

Genetically decreased or increased, mouse smooth muscle-specific α2-Na+ pump expression effects on blood pressure are attenuated by compensatory changes in arterial Ca2+ transporter expression. This implies that the increased Ca2+ transporter expression observed in many hypertension models is not compensatory but, rather, contributes significantly to the blood pressure elevation.
Keywords: Na+/Ca+ exchanger 1, α2-Na+ pump, angiotensin II, salt, sarco(endo)plasmic reticulum Ca2+-ATPase 2, blood pressure
Abstract
Arterial myocytes express α1-catalytic subunit isoform Na+ pumps (75–80% of total), which are ouabain resistant in rodents, and high ouabain affinity α2-Na+ pumps. Mice with globally reduced α2-pumps (but not α1-pumps), mice with mutant ouabain-resistant α2-pumps, and mice with a smooth muscle (SM)-specific α2-transgene (α2SM-Tg) that induces overexpression all have altered blood pressure (BP) phenotypes. We generated α2SM-DN mice with SM-specific α2 (not α1) reduction (>50%) using nonfunctional dominant negative (DN) α2. We compared α2SM-DN and α2SM-Tg mice to controls to determine how arterial SM α2-pumps affect vasoconstriction and BP. α2SM-DN mice had elevated basal mean BP (mean BP by telemetry: 117 ± 4 vs. 106 ± 1 mmHg, n = 7/7, P < 0.01) and enhanced BP responses to chronic ANG II infusion (240 ng·kg−1·min−1) and high (6%) NaCl. Several arterial Ca2+ transporters, including Na+/Ca2+ exchanger 1 (NCX1) and sarcoplasmic reticulum and plasma membrane Ca2+ pumps [sarco(endo)plasmic reticulum Ca2+-ATPase 2 (SERCA2) and plasma membrane Ca2+-ATPase 1 (PMCA1)], were also reduced (>50%). α2SM-DN mouse isolated small arteries had reduced myogenic reactivity, perhaps because of reduced Ca2+ transporter expression. In contrast, α2SM-Tg mouse aortas overexpressed α2 (>2-fold), NCX1, SERCA2, and PMCA1 (43). α2SM-Tg mice had reduced basal mean BP (104 ± 1 vs. 109 ± 2 mmHg, n = 15/9, P < 0.02) and attenuated BP responses to chronic ANG II (300–400 ng·kg−1·min−1) with or without 2% NaCl but normal myogenic reactivity. NCX1 expression was inversely related to basal BP in SM-α2 engineered mice but was directly related in SM-NCX1 engineered mice. NCX1, which usually mediates arterial Ca2+ entry, and α2-Na+ pumps colocalize at plasma membrane-sarcoplasmic reticulum junctions and functionally couple via the local Na+ gradient to help regulate cell Ca2+. Altered Ca2+ transporter expression in SM-α2 engineered mice apparently compensates to minimize Ca2+ overload (α2SM-DN) or depletion (α2SM-Tg) and attenuate BP changes. In contrast, Ca2+ transporter upregulation, observed in many rodent hypertension models, should enhance Ca2+ entry and signaling and contribute significantly to BP elevation.
NEW & NOTEWORTHY
Genetically decreased or increased, mouse smooth muscle-specific α2-Na+ pump expression effects on blood pressure are attenuated by compensatory changes in arterial Ca2+ transporter expression. This implies that the increased Ca2+ transporter expression observed in many hypertension models is not compensatory but, rather, contributes significantly to the blood pressure elevation.
arterial smooth muscle (SM) cells (ASMCs) from rodents and humans express two types of Na+ pumps (11, 28, 57). The majority of pumps (∼75–80%) have an α1-catalytic subunit, which, in rodents, has unusually low ouabain affinity (3, 36); the remainder are high ouabain affinity α2-Na+ pumps. α1-Pumps (“housekeepers”) maintain the low intracellular Na+ concentration in most of the cytoplasm (1). The more sparse α2-pumps colocalize with plasma membrane (PM) Na+/Ca2+ exchanger 1 (NCX1) and sarcoplasmic reticulum (SR) Ca2+ pumps [sarco(endo)plasmic reticulum Ca2+-ATPase 2 (SERCA2)] at PM-SR junctions in ASMCs (21, 28, 42, 51). As a result, the functional coupling of these transporters helps regulate local cytosolic Na+ and Ca2+ concentrations and Ca2+ signaling (1, 25, 31, 40, 42).
A number of studies have linked the α2-Na+ pump and its high-affinity ligand, endogenous ouabain (EO), to blood pressure (BP) regulation. In mice with one null mutant α2-allele (α2+/−) (20), arterial α2-Na+ pump expression is reduced by ∼50%, but the total Na+ pump number (mostly α1) is still ∼90% of normal (50, 57); these mice have modestly elevated basal BP (57). In contrast, mice with one null α1-allele (α1+/−) express many fewer total Na+ pumps in ASMCs (∼60% of normal) but have normal basal BP (57). In addition, plasma EO is elevated in many humans with essential hypertension or mineralocorticoid hypertension (33, 39, 49), in several rodent hypertension models (14, 15, 24, 27), and in pregnant rats (19). Indeed, prolonged ouabain administration induces hypertension in rodents (11, 12, 34). Furthermore, genetically engineered mice with ouabain-resistant α2-Na+ pumps (α2R/R) are resistant to adrenocorticotropic hormone-induced hypertension (10, 30) and ouabain-induced hypertension (11). In addition, α2R/R dams have unusually low BP during late pregnancy (38). Thus, there is ample reason to characterize the influence of arterial α2-Na+ pumps on BP. To this end, we studied mice that were genetically engineered to either decrease or increase expression of α2-Na+ pumps in SM.
