ hybridization.
2.3 In situ hybridization
Fresh-frozen brains (n = 5–6 per condition) were serially sectioned (at 10-μm thickness) throughout the PFC and striatal levels using a cryostat, thaw-mounted onto Superfrost plus microslides (VWR, Batavia, IL), and stored at −20°C until hybridization. Semi-adjacent sections were processed for the in situ hybridization localization of BDNF, trkB, NRG-1 and ErbB4 mRNAs using lineralized cDNA plasmids and were labeled using the proper polymerase (T3 for BDNF and T7 for NRG-1, ErbB4 and trkB) and 35S-UTP (PerkinElmer, Boston, MA), as previously described (Seroogy and Herman, 1997; Numan et al., 2005; Dickerson et al., 2009; Hemmerle et al., 2012). The BDNF cRNA (a gift from Drs. Christine Gall and Julie Lauterborn, University of California-Irvine) included 384 bases complementary to the rat BDNF mRNA coding region (nucleotides 388–771; Isackson et al., 1991; Gall et al., 1992). The trkB riboprobe detects the kinase-specific, full-length form of the rat trkB receptor (nucleotides 1358–1558; Middlemas et al., 1991; Goodness et al., 1997). The pan-NRG-1 plasmid was contained in a pCR-TOPO vector and consisted of 500 bp. The ErbB4 cRNA (kindly provided by Dr. Harley Kornblum, UCLA) was transcribed from a PCR 2.1 vector containing a 1.8 kb fragment complementary to a region of the ErbB4 receptor extracellular domain (Kornblum et al., 2000).
For hybridization pretreatment, slides were brought to room temperature and placed in 4% paraformaldehyde (pH 7.4) for 10 minutes to fix tissue. The slides were then placed in a series of five-minute washes involving phosphate-buffered saline (PBS) (twice), followed by 0.1 M PBS/0.2% glycine (twice), and again 0.1 M PBS (twice). All washes were made with diethyl pyrocarbonate (DEPC)-treated water. Triethanolamine (pH 8.0) containing 0.25% acetic anhydride was then used for 10 minutes for acetylation of the slides. Finally, slides were dehydrated in a series of ethanol washes, delipidated in chloroform and air-dried prior to hybridization.
Following pretreatment, the sections were hybridized with 50 μl of hybridization solution and subsequently coverslipped. The hybridization solution for each probe included the following: 20mM Tris-HCl, 10mM EDTA, 1X Denhardt’s, 335mM NaCl, 50% deionized formamide, 10% dextran sulfate, 0.3 mg/ml denatured salmon sperm DNA, 0.15 mg/ml tRNA, 40mM dithiothreitol, DEPC H2O, and the appropriate 35S-labeled cRNA probe. The resulting concentration of the hybridization solution was 1 x 106 cpm/slide. The slides were incubated inside a sealed, humidified chamber at 60°C for 16 hours. After hybridization, the coverslips were removed and the slides were washed in a series of standard sodium citrate washes of decreasing concentration and RNase buffer and air-dried. After the post-hybridization washes, the hybridized slides were exposed to BioMax MR Film (Kodak, Rochester, NY) for appropriate periods for each probe (NRG-1: 12 days, ErbB4: 9 days, BDNF: 7 days, trkB: 4 days). Kodak GPX developer and fixer was then used to develop the films.
2.4 Analysis
Film autoradiograms were analyzed by densitometry using Scion Image software (NIH) to compare optic densities (OD) of hybridization (mean corrected gray level) in select brain regions of the MIA versus control offspring. The following brain regions were examined: PFC, anterior, frontal, and parietal cortices, striatum, piriform cortex, and hippocampus. Control measurements for background were taken from unlabeled regions on each section and subtracted from each OD value to give a corrected OD value. Densitometric measurements were taken of each area from at least six sections per animal for each probe and averaged to obtain the mean corrected OD. The MIA offspring data are presented as a percentage of the control offspring values. Group differences were determined by t-test (GraphPad Prism) and considered significant when p < 0.05.
3. Results
3.1 NRG-1
The PFC of MIA offspring exhibited a slight, but significant decrease in NRG-1 expression at P60 compared to saline-treated animals (t(10) = 3.069, p < 0.05) (Fig. 1). No alterations were observed in NRG-1 expression in the other cortical regions examined, in the striatum or in the hippocampus (see Table 1).
Figure 1.
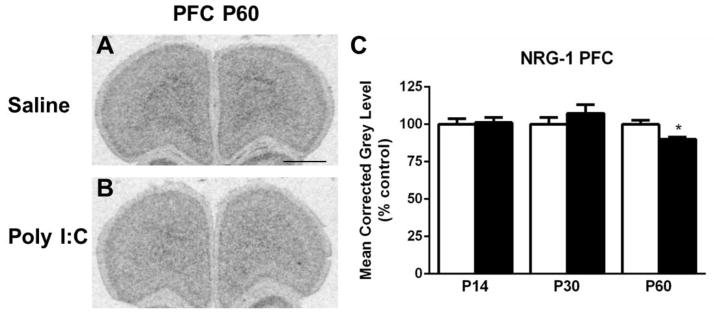
A–B. Representative autoradiograms of the prefrontal cortex (PFC) at P60 showing cRNA-labeling of neuregulin-1 (NRG-1) under saline (control) and MIA offspring conditions. C. Quantification of hybridization signal revealed that levels of NRG-1 mRNA are decreased in the PFC at P60. Open bar, control offspring; black bar, MIA offspring. Data are expressed as mean ± SEM. *p < 0.05 compared to respective control values. Scale bar in A = 1000 μm for A,B.
Table 1.
Summary of Poly I:C-Induced Alterations in Neurotrophic Factor/Receptor Expression by Forebrain Region
| Brain Region | Neurotrophic Factor/Receptor
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BDNF
|
trkB
|
NRG-1
|
ErbB4
|
|||||||||
| P14 | P30 | P60 | P14 | P30 | P60 | P14 | P30 | P60 | P14 | P30 | P60 | |
| Prefrontal Cortex | ↔ | ↔ | ↑ | ↔ | ↔ | ↔ | ↔ | ↔ | ↓ | ↑ | ↔ | ↔ |
|
| ||||||||||||
| Anterior Cingulate Cortex | ↓ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↑ | ↑ |
|
| ||||||||||||
| Frontal Cortex | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↑ |

|
|
| ||||||||||||
| Parietal Cortex | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↑ |
|
|
| ||||||||||||
| Piriform Cortex | ↔ | ↔ | ↔ | ↑ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↓ | ↔ |
|
| ||||||||||||
| Striatum | NA | NA | NA | ↔ | ↔ | ↑ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ |
|
| ||||||||||||
| Hippocampal Region CA1 | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ |
|
| ||||||||||||
| Hippocampal Region CA3 | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ |
|
| ||||||||||||
| Dentate Gyrus | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ |
Abbreviations: NA, not applicable, i.e., no measurable expression found in this region; BDNF, brain-derived neurotrophic factor; trkB, tropomyosin receptor kinase B; NRG-1, neuregulin-1; ↑, increased expression;
, strong trend towards increased expression; ↓, decreased expression; ↔ no change in expression.
