

Abstract
β-Site APP-cleaving enzyme 1 (BACE1) initiates the generation of amyloid-β (A β), thus representing a prime therapeutic target for Alzheimer’s disease (AD). Previous work including ours has used BACE1 haploinsufficiency (BACE1+/−; i.e., 50% reduction) as a therapeutic relevant model to evaluate the efficacy of partial β-secretase inhibition. However, it is unclear whether the extent of Aβ reductions in amyloid precursor protein (APP) transgenic mice with BACE1+/− gene ablation may vary with sex or disease progression. Here, we compared the impacts of BACE1 haploinsufficiency on Aβ concentrations and APP processing in 5XFAD Alzheimer mice (1) between males and females and (2) between different stages with moderate and robust Aβ accumulation. First, male and female 5XFAD mice at 6–7 months of age showed equivalent levels of Aβ, BACE1, full-length APP and its metabolites. BACE1 haploinsufficiency significantly lowered soluble Aβ oligomers, total Aβ42 levels and plaque burden in 5XFAD mouse brains irrespective of sex. Furthermore, there was no sex difference in reductions of β-cleavage products of APP (C99 and sAPPβ) found in BACE1+/−·5XFAD mice relative to BACE1+/+·5XFAD controls. Meanwhile, APP and sAPPα levels in BACE1+/−·5XFAD mice were higher than those of 5XFAD controls regardless of sex. Based on these observations, we next combined male and female data to examine the effects of BACE1 haploinsufficiency in 5XFAD mice at 12–14 months of age, as compared with those in 6–7-month-old 5XFAD mice. Oligomeric Aβ and C99 levels were dramatically elevated in older 5XFAD mice. Although the β-metabolites of APP were significantly reduced by BACE1 haploinsufficiency in both age groups, high levels of these toxic amyloidogenic fragments remained in 12–14-month-old BACE1+/−·5XFAD mice. The present findings are consistent with our previous behavioral data showing that BACE1 haploinsufficiency rescues memory deficits in 5XFAD mice irrespective of sex but only in the younger age group.
Keywords: BACE1, haploinsufficiency, Aβ oligomers, C99, sex, 5XFAD
INTRODUCTION
BACE1 is an aspartyl protease that is responsible for cleaving amyloid precursor protein (APP) at the β-secretase cleavage site, the first and rate-limiting step in the production of toxic amyloid-β (Aβ) peptides (Vassar, 2001). Given increasing consensus that Aβ accumulates to high levels in the brain and triggers a pathogenic cascade ultimately leading to neuron death and memory deficits in Alzheimer’s disease (AD) (Hardy and Higgins, 1992, Hardy and Selkoe, 2002), BACE1 is a prime target to prevent or treat this devastating neurodegenerative disorder (Ohno, 2006, Cole and Vassar, 2007, Ohno, 2008, Vassar et al., 2009). This view is strongly supported by experimental data demonstrating that BACE1 gene deletion (BACE1−/−) dramatically reduces cerebral Aβ levels and prevents the development of AD-like pathologies and memory impairments in different lines of APP transgenic mice (Ohno et al., 2004, Laird et al., 2005, Ohno et al., 2006, Ohno et al., 2007). However, a growing number of BACE1 substrates besides APP and some adverse phenotypes in BACE1−/− knockout mice imply that chronic over-inhibition of β-secretase activities may induce potential mechanism-based side effects (Ohno, 2006, Ohno, 2008, Vassar, 2014, Vassar et al., 2014, Yan and Vassar, 2014). In fact, a recent investigation clearly demonstrates that chronic oral administration of bioavailable BACE1 inhibitors at the high dosage to normal adult mice impairs cognition as well as structural and functional synaptic plasticity, the effects that were occluded in BACE1−/− mice (Filser et al., 2015). Although clinical relevance of these findings remains to be determined, the mouse model studies suggest a need for the successful balance between tolerable side effects and sufficient Aβ reductions for efficacy in therapeutic BACE1 inhibition.
Our laboratory and others have used BACE1 haploinsufficiency (BACE1+/−; i.e., 50% reduction) as a therapeutic relevant model to evaluate the efficacy of partial inhibition of this enzyme (Laird et al., 2005, McConlogue et al., 2007, Devi and Ohno, 2010a, Devi and Ohno, 2010b, Kimura et al., 2010, Rabe et al., 2011, Chabrier et al., 2012, Devi and Ohno, 2012, Devi and Ohno, 2013, Sadleir et al., 2015). In these studies, BACE1+/− reduction is sufficient to lower brain Aβ concentrations and prevent AD-like phenotypes such as amyloid plaque and tau pathologies, cholinergic neuron death, mitochondrial dysfunction, hippocampal CA1 synaptic failure, and memory deficits in transgenic mouse models. However, it is noted that the degree of Aβ lowering associated with BACE1 haploinsufficiency may vary depending on sex, age or disease progression in different APP mice used. To address this issue more comprehensively, we compared the effects of BACE1+/− ablation on cerebral Aβ levels and APP processing in (1) male vs. female 5XFAD and (2) younger vs. older 5XFAD mice.
EXPERIMENTAL PROCEDURES
Animals
We used 5XFAD mice (Tg6799 line) that co-overexpress familial AD (FAD) mutant forms of human APP (the Swedish mutation: K670N, M671L; the Florida mutation: I716V; the London mutation: V717I) and presenilin 1 (PS1) (M146L; L286V) transgenes under transcriptional control of the neuron-specific mouse Thy-1 promoter (Oakley et al., 2006, Ohno et al., 2006, Ohno et al., 2007). Hemizygous 5XFAD transgenic mice (backcrossed to C57Bl/6 with >10 generations) were crossbred to heterozygous BACE1 knockout (BACE1+/−) mice (C57Bl/6 background, The Jackson Laboratory, Bar Harbor, ME, USA) (Cai et al., 2001, Laird et al., 2005). Genotyping was performed by PCR analysis of tail DNA. Male and female mice at 6–7 and 12–14 months of age, which showed moderate and massive levels of Aβ deposition in 5XFAD controls, respectively (Oakley et al., 2006, Ohno et al., 2007, Kimura et al., 2010), were used for the experiments. All animal procedures were approved by the Nathan Kline Institute Animal Care and Use Committee and conducted in accordance with National Institutes of Health guidelines.