The present report describes a new mouse model: SM-specific knockdown of α2-Na+ pumps, by ∼60% in ASMCs, generated with a dominant negative (DN), nonfunctional α2-transgene (α2SM-DN mice). We then compared basal BP and BP responses to chronic ANG II infusion and high dietary salt (HS) in these α2SM-DN mice and in mice that markedly overexpress α2-Na+ pumps in their ASMCs due to a SM-specific normal α2-transgene (α2SM-Tg) (43). The results reveal that basal BP and the responses to ANG II and HS are significantly, albeit modestly, increased in α2SM-DN mice and modestly decreased when α2-Na+ pump expression is increased in ASMCs.
We attribute the relatively small magnitude of these BP changes to observed concurrent modifications in the expression of ASMC NCX1 and other Ca2+ transporters that likely compensate for the alterations in α2 expression by minimizing changes in cell Ca2+ homeostasis. This view is bolstered by reports that basal BP is inversely correlated with ASMC NCX1 expression in mice with SM-specific genetic modification of NCX1 expression (18, 53, 58). We discuss how this helps explain the finding that the BP phenotypes in SM-α2 engineered mice are only modestly different from those of wild-type (WT) control mice even though α2-Na+ pump activity apparently has a major influence on Ca2+ homeostasis and contractile regulation. This illustrates why attempts to deduce details about the significance of individual proteins in BP regulation from single gene-engineered mouse models may be misleading unless related compensatory changes are also considered. Finally, we describe how an understanding of these compensatory responses to primary arterial defects in α2-Na+ pumps provides insights into the significance of arterial myocyte Ca2+ transporter protein reprogramming that is observed in many hypertension models.
MATERIALS AND METHODS
Experimental Animals
Ethical approval.
All mouse protocols were approved by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine. The protocol used for the generation of α2SM-DN mice was approved by the Institutional Animal Care and Use Committee of Cornell University College of Veterinary Medicine.
Mice were housed in an Association for Assessment and Accreditation of Laboratory Animal Care-approved facility with a 12:12-h light-dark cycle. Mice were unrestrained and had free access to food and water.
Generation of Na+ pump α2-subunit NH2-terminal constructs.
We generated the DNA sequence corresponding to the NH2-terminal 1–120 amino acids (including the cytoplasmic domain and first transmembrane helix; see Fig. 1A) of the α2-isoform of the Na+ pump catalytic subunit with a COOH-terminal Flag epitope tag (Fig. 1B). This sequence was cloned into pFlag-CMV-5a vector (Sigma Chemical, St. Louis, MO) by a PCR-based approach. A COOH-terminal Flag tag was then also cloned in using primer pairs (5′-TTAAAGCTTCGCCACCATGGGTCGTGGGGCA-3′ and 5′-CGCCGAATTCCCTACTTGTCATCGTCGTCCT-3′) to create a DN construct with a NH2-terminal Kozak sequence [α2(1–120)flag; Fig. 1]. Expression of this nonfunctional construct, which contains the normal α2 sorting sequence, replaces native, functional α2-Na+ pumps in arterial myocytes and is therefore DN (51).
Fig. 1.
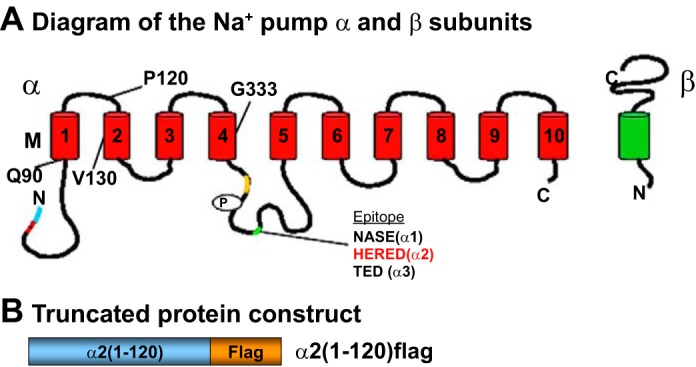
A: Na+ pump α- and β-subunit organization in the plasma membrane (PM). The locations of the antibody epitope amino acid sequences used for the generation of isoform-specific antibodies are indicated on the α-subunit. They are all located within a short segment on the large cytoplasmic loop between transmembrane segments TM4 and TM5 (41). B: structure of the Flag-tagged Na+ pump α2-subunit NH2-terminal segment used to generate α2 dominant negative (DN) mice. This construct is expressed in the PM and causes greatly reduced PM expression of native α2-Na+ pumps (the DN effect) in arterial