3.2 ErbB4
Several regions exhibited changes in ErbB4 expression in the MIA offspring group compared to controls. Hybridization for ErbB4 mRNA in the PFC was increased at P14 in MIA offspring (t(10) =2.923, p < 0.05) (Fig. 2). In the anterior cingulate cortex (AC Ctx), ErbB4 mRNA expression was significantly elevated at both P30 (t(10) = 3.285, p < 0.05) and P60 (t(10) = 2.777, p < 0.05) when compared to the control offspring (Fig. 3A–E). Additionally at P30, levels of ErbB4 mRNA expression in the frontal (t(10) = 3.184, p < 0.05) and parietal (t(10) = 3.015, p < 0.05) cortices were increased in the MIA offspring (Fig. 3A–B, F–G), whereas there was a trend towards an increase in mRNA expression at P60 in these two regions (t(10) = 2.170, P = 0.08) (frontal); t(10) = 2.024, p =0.09 (parietal)) (Fig. 3C–D, F–G). Finally, in the piriform cortex, a decrease in mRNA expression was noted at P30 in the MIA offspring group compared to the control offspring (t(10) = 2.379, p < 0.05) (Fig. 4). No alterations in ErbB4 hybridization were observed in the striatum or hippocampus (Table 1).
Figure 2.
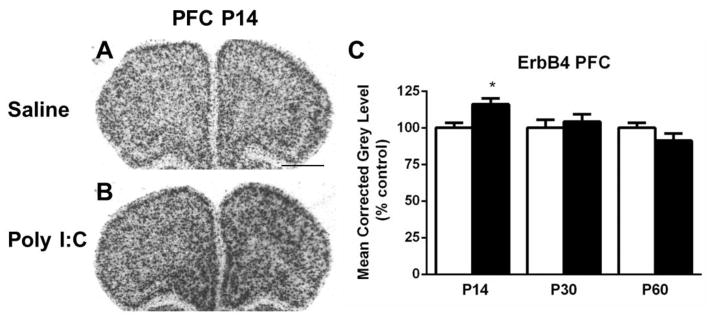
A–B. Representative autoradiograms of the prefrontal cortex (PFC) at P14 showing labeling of ErbB4 mRNA under saline (control) and MIA offspring conditions. C. Quantification of ErbB4 mRNA expression in the PFC demonstrates increased hybridization signal at P14. Open bar, control offspring; black bar, MIA offspring. Data are expressed as mean ± SEM. *p < 0.05 compared to respective control values. Scale bar in A = 1000 μm for A,B.
Figure 3.

A–D. Representative autoradiograms of ErbB4 mRNA expression in the anterior cingulate cortex (AC Ctx), frontal cortex (Fr Ctx) and parietal cortex (Par Ctx) at P30 (A–B) and P60 (C–D) under saline (control) and MIA offspring conditions. E. Quantification of the hybridization signal revealed increased expression of ErbB4 at P30 and P60 in the AC Ctx in MIA offspring F. Levels of ErbB4 mRNA expression in the Fr Ctx increased at P30, and exhibited a trend towards an increase at P60. G. In the Par Ctx there is an increase of mRNA expression at P30, also with a trend towards elevation at P60. Open bar, control offspring; black bar, MIA offspring. Data are expressed as mean ± SEM. #p=0.08, *p < 0.05, **p < 0.01 compared to respective control values. Scale bar in A = 1000 μm for A–D.
Figure 4.
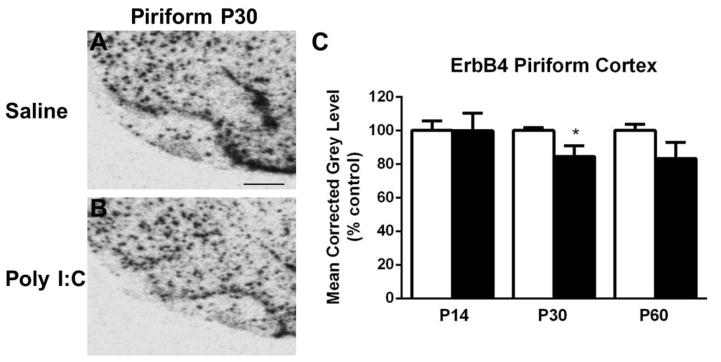
A–B. Representative autoradiograms of the piriform cortex (Pir Ctx) at P30 showing cRNA-labeling of ErbB4 mRNA under saline (control) and MIA offspring conditions. C. In the Pir Ctx a decrease in mRNA expression was detected at P30. Open bar, control offspring; black bar, MIA offspring. Data are expressed as mean ± SEM. *p < 0.05 compared to respective control values. Scale bar in A = 500 μm for A,B.
3.3 BDNF
Two regions examined exhibited alterations in BDNF expression in MIA compared to the control offspring. In the PFC, BDNF expression in the MIA offspring was elevated at the P60 time point (t(9) = 3.094, p < 0.05) (Fig. 5A–B, E). In contrast, the BDNF mRNA levels were decreased in the AC Ctx of MIA offspring at P14 (t(9) = 2.618, p < 0.05) (Fig. 5C–D, F). There were no changes in expression found in the frontal, parietal, and piriform cortices, hippocampus or striatum (Table 1).
Figure 5.
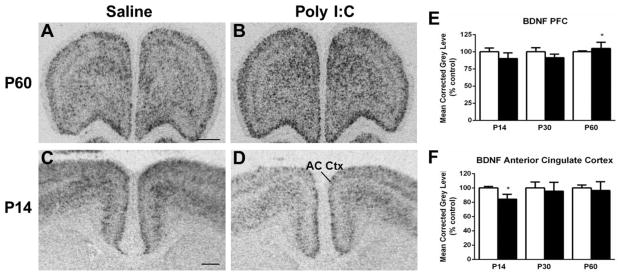
Representative autoradiograms of BDNF mRNA expression in the prefrontal cortex (PFC) at P60 (A–B) and in the anterior cingulate cortex (AC Ctx) at P14 (C–D) under saline (control) and MIA offspring conditions. E. Quantification of BDNF mRNA expression in the prefrontal cortex (PFC) indicated an increased level of expression at P60. F. There was a decrease of BDNF mRNA in the anterior cingulate cortex (AC Ctx) of Poly I:C-treated rats at P14. Open bar, control offspring; black bar, MIA offspring. Data are expressed as mean ± SEM. *p < 0.05 compared to respective control values. Scale bar in A = 1000 μm for A,B; C = 500 μm for C,D.
3.4 trkB
Hybridization levels of trkB were elevated in select regions. In the piriform cortex, compared to controls, trkB expression in MIA offspring was increased at P14 (t(9) = 2.555, p < 0.05) (Fig. 6A–B, E). Levels of trkB mRNA were also upregulated in the striatum of MIA in comparison to control offspring at P60 (t(9) = 2.459, p < 0.05) (Fig. 6C–D, F). No significant changes were observed in the prefrontal, frontal, or parietal cortices or in the hippocampus (Table 1).
Figure 6.