Western blotting
Hemibrain samples were taken from the mice under deep isoflurane anesthesia and were snapfrozen for biochemical assays. For immunoblot analysis, each sample was homogenized in 8-fold volumes of cold homogenization medium containing 70 mM sucrose, 210 mM mannitol, 2 mM HEPES, 0.1 mM EDTA and protease/phosphatase inhibitor cocktail, and centrifuged at 10,000 g for 10 min to remove any insoluble material. Protein concentrations were determined by a BCA protein assay kit (Pierce, Rockford, IL, USA), and 10–50 μg of protein was run on NuPAGE 4–12% or 10% Bis-Tris gels (Invitrogen, Carlsbad, CA, USA) and transferred to nitrocellulose membrane. After blocking, membranes were probed with the following primary antibodies: anti-BACE1 (1:1,000, B0681, Sigma-Aldrich, St. Louis, MO, USA), an antibody that recognizes C-terminal epitope in APP (1:1,000, C1/6.1, kindly provided by Dr. Paul Mathews, Nathan Kline Institute) to detect full-length APP/C-terminal fragments, an antibody specific for the β-secretase-cleaved soluble ectodomain of APP with the Swedish mutation (sAPPβ-Swe; 6A1) (1:1,000, 10321, IBL America, Minneapolis, MN, USA), anti-sAPPα (1:500, 11088, Immuno-Biological Laboratories, Minneapolis, MN, USA), and anti-β-actin (1:15,000, AC-15, Sigma-Aldrich). The membranes were then incubated with horseradish peroxidase-conjugated secondary IgG. Immunoblot signals were visualized by an ECL chemiluminescence substrate reagent kit (Pierce) and quantified by densitometric scanning and image analysis using Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
ELISAs of soluble Aβ oligomers and total Aβ42
To measure the concentrations of soluble Aβ oligomers, each hemibrain sample was homogenized in 8-fold volumes of homogenization medium, as described above. To quantitate total levels of Aβ42, each hemibrain was extracted in 8-fold volumes of cold 5 M guanidine HCl plus 50 mM Tris HCl (pH 8.0) buffer and centrifuged at 20,000 g for 1 h at 4°C to remove insoluble material. Final guanidine HCl concentrations were below 0.1 M. Protein concentrations were determined by a BCA protein assay kit (Pierce). Supernatant fractions were analyzed by well-established human Aβ ELISA kits specific to oligomeric forms of Aβ (27725, IBL America, Minneapolis, MN, USA) and Aβ42 (KHB3441, Invitrogen) according to the protocols of the manufacturers. Optical densities at 450 nm of each well were read on a VersaMax tunable microplate reader (Molecular Devices, Sunnyvale, CA, USA), and sample Aβ oligomer and Aβ42 concentrations were determined by comparison with the respective standard curves. Aβ oligomer and Aβ42 concentration values were normalized to total brain protein concentrations and expressed in picograms and nanograms per milligram of total protein, respectively.
Aβ immunohistochemistry
Mice were transcardially perfused with 0.1 M phosphate buffered saline (PBS, pH7.4), followed by 4% paraformaldehyde in PBS under deep isoflurane anesthesia. Brains were postfixed for 24 h in 4% paraformaldehyde in PBS at 4°C and transferred to PBS. The brain was sectioned coronally at 30 μm using a vibratome (VT1200, Leica Microsystems, Wetzlar, Germany), and successive sections were stored in PBS containing 0.05% sodium azide at 4°C. Brain sections taken at levels between −1.7 and −1.9 mm to bregma according to the mouse brain atlas of Franklin and Paxinos (2008) were stained by the avidin-biotin peroxidase complex (ABC) method as described previously (Kimura et al., 2010, Devi and Ohno, 2013, Devi and Ohno, 2015). Briefly, the sections were incubated overnight at 4°C with mouse monoclonal anti-Aβ1–16 (6E10) antibody (1:200, SIG-39347, Covance, Princeton, NJ, USA). The ABC kit (PK-2200, Vector Laboratories, Burlingame, CA, USA) was utilized with 3,3′-diaminobenzidine tetrahydrochloride as a chromogen to visualize the reaction product. The sections were then mounted on charged slides, dehydrated in a series of alcohol, cleared in xylene and covered with a coverslip. Light microscopy was conducted on an Axioskop 2 microscope equipped with an AxioCaM HRc digital camera (Zeiss, Oberkochen, Germany) for capturing images. Semi-quantitative analysis was performed using AxioVision imaging software with the AutoMeasure module (Zeiss). The threshold optical density that discriminated staining from background was determined and held constant for all quantifications. Identified objects were individually inspected by the same investigator to confirm the object as a plaque or not in a blinded manner. Percentage area occupied by Aβ deposits in the hippocampus was assessed bilaterally to compare plaque burden between BACE1+/−·5XFAD mice and 5XFAD controls.
Data analysis
Data were analyzed by a two-way analysis of variance (ANOVA) and post-hoc Bonferroni comparisons were performed to determine the significance of differences between the groups when appropriate. Data were presented as mean ± S.E.M. and the level of significance was set for p value less than 0.05.
RESULTS
Comparison of changes in Aβ levels and APP processing between male and female BACE1+/−·5XFAD mice at 6–7 months of age
We used 5XFAD APP/PS1 transgenic mice; a rapid-onset and aggressive amyloid model based on a combination of five FAD mutations and consequently accelerated production of Aβ42 that is more toxic or prone to aggregation (Oakley et al., 2006, Ohno et al., 2006, Ohno et al., 2007). A series of investigations from our laboratory as well as others has demonstrated that 5XFAD mice begin to develop visible amyloid deposition as early as 2 months of age and exhibit significant cognitive impairments on hippocampus-dependent behavioral tasks around 6 months concomitant with moderate Aβ accumulation and the onset of Schaffer collateral-CA1 synaptic dysfunction (Oakley et al., 2006, Ohno et al., 2006, Kimura and Ohno, 2009, Ohno, 2009, Devi and Ohno, 2010a, Hongpaisan et al., 2011, Chen et al., 2012, Jawhar et al., 2012, Seo et al., 2014, Zhang et al., 2014, Devi and Ohno, 2015). We first compared the effects of BACE1 haploinsufficiency on soluble Aβ oligomers, a plausible mediator of synaptic and memory failure in AD (Haass and Selkoe, 2007, Ferreira and Klein, 2011, Zahs and Ashe, 2013), in male and female 5XFAD mice at 6–7 months of age (Fig. 1A). ELISA assays that specifically detect oligomeric forms of Aβ showed equivalent baseline levels of Aβ oligomers in male and female 5XFAD mice. A two-way ANOVA revealed a significant main effect of BACE1+/− mutation (p < 0.05) in the absence of a main effect of sex or a significant genotype X sex interaction. Post-hoc Bonferroni tests indicated that Aβ oligomers were significantly and equivalently reduced in male and female BACE1+/−·5XFAD mouse brains as compared with their respective 5XFAD controls (p < 0.05). Similarly, a two-way ANOVA for total Aβ42 levels in guanidine extracts (Fig. 1B) and hippocampal plaque loads (Fig. 1C) also revealed only significant main effects of BACE1+/− genotype (p < 0.05) without a main effect of sex or a significant genotype X sex interaction. Levels of soluble Aβ oligomers, total Aβ42 and plaque burden in male and female BACE1+/−·5XFAD mice relative to their respective 5XFAD controls are summarized in Table 1.
