

Summary
Although genetically engineered mouse (GEM) models are often used to evaluate cancer therapies, extrapolation of such preclinical data to human cancer can be challenging. Here we introduce an approach that uses drug perturbation data from GEM models to predict drug efficacy in human cancer. Network-based analysis of expression profiles from in vivo treatment of GEM models identified drugs and drug combinations that inhibit the activity of FOXM1 and CENPF, which are master regulators of prostate cancer malignancy. Validation of mouse and human prostate cancer models confirmed the specificity and synergy of a predicted drug combination to abrogate FOXM1/CENPF activity and inhibit tumorigenicity. Network-based analysis of treatment signatures from GEM models identified treatment-responsive genes in human prostate cancer that are potential biomarkers of patient response. More generally, this approach allows systematic identification of drugs that inhibit tumor dependencies, thereby improving the utility of GEM models for prioritizing drugs for clinical evaluation.
Graphical abstract
Introduction
Recent large-scale genomic analyses have led to the identification of “actionable” driver genes of specific cancers that are therapeutically accessible, including oncogene and non-oncogene dependencies (Al-Lazikani et al., 2012; Garraway and Lander, 2013; Luo et al., 2009; Rubio-Perez et al., 2015). However, the accurate and efficient identification of drugs and drug combinations that inhibit such drivers within specific tumor contexts represents a major challenge, particularly for transcriptional regulators that, in general, are pharmacologically inaccessible. Genetically engineered mouse (GEM) models are well-suited to empower investigations of targeted inhibitors in the context of the native tumor microenvironment in vivo (Abate-Shen and Pandolfi, 2013; Politi and Pao, 2011; Sharpless and Depinho, 2006). However, species differences with respect to tumor histology, physiology, pharmacology, and metabolism often preclude direct extrapolation of preclinical findings from mouse models to human cancer.
In the current study, we introduce an innovative regulatory network-based method that uses expression profiles from drug-treated GEM models to predict drugs and drug combinations that specifically inhibit the activity of established human cancer dependencies. We focus this proof-of-concept study on prostate cancer, a disease characterized by heterogeneity of its causal mechanisms and range of disease outcomes (Chang et al., 2014; Cooperberg et al., 2005; Roychowdhury and Chinnaiyan, 2013; Shen and Abate-Shen, 2010). In particular, while most locally invasive prostate tumors are curable, recurrent or aggressive tumors initially respond to androgen deprivation therapy but ultimately relapse to castration-resistant metastatic disease, which is nearly always fatal (Ryan and Tindall, 2011; Scher and Sawyers, 2005). While treatment options for castration-resistant metastatic prostate cancer have significantly improved in recent years (Mukherji et al., 2014; Rathkopf and Scher, 2013; Wong et al., 2014), none of the available treatments are as yet curative.
We have recently generated genome-wide reverse engineered regulatory networks (henceforth interactomes) for both mouse and human prostate cancer (Aytes et al., 2014). Interrogation of these interactomes identified FOXM1 and CENPF as master regulators (i.e., key driver genes), which function synergistically to elicit synthetic lethality and are robust predictors of poor patient outcome (Aytes et al., 2014). Here, we show that interrogation of in vivo drug perturbation signatures from GEM models represents an effective strategy for systematic identification of specific drugs and drug combinations that inhibit the transcriptional activity of FOXM1/CENPF. Strikingly, drug combinations that revert transcriptional activity of these proteins are highly effective in abrogating tumorigenesis in vivo and well-correlated with patient outcome. We propose that this computational method can be generalized for more effective utilization of preclinical data from GEM models to predict optimal drug and drug-combinations and thereby dramatically improve the utilization of GEM models to prioritize compounds for clinical investigation.
Results
Systematic inference of FOXM1/CENPF inhibitors in vivo
The current methodology is predicated on our previous analyses showing that expression of the target genes of a given master regulator (MR) (its regulon) represents an effective reporter to predict the activity of the MR for a given cancer phenotype (Aytes et al., 2014; Carro et al., 2010; Chen et al., 2014). Here we have extended this concept to evaluate whether such regulon can be used as a reporter to quantitatively measure the ability of a drug or drug combination to inhibit the activity of the corresponding MR. In general, reversion of MR activity would correspond to the ability of a given drug to down-regulate its activated target genes and up-regulate its repressed targets (Fig. 1A). As a proof-of-concept for this approach, we evaluated drugs for their ability to inhibit the master regulator pair, FOXM1/CENPF, which we have previously established to be a key synthetic lethal dependency of prostate tumor malignancy (Aytes et al., 2014). In particular, we tested whether candidate therapeutic agents could be prioritized based on in vivo perturbation by assessing their ability to “reverse” the FOXM1/CENPF regulon. We focused on the activated targets shared of FOXM1/CENPF, since the number of repressed targets is too few for analysis. However, both activated and repressed targets may be used in general.
Figure 1. Computational prediction of drugs that inhibit FOXM1/CENPF activity in vivo.