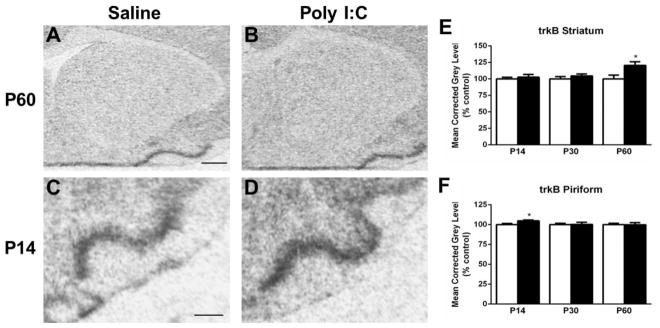
Representative autoradiograms of trkB mRNA expression in the striatum at P60 (A–B) and in the piriform cortex at P14 (C–D) under saline (control) and MIA offspring conditions. E. Measurement of hybridization signal in the striatum revealed an increase in trkB mRNA levels in Poly I:C-treated animals at P60. F. Levels of trkB mRNA in the piriform cortex are elevated at P14 in Poly I:C animals compared with saline-treated animals. Open bar, control offspring; black bar, MIA offspring. Data are expressed as mean ± SEM. *p < 0.05 compared to respective control values. Scale bar in A = 1000 μm for A,B; C = 500 μm for C,D.
4. Discussion
Expression of schizophrenia-related genes after MIA was altered in multiple forebrain regions throughout brain development (Table 1). These regions include the prefrontal, anterior cingulate, frontal, parietal and piriform cortices. The subcortical striatum also exhibited modifications in gene expression, but only in adulthood. In contrast, no changes in gene expression were noted in the hippocampus. Our study encompassed multiple periods of rat neurodevelopment with postnatal (P14), adolescent (P30) and adult (P60) (McCormick et al., 2010) time points examined. This allowed us to observe how gene expression was altered throughout the major developmental stages including equivalent time spans of behavioral symptom manifestation in adolescence and adulthood. This is especially important in this model, as many of the behavioral and cognitive adaptations do not emerge until the animals reach adulthood (Meyer and Feldon, 2010). The present altered levels of schizophrenia-related genes in juveniles and adults may have implications for the later behavioral deficits observed in animals prenatally exposed to immune activation.
Several candidate genes for increased schizophrenia risk include genes associated with neuronal plasticity and synaptic formation or impairment thereof (Le Strat et al., 2009). The genes examined in this study are important in neuronal maturation, including migration, axon formation, and myelination and the continued neuronal plasticity observed in adulthood. Clinical studies have found the expression of BDNF, trkB, NRG-1, and ErbB4 altered in the post-mortem tissue of schizophrenic patients (Weickert et al., 2003; Law et al., 2006; Chong et al., 2008; Joshi et al., 2014; Ray et al., 2014). In addition to the ligands and the receptors themselves, the signal cascades stimulated by ErbB4 and trkB receptor activation, including the PI3K-Akt, PLCγ and Raf-MAP kinase pathways, are specifically implicated in the pathology of schizophrenia (Stornetta and Zhu, 2011; Zheng et al., 2012). Upregulation of neurotrophic factors is often viewed as a beneficial compensatory signaling mechanism. However, alteration in trophic factor expression can paint a complicated picture, as increased levels are not always an advantageous outcome. For example, in the forced swim test, elevation of BDNF in the mesolimbic pathway of rats contributes to behavioral alterations of relevance to depression (Eisch et al., 2003).
This study is believed to be the first to examine NRG-1/ErbB4 gene expression in offspring exposed to MIA. ErbB4 is a receptor tyrosine kinase that can form both homo- and heterodimers with other receptors in its family after ligand binding. Of the multiple members of the neuregulin family that bind with ErbB4, NRG-1 is the most strongly linked to schizophrenia at this time (Buonanno, 2010), though some studies indicate mutations of NRG-3 may increase risk as well (Kao et al., 2010). Neuregulin-1 has numerous different isoforms via various promoters and alternative splicing (Buonanno and Fischbach, 2001; Deng et al., 2013; Seroogy et al., 2013), and in our study we used a pan-NRG-1 probe to detect expression of multiple forms of NRG-1. It is possible that a riboprobe targeting more specific isoforms would uncover more subtle variations in NRG-1 gene expression. Whereas there was only modulation of NRG-1 mRNA in the PFC at P60, this change occurred long after exposure to the maternal immune response had taken place, suggesting that prenatal exposure to the immune activation may result in long-term plasticity not evident until adulthood. ErbB4 appears to be more malleable in this model, with alterations observed in various regions, including the PFC and AC Ctx, at all three developmental time points.
Activation of the ErbB4 receptor via NRG-1 is essential in the functional development of major neurotransmitter systems linked to schizophrenia, including dopamine, glutamate and GABA (Laruelle et al., 2003; Lewis et al., 2005) and interference in ErbB4 signaling in these systems can have detrimental effects (Stefansson et al., 2002). Additionally, ErbB4 activation is increased in the PFC of schizophrenic brains and may be specifically related to deficits in NMDA receptor (NMDAR) signaling in schizophrenia patients (Hahn et al., 2006). Interestingly, in our model, an increase in ErbB4 expression was found in cortical regions during later developmental time points, comparable to the time period of symptom manifestation in humans. Consonant with our findings, elevated expression of ErbB4 mRNA has been shown in the PFC of schizophrenic brains (Joshi et al., 2014). Research has suggested that NRG-1 signaling via ErbB4 can result in a decrease in NMDAR activation (Huang et al., 2000; Gu et al., 2005), relating to the glutamatergic hypoactivity hypothesis of schizophrenia. However, knockout of ErbB4 in neurons/glia produced lower motor activity and reduced grip strength, which may correlate with schizophrenia symptoms (Golub et al., 2004; Esper et al., 2006). These studies present an imprecise picture of the relationship between ErbB4 levels and behavioral changes, but it is possible that there is a distinct pattern of changes in ErbB4 expression found depending on the brain region.
The other major neurotrophic factor examined, BDNF, is a member of the neurotrophin family and BDNF/trkB signaling is believed to play a large role in the normal function of the nervous system, including having a major role in synaptic efficacy and plasticity (Thoenen, 1995; Poo, 2001; Waterhouse and Xu, 2009). Like ErbB4, the expression of BDNF is vital to the function of forebrain GABAergic, dopaminergic, and glutamatergic circuits (Itami et al., 2000; Glorioso et al., 2006; Pillai, 2008). Schizophrenia patients display reduction of BDNF serum and plasma levels in several studies (Palomino et al., 2006; Grillo et al., 2007), though how this precisely correlates with brain BDNF levels is unknown at this time. Postmortem brains of schizophrenia patients show increases of BDNF protein in the frontal and parietal cortices and decreases in BDNF protein and mRNA in the PFC (Durany et al., 2001; Weickert et al., 2003; Pillai, 2008). Loss of trkB signaling in the PFC of schizophrenia patients appears to be a factor in the dysfunction of GABAergic neurons (Hashimoto et al., 2005). Similar to findings in ErbB4 studies, there is inconsistent data on BDNF alterations in the hippocampus and cortex of schizophrenia patients (Angelucci et al., 2005; Autry and Monteggia, 2012). Interestingly, other studies have described an interaction between trkB and ErbB4 signaling pathways in cortical neurons, with trkB being necessary for NRG-1 phosphorylation of NRB2 prior to synaptogenesis (Pandya and Pillai, 2014). Moreover, NRG-1 is able to increase the release of BDNF (Pandya and Pillai, 2014). Reduction of trkB/ErbB4 interaction is also observed in the prefrontal cortex of schizophrenia patients (Pandya and Pillai, 2014). In our study, most areas with alterations in BDNF/trkB expression did not also exhibit changes in NRG-1/ErbB4 and vice versa, a major exception being the PFC where BDNF was upregulated and ErbB4 was decreased at P60.