Fig. 1.
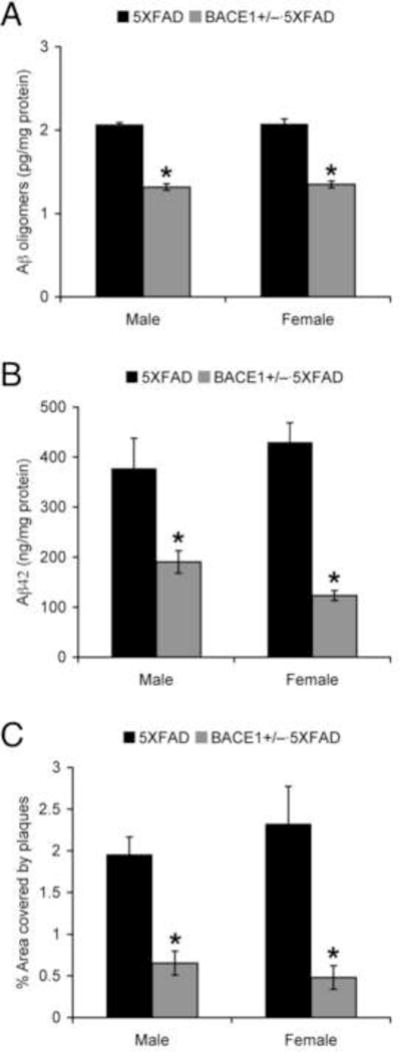
Effects of BACE1 haploinsufficiency on Aβ concentrations and plaque burden in male and female 5XFAD mice at 6–7 months of age. (A, B) Levels of soluble Aβ oligomers in hemibrain samples (A) and total Aβ42 in guanidine extracts (B) were quantified by sandwich ELISAs and expressed in picograms and nanograms per milligram of total protein, respectively (n = 3–8 mice per group). (C) Percentage area occupied by plaques, immunostained with the 6E10 anti-Aβ antibody in the hippocampus, was measured for quantification (n = 5–6 mice per group). All of these measures for β-amyloidosis were equivalent in male and female 5XFAD controls and significantly reduced regardless of sex in BACE1+/−·5XFAD mice (*p < 0.05 vs. 5XFAD). All data are presented as mean ± S.E.M.
Table 1.
Summary of percent levels of Aβ, BACE1 and APP metabolites in male and female BACE1+/−·5XFAD mice at 6–7 months of age as compared with their respective 5XFAD controls
| % relative to 5XFAD controls (mean ± S.E.M.)
|
||
|---|---|---|
| Male BACE1+/−·5XFAD | Female BACE1+/−·5XFAD | |
| Aβ oligomers | 63.8 ± 1.8 | 65.1 ± 2.0 |
| Total Aβ42 | 50.5 ± 5.9 | 28.7 ± 2.4 |
| Plaque load | 33.3 ± 7.4 | 20.6 ± 6.0 |
| BACE1 | 38.0 ± 3.4 | 57.9 ± 9.2 |
| fl-APP | 106.9 ± 2.6 | 125.2 ± 5.1 |
| C99 | 51.0 ± 3.5 | 44.9 ± 2.2 |
| C83 | 74.7 ± 5.0 | 72.5 ± 15.9 |
| sAPPβ-Swe | 56.7 ± 3.7 | 43.1 ± 4.7 |
| sAPPα | 123.0 ± 5.6 | 153.7 ± 5.3 |
Next, we performed western blot analysis and compared changes in APP processing between male and female 5XFAD mice (Fig. 2A). Two-way ANOVAs for BACE1 expression (Fig. 2B) or β-metabolites of APP such as the β-secretase-cleaved C-terminal fragment of APP (C99) (Fig. 2D) and its counterpart sAPPβ (Fig. 2F) revealed significant main effects of BACE1 haploinsufficiency (p < 0.05) without main effects of sex or significant genotype X sex interactions. Post-hoc Bonferroni tests showed that all of these parameters associated with the β-cleavage of APP were indistinguishable between male and female 5XFAD controls and significantly reduced to the same extent in male and female BACE1+/−·5XFAD mouse brains (p < 0.05). Meanwhile, two-way ANOVAs for full-length APP (Fig. 2C) and the α-secretase-cleaved soluble ectodomain of APP (sAPPα) (Fig. 2G) revealed significant elevations in association with BACE1+/− deletion (p < 0.05) in the absence of main effects of sex or significant genotype X sex interactions. Post-hoc tests indicated significant increases of full-length APP and sAPPα in female BACE1+/−·5XFAD mice (p < 0.05) and a trend toward increase of sAPPα in male BACE1+/−·5XFAD mice (p = 0.052) as compared with respective 5XFAD controls. Intriguingly, a trend toward decrease rather than increase in the other α-cleavage product C83 was found in BACE1+/−·5XFAD mice (p = 0.079; two-way ANOVA), a change in males reaching statistical significance (p < 0.05) (Fig. 2E). Levels of the parameters for APP processing in male and female BACE1+/−·5XFAD mice as compared to the respective 5XFAD controls are summarized in Table 1.
Fig. 2.