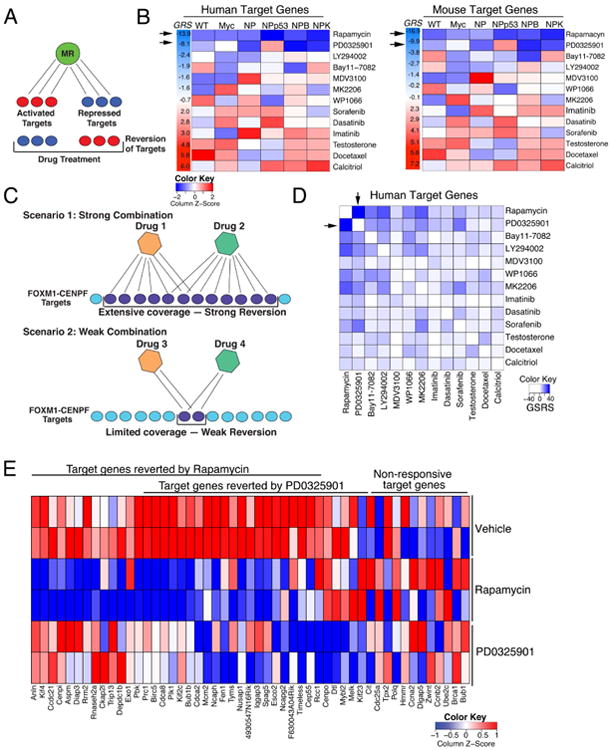
(A) Shown is the strategy for prediction of single drugs. Drug reversion scores were calculated based on the degree to which target genes that are activated (red) by a master regulator (MR) are inhibited (blue) following drug treatment, and conversely the degree to which target genes that are repressed (blue) by the MR are activated (red) following drug treatment (see Detailed Experimental Procedures). (B) Heat-map representations of GSEA used to calculate drug reversion scores across a series of GEM models with a series of drugs, as indicated (see Detailed Experimental Procedures). GSEA were done using the mouse in vivo drug perturbation signatures as the reference and human or mouse FOXM1/CENPF target genes inferred from their respective prostate cancer interactomes, as indicated, as the query gene set. Global reversion scores (GRS) were calculated for each drug by combining the individual NES for each GEM model using a metric based on the Stouffer integration formulation (see Detailed Experimental Procedures). Arrows point to the two drugs with the highest GRSs. (C) Shown is the strategy for prediction of drug synergy. Pairwise combinations of data from individual drug treatments (as in A) were assessed to predict drugs that effectively revert FOXM1/CENPF target genes when used in combination. Scenario 1 illustrates two drugs that inhibit (i.e., revert) many target genes, thereby resulting in strong reversion. Scenario 2 illustrates two other drugs that inhibit (i.e., revert) relatively few target genes, thereby resulting in weak reversion. (D) Heat-map representation depicting global synergistic reversion scores (GSRS) for each possible pair of drugs across the series of GEM models based on FOXM1/CENPF human target genes. GSRSs were calculated by combining the synergistic reversion scores for targets affected by the drug combinations (see Detailed Experimental Procedures). Heat-map intensity (blue) represents the predicted degree of reversion (GSRS); arrows indicate drug pairs with the highest combined GSRS. (E) Heat-map showing the relative expression levels of FOXM1/CENPF target genes reverted by treatment with rapamycin or PD0325901 versus vehicle; also shown are genes that are not reverted (i.e., non-responsive) to these drugs.
Figure S1 is related to Figure 1; computational predictions of GRS and GSRS scores are provided in Tables S1 and S2, respectively.
To assess this strategy, we used a drug perturbation dataset that includes drugs with known to prostate cancer-relevance, such as those that inhibit the androgen receptor, or key signaling pathways such as PI3-kinase/mTOR or MAP-kinase, or standard chemotherapy (see (Aytes et al., 2014) and Detailed Experimental Procedures). In vivo drug perturbation studies were performed using multiple GEM models representative of advanced prostate cancer (see (Aytes et al., 2014) and Detailed Experimental Procedures) to avoid potential bias introduced by any individual model. The in vivo drug perturbation data were analyzed by Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) to assess the inhibition (i.e., reversion) of FOXM1/CENPF shared target genes; analyses were performed separately for the mouse and human targets (Fig. 1B). Using GSEA, we obtained a Normalized Enrichment Score (NES) for each drug signature and each GEM model, which we define as the Reversion Score (RSFOXM1/CENPF), to assess the compound's ability to inhibit FOXM1/CENPF activity in a specific GEM model (Table S1). From these analyses, a Global Reversion Score (GRSFOXM1/CENPF) was assigned for each drug by integrating each of the GEM-specific RSFOXM1/CENPF scores, using a metric based on Stouffer's integration formulation (Whitlock, 2005) (Fig. 1B; Fig. S1; see Detailed Experimental Procedures). Thus, drugs that most effectively inhibit FOXM1/CENPF activity are those having the most negative GRSFOXM1/CENPF. Notably, FOXM1/CENPF target genes from either mouse or human yielded equivalent GRSFOXM1/CENPF (Fig. 1B; Table S1), indicating conservation of the predicted drug response between mouse and human prostate cancer.
Among the individual drugs tested in the GEM models, the two with the most significant negative GRSFOXM1/CENPF were rapamycin and PD0325901. These drugs inhibit the PI3-Kinase/mTOR and MAP-kinase signaling pathways, respectively, which are frequently dysregulated in advanced prostate cancer (Aytes et al., 2013; Kinkade et al., 2008; Taylor et al., 2010). Specifically, the global reversion scores for rapamycin were GRSH = -13.9 (human targets) and GRSM = -16.9 (mouse targets) and for PD0325901 were GRSH = -8.1 and GRSM = -9.9 (Fig. 1B; Table S1). In contrast to rapamycin and PD0325901, other drugs including docetaxel, a standard of care chemotherapy for advanced prostate cancer (Pienta and Smith, 2005), were not predicted to be effective for inhibiting the FOXM1/CENPF regulon (GRSH = 5.8 and GRSM = 5.6; Fig. 1B; Fig. S1).
Systematic inference of drug synergy
Next, we tested whether this computational approach could be extended to infer drug combinations that cooperate to inhibit MR activity, again using FOXM1 and CENPF as a proof-of-concept. These analyses are based on the hypothesis that effective drug combinations should induce a more significant reversal of MR-specific regulon expression, compared to the individual drugs (Fig. 1C; see Detailed Experimental Procedures). Notably, such logic can be implemented based on individual drug signatures, without requiring in vivo signatures from drug combinations, which vastly increases the experimental efficiency for prioritizing drug combinations based on in vivo preclinical data.
To estimate a global synergistic reversion score (GSRS) for each drug-pair, we assessed the predicted reversion score for all possible combinations of two drug treatments across each of the GEM models. First, the synergistic reversion score (SRS) was calculated for each GEM model as an F-score that first maximizes the number of unique targets affected by each drug, and then the total number of targets affected by both drugs (Fig. 1C; see Detailed Experimental Procedures). These analyses identified several combinations, most of which included rapamycin or PD0325901, which were predicted to be more effective than the individual compounds based on their GSRSs (Fig. 1D; Table S2). In particular, the rapamycin + PD0325901 combination was predicted to have the strongest global inhibition of the FOXM1/CENPF regulon, both with respect to total number of targets affected by both drugs and the number of unique targets affected by each drug, resulting in the most significant negative global synergistic reversion score (GSRSH = —23.4; p-value < 0.001 compared to a random model; see Detailed Experimental Procedures). This theoretical prediction was validated by assessment of FOXM1/CENPF target genes that were reverted by rapamycin or PD0325901 following drug treatment in vivo (Fig 1E).