In previous studies, animals receiving Poly I:C acutely in adulthood displayed alterations in BDNF mRNA levels in the cortex and hippocampus, potentially linked to behavioral changes seen in the animals (Kranjac et al., 2012; Gibney et al., 2013). Decreases in BDNF within the frontal cortex are found in additional developmental models of relevance to schizophrenia including maternal stress and genetic models (Pillai and Mahadik, 2008; Matrisciano et al., 2012). Other studies utilizing the MIA model have identified decreased BDNF in the placentas of treated dams, whereas whole brain modification of BDNF protein content was undetectable on P1 (Gilmore et al., 2005). Locating regions of BDNF perturbation is important, as studies of behaviors of relevance to schizophrenia in BDNF knockout models suggest that BDNF contributes to the development of positive symptoms like hyperactivity and elevated responsiveness to stimulant drugs (Autry and Monteggia, 2012). In the current study, we found changes in BDNF and trkB expression in cortical regions and the striatum at P14 or P60, but no changes were observed in the hippocampus. This variance from other studies could be explained by the use of alternative models, as several involved injections of Poly I:C into the adult animals themselves (Kranjac et al., 2012; Gibney et al., 2013).
Mechanisms underlying this developmental modulation of schizophrenia-related genes is not known, but may be related to the cytokine release resulting from the MIA. After administration of Poly I:C to pregnant dams, cytokines, including the pro-inflammatory cytokines IL-1α, IL-1β, IL-6, tumor neurosis factor-α (TNF-α), and interferon-γ, are altered in the offspring in a region-specific manner throughout development (Meyer et al., 2008; Garay et al., 2013; Pratt et al., 2013). Some anti-inflammatory cytokines and chemokines appear to be altered as well (Garay et al., 2013). The enduring modulation of cytokine levels in the MIA model may have clinical relevance, as increased cytokine levels have been reported in schizophrenia patients, including IL-6 and TNF-α (Strous and Shoenfeld, 2006; Miller et al., 2011). The release of cytokines, particularly IL-6, appears to be essential for the behavioral and physiological effects observed in the MIA model (Smith et al., 2007). There are, however, conflicting data as to whether the number and density of microglia themselves are altered (Garay et al., 2013; Van den Eynde et al., 2014).
While we did not measure regional alterations of cytokines in the present experiments, there is strong evidence linking cytokine levels to regulation of the neurotrophic factors examined in this study (Imai et al., 2007; Noga et al., 2007; Marballi et al., 2010; Skaper et al., 2012; Calabrese et al., 2014). Other prenatal models of relevance to schizophrenia, including prenatal restraint stress, also detect changes in forebrain expression of BDNF and ErbB4 (Matrisciano et al., 2012; Stevens et al., 2013). Neurotrophic factor alterations could be a fundamental indication of processes underlying the deficits observed in different models. Cytokine-induced alterations of trophic factors such as BDNF may therefore identify a mechanism linking immune activation to the brain plasticity underlying behavioral deficits in these developmental models (Calabrese et al., 2014).
Why particular brain regions are more susceptible to neurotrophic factor modifications is unknown at this time. It is believed that the neuroanatomical changes observed in developmental models are a result of alterations in neurodevelopment rather than neurodegeneration (Meyer and Feldon, 2010). Given prior studies identifying brain parenchymal alterations in the MIA model, it is possible that differential loss of certain cell types may contribute to the altered mRNA expression observed in our study. Reductions in volume of the hippocampus, prefrontal cortex, and striatum have been consistently observed in MIA offspring. Striatal and hippocampal volume reductions preceded the onset of behavioral abnormalities in both sexes, with structural abnormalities appearing developmentally earlier in males (Piontkewitz et al., 2009,2011a,b). Future studies utilizing co-localization of cell-specific markers could help to further elucidate this potential mechanism.
One caveat in the relevance of the MIA neurodevelopmental model to schizophrenia is that gene expression in our study was examined in prenatally challenged Sprague Dawley rats presumably devoid of schizophrenia risk-allele genes. Previous studies demonstrate significant increase in schizophrenia risk in human MIA offspring with a family history of psychotic disorder, whereas enhancement of schizophrenia risk was not detectable in offspring lacking a family genetic risk for the illness (Clarke et al., 2009). Thus, the interaction between the environmental influence of MIA and schizophrenia susceptibility alleles is likely a requirement for elevation of schizophrenia risk. Evidence supporting this gene/environment interaction has also been demonstrated in other rodent animal models, where activation of the prenatal and neonatal immune system by Poly I:C in DISC1 mutant mice results in offspring with behavior and cellular effects worsened compared to mutant animals alone (Abazyan et al., 2010; Ibi et al., 2010; Nagai et al., 2011). In heterozygous knockouts of NRG-1, it appears that NRG-1 is necessary for the reduced sociability and spatial working memory deficits observed in MIA offspring, whereas PPI deficits (reflecting sensorimotor gating impairment) were observed in both knockout and wild-type mice (O’Leary et al., 2014). This suggests that NRG-1 plays a key role in the manifestation of many, but not all, of the behavioral deficits observed in the MIA model. To draw a more complete picture, it would be important to examine alterations of neurotrophic factors in genetic models, like DISC1 mutant mice, after exposure to MIA to determine if the gene expression changes are exacerbated compared to wild-type animals.
An additional consideration for data interpretation in our study is that alterations in mRNA expression may not directly correlate with protein expression, the ultimate effectors of biological action. While mRNA and protein expression vary in a synchronous fashion in some systems (e.g. Wang et al., 1995; Ohnuma et al., 2003; Dickerson et al., 2009), there are other examples of genes for which mRNA and protein expression do not demonstrate complete correlation (e.g. Conner et al., 1997; Gygi et al., 1999). Protein expression may also vary at the subregional level as well (see Weickert et al., 2003). The differences in mRNA expression observed in our data set should also be considered preliminary because we did not correct for multiple comparisons. While the data presented therefore provide a framework for trophic factor plasticity in the MIA model, future confirmatory studies would ideally analyze both mRNA and protein levels to provide a more complete understanding of cellular mechanisms. It will likewise be intriguing to examine the potential linkage of gene expression alterations in regions like the PFC and piriform cortex to behavioral dysfunctions observed in MIA models such as PPI and novel object recognition (Ozawa et al., 2006; Lukasz et al., 2013).
In conclusion, animals exposed to MIA via Poly I:C administration display region-specific, aberrant expression of key schizophrenia-related genes at different stages of postnatal development. These modifications appear to predominately affect neocortical regions of the brain compared to the hippocampus, with ErbB4 displaying the most plastic properties. Further study is needed to determine contributions of these effects to development of behavioral and neurochemical alterations of relevance to neuropsychiatric disease states. Deciphering these alterations will aid in uncovering the biological basis of schizophrenia-related behavioral symptoms and allow development of more targeted therapies.
Acknowledgments
Role of funding sources:
Funding sources had no involvement in study design, in the writing of the report, or in the decision to submit the manuscript for publication.
This work was supported by the Morris Braun Foundation, Selma Schottenstein Harris Lab for Research in Parkinson’s, Gardner Family Center for Parkinson’s Disease and Movement Disorders, National Institute on Drug Abuse (R01 DA016778-01 and R21 DA031876-02), National Institute of Mental Health (R21 MH083192-01) and the Department of Veterans Affairs Medical Research Service. AMH was supported by National Institutes of Health T32 DK059803. We thank Ben Packard and Dr. Jon Dickerson for help in generating the NRG-1 plasmid.