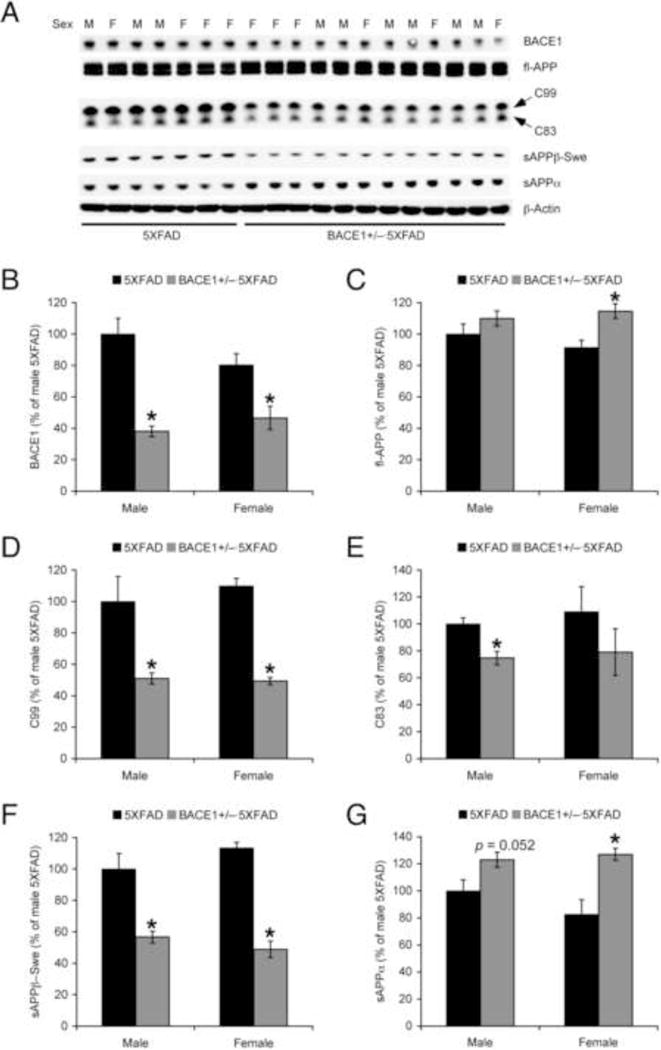
Effects of BACE1 haploinsufficiency on APP processing in male and female 5XFAD mice at 6–7 months of age. (A) Representative immunoblots of protein extracts from hemibrain homogenates of male (M) and female (F) mice. (B–G) Immunoreactive bands were quantified and expressed as the percentage of male 5XFAD controls (n = 3–6 mice per group). BACE1 haploinsufficiency significantly reduced BACE1, C99 and sAPPβ with the Swedish mutation (sAPPβ-Swe) in 5XFAD mice, whereas it increased full-length APP and sAPPα (*p < 0.05 vs. 5XFAD). There was no significant sex difference in these changes observed in BACE1+/−·5XFAD mice as well as baseline values in 5XFAD controls. All data are presented as mean ± S.E.M.
Collectively, reductions in Aβ levels and pathology as well as changes in APP processing occurred equivalently in male and female BACE1+/−·5XFAD mice as compared with those of respective 5XFAD controls.
Comparison of changes in soluble Aβ oligomers and APP processing between younger and older BACE1+/−·5XFAD mice
Since younger 5XFAD mice showed no sex difference in their response to BACE1 haploinsufficiency, we next combined male and female data together to compare the effects of BACE1+/− reduction in 5XFAD mice at 6–7 and 12–14 months of age that develop moderate and massive Aβ plaque pathology, respectively (Fig. 3). ELISA data showed that levels of soluble Aβ oligomers were greatly increased with age in 5XFAD mice (p < 0.05; 385.6% relative to younger 5XFAD), while the degree of Aβ oligomer reductions in BACE1+/−·5XFAD mice compared with 5XFAD controls (p < 0.05) were indistinguishable between younger and older age groups (64.5% and 63.1%, respectively) (Fig. 3A). Consequently, residual levels of Aβ oligomers in 12–14-month-old BACE1+/−·5XFAD mice were significantly higher than those of 6-month-old 5XFAD mice (p < 0.05). Similar patterns of changes were observed in C99 levels as assessed by immunoblot analysis of brain samples (Fig. 3B, D). Levels of C99 in younger and older BACE1+/−·5XFAD mice compared to the age-matched 5XFAD controls were 43.3% (p < 0.05) and 79.2% (p < 0.05), respectively.
Fig. 3.
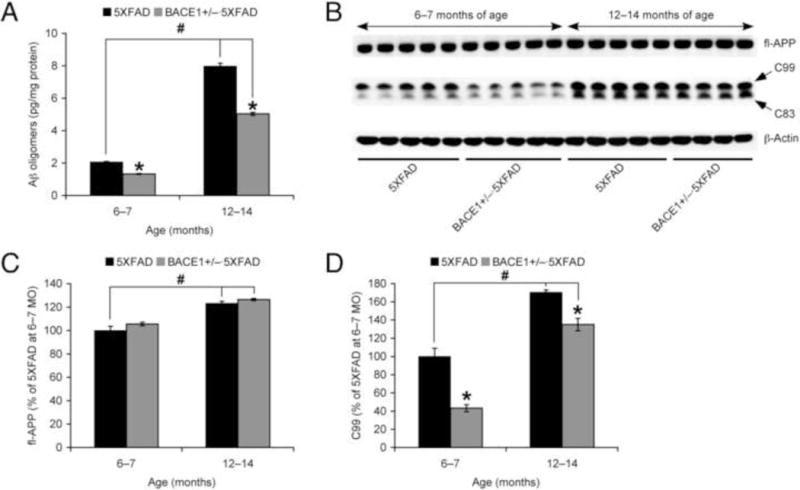
Effects of BACE1 haploinsufficiency on soluble Aβ oligomers and APP processing in 5XFAD mice during different disease stages. (A) Levels of soluble Aβ oligomers in hemibrain samples were quantified by sandwich ELISA and expressed in picograms per milligram of total protein (n = 6–9 mice per group). (B) Representative immunoblots of protein extracts from hemibrain homogenates of mice. (C, D) Immunoreactive bands were quantified and expressed as the percentage of 5XFAD controls at 6–7 months of age (n = 4–5 mice per group). While BACE1 haploinsufficiency significantly reduced Aβ oligomer and C99 levels in 5XFAD mice irrespective of age (*p < 0.05 vs. 5XFAD controls), both toxic β-cleavage products of APP were elevated with age in 5XFAD mice and their remaining levels in 12–14-month-old BACE1+/−·5XFAD mice were significantly higher than those of 6–7-month-old 5XFAD controls (#p < 0.05). Furthermore, BACE1 haploinsufficiency had no effect on age-dependent increases in full-length APP expression in 5XFAD mice. All data are presented as mean ± S.E.M.
As a mechanism underlying accelerated β-amyloidogenesis in 5XFAD mice with aging, we previously demonstrated that BACE1 expression is significantly elevated (~2-fold relative to wild-type controls) in older 5XFAD mice (≥12 months of age) (Devi and Ohno, 2010b, Devi and Ohno, 2013). In these studies, haploinsufficiency lowered BACE1 expression by ~50% regardless of age in concordance with a single BACE1 allele ablation, whereas BACE1 levels equivalent to wild-type controls remained in older BACE1+/−·5XFAD mice (i.e., twice the gene copy number). In addition, the present results indicated that full-length APP was also age-dependently increased in 5XFAD mouse brains (p < 0.05) (Fig. 3B, C). Furthermore, BACE1 haploinsufficiency did not affect the upregulation of APP expression in 12–14-month-old 5XFAD mice. Together, these results suggest that older BACE1+/−·5XFAD mice have persistent elevations of APP as well as BACE1, thus suffering from high residual levels of toxic β-amyloidogenic peptides (not only Aβ but also C99) in brains.