Experimental validation of drug specificity and synergy in cell culture
Based on these computational predictions, we performed experimental validation to assess whether rapamycin and/or PD0325901 specifically inhibit the FOXM1/CENPF regulon in relevant mouse and human prostate cancer cell culture models, and if so, whether these drugs affect cell growth and tumorigenicity in a FOXM1/CENPF-dependent manner. First, we validated the underlying computational prediction that treatment with rapamycin and PD0325901 reverts the expression of shared target genes of FOXM1/CENPF. Using real-time PCR, we found that treatment with rapamycin and PD0325901, but not docetaxel, inhibited expression of both FOXM1 and CENPF as well as their shared target genes in several human and mouse prostate cancer (Fig. 2A; Fig. S2A). This inhibition of target genes was coincident with inhibition of the corresponding signaling pathways, namely PI3-Kinase/mTOR and MAP-kinase in the mouse and human cells (Fig. S2B, C). Notably, inhibition of colony formation was significantly greater when the drugs were combined than when used individually (Fig. 2B, C), which supports the computational prediction of rapamycin + PD0325901 synergy.
Figure 2. Validation of drug efficacy, synergy, and specificity in prostate cancer cells.
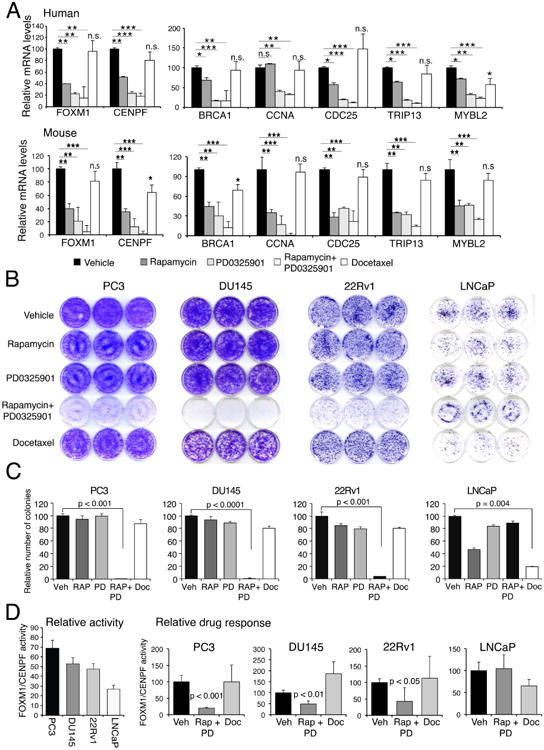
(A) Real-time PCR of mRNA expression levels of FOXM1 and CENPF and their shared target genes following treatment with rapamycin and/or PD0325901, or docetaxel in DU145 human prostate cancer cells (top) or NPK mouse prostate tumors (bottom). (B, C) Colony formation assays in the indicated human prostate cancer cells, PC3, DU145, 22Rv1, and LNCaP, following treatment with rapamycin (Rap) and/or PD0325901 (PD), or docetaxel (Doc). (B) Representative colony formation assays. (C) Quantification of independent assays performed in triplicate. (D) (Left) Relative Activity of FOXM1/CENPF in human prostate cancer cells lines. Activity levels were calculated based on expression levels of 10 FOXM1/CENPF target genes (see Fig. S2F). (Right) Relative drug response assessed for FOXM1/CENPF activity levels in the human prostate cancer cell lines following treatment with rapamycin + PD0325901 (Rap + PD), or docetaxel (Doc). Differences between treatment groups were assessed using Student's t-test. When indicated, p values are represented as * <0.01, ** < 0.001, and ***, <0.0001. Bars represent mean +/- standard deviation (SD).
Figures S2 and S3 are related to Figure 2.
To address the specificity of the rapamycin + PD0325901 drug combination for inhibition of FOXM1/CENPF activity, we assessed whether this combination was preferentially more potent in contexts having high levels of FOXM1/CENPF activity. First, we surveyed the expression and activity of FOXM1/CENPF in a series of human and mouse cell lines; “activity” was determined experimentally by analyses of the expression of FOXM1/CENPF shared target genes (Fig. 2D; Fig. S2D-F). These studies revealed that PC3 cells have the highest levels of FOXM1/CENPF activity whereas LNCaP cells have lower levels (Fig. 2D). Correspondingly, human prostate cancer cells with higher levels of FOXM1/CENPF activity had greater response to rapamycin + PD0325901 treatment, as evident from the strong inhibition of activity and colony formation, whereas LNCaP cells, which have low levels of FOXM1/CENPF activity, had a modest response to rapamycin + PD0325901 (Fig. 2B-D). In contrast, this relationship to FOXM1/CENPF activity was not observed following docetaxel treatment of these cells (Fig. 2B-D). Similar findings were observed in mouse prostate cancer cells wherein response to rapamycin + PD0325901 treatment was correlated with the relative levels of FoxM1/Cenpf activity (Fig. S2D, E).
Moreover, the dependence on FOXM1/CENPF in the human prostate cancer cells was evident by the reduction in the IC50 for rapamycin + PD0325901, but not docetaxel, following the silencing of both FOXM1 and CENPF in human prostate cancer cell lines with high levels of activity (Fig. S3). Conversely, overexpression of FOXM1 and CENPF in a non-prostate cancer cell line, HEK293, resulted in an increase in the IC50 for rapamycin + PD0325901, but not docetaxel (Fig. S3). Taken together, these findings validate the computational prediction that FOXM1/CENPF activity is specifically inhibited by rapamycin + PD0325901.
Experimental validation of drug efficacy and specificity in vivo
The synergistic effects of combined treatment with rapamycin + PD0325901 were even more dramatic in vivo. In particular, we performed preclinical studies using NPK mice (Nkx3.1CreERt2; Ptenflox/flox; KrasLSL-G12D/+), which model aggressive, metastatic prostate cancer that is dependent on FOXM1/CENPF activity (Aytes et al., 2013; Aytes et al., 2014). Tumor-bearing NPK mice were treated with rapamycin and/or PD0325901, or docetaxel, for five days (i.e., the dynamic response cohort) or one month (i.e., the therapeutic response cohort) (Fig. 3A; Table S3). Mice were then either sacrificed for analysis or monitored for the effects of drug treatment on survival and metastasis (i.e., the survival response cohort) (Fig. 3A; Table S3).
Figure 3. Validation of drug synergy and specificity in vivo.