Footnotes
Author contributions:
KBS and NMR designed the study, wrote the protocols, and supervised the project. AMH, RA, SLB and KHL performed the research. AMH, NMR and KBS wrote the first draft of the manuscript. All authors contributed to and have approved the final manuscript.
Conflict of interest:
The authors disclose the following relationships:
Dr. N. Richtand is a member of the speaker’s bureaus of Otsuka America Pharmaceutical and Sunovion Pharmaceuticals and serves on the advisory board of Otsuka America Pharmaceutical, outside the submitted work. All other authors declare that they have no conflicts of interest in relation to this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abazyan B, Nomura J, Kannan G, Ishizuka K, Tamashiro KL, Nucifora F, Pogorelov V, Ladenheim B, Yang C, Krasnova IN, Cadet JL, Pardo C, Mori S, Kamiya A, Vogel MW, Sawa A, Ross CA, Pletnikov MV. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry. 2010;68(12):1172–1181. doi: 10.1016/j.biopsych.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci F, Brene S, Mathe AA. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry. 2005;10(4):345–352. doi: 10.1038/sj.mp.4001637. [DOI] [PubMed] [Google Scholar]
- Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev. 2012;64(2):238–258. doi: 10.1124/pr.111.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakos J, Duncko R, Makatsori A, Pirnik Z, Kiss A, Jezova D. Prenatal immune challenge affects growth, behavior, and brain dopamine in offspring. Ann NY Acad Sci. 2004;1018:281–287. doi: 10.1196/annals.1296.033. [DOI] [PubMed] [Google Scholar]
- Bronson SL, Ahlbrand R, Horn PS, Kern JR, Richtand NM. Individual differences in maternal response to immune challenge predict offspring behavior: contribution of environmental factors. Behav Brain Res. 2011;220(1):55–64. doi: 10.1016/j.bbr.2010.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull. 2006;32(2):200–202. doi: 10.1093/schbul/sbj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167(3):261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V, Perrin M, Gorman JM, Susser ES. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry. 2004;161(5):889–895. doi: 10.1176/appi.ajp.161.5.889. [DOI] [PubMed] [Google Scholar]
- Brown AS, Patterson PH. Maternal infection and schizophrenia: implications for prevention. Schizophr Bull. 2011;37(2):284–290. doi: 10.1093/schbul/sbq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonanno A. The neuregulin signaling pathway and schizophrenia: from genes to synapses and neural circuits. Brain Res Bull. 2010;83(3–4):122–131. doi: 10.1016/j.brainresbull.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonanno A, Fischbach GD. Neuregulin and ErbB receptor signaling pathways in the nervous system. Curr Opin Neurobiol. 2001;11(3):287–296. doi: 10.1016/s0959-4388(00)00210-5. [DOI] [PubMed] [Google Scholar]
- Calabrese F, Rossetti AC, Racagni G, Gass P, Riva MA, Molteni R. Brain-derived neurotrophic factor: a bridge between inflammation and neuroplasticity. Front Cell Neurosci. 2014;8(430) doi: 10.3389/fncel.2014.00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong VZ, Thompson M, Beltaifa S, Webster MJ, Law AJ, Weickert CS. Elevated neuregulin-1 and ErbB4 protein in the prefrontal cortex of schizophrenic patients. Schizophr Res. 2008;100(1–3):270–280. doi: 10.1016/j.schres.2007.12.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke MC, Tanskanen A, Huttunen M, Whittaker JC, Cannon M. Evidence for an interaction between familial liability and prenatal exposure to infection in the causation of schizophrenia. Am J Psychiatry. 2009;166(9):1025–1030. doi: 10.1176/appi.ajp.2009.08010031. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17(7):2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Pan B, Engel M, Huang XF. Neuregulin-1 signalling and antipsychotic treatment: potential therapeutic targets in a schizophrenia candidate signalling pathway. Psychopharmacology (Berl) 2013;226(2):201–215. doi: 10.1007/s00213-013-3003-2. [DOI] [PubMed] [Google Scholar]
- Dickerson JW, Hemmerle AM, Numan S, Lundgren KH, Seroogy KB. Decreased expression of ErbB4 and tyrosine hydroxylase mRNA and protein in the ventral midbrain of aged rats. Neuroscience. 2009;163(1):482–489. doi: 10.1016/j.neuroscience.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durany N, Michel T, Zochling R, Boissl KW, Cruz-Sanchez FF, Riederer P, Thome J. Brain-derived neurotrophic factor and neurotrophin 3 in schizophrenic psychoses. Schizophr Res. 2001;52(1–2):79–86. doi: 10.1016/s0920-9964(00)00084-0. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Bolanos CA, de Wit J, Simonak RD, Pudiak CM, Barrot M, Verhaagen J, Nestler EJ. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: a role in depression. Biol Psychiatry. 2003;54(10):994–1005. doi: 10.1016/j.biopsych.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Escobar M, Crouzin N, Cavalier M, Quentin J, Roussel J, Lanté F, Batista-Novais AR, Cohen-Solal C, De Jesus Ferreira MC, Guiramand J, Barbanel G, Vignes M. Early, time-dependent disturbances of hippocampal synaptic transmission and plasticity after in utero immune challenge. Biol Psychiatry. 2011;70(10):992–999. doi: 10.1016/j.biopsych.2011.01.009. [DOI] [PubMed] [Google Scholar]
- Esper RM, Pankonin MS, Loeb JA. Neuregulins: versatile growth and differentiation factors in nervous system development and human disease. Brain Res Rev. 2006;51(2):161–175. doi: 10.1016/j.brainresrev.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Fortier ME, Joober R, Luheshi GN, Boksa P. Maternal exposure to bacterial endotoxin during pregnancy enhances amphetamine-induced locomotion and startle responses in adult rat offspring. J Psychiatr Res. 2004;38(3):335–345. doi: 10.1016/j.jpsychires.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Gall CM, Gold SJ, Isackson PJ, Seroogy KB. Brain-derived neurotrophic factor and neurotrophin-3 mRNAs are expressed in ventral midbrain regions containing dopaminergic neurons. Mol Cell Neurosci. 1992;3(1):56–63. doi: 10.1016/1044-7431(92)90009-q. [DOI] [PubMed] [Google Scholar]
- Garay PA, Hsiao EY, Patterson PH, McAllister AK. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun. 2013;31:54–68. doi: 10.1016/j.bbi.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney SM, McGuinness B, Prendergast C, Harkin A, Connor TJ. Poly I:C-induced activation of the immune response is accompanied by depression and anxiety-like behaviours, kynurenine pathway activation and reduced BDNF expression. Brain Behav Immun. 2013;28:170–181. doi: 10.1016/j.bbi.2012.11.010. [DOI] [PubMed] [Google Scholar]
- Gilmore JH, Jarskog LF, Vadlamudi S. Maternal poly I:C exposure during pregnancy regulates TNF alpha, BDNF, and NGF expression in neonatal brain and the maternal-fetal unit of the rat. J Neuroimmunol. 2005;159(1–2):106–112. doi: 10.1016/j.jneuroim.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Glorioso C, Sabatini M, Unger T, Hashimoto T, Monteggia LM, Lewis DA, Mirnics K. Specificity and timing of neocortical transcriptome changes in response to BDNF gene ablation during embryogenesis or adulthood. Mol Psychiatry. 2006;11(7):633–648. doi: 10.1038/sj.mp.4001835. [DOI] [PubMed] [Google Scholar]