DISCUSSION
Our previous investigations have demonstrated beneficial effects of BACE1 haploinsufficiency, including the prevention of cognitive and synaptic dysfunctions, in 5XFAD mice at ~6 months of age harboring moderate levels of Aβ accumulation (Devi and Ohno, 2010b, Kimura et al., 2010, Devi and Ohno, 2012, Devi and Ohno, 2013). The findings are based on the combined data from male and female animals because of the absence of significant sex difference. Consistent with these results, we showed that male and female 5XFAD mouse brains at 6–7 months of age have equivalent levels of total Aβ42, plaque deposition, and soluble Aβ oligomers, critical forms of Aβ proposed to cause synaptic and memory failure in AD (Haass and Selkoe, 2007, Ferreira and Klein, 2011, Zahs and Ashe, 2013). Importantly, the present study demonstrated significant reductions of these Aβ species in BACE1+/−·5XFAD mice irrespective of sex. Furthermore, BACE1 haploinsufficiency significantly lowered direct β-cleavage metabolites of APP (C99 and sAPPβ) in 5XFAD mouse brains, changes that were also indistinguishable between males and females. Therefore, these data indicate that BACE1 reduction by 50% is sufficient to suppress de novo Aβ production in both male and female 5XFAD transgenic mice and consequently lower cerebral Aβ, as measured by soluble oligomeric species as well as by guanidine-extracted total amounts of Aβ42 or amyloid plaque load.
In contrast to our findings, Sadleir et al. (2015) recently reported that Aβ levels were significantly reduced in female but not in male BACE1+/−·5XFAD mice at 4, 6 and 9 months of age. Intriguingly, baseline levels of Aβ42, C99 and sAPPβ were significantly higher in female 5XFAD mouse brains than those of age-matched male 5XFAD controls. It is hypothesized that the greater amounts of β-products of APP may be accounted for by the higher transgenic APP levels in female 5XFAD mice most likely due to the presence of estrogen responsive element in their murine Thy-1 promotor sequence. As a consequence, they argue that BACE1 expression is not in excess over APP in female 5XFAD mice with higher transgene overexpression (~200% relative to endogenous APP level), rendering Aβ and other β-product levels responsive to reductions by BACE1 haploinsufficiency. Meanwhile, BACE1 may be in excess over APP in 5XFAD males with lower transgenic APP overexpression (~160%) so that 50% BACE1 reduction has little effect on lowering Aβ. In this study, we found no difference in APP expression levels between male and female 5XFAD mice, and APP overexpression relative to wild-type control levels in the 5XFAD model (Tg6799 line) was much higher (300–500%) in a series of previous investigations including ours (Oakley et al., 2006, Ohno et al., 2007, Hong et al., 2013, Devi and Ohno, 2014, Py et al., 2014, Devi et al., 2015). The results using well-established anti-APP antibodies against the C-terminus (C1/6.1) and N-terminus (22C11) are consistent in these studies. Collectively, although it is unclear why APP overexpression was so low in a cohort of 5XFAD males tested by Sadleir et al. (2015) with anti-C-terminal APP (Y188), it seems reasonable to conceive that levels of β-cleaved metabolites are consistently sensitive to reductions by BACE1 haploinsufficiency if APP levels are highly overexpressed in 5XFAD mice regardless of sex. This is in accordance with the observations in other AD transgenic mice with high APP overexpression showing that BACE1+/− mutation-associated reductions of Aβ, C99 and sAPPβ are significant and indistinguishable between males and females (Laird et al., 2005, Rabe et al., 2011). Moreover, it should be noted that BACE1+/− knockout mice show no or only marginal reductions (10–20%) of endogenous Aβ40 as compared with wild-type controls (Pastorino et al., 2004, Nishitomi et al., 2006, Sankaranarayanan et al., 2008, Rabe et al., 2011). In this regard, while pharmacological data demonstrate that chronic treatments with bioavailable small-molecule BACE1 inhibitors (e.g., GRL-8234 and TAK-070) in preventive settings improve memory impairments concomitant with significant cerebral Aβ reductions in 5XFAD as well as Tg2576 mice (Fukumoto et al., 2010, Chang et al., 2011, Devi et al., 2015), it would be important to further evaluate their therapeutic efficacy in APP knock-in mice that circumvent APP overexpression (Webster et al., 2013, Li et al., 2014, Saito et al., 2014) and thus may be less responsive to rescue by partial BACE1 inhibition.
Whereas β-metabolites of APP were reduced, levels of full-length APP and sAPPβ were increased, to a lesser extent, in BACE1+/−·5XFAD mice at 6–7 months of age without a significant sex difference. These results are in agreement with previous observations in different APP transgenic mice including male and female APP23 as well as 5XFAD (Rabe et al., 2011, Sadleir et al., 2015), indicative of elevated steady-state APP levels and some compensatory activation of the α-secretory pathway (i.e., larger increase rates in sAPPα than APP) as a consequence of robust reduction in the β-cleavage of APP. Nevertheless, we unexpectedly found a trend toward decrease rather than increase in the other α-cleavage product C83 in BACE1+/−·5XFAD mice (p = 0.079). What mechanisms can reconcile this observation with an increase in the direct α-cleavage metabolite sAPPα? It is theoretically possible that C83 may be cleaved more (thus reduced) to compensate for C99 decrease and substitute for C99 to produce APP intracellular domain (AICD). However, this mechanism seems unlikely given a growing body of data showing that AICD is preferentially derived from C99 in the amyloidogenic pathway rather than from C83 in the nonamyloidogenic pathway (Goodger et al., 2009, Belyaev et al., 2010, Flammang et al., 2012). Meanwhile, it is interesting to note experimental evidence that the α-secretase-mediated proteolytic conversion of C99 to C83 may occur in AD conditions (Jager et al., 2009, Flammang et al., 2012). Since C83 signals are accompanied by more extensive C99 bands in 5XFAD mice overexpressing human APP with the Swedish mutation, it seems most likely that some component of C83 could derive from the α-cleavage of C99 in this model. We hypothesize that the C83 reduction found in BACE1+/−·5XFAD mice may reflect a secondary change that occurs as a consequence of prior C99 reduction. This view is supported by our recent pharmacological data demonstrating that administration of the β-secretase inhibitor GRL-8234 to 5XFAD mice induced C83 lowering concomitant with C99 reductions (Devi et al., 2015). The results are also consistent with clinical observations that the C-terminally truncated short fragments of Aβ, which are released by sequential β- and α-cleavage of APP, are elevated in AD compared to non-demented controls and their concentrations change in accordance with the availability of the substrate C99 (e.g., increase by γ-secretase inhibition) (Portelius et al., 2006, Portelius et al., 2010, Portelius et al., 2011).