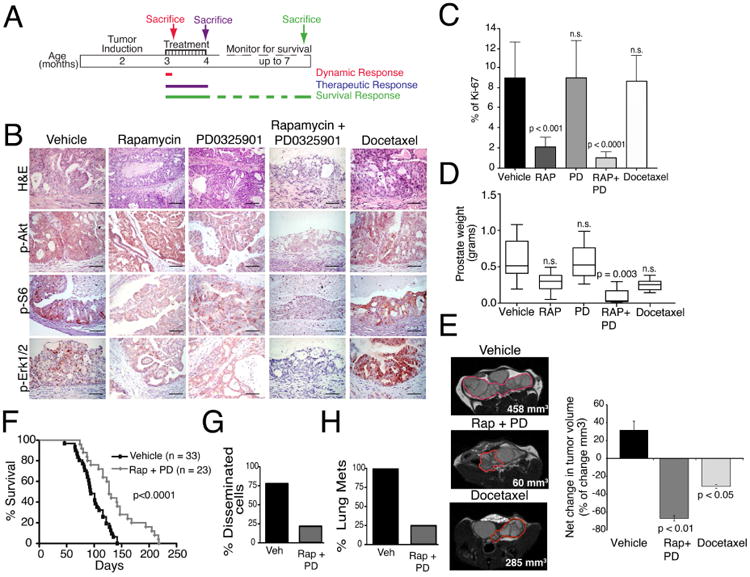
(A) Shown is the design of preclinical studies. NPK mice were induced to form tumors by delivery of tamoxifen at 2 months of age as in (Aytes et al., 2013). Mice were treated with rapamycin and/or PD032590, or docetaxel for 5 days (dynamic response cohort) or one month, following which mice were sacrificed for analyses (therapeutic response cohort) or monitored for survival (survival response cohort). (B-E) Analysis of the therapeutic response cohort following treatment with rapamycin (Rap) and/or PD0325901 (PD), or docetaxel (Doc) as indicated (n = 5 mice/treatment group). (B) Representative sections of hematoxylin and eosin (H&E) staining or immunohistochemical staining for the indicated markers of the PI3K/mTOR or MAP-Kinase signaling pathways; scale bars represent 100 μm. (C) Relative cellular proliferation following drug treatment as determined by the percent of Ki67 positive cells relative to total epithelial cells. (D) Prostate weight (in grams) following drug treatment. (E) Longitudinal MRI imaging showing representative MRI images following drug treatment with tumor volumes indicated. The panel to the right represents the net change in tumor volume following 1 month of drug treatment. (F-H) Shown is analysis of the survival response cohort. (F) Survival analysis showing the improvement in survival following treatment with rapamycin and PD0325901 (Rap + PD) compared with the vehicle-treated mice. (G) Percentage of mice with disseminated cells in the bone marrow and (H) percentage of mice with lung metastases following treatment with vehicle (Veh) or rapamycin + PD0325901 (Rap + PD) (n = 10 mice/treatment group). Differences between groups were assessed using Student's t-test; bars represent mean +/-standard deviation (SD). On panel F, p-value corresponds to a Log-rank test.
Whereas treatment with either drug individually had a modest therapeutic benefit at the various endpoints, the combination of rapamycin + PD0325901 had a profound effect at all tumor endpoints in the therapeutic response cohort (Fig. 3B-E). In particular, treatment with rapamycin + PD0325901, but not docetaxel, resulted in profound abrogation of the histological phenotype, coincident with inhibition of relevant signaling pathways, as evident by immunohistochemistry (Fig. 3B). Moreover, tumors treated with rapamycin + PD0325901, but not docetaxel, displayed a significant decrease in cellular proliferation (p < 0.0001) (Fig. 3C), as well as significant reduction in tumor burden, as measured by tumor weight (p = 0.003) and tumor volume using MRI (p < 0.01) (Fig. 3D,E). Furthermore, these effects on phenotype and tumor burden were accompanied by a significant improvement in survival (p < 0.0001) (Fig. 3F), as well as a 3-fold reduction in the incidence of disseminated tumor cells in the bone marrow and a 4-fold reduction in the incidence of lung metastases (Fig. 3G,H). Together, these findings validate the concept that treatment with rapamycin + PD0325901 inhibits growth of FOXM1/CENPF-dependent tumors.
Relationship of mouse drug treatment signatures to human cancer
Given the striking reduction in tumor and metastatic burden following treatment with rapamycin + PD0325901, we evaluated whether this combination might be sufficient to broadly inhibit molecular processes associated with advanced, FOXM1/CENPF-dependent prostate cancer. We addressed this question by analyzing signatures obtained by differential gene expression analysis of NPK prostate tumors treated with vehicle or rapamycin + PD0325901 for 1 month (i.e., the therapeutic response cohort; Table S3), which resulted in extensive abrogation of the tumor phenotype (see Fig. 3). We compared this “therapeutic response” signature to a reference mouse “tumor” signature, corresponding to differential gene expression between phenotypically wild-type prostates and NPK prostate tumors, which captures the transition from normal prostate to fully malignant prostate cancer (Table S3). Strikingly, genes that were differentially expressed in the “therapeutic response” signature were strongly negatively-enriched in the mouse “tumor” signature (NES= -8.58; p < 0.001) (Fig. S4A). Further evidence that rapamycin + PD0325901 treatment results in broad inhibition beyond their respective target signaling pathways was provided by biological pathway analysis. In particular, pathways that were significantly inhibited (i.e., reverted) following treatment of the NPK tumors with rapamycin + PD0325901, but not docetaxel, include several that are important for tumor progression and are not directly related to mTOR/PI3K/MAP kinase signaling (Fig. 4A; Table S4).
Figure 4. Cross-species analyses of drug treatment response.