- Golub MS, Germann SL, Lloyd KC. Behavioral characteristics of a nervous system-specific erbB4 knock-out mouse. Behav Brain Res. 2004;153(1):159–170. doi: 10.1016/j.bbr.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Goodness TP, Albers KM, Davis FE, Davis BM. Overexpression of nerve growth factor in skin increases sensory neuron size and modulates Trk receptor expression. Eur J Neurosci. 1997;9(8):1574–1585. doi: 10.1111/j.1460-9568.1997.tb01515.x. [DOI] [PubMed] [Google Scholar]
- Grillo RW, Ottoni GL, Leke R, Souza DO, Portela LV, Lara DR. Reduced serum BDNF levels in schizophrenic patients on clozapine or typical antipsychotics. J Psychiatr Res. 2007;41(1–2):31–35. doi: 10.1016/j.jpsychires.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Gu Z, Jiang Q, Fu AK, Ip NY, Yan Z. Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J Neurosci. 2005;25(20):4974–4984. doi: 10.1523/JNEUROSCI.1086-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19(3):1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, Bakshi K, Kamins J, Borgmann-Winter KE, Siegel SJ, Gallop RJ, Arnold SE. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12(7):824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Bergen SE, Nguyen QL, Xu B, Monteggia LM, Pierri JN, Sun Z, Sampson AR, Lewis DA. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J Neurosci. 2005;25(2):372–383. doi: 10.1523/JNEUROSCI.4035-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmerle AM, Dickerson JW, Herring NR, Schaefer TL, Vorhees CV, Williams MT, Seroogy KB. (±)3,4-methylenedioxymethamphetamine (“ecstasy”) treatment modulates expression of neurotrophins and their receptors in multiple regions of adult rat brain. J Comp Neurol. 2012;520(11):2459–2474. doi: 10.1002/cne.23048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YZ, Won S, Ali DW, Wang Q, Tanowitz M, Du QS, Pelkey KA, Yang DJ, Xiong WC, Salter MW, Mei L. Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron. 2000;26(2):443–455. doi: 10.1016/s0896-6273(00)81176-9. [DOI] [PubMed] [Google Scholar]
- Ibi D, Nagai T, Kitahara Y, Mizoguchi H, Koike H, Shiraki A, Takuma K, Kamei H, Noda Y, Nitta A, Nabeshima T, Yoneda Y, Yamada K. Neonatal polyI:C treatment in mice results in schizophrenia-like behavioral and neurochemical abnormalities in adulthood. Neurosci Res. 2009;64(3):297–305. doi: 10.1016/j.neures.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Ibi D, Nagai T, Koike H, Kitahara Y, Mizoguchi H, Niwa M, Jaaro-Peled H, Nitta A, Yoneda Y, Nabeshima T, Sawa A, Yamada K. Combined effect of neonatal immune activation and mutant DISC1 on phenotypic changes in adulthood. Behav Brain Res. 2010;206(1):32–37. doi: 10.1016/j.bbr.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai F, Suzuki H, Oda J, Ninomiya T, Ono K, Sano H, Sawada M. Neuroprotective effect of exogenous microglia in global brain ischemia. J Cereb Blood Flow Metab. 2007;27(3):488–500. doi: 10.1038/sj.jcbfm.9600362. [DOI] [PubMed] [Google Scholar]
- Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6(6):937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- Itami C, Mizuno K, Kohno T, Nakamura S. Brain-derived neurotrophic factor requirement for activity-dependent maturation of glutamatergic synapse in developing mouse somatosensory cortex. Brain Res. 2000;857(1–2):141–150. doi: 10.1016/s0006-8993(99)02352-5. [DOI] [PubMed] [Google Scholar]
- Joshi D, Fullerton JM, Weickert CM. Elevated ErbB4 mRNA is related to interneuron deficit in prefrontal cortex in schizophrenia. J Psychiatr Res. 2014;53:125–132. doi: 10.1016/j.jpsychires.2014.02.014. [DOI] [PubMed] [Google Scholar]
- Kao WT, Wang Y, Kleinman JE, Lipska BK, Hyde TM, Weinberger DR, Law AJ. Common genetic variation in Neuregulin 3 (NRG3) influences risk for schizophrenia and impacts NRG3 expression in human brain. Proc Natl Acad Sci USA. 2010;107(35):15619–15624. doi: 10.1073/pnas.1005410107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knusel B, Winslow JW, Rosenthal A, Burton LE, Seid DP, Nikolics K, Hefti F. Promotion of central cholinergic and dopaminergic neuron differentiation by brain-derived neurotrophic factor but not neurotrophin 3. Proc Natl Acad Sci USA. 1991;88(3):961–965. doi: 10.1073/pnas.88.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblum HI, Yanni DS, Easterday MC, Seroogy KB. Expression of the EGF receptor family members ErbB2, ErbB3, and ErbB4 in germinal zones of the developing brain and in neurosphere cultures containing CNS stem cells. Dev Neurosci. 2000;22(1–2):16–24. doi: 10.1159/000017423. [DOI] [PubMed] [Google Scholar]
- Kranjac D, McLinden KA, Koster KM, Kaldenbach DL, Chumley MJ, Boehm GW. Peripheral administration of poly I:C disrupts contextual fear memory consolidation and BDNF expression in mice. Behav Brain Res. 2012;228(2):452–457. doi: 10.1016/j.bbr.2011.12.031. [DOI] [PubMed] [Google Scholar]
- Lante F, Meunier J, Guiramand J, Maurice T, Cavalier M, de Jesus Ferreira MC, Aimar R, Cohen-Solal C, Vignes M, Barbanel G. Neurodevelopmental damage after prenatal infection: role of oxidative stress in the fetal brain. Free Radic Biol Med. 2007;42(8):1231–1245. doi: 10.1016/j.freeradbiomed.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Kegeles LS, Abi-Dargham A. Glutamate, dopamine, and schizophrenia: from pathophysiology to treatment. Ann NY Acad Sci. 2003;1003:138–158. doi: 10.1196/annals.1300.063. [DOI] [PubMed] [Google Scholar]
- Law AJ, Lipska BK, Weickert CS, Hyde TM, Straub RE, Hashimoto R, Harrison PJ, Kleinman JE, Weinberger DR. Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5′ SNPs associated with the disease. Proc Natl Acad Sci USA. 2006;103(17):6747–6752. doi: 10.1073/pnas.0602002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Strat Y, Ramoz N, Gorwood P. The role of genes involved in neuroplasticity and neurogenesis in the observation of a gene-environment interaction (GxE) in schizophrenia. Curr Mol Med. 2009;9(4):506–518. doi: 10.2174/156652409788167104. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6(4):312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lukasz B, O’Sullivan NC, Loscher JS, Pickering M, Regan CM, Murphy KJ. Peripubertal viral-like challenge and social isolation mediate overlapping but distinct effects on behaviour and brain interferon regulatory factor 7 expression in the adult Wistar rat. Brain Behav Immun. 2013;27(1):71–79. doi: 10.1016/j.bbi.2012.09.011. [DOI] [PubMed] [Google Scholar]