Previous work from our laboratory and others has shown that partial BACE1 gene suppression (e.g., haploinsufficiency and siRNA) reduces Aβ/C99 levels and mitigates AD-like pathologies and memory impairments in transgenic mouse models (Singer et al., 2005, McConlogue et al., 2007, Devi and Ohno, 2010a, Devi and Ohno, 2010b, Kimura et al., 2010, Chabrier et al., 2012, Devi and Ohno, 2012, Devi and Ohno, 2013), while some reports indicate the decreased therapeutic efficacies with progression of disease. In 5XFAD mice at advanced age (≥12 months), BACE1 haploinsufficiency is no longer able to rescue memory deficits and has only marginal effects on highly increased levels of total Aβ42 and plaque burden (Devi and Ohno, 2010b, Devi and Ohno, 2012, Devi and Ohno, 2013). Likewise, chronic administration of BACE1 inhibitors ameliorates memory impairments concomitant with cerebral Aβ/C99 lowering in Tg2576 and 5XFAD mice only if the treatment is started at relatively earlier (or prepathological) stages (Fukumoto et al., 2010, Chang et al., 2011, Devi et al., 2015). In this study, we demonstrated that toxic β-cleaved fragments of APP, i.e., soluble Aβ oligomers (Haass and Selkoe, 2007, Ferreira and Klein, 2011, Zahs and Ashe, 2013) and C99 (Nalbantoglu et al., 1997, Lauritzen et al., 2012, Tamayev and D’Adamio, 2012, Tamayev et al., 2012) both of which are responsible for synaptic/cognitive dysfunctions and neurodegeneration in AD, were dramatically elevated in 5XFAD mouse brains during advanced stages. A combination of BACE1 elevations as reported in our previous studies (Devi and Ohno, 2010b, Devi and Ohno, 2013) and increased expression of its substrate APP shown in the present study seems to underlie the detrimental acceleration of β-amyloidogenic processing of APP in older 5XFAD mice. Interestingly, recent evidence indicates that both BACE1 and APP become elevated in neurons surrounding amyloid plaques in 5XFAD mice (Kandalepas et al., 2013), in brains of Aβ25–35 injected animals (Lin et al., 2009) and in primary cultured neurons following exposure to Aβ42 (Sadleir et al., 2014), representing a detrimental feed-forward link between Aβ accumulation and BACE1/APP elevations during AD progression. Mechanistically, although transgenic APP overexpression is under transcriptional control of the neuron-specific Thy-1 promoter in 5XFAD mice (Oakley et al., 2006), further posttranscriptional mechanisms (e.g., overactivation of the PERK-dependent translation initiation factor eIF2α phosphorylation pathway) are implicated in elevations of APP as well as BACE1 in this model during advanced disease stages (O’Connor et al., 2008, Devi and Ohno, 2010b, Devi and Ohno, 2013, Devi and Ohno, 2014).
Notably, BACE1 haploinsufficiency did not reverse the upregulation of APP (this study) or BACE1 (Devi and Ohno, 2013) in 5XFAD mice at later age. Similarly, neither APP nor BACE1 elevations were affected by treatments with the BACE1 inhibitor GRL-8234 (Devi et al., 2015). Consequently, BACE1+/−·5XFAD mice and GRL8234-treated 5XFAD mice at ≥12 months of age have high residual levels of toxic Aβ oligomers and C99 (even if partially reduced), which render these mice no longer responsive to cognitive rescue by partial BACE1 inhibition. To avoid potential mechanism-based adverse effects that may be caused by direct over-suppression of β-secretase activities such as chronic exposure to a highest dose of inhibitor drugs (Filser et al., 2015), our results suggest that BACE1 inhibitors in combination with APP-lowering drugs (Rosenkranz et al., 2013, Asuni et al., 2014) or agents that can block disease-specific BACE1-elevating mechanisms (Medeiros et al., 2012, Ly et al., 2013, Devi and Ohno, 2014) may represent safer and more efficacious interventions to treat memory deficits in diagnosed AD with established amyloid pathology.
Highlights.
BACE1+/− mutation significantly reduces Aβ levels irrespective of sex in 5XFAD mice.
BACE1 haploinsufficiency also affects APP processing regardless of sex in 5XFAD mice.
Both Aβ oligomers and C99 levels dramatically increase with age in 5XFAD mice.
These β-products of APP are also partially reduced in older BACE1+/−·5XFAD mice.
BACE1+/−·5XFAD mice at later age harbor high residual levels of Aβ oligomers and C99.
Acknowledgments
This work was supported by the Alzheimer’s Art Quilt Initiative Grant (M.O.) and the National Institutes of Health Grant AG044703 (M.O.).
Abbreviations
- Aβ
amyloid-β
- AD
Alzheimer’s disease
- ANOVA
analysis of variance
- APP
amyloid precursor protein
- BACE1
β-site APP-cleaving enzyme 1
- FAD
familial Alzheimer’s disease
- PS1
presenilin 1
- sAPPα
α-secretase-cleaved soluble ectodomain of APP
- sAPPβ
β-secretase-cleaved soluble ectodomain of APP
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Asuni AA, Guridi M, Pankiewicz JE, Sanchez S, Sadowski MJ. Modulation of amyloid precursor protein expression reduces β-amyloid deposition in a mouse model. Ann Neurol. 2014;75:684–699. doi: 10.1002/ana.24149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyaev ND, Kellett KA, Beckett C, Makova NZ, Revett TJ, Nalivaeva NN, Hooper NM, Turner AJ. The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a β-secretase-dependent pathway. J Biol Chem. 2010;285:41443–41454. doi: 10.1074/jbc.M110.141390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Chabrier MA, Blurton-Jones M, Agazaryan AA, Nerhus JL, Martinez-Coria H, Laferla FM. Soluble Aβ promotes wild-type tau pathology in vivo. J Neurosci. 2012;32:17345–17350. doi: 10.1523/JNEUROSCI.0172-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WP, Huang X, Downs D, Cirrito JR, Koelsch G, Holtzman DM, Ghosh AK, Tang J. β-Secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J. 2011;25:775–784. doi: 10.1096/fj.10-167213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang YP, Teng Z, Chen C. Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep. 2012;2:1329–1339. doi: 10.1016/j.celrep.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SL, Vassar R. The Alzheimer’s disease β-secretase enzyme, BACE1. Mol Neurodegener. 2007;2:22. doi: 10.1186/1750-1326-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. Genetic reductions of β-site amyloid precursor protein-cleaving enzyme 1 and amyloid-β ameliorate impairment of conditioned taste aversion memory in 5XFAD Alzheimer’s disease model mice. Eur J Neurosci. 2010a;31:110–118. doi: 10.1111/j.1460-9568.2009.07031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. Phospho-eIF2α level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS One. 2010b;5:e12974. doi: 10.1371/journal.pone.0012974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. Mitochondrial dysfunction and accumulation of the β-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol Dis. 2012;45:417–424. doi: 10.1016/j.nbd.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. Mechanisms that lessen benefits of β-secretase reduction in a mouse model of Alzheimer’s disease. Transl Psychiatry. 2013;3:e284. doi: 10.1038/tp.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. PERK mediates elF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2014;35:2272–2281. doi: 10.1016/j.neurobiolaging.2014.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. TrkB reduction exacerbates Alzheimer’s disease-like signaling aberrations and memory deficits without affecting β-amyloidosis in 5XFAD mice. Transl Psychiatry. 2015;5:e562. doi: 10.1038/tp.2015.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Tang J, Ohno M. Beneficial effects of the β-secretase inhibitor GRL-8234 in 5XFAD Alzheimer’s transgenic mice lessen during disease progression. Curr Alzheimer Res. 2015;12:13–21. doi: 10.2174/1567205012666141218125042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ST, Klein WL. The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem. 2011;96:529–543. doi: 10.1016/j.nlm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filser S, Ovsepian SV, Masana M, Blazquez-Llorca L, Brandt Elvang A, Volbracht C, Muller MB, Jung CK, Herms J. Pharmacological inhibition of BACE1 impairs synaptic plasticity and cognitive functions. Biol Psychiatry. 2015;77:729–739. doi: 10.1016/j.biopsych.2014.10.013. [DOI] [PubMed] [Google Scholar]
- Flammang B, Pardossi-Piquard R, Sevalle J, Debayle D, Dabert-Gay AS, Thevenet A, Lauritzen I, Checler F. Evidence that the amyloid-β protein precursor intracellular domain, AICD, derives from β-secretase-generated C-terminal fragment. J Alzheimers Dis. 2012;30:145–153. doi: 10.3233/JAD-2012-112186. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. New York: Academic Press; 2008. [Google Scholar]
- Fukumoto H, Takahashi H, Tarui N, Matsui J, Tomita T, Hirode M, Sagayama M, Maeda R, Kawamoto M, Hirai K, Terauchi J, Sakura Y, Kakihana M, Kato K, Iwatsubo T, Miyamoto M. A noncompetitive BACE1 inhibitor TAK-070 ameliorates Aβ pathology and behavioral deficits in a mouse model of Alzheimer’s disease. J Neurosci. 2010;30:11157–11166. doi: 10.1523/JNEUROSCI.2884-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodger ZV, Rajendran L, Trutzel A, Kohli BM, Nitsch RM, Konietzko U. Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. J Cell Sci. 2009;122:3703–3714. doi: 10.1242/jcs.048090. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hong I, Kang T, Yoo Y, Park R, Lee J, Lee S, Kim J, Song B, Kim SY, Moon M, Yun KN, Kim JY, Mook-Jung I, Park YM, Choi S. Quantitative proteomic analysis of the hippocampus in the 5XFAD mouse model at early stages of Alzheimer’s disease pathology. J Alzheimers Dis. 2013;36:321–334. doi: 10.3233/JAD-130311. [DOI] [PubMed] [Google Scholar]
- Hongpaisan J, Sun MK, Alkon DL. PKC ɛ activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J Neurosci. 2011;31:630–643. doi: 10.1523/JNEUROSCI.5209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Leuchtenberger S, Martin A, Czirr E, Wesselowski J, Dieckmann M, Waldron E, Korth C, Koo EH, Heneka M, Weggen S, Pietrzik CU. α-Secretase mediated conversion of the amyloid precursor protein derived membrane stub C99 to C83 limits Aβ generation. J Neurochem. 2009;111:1369–1382. doi: 10.1111/j.1471-4159.2009.06420.x. [DOI] [PubMed] [Google Scholar]
- Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging. 2012;33:196.e129–196.e140. doi: 10.1016/j.neurobiolaging.2010.05.027. [DOI] [PubMed] [Google Scholar]
- Kandalepas PC, Sadleir KR, Eimer WA, Zhao J, Nicholson DA, Vassar R. The Alzheimer’s β-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 2013;126:329–352. doi: 10.1007/s00401-013-1152-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, Devi L, Ohno M. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. J Neurochem. 2010;113:248–261. doi: 10.1111/j.1471-4159.2010.06608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, Ohno M. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol Dis. 2009;33:229–235. doi: 10.1016/j.nbd.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen I, Pardossi-Piquard R, Bauer C, Brigham E, Abraham JD, Ranaldi S, Fraser P, St-George-Hyslop P, Le Thuc O, Espin V, Chami L, Dunys J, Checler F. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J Neurosci. 2012;32:16243–16255a. doi: 10.1523/JNEUROSCI.2775-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Guo Q, Inoue T, Polito VA, Tabuchi K, Hammer RE, Pautler RG, Taffet GE, Zheng H. Vascular and parenchymal amyloid pathology in an Alzheimer disease knock-in mouse model: interplay with cerebral blood flow. Mol Neurodegener. 2014;9:28. doi: 10.1186/1750-1326-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HB, Yang XM, Li TJ, Cheng YF, Zhang HT, Xu JP. Memory deficits and neurochemical changes induced by C-reactive protein in rats: implication in Alzheimer’s disease. Psychopharmacology (Berl) 2009;204:705–714. doi: 10.1007/s00213-009-1499-2. [DOI] [PubMed] [Google Scholar]
- Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, Song W. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest. 2013;123:224–235. doi: 10.1172/JCI64516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConlogue L, Buttini M, Anderson JP, Brigham EF, Chen KS, Freedman SB, Games D, Johnson-Wood K, Lee M, Zeller M, Liu W, Motter R, Sinha S. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J Biol Chem. 2007;282:26326–26334. doi: 10.1074/jbc.M611687200. [DOI] [PubMed] [Google Scholar]
- Medeiros R, Kitazawa M, Chabrier MA, Cheng D, Baglietto-Vargas D, Kling A, Moeller A, Green KN, LaFerla FM. Calpain inhibitor A-705253 mitigates Alzheimer’s disease-like pathology and cognitive decline in aged 3xTgAD mice. Am J Pathol. 2012;181:616–625. doi: 10.1016/j.ajpath.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Nalbantoglu J, Tirado-Santiago G, Lahsaini A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997;387:500–505. doi: 10.1038/387500a0. [DOI] [PubMed] [Google Scholar]
- Nishitomi K, Sakaguchi G, Horikoshi Y, Gray AJ, Maeda M, Hirata-Fukae C, Becker AG, Hosono M, Sakaguchi I, Minami SS, Nakajima Y, Li HF, Takeyama C, Kihara T, Ota A, Wong PC, Aisen PS, Kato A, Kinoshita N, Matsuoka Y. BACE1 inhibition reduces endogenous Abeta and alters APP processing in wild-type mice. J Neurochem. 2006;99:1555–1563. doi: 10.1111/j.1471-4159.2006.04178.x. [DOI] [PubMed] [Google Scholar]