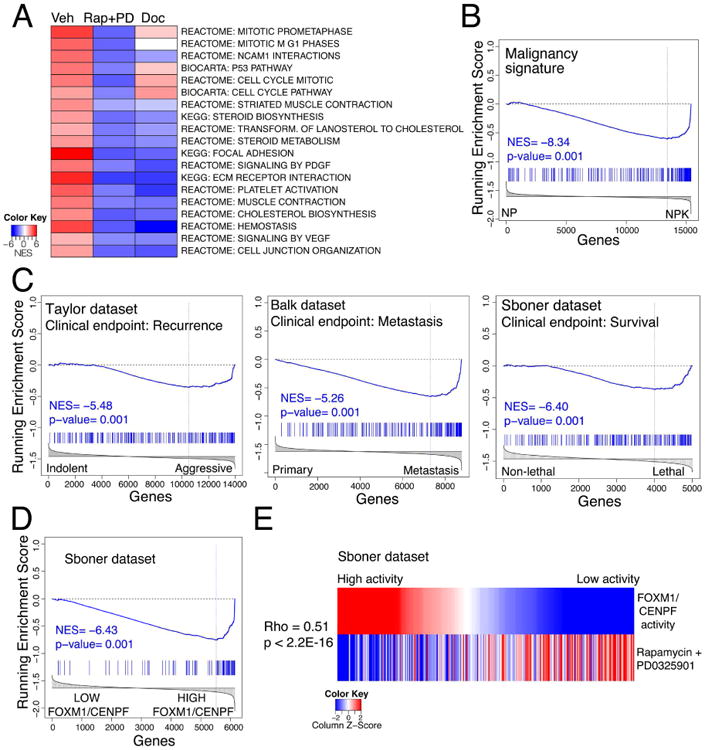
(A) Heat-map depiction showing representative pathways that are significantly changed following treatment with rapamycin + PD0325901 (Rap + PD) or docetaxel (Doc) relative to vehicle treatment (Veh). Pathway analysis was done by GSEA using a “humanized” version of the dynamic response allograft tumor signature (see Table S3 and Detailed Experimental Procedures). A complete list of pathways is provided in Table S4. (B-D) GSEA using as the query gene set the mouse rapamycin + PD0325901 dynamic treatment response signature (Panel B) or a “humanized” version of this signature (Panels C and D); normalized enrichment score (NES) and associated p-values are shown. In panel B, the reference is mouse “malignancy” signature, which represents differentially expressed genes from NP versus NPK mouse tumors as reported in (Aytes et al., 2013). In panel C, the references are three independent human tumor signatures (i.e., Taylor, Balk, or Sboner), each of which compare differentially expressed genes representing less aggressive versus more aggressive prostate cancer specimens (Table S3). In panel D, the reference signature represents differentially expressed genes in patients from the Sboner dataset having low versus high levels of FOXM1/CENPF activity, which was inferred using single sample MARINa (ssMARINa) (see Detailed Experimental Procedures). (E) Heat-map showing the correlation in human patients from the Sboner dataset of FOXM1/CENPF activity levels (top) with the corresponding predicted drug treatment response for rapamycin + PD0325901 (bottom). As above, FOXM1/CENPF activity was estimated for each patient using ssMARINa. The treatment response for each patient was inferred using a “humanized” version of the mouse dynamic treatment signature (see Detailed Experimental Procedures). Correlation (rho) and associated p-value were estimated using Spearman's correlation coefficient.
Figure S4, and Tables S3 and S4 are related to Figure 4.
To evaluate molecular processes that are inhibited immediately following drug treatment, we analyzed a “dynamic response” signature, representing a time point wherein the drugs are active but the tumor phenotype has not yet been abrogated (Fig. 3A; Table S3; and data not shown). In particular, this short-term treatment with rapamycin + PD0325901 resulted in reversion of FOXM1/CENPF targets, as predicted by our computational approach (Fig. S4B and see Fig. 1E). Comparison of this “dynamic response” signature to a reference mouse “malignancy signature,” based on comparison of non-malignant prostate tumors from NP mice to fully malignant NPK tumors (Aytes et al., 2013), revealed a striking negative enrichment (i.e., strong reversion) (NES= -8.34; p < 0.001) (Fig. 4B), suggesting that the rapamycin + PD0325901 combination inhibits molecular processes associated with NPK tumor malignancy even prior to their overt effects on the tumor phenotype.
To assess conservation of these molecular changes with human prostate cancer, we performed GSEA to compare a humanized version of the mouse “dynamic response” signature with human prostate cancer signatures (see Detailed Experimental Procedures). We used three independent human prostate cancer signatures, each of which is based on distinct clinical endpoints (Table S3): (i) a malignancy signature based on the Taylor dataset (Taylor et al., 2010), which compares patients having low Gleason score and no biochemical recurrence (n = 39) to those with high Gleason score and a short time to biochemical recurrence (n = 10) (Aytes et al., 2013); (ii) a metastasis signature based on the Balk dataset (Stanbrough et al., 2006), which compares hormone-naïve prostate tumors (n = 22) to bone metastases from castration-resistant prostate cancer (n = 29) (Aytes et al., 2014); and (iii) a survival signature based on the Sboner dataset (Sboner et al., 2010), which compares transurethral resections from patients who survived for nearly 200 months (n=12) to those who died of prostate cancer within 12 months (n=6) (Wang et al., 2013). Strikingly, the mouse “dynamic response” signature was strongly negatively enriched when compared with each of these human signatures, indicating that genes that are consistently overexpressed in aggressive prostate cancer are inhibited following drug treatment: Taylor signature (NES = —5.48, p < 0.001), Balk signature (NES = —5.26, p < 0.001), and Sboner signature (NES = —6.40, p < 0.001) (Fig. 4C). In contrast, the docetaxel treatment response signature was either minimally or not negatively enriched in these human signatures (Fig. S4C).
We then asked whether the mouse “dynamic response” signature could reverse a “FOXM1/CENPF activity” signature in human prostate cancer. This “FOXM1/CENPF activity” signature, defined using Sboner dataset (Sboner et al., 2010), corresponds to differential gene expression between patient samples having low- versus high-levels of FOXM1/CENPF activity, which was measured by enrichment of the FOXM1/CENPF regulon in each patient using single-sample MAster Regulator INference algorithm (ssMARINa) as in ((Aytes et al., 2014) and see Detailed Experimental Procedures). GSEA comparing the “FOXM1/CENPF” activity signature with the mouse “dynamic response” signature showed strong negative enrichment (NES = -6.43, p < 0.001) (Fig 4D), which supports the concept that patients with the high levels of FOXM1/CENPF activity should respond more effectively to rapamycin + PD0325901 treatment. Notably, similar comparison with a docetaxel treatment response signature did not indicate such relationship (NES = 0.37, p = 0.77) (Fig. S4D).