- Marballi K, Quinones MP, Jimenez F, Escamilla MA, Raventos H, Soto-Bernardini MC, Ahuja SS, Walss-Bass C. In vivo and in vitro genetic evidence of involvement of neuregulin 1 in immune system dysregulation. J Mol Med (Berl) 2010;88(11):1133–1141. doi: 10.1007/s00109-010-0653-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrisciano F, Tueting P, Maccari S, Nicoletti F, Guidotti A. Pharmacological activation of group-II metabotropic glutamate receptors corrects a schizophrenia-like phenotype induced by prenatal stress in mice. Neuropsychopharmacology. 2012;37(4):929–938. doi: 10.1038/npp.2011.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick CM, Mathews IZ, Thomas C, Waters P. Investigations of HPA function and the enduring consequences of stressors in adolescence in animal models. Brain Cogn. 2010;72(1):73–85. doi: 10.1016/j.bandc.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol. 2010;90(3):285–326. doi: 10.1016/j.pneurobio.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev. 2005;29(6):913–947. doi: 10.1016/j.neubiorev.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Meyer U, Murray PJ, Urwyler A, Yee BK, Schedlowski M, Feldon J. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol Psychiatry. 2008;13(2):208–221. doi: 10.1038/sj.mp.4002042. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Fatemi SH. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev. 2009;33(7):1061–1079. doi: 10.1016/j.neubiorev.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Middlemas DS, Lindberg RA, Hunter T. trkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol. 1991;11(1):143–153. doi: 10.1128/mcb.11.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B. Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry. 2011;70(7):663–671. doi: 10.1016/j.biopsych.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missault S, Van den Eynde K, Vanden Berghe W, Fransen E, Weeren A, Timmermans JP, Kumar-Singh S, Dedeurwaerdere S. The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav Immun. 2014;42:138–146. doi: 10.1016/j.bbi.2014.06.013. [DOI] [PubMed] [Google Scholar]
- Nagai T, Kitahara Y, Ibi D, Nabeshima T, Sawa A, Yamada K. Effects of antipsychotics on the behavioral deficits in human dominant-negative DISC1 transgenic mice with neonatal polyI:C treatment. Behav Brain Res. 2011;225(1):305–310. doi: 10.1016/j.bbr.2011.07.049. [DOI] [PubMed] [Google Scholar]
- Nicodemus KK, Luna A, Vakkalanka R, Goldberg T, Egan M, Straub RE, Weinberger DR. Further evidence for association between ErbB4 and schizophrenia and influence on cognitive intermediate phenotypes in healthy controls. Mol Psychiatry. 2006;11(12):1062–1065. doi: 10.1038/sj.mp.4001878. [DOI] [PubMed] [Google Scholar]
- Noga O, Peiser M, Altenahr M, Knieling H, Wanner R, Hanf G, Grosse R, Suttorp N. Differential activation of dendritic cells by nerve growth factor and brain-derived neurotrophic factor. Clin Exp Allergy. 2007;37(11):1701–1708. doi: 10.1111/j.1365-2222.2007.02832.x. [DOI] [PubMed] [Google Scholar]
- Norton N, Moskvina V, Morris DW, Bray NJ, Zammit S, Williams NM, Williams HJ, Preece AC, Dwyer S, Wilkinson JC, Spurlock G, Kirov G, Buckland P, Waddington JL, Gill M, Corvin AP, Owen MJ, O’Donovan MC. Evidence that interaction between neuregulin 1 and its receptor erbB4 increases susceptibility to schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(1):96–101. doi: 10.1002/ajmg.b.30236. [DOI] [PubMed] [Google Scholar]
- Numan S, Gall CM, Seroogy KB. Developmental expression of neurotrophins and their receptors in postnatal rat ventral midbrain. J Mol Neurosci. 2005;27(2):245–260. doi: 10.1385/JMN:27:2:245. [DOI] [PubMed] [Google Scholar]
- Ohnuma T, Kato H, Arai H, McKenna PJ, Emson PC. Expression of Fyn, a non-receptor tyrosine kinase in prefrontal cortex from patients with schizophrenia and its correlation with clinical onset. Brain Res Mol Brain Res. 2003;112(1–2):90–94. doi: 10.1016/s0169-328x(03)00051-2. [DOI] [PubMed] [Google Scholar]
- O’Leary C, Desbonnet L, Clarke N, Petit E, Tighe O, Lai D, Harvey R, Waddington JL, O’Tuathaigh C. Phenotypic effects of maternal immune activation and early postnatal milieu in mice mutant for the schizophrenia risk gene neuregulin-1. Neuroscience. 2014;277:294–305. doi: 10.1016/j.neuroscience.2014.06.028. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, Iyo M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry. 2006;59(6):546–554. doi: 10.1016/j.biopsych.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Palomino A, Vallejo-Illarramendi A, Gonzalez-Pinto A, Aldama A, Gonzalez-Gomez C, Mosquera F, Gonzalez-Garcia G, Matute C. Decreased levels of plasma BDNF in first-episode schizophrenia and bipolar disorder patients. Schizophr Res. 2006;86(1–3):321–322. doi: 10.1016/j.schres.2006.05.028. [DOI] [PubMed] [Google Scholar]
- Pandya CD, Kutiyanawalla A, Pillai A. BDNF-TrkB signaling and neuroprotection in schizophrenia. Asian J Psychiatr. 2013;6(1):22–28. doi: 10.1016/j.ajp.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya CD, Pillai A. TrkB interacts with ErbB4 and regulates NRG1-induced NR2B phosphorylation in cortical neurons before synaptogenesis. Cell Commun Signal. 2014;12(1):47. doi: 10.1186/s12964-014-0047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection: window on neuroimmune interactions in fetal brain development and mental illness. Curr Opin Neurobiol. 2002;12(1):115–118. doi: 10.1016/s0959-4388(02)00299-4. [DOI] [PubMed] [Google Scholar]
- Pillai A. Brain-derived neurotropic factor/TrkB signaling in the pathogenesis and novel pharmacotherapy of schizophrenia. Neurosignals. 2008;16(2–3):183–193. doi: 10.1159/000111562. [DOI] [PubMed] [Google Scholar]
- Pillai A, Mahadik SP. Increased truncated TrkB receptor expression and decreased BDNF/TrkB signaling in the frontal cortex of reeler mouse model of schizophrenia. Schizophr Res. 2008;100(1–3):325–333. doi: 10.1016/j.schres.2007.11.030. [DOI] [PubMed] [Google Scholar]
- Piontkewitz Y, Assaf Y, Weiner I. Clozapine administration in adolescence prevents postpubertal emergence of brain structural pathology in an animal model of schizophrenia. Biol Psychiatry. 2009;66(11):1038–1046. doi: 10.1016/j.biopsych.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Piontkewitz Y, Arad M, Weiner I. Abnormal trajectories of neurodevelopment and behavior following in utero insult in the rat. Biol Psychiatry. 2011a;70(9):842–851. doi: 10.1016/j.biopsych.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Piontkewitz Y, Arad M, Weiner I. Risperidone administered during asymptomatic period of adolescence prevents the emergence of brain structural pathology and behavioral abnormalities in an animal model of schizophrenia. Schizophr Bull. 2011b;37(6):1257–1269. doi: 10.1093/schbul/sbq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2(1):24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Pratt L, Ni L, Ponzio NM, Jonakait GM. Maternal inflammation promotes fetal microglial activation and increased cholinergic expression in the fetal basal forebrain: role of interleukin-6. Pediatr Res. 2013;74(4):393–401. doi: 10.1038/pr.2013.126. [DOI] [PubMed] [Google Scholar]
- Ray MT, Shannon Weickert C, Webster MJ. Decreased BDNF and TrkB mRNA expression in multiple cortical areas of patients with schizophrenia and mood disorders. Transl Psychiatry. 2014;4:e389. doi: 10.1038/tp.2014.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richtand NM, Ahlbrand R, Horn P, Stanford K, Bronson SL, McNamara RK. Effects of risperidone and paliperidone pre-treatment on locomotor response following prenatal immune activation. J Psychiatr Res. 2011;45(9):1194–1201. doi: 10.1016/j.jpsychires.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richtand NM, Ahlbrand R, Horn PS, Chambers B, Davis J, Benoit S. Effects of prenatal immune activation and peri-adolescent stress on amphetamine-induced conditioned place preference in the rat. Psychopharmacology (Berl) 2012;222(2):313–324. doi: 10.1007/s00213-012-2646-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roenker N, Gudelsky GA, Ahlbrand RL, Bronson SL, Kern JR, Waterman H, Richtand NM. Effect of paliperidone and risperidone on extracellular glutamate in the prefrontal cortex of rats exposed to prenatal immune activation or MK-801. Neurosci Lett. 2011;500(3):167–171. doi: 10.1016/j.neulet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero E, Guaza C, Castellano B, Borrell J. Ontogeny of sensorimotor gating and immune impairment induced by prenatal immune challenge in rats: implications for the etiopathology of schizophrenia. Mol Psychiatry. 2010;15(3):372–383. doi: 10.1038/mp.2008.44. [DOI] [PubMed] [Google Scholar]
- Rybakowski JK. BDNF gene: functional Val66Met polymorphism in mood disorders and schizophrenia. Pharmacogenomics. 2008;9(11):1589–1593. doi: 10.2217/14622416.9.11.1589. [DOI] [PubMed] [Google Scholar]
- Seroogy KB, Dickerson JW, Cassella SN, Zhang-Auberson L. Neuregulins. In: Kastin AJ, editor. Handbook of Biologically Active Peptides. 2. Elsevier Academic Press; Amsterdam: 2013. pp. 1633–1638. [Google Scholar]
- Seroogy KB, Herman JP. In situ hybridization approaches to the study of the nervous system. In: Turner A, Bachelard H, editors. Neurochemistry - A practical approach. Oxford University Press; Oxford: 1997. pp. 121–150. [Google Scholar]
- Shamir A, Kwon OB, Karavanova I, Vullhorst D, Leiva-Salcedo E, Janssen MJ, Buonanno A. The importance of the NRG-1/ErbB4 pathway for synaptic plasticity and behaviors associated with psychiatric disorders. J Neurosci. 2012;32(9):2988–2997. doi: 10.1523/JNEUROSCI.1899-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23(1):297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Tu N, Patterson PH. Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in the fetus. Int J Dev Neurosci. 2005;23(2–3):299–305. doi: 10.1016/j.ijdevneu.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Silberberg G, Darvasi A, Pinkas-Kramarski R, Navon R. The involvement of ErbB4 with schizophrenia: association and expression studies. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(2):142–148. doi: 10.1002/ajmg.b.30275. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Giusti P, Facci L. Microglia and mast cells: two tracks on the road to neuroinflammation. FASEB J. 2012;26(8):3103–3117. doi: 10.1096/fj.11-197194. [DOI] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27(40):10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, Brynjolfsson J, Gunnarsdottir S, Ivarsson O, Chou TT, Hjaltason O, Birgisdottir B, Jonsson H, Gudnadottir VG, Gudmundsdottir E, Bjornsson A, Ingvarsson B, Ingason A, Sigfusson S, Hardardottir H, Harvey RP, Lai D, Zhou M, Brunner D, Mutel V, Gonzalo A, Lemke G, Sainz J, Johannesson G, Andresson T, Gudbjartsson D, Manolescu A, Frigge ML, Gurney ME, Kong A, Gulcher JR, Petursson H, Stefansson K. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71(4):877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Thorgeirsson TE, Gulcher JR, Stefansson K. Neuregulin 1 in schizophrenia: out of Iceland. Mol Psychiatry. 2003;8(7):639–640. doi: 10.1038/sj.mp.4001384. [DOI] [PubMed] [Google Scholar]
- Stevens HE, Su T, Yanagawa Y, Vaccarino FM. Prenatal stress delays inhibitory neuron progenitor migration in the developing neocortex. Psychoneuroendocrinology. 2013;8(4):509–521. doi: 10.1016/j.psyneuen.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Zhu JJ. Ras and Rap signaling in synaptic plasticity and mental disorders. Neuroscientist. 2011;17(1):54–78. doi: 10.1177/1073858410365562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strous RD, Shoenfeld Y. Schizophrenia, autoimmunity and immune system dysregulation: a comprehensive model updated and revisited. J Autoimmun. 2006;27(2):71–80. doi: 10.1016/j.jaut.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Taylor P. Practical Teratology. Academic Press; London: 1986. [Google Scholar]
- Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270(5236):593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- Van den Eynde K, Missault S, Fransen E, Raeymaekers L, Willems R, Drinkenburg W, Timmermans JP, Kumar-Singh S, Dedeurwaerdere S. Hypolocomotive behaviour associated with increased microglia in a prenatal immune activation model with relevance to schizophrenia. Behav Brain Res. 2014;258:179–186. doi: 10.1016/j.bbr.2013.10.005. [DOI] [PubMed] [Google Scholar]
- Vuillermot S, Weber L, Feldon J, Meyer U. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. J Neurosci. 2010;30(4):1270–1287. doi: 10.1523/JNEUROSCI.5408-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Runyan S, Yadin E, Friedman E. Prenatal exposure to cocaine selectively reduces D1 dopamine receptor-mediated activation of striatal Gs proteins. J Pharmacol Exp Ther. 1995;273(1):492–498. [PubMed] [Google Scholar]
- Waterhouse EG, Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol Cell Neurosci. 2009;42(2):81–89. doi: 10.1016/j.mcn.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Hyde TM, Lipska BK, Herman MM, Weinberger DR, Kleinman JE. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry. 2003;8(6):592–610. doi: 10.1038/sj.mp.4001308. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Ligons DL, Romanczyk T, Ungaro G, Hyde TM, Herman MM, Weinberger DR, Kleinman JE. Reductions in neurotrophin receptor mRNAs in the prefrontal cortex of patients with schizophrenia. Mol Psychiatry. 2005;10(7):637–650. doi: 10.1038/sj.mp.4001678. [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Yoon BH, Jun JK, Romero R, Park KH, Gomez R, Choi JH, Kim IO. Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1beta, and tumor necrosis factor-alpha), neonatal brain white matter lesions, and cerebral palsy. Am J Obstet Gynecol. 1997;177(1):19–26. doi: 10.1016/s0002-9378(97)70432-0. [DOI] [PubMed] [Google Scholar]
- Zheng W, Wang H, Zeng Z, Lin J, Little PJ, Srivastava LK, Quirion R. The possible role of the Akt signaling pathway in schizophrenia. Brain Res. 2012;1470:145–158. doi: 10.1016/j.brainres.2012.06.032. [DOI] [PubMed] [Google Scholar]