- O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hebert SS, De Strooper B, Haass C, Bennett DA, Vassar R. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M. Genetic and pharmacological basis for therapeutic inhibition of β- and γ-secretases in mouse models of Alzheimer’s memory deficits. Rev Neurosci. 2006;17:429–454. doi: 10.1515/revneuro.2006.17.4.429. [DOI] [PubMed] [Google Scholar]
- Ohno M. β-Secretase as a prime therapeutic target for Alzheimer’s disease: a perspective from mouse model studies. In: Araki W, editor. Recent advances in the biology of secretases, key proteases in Alzheimer’s disease. Kerala: Research Signpost; 2008. pp. 1–25. [Google Scholar]
- Ohno M. Failures to reconsolidate memory in a mouse model of Alzheimer’s disease. Neurobiol Learn Mem. 2009;92:455–459. doi: 10.1016/j.nlm.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL, Vassar R, Disterhoft JF. Temporal memory deficits in Alzheimer’s mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci. 2006;23:251–260. doi: 10.1111/j.1460-9568.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis. 2007;26:134–145. doi: 10.1016/j.nbd.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Ikin AF, Lamprianou S, Vacaresse N, Revelli JP, Platt K, Paganetti P, Mathews PM, Harroch S, Buxbaum JD. BACE (β-secretase) modulates the processing of APLP2 in vivo. Mol Cell Neurosci. 2004;25:642–649. doi: 10.1016/j.mcn.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Portelius E, Dean RA, Gustavsson MK, Andreasson U, Zetterberg H, Siemers E, Blennow K. A novel Aβ isoform pattern in CSF reflects γ-secretase inhibition in Alzheimer disease. Alzheimers Res Ther. 2010;2:7. doi: 10.1186/alzrt30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portelius E, Price E, Brinkmalm G, Stiteler M, Olsson M, Persson R, Westman-Brinkmalm A, Zetterberg H, Simon AJ, Blennow K. A novel pathway for amyloid precursor protein processing. Neurobiol Aging. 2011;32:1090–1098. doi: 10.1016/j.neurobiolaging.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Portelius E, Zetterberg H, Andreasson U, Brinkmalm G, Andreasen N, Wallin A, Westman-Brinkmalm A, Blennow K. An Alzheimer’s disease-specific β-amyloid fragment signature in cerebrospinal fluid. Neurosci Lett. 2006;409:215–219. doi: 10.1016/j.neulet.2006.09.044. [DOI] [PubMed] [Google Scholar]
- Py NA, Bonnet AE, Bernard A, Marchalant Y, Charrat E, Checler F, Khrestchatisky M, Baranger K, Rivera S. Differential spatio-temporal regulation of MMPs in the 5xFAD mouse model of Alzheimer’s disease: evidence for a pro-amyloidogenic role of MT1-MMP. Front Aging Neurosci. 2014;6:247. doi: 10.3389/fnagi.2014.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe S, Reichwald J, Ammaturo D, de Strooper B, Saftig P, Neumann U, Staufenbiel M. The Swedish APP mutation alters the effect of genetically reduced BACE1 expression on the APP processing. J Neurochem. 2011;119:231–239. doi: 10.1111/j.1471-4159.2011.07412.x. [DOI] [PubMed] [Google Scholar]
- Rosenkranz SC, Geissen M, Harter K, Szalay B, Ferrer I, Vogel J, Smith S, Glatzel M. Amyloid-precursor-protein-lowering small molecules for disease modifying therapy of Alzheimer’s disease. PLoS One. 2013;8:e82255. doi: 10.1371/journal.pone.0082255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadleir KR, Eimer WA, Cole SL, Vassar R. Aβ reduction in BACE1 heterozygous null 5XFAD mice is associated with transgenic APP level. Mol Neurodegener. 2015;10:1. doi: 10.1186/1750-1326-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadleir KR, Eimer WA, Kaufman RJ, Osten P, Vassar R. Genetic inhibition of phosphorylation of the translation initiation factor eIF2α does not block Aβ-dependent elevation of BACE1 and APP levels or reduce amyloid pathology in a mouse model of Alzheimer’s disease. PLoS One. 2014;9:e101643. doi: 10.1371/journal.pone.0101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci. 2014;17:661–663. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Price EA, Wu G, Crouthamel MC, Shi XP, Tugusheva K, Tyler KX, Kahana J, Ellis J, Jin L, Steele T, Stachel S, Coburn C, Simon AJ. In vivo β-secretase 1 inhibition leads to brain Aβ lowering and increased α-secretase processing of amyloid precursor protein without effect on neuregulin-1. J Pharmacol Exp Ther. 2008;324:957–969. doi: 10.1124/jpet.107.130039. [DOI] [PubMed] [Google Scholar]
- Seo J, Giusti-Rodriguez P, Zhou Y, Rudenko A, Cho S, Ota KT, Park C, Patzke H, Madabhushi R, Pan L, Mungenast AE, Guan JS, Delalle I, Tsai LH. Activity-dependent p25 generation regulates synaptic plasticity and Aβ-induced cognitive impairment. Cell. 2014;157:486–498. doi: 10.1016/j.cell.2014.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci. 2005;8:1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- Tamayev R, D’Adamio L. Inhibition of γ-secretase worsens memory deficits in a genetically congruous mouse model of Danish dementia. Mol Neurodegener. 2012;7:19. doi: 10.1186/1750-1326-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayev R, Matsuda S, Arancio O, D’Adamio L. β- but not γ-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol Med. 2012;4:171–179. doi: 10.1002/emmm.201100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R. The β-secretase, BACE: a prime drug target for Alzheimer’s disease. J Mol Neurosci. 2001;17:157–170. doi: 10.1385/JMN:17:2:157. [DOI] [PubMed] [Google Scholar]
- Vassar R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res Ther. 2014;6:89. doi: 10.1186/s13195-014-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC. The β-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Kuhn PH, Haass C, Kennedy ME, Rajendran L, Wong PC, Lichtenthaler SF. Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J Neurochem. 2014;130:4–28. doi: 10.1111/jnc.12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SJ, Bachstetter AD, Van Eldik LJ. Comprehensive behavioral characterization of an APP/PS-1 double knock-in mouse model of Alzheimer’s disease. Alzheimers Res Ther. 2013;5:28. doi: 10.1186/alzrt182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R, Vassar R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014;13:319–329. doi: 10.1016/S1474-4422(13)70276-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahs KR, Ashe KH. β-Amyloid oligomers in aging and Alzheimer’s disease. Front Aging Neurosci. 2013;5:28. doi: 10.3389/fnagi.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Liu X, Schroeder JP, Chan CB, Song M, Yu SP, Weinshenker D, Ye K. 7,8-Dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2014;39:638–650. doi: 10.1038/npp.2013.243. [DOI] [PMC free article] [PubMed] [Google Scholar]