We further evaluated the correlation between FOXM1/CENPF activity levels and predicted treatment response in each patient in the Sboner dataset using ssMARINa and GSEA, respectively. We found that inferred FOXM1/CENPF activity levels and predicted treatment response were strongly correlated (Spearman's rho = 0.51, p < 2.2 × 10-16) (Fig 4E), which was not the case for the docetaxel treatment response (Fig. S4E). Taken together, these computational analyses suggest that the molecular programs (i.e., genes and pathways) specifically inhibited (reverted) by rapamycin + PD0325901 in the mouse model are conserved with those that drive aggressive human prostate cancer, and in particular in patients having high levels of FOXM1/CENPF activity.
Conservation of treatment response in mouse and human prostate cancer
Given the conservation in the molecular programs affected by drug treatment in the GEM models and human prostate cancer, we next asked whether we could use the mouse treatment response signature to identify genes predicted to be associated with treatment response in humans. First, we identified candidate rapamycin + PD0325901-responsive genes by interrogating the mouse prostate cancer interactome (Aytes et al., 2014) with the “dynamic response” signature using the standard MARINa algorithm to identify master regulators (MRs) of treatment responses in the mouse (Lefebvre et al., 2010). We then compared these MRs with the orthologous human genes to identify those predicted both to be regulated by FOXM1/CENPF in human prostate cancer and to be down-regulated by drug treatment; we refer to these as “predicted treatment-responsive genes” and distinguish them from other FOXM1/CENPF target genes that are not predicted to be responsive to the treatment (Fig. 5A). Notably, real-time PCR analyses confirmed that the expression levels of these predicted treatment-responsive genes were indeed inhibited by rapamycin + PD0325901 in human prostate cancer cell lines, whereas the expression levels of the predicted non-responsive genes was not inhibited by such treatment (Fig. 5B).
Figure 5. Conservation of treatment responsive genes in human prostate cancer.

(A) FOXM1/CENPF sub-network of human target genes predicted to be responsive or non-responsive to treatment with rapamycin + PD0325901 based on comparison with treatment response for the mouse model. Relative change in activity following drug treatment is indicated by levels of “blue” for genes predicted to be reverted by the drugs and levels of “red” for those predicted to be activated or unaffected (i.e., non-responsive). (B) Real-time PCR showing the actual change in expression levels of FOXM1/CENPF target genes following treatment with vehicle or rapamycin + PD0325901. The “predicted treatment responsive genes” correspond to those represented by the blue circles in panel A, and the “predicted non-responsive genes” to the other genes. PCR was done using DU145 cells; differences were assessed using t-test (p-values are represented as * <0.01, ** < 0.001, and ***, <0.0001) and bars represent mean +/-standard deviation (SD). (C-E) Association of predicted treatment responsive genes with lethal prostate cancer and disease outcome. (C) Summary table showing the significance of elevated expression levels in metastases versus primary tumors in the Taylor and Balk datasets (columns on the left; p-value was calculated using t-test). The column on the right shows a COX regression model indicating the association based on master regulator activity levels of the predicted treatment responsive genes with prostate cancer-specific survival estimated for patients in the Sboner dataset; COX p-value was calculated using Wald test. (NA, sufficient targets not represented; NS, not significant). (D) Oncoprint visualization from cBioportal showing the percent of alterations of the predicted treatment responsive genes in metastases samples from the Taylor dataset. (E) Heat-map showing the master regulator activity levels of treatment responsive genes in primary tumors versus bone metastases from the Balk dataset. (F) Heat-map comparing the master regulator activity levels of the treatment responsive genes (upper rows) across each patient in the Sboner dataset with inferred treatment response for each patient (Rap +PD, bottom row). The activity levels and the treatment response for each patient were estimated using single-sample MARINa (ssMARINa) and GSEA, respectively (see Detailed Experimental Procedures). The correlation between the average activity levels of all treatment responsive genes and the predicted response was estimated using Spearman's correlation coefficient. (G) Kaplan-Meier survival analysis based on the master regulator activity levels of predicted treatment responsive genes in the Sboner dataset using prostate cancer-specific survival as the endpoint. The p-value was estimated using a log-rank test of the difference in outcome between patients with higher activity levels (red) and those with lower/no activity (blue).
These treatment-responsive genes (including TOP2A, UHRF1, ASF1B, MCM4, WHSC1, MCM2, SUV39H1, BLM, BRCA1, CCNA2, E2F1, and MYBL2) have known functions in DNA repair, epigenetic modifications, cell cycle, proliferation, and/or survival, which are all associated with cancer malignancy. Notably, each of these is overexpressed in advanced human prostate cancer, and their activity levels are associated with disease outcome, as shown by univariate analyses using a COX proportional hazard model on the Sboner dataset (Fig. 5C,D). Moreover, analyses based on the Balk dataset revealed robust activity levels of the treatment-responsive genes in metastatic samples compared to primary tumors (Fig. 5E).
We further demonstrated the association of the activity levels of the treatment-responsive genes with drug response on a patient-by-patient basis using ssMARINa on the Sboner dataset (Fig 5F). In particular, the average activity levels of treatment-responsive genes were strikingly correlated with the rapamycin + PD0325901 drug response (Spearman rho = 0.57, p < 2.2 × 10-16) (Fig. 5F), similar to that observed for the FOXM1/CENPF activity (see Fig. 4D). Most notably, multivariate Kaplan-Meier survival analysis using the Sboner dataset to evaluate disease-specific survival revealed that patients with higher activity levels of the treatment-responsive genes have a shorter time to prostate cancer-specific death compared to patients with lower activity levels (Long-Rank p-value = 1.7 × 10-5) (Fig. 5G). Importantly, the activity of the treatment-responsive sub-network of the FOXM1/CENPF regulon was more significant than the FOXM1/CENPF regulon (Log-Rank p-value=1.3 × 10-4) and also outperformed a random comparable set of genes with respect to the Cox proportional hazard model (p-value for improvement <0.001) and Kaplan-Meier survival analysis (p-value for improvement <0.015) (see Detailed Experimental Procedures). Taken together, these findings suggest that computationally-predicted treatment-responsive genes can be used to identify patients that are likely to benefit from treatment with drugs that co-target the PI3-kinase/mTOR and MAP-kinase signaling pathways, and provides a proof-of-concept for the overall approach.
Discussion
In this study, we introduce a generalizable computational approach to extrapolate in vivo preclinical treatment data from GEM models to inform on human cancer treatment. Our method infers drug efficacy based on the ability of a given drug to revert the transcriptional regulon of key dependencies that drive the tumor phenotype. Importantly, we show that this method can be used to prioritize drug combinations based on analysis of individual compounds, which greatly enhances the value of in vivo preclinical analyses of compounds in mice. We demonstrate this approach with a proof-of-concept study based on identification of drugs and drug combinations that inhibit the activity of FOXM1/CENPF, which were chosen for their established relevance for lethal prostate cancer (Aytes et al., 2014). However, this approach should be applicable to identify candidate drugs and drug combinations for many other driver gene(s) of interest and not limited to prostate cancer. Notably, the molecular programs affected by drug treatment in the GEM model are well-conserved with human prostate cancer, which supports the concept that analyses of drug treatment data from mouse models can be used to identify treatment responsive genes for human prostate cancer. Thus, we have described a method to identify drugs and drug combinations that specifically inhibit cancer driver genes, as well as to identify potential biomarkers to predict the efficacy of drug treatments for individual patients.
Several features of our approach distinguish it from other strategies previously used to screen for drug response in human cancer. First, most other approaches have been based on analyses of cancer cell lines in culture (e.g., (Barretina et al., 2012; Garnett et al., 2012)), whereas our study is based on drug perturbation of GEM models in vivo. Thus, we evaluate drug efficacy in the context of the native tumor microenvironment and intact immune system, which are now widely recognized as being essential for drug response in vivo, particularly given recent advances in immunotherapy. Although the tumor context of any individual GEM model is unlikely to fully recapitulate that of human cancer, we address this limitation by analyzing multiple distinct GEM models to avoid idiosyncratic GEM-specific biology. Indeed, we have observed a remarkable concordance of the molecular consequences of drug treatment between our “consensus” analyses of mouse models and human prostate cancer.
A second distinguishing feature of our approach is its ability to identify synergistic drug combinations based on single agent treatment data. From a practical standpoint, the number of drugs that can be feasibly evaluated using in vivo perturbations in a series of GEM models is limited. Therefore, the ability to evaluate the efficacy of drug combinations by profiling a relatively small number of single drugs (e.g., the 100 most relevant compounds) would allow assessment of a very large potential combination therapy space (e.g., 4,950 combinations), thus affording significant economy of scale.
A third important feature is that our computational method identifies drugs based on their ability to inhibit specific drivers of the tumor phenotype, rather than on overall toxicity or inhibition of more general tumor-related parameters. In particular, the method evaluates the efficacy of drug response based on inhibition of the transcriptional regulon of specific master regulators of interest. Furthermore, our computational analysis of treatment response in the GEM models in vivo has also identified treatment-responsive genes that are conserved in human prostate cancer. We propose that such treatment-responsive genes may serve as surrogate biomarkers to infer the potential efficacy of drug treatments in patients. In particular, our current findings suggest that previous analyses may have underestimated the value of molecular inference of preclinical data from GEM models for not only predicting optimal drug combinations but also for identifying molecular markers for predicting treatment response to such drugs.
The PI3-kinase/mTOR and MAP-kinase signaling pathways are known to be dysregulated in many advanced prostate cancers (Aytes et al., 2013; Kinkade et al., 2008; Taylor et al., 2010). Currently, drugs that target these pathways (albeit not rapamycin and PD0325901) are being evaluated in numerous clinical trials for prostate cancer and many other solid tumors, including combination therapy regimes. Results from the current study as well as previous work (Aytes et al., 2014) suggest that aberrant levels of FOXM1 and CENPF, as assessed by immunostaining of tumor samples, may identify patients who would likely benefit from treatment with agents that target the PI3-kinase/mTOR and/or MAP-kinase signaling pathways. In addition, our study suggests that the treatment-responsive genes we have identified could provide intermediate biomarkers to assess short-term efficacy of combination therapy in patients, a strategy that can be readily generalized to other targets and therapies. Thus, our studies may inform or modulate the design of clinical trials or help provide a mechanistic basis for clinical findings.
Beyond prostate cancer, our computational methodology may be beneficial to identify drugs that target key actionable targets in vivo for a wide range of tumor types, oncogene and non-oncogene dependencies, and therapeutic agents, including both FDA-approved and experimental compounds. Since many cancer types now have relevant GEM models that are being used in many preclinical studies, it would be advantageous to use our approach to apply these preclinical data from GEMs to analyze treatment response in human cancer.
Experimental Procedures
Computational prediction of drug synergy
Computational inference of drugs that inhibit FOXM1 and CENPF activity was done using their shared target genes predicted from the mouse or human prostate cancer interactomes and using in vivo drug perturbation data, which were described in (Aytes et al., 2014). Target gene reversion (i.e., inhibition) was assessed using GSEA for each drug across each GEM model. Global reversion scores (GRS) for each drug were then inferred by integrating the reversion scores across each GEM model using a metric based on the Stouffer integration formulation (Whitlock, 2005). Optimal drug combinations were predicted from the single-agent in vivo drug perturbation data by determining the synergistic reversion scores (SRS) for each drug using an F1 statistical measure, which maximizes the number of unique targets affected by each drug as well as the total number of targets affected by two drugs. Global synergistic reversion scores (GSRSs) were then estimated as an average SRS weighted by the number of mouse models in which a drug pair was estimated to be effective (i.e, to share a non-zero SRS). Details of the computational methods used to compute GRS and GSRS are provided in the Detailed Experimental Procedures, and the data in Tables S1 and S2.
Efficacy of drug treatment
Cell culture studies were done as described previously (Aytes et al., 2014) using human prostate cancer cell lines obtained from ATCC and mouse cell lines derived from the GEM models used herein ((Aytes et al., 2013) and manuscript in preparation). Rapamycin and docetaxel were purchased from LC labs; PD0325901 was provided by Pfizer. Cell culture assays were performed in a minimum of two independent experiments each done in triplicate; data are represented by the mean ± SD. For in vivo studies, tumor-bearing NPK mice (Aytes et al., 2013) or allografted NPK tumors were treated with vehicle or rapamycin (10 mg/kg) and/or PD0325901 (10 mg/kg), or docetaxel (10 mg/kg) as described (Kinkade et al., 2008). At the time of sacrifice, tissues were collected for histopathological and molecular analysis as described (Aytes et al., 2013; Kinkade et al., 2008). GraphPad Prism software (Version 5.0) was used for statistical analyses and to generate data plots. A complete list of primers used in this study is provided in Table S5.
Cross-species computational analysis of drug treatments signatures
Gene expression profiles based on Illumina expression arrays as in (Aytes et al., 2014) were used to generate drug treatment signatures for the mouse tumors or allografts, as detailed in Table S3. For comparison of mouse treatment signatures with human signatures, the mouse genes were mapped to their corresponding human orthologs. Single-sample computation of FOXM1/CENPF activity levels or drug treatment across human patients was inferred for each patient sample using single sample MaRINA (ssMARINa) as described (Aytes et al., 2014) and see Detailed Experimental Procedures. COX proportional hazard model and Kaplan-Meier analysis were done using the “surv” and “coxph” functions from the survcomp package in R v2.14.0.
Supplementary Material
Acknowledgments
This work was supported by grants CA173481 (to CAS), CA084294 (to CAS, MMS and AC), U54 CA121852 (to AC, CAS, MMS), CA154293 (to MMS and CAS), DK076602 (to MMS), and Silico Research Centre of Excellence NCI-caBIG, SAIC 29XS192 (to AC). AA was a recipient of a Marie Curie International Outgoing Fellowship (PIOF-GA-2009-253290), co-sponsored with the Catalan Institute of Oncology-Bellvitge Institute for Biomedical Research, Barcelona, Spain, and a recipient of a pilot award from the Irving Institute for Clinical and Translational Research at Columbia University supported by the National Center for Advancing Translational Sciences, NIH (UL1 TR000040). AM is a recipient of a Prostate Cancer Foundation Young Investigator Award. CAS is an American Cancer Society Research Professor supported in part by a generous gift from the F.M. Kirby Foundation.
Footnotes
Accession numbers: Gene expression profiling data reported in this study are provided in Geo: GSE69211 and Geo: GSE69213.
Author contributions: AM performed all of the computational analyses and AA performed all of the experimental analyses. MZ provided the new mouse cell lines described herein. AM, AA, MMS, CAS, and AC designed experiments, analyzed the data, and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abate-Shen C, Pandolfi PP. Effective utilization and appropriate selection of genetically engineered mouse models for translational integration of mouse and human trials. Cold Spring Harbor protocols. 2013;2013 doi: 10.1101/pdb.top078774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nature biotechnology. 2012;30:679–692. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- Aytes A, Mitrofanova A, Kinkade CW, Lefebvre C, Lei M, Phelan V, LeKaye HC, Koutcher JA, Cardiff RD, Califano A, et al. ETV4 promotes metastasis in response to activation of PI3-kinase and Ras signaling in a mouse model of advanced prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E3506–3515. doi: 10.1073/pnas.1303558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytes A, Mitrofanova A, Lefebvre C, Alvarez MJ, Castillo-Martin M, Zheng T, Eastham JA, Gopalan A, Pienta KJ, Shen MM, et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer cell. 2014;25:638–651. doi: 10.1016/j.ccr.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AJ, Autio KA, Roach M, 3rd, Scher HI. High-risk prostate cancer-classification and therapy. Nature reviews Clinical oncology. 2014;11:308–323. doi: 10.1038/nrclinonc.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Alvarez MJ, Talos F, Dhruv H, Rieckhof GE, Iyer A, Diefes KL, Aldape K, Berens M, Shen MM, Califano A. Identification of Causal Genetic Drivers of Human Disease through Systems-Level Analysis of Regulatory Networks. Cell. 2014;159:402–414. doi: 10.1016/j.cell.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooperberg MR, Moul JW, Carroll PR. The changing face of prostate cancer. J Clin Oncol. 2005;23:8146–8151. doi: 10.1200/JCO.2005.02.9751. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon-Cardo C, Abate-Shen C. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. The Journal of clinical investigation. 2008;118:3051–3064. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre C, Rajbhandari P, Alvarez MJ, Bandaru P, Lim WK, Sato M, Wang K, Sumazin P, Kustagi M, Bisikirska BC, et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol Syst Biol. 2010;6:377. doi: 10.1038/msb.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji D, Omlin A, Pezaro C, Shamseddine A, de Bono J. Metastatic castration-resistant prostate cancer (CRPC): preclinical and clinical evidence for the sequential use of novel therapeutics. Cancer metastasis reviews. 2014;33:555–566. doi: 10.1007/s10555-013-9473-1. [DOI] [PubMed] [Google Scholar]
- Pienta KJ, Smith DC. Advances in prostate cancer chemotherapy: a new era begins. CA Cancer J Clin. 2005;55:300–318. doi: 10.3322/canjclin.55.5.300. quiz 323-305. [DOI] [PubMed] [Google Scholar]
- Politi K, Pao W. How genetically engineered mouse tumor models provide insights into human cancers. J Clin Oncol. 2011;29:2273–2281. doi: 10.1200/JCO.2010.30.8304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathkopf D, Scher HI. Androgen receptor antagonists in castration-resistant prostate cancer. Cancer journal. 2013;19:43–49. doi: 10.1097/PPO.0b013e318282635a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury S, Chinnaiyan AM. Advancing precision medicine for prostate cancer through genomics. J Clin Oncol. 2013;31:1866–1873. doi: 10.1200/JCO.2012.45.3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Perez C, Tamborero D, Schroeder MP, Antolin AA, Deu-Pons J, Perez-Llamas C, Mestres J, Gonzalez-Perez A, Lopez-Bigas N. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer cell. 2015;27:382–396. doi: 10.1016/j.ccell.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Ryan CJ, Tindall DJ. Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. J Clin Oncol. 2011;29:3651–3658. doi: 10.1200/JCO.2011.35.2005. [DOI] [PubMed] [Google Scholar]
- Sboner A, Demichelis F, Calza S, Pawitan Y, Setlur SR, Hoshida Y, Perner S, Adami HO, Fall K, Mucci LA, et al. Molecular sampling of prostate cancer: a dilemma for predicting disease progression. BMC Med Genomics. 2010;3:8. doi: 10.1186/1755-8794-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nature reviews Drug discovery. 2006;5:741–754. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes & development. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer research. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA, Mitrofanova A, Bergren SK, Abate-Shen C, Cardiff RD, Califano A, Shen MM. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nature cell biology. 2013;15:274–283. doi: 10.1038/ncb2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher's approach. Journal of evolutionary biology. 2005;18:1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- Wong YN, Ferraldeschi R, Attard G, de Bono J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nature reviews Clinical oncology. 2014;11:365–376. doi: 10.1038/nrclinonc.2014.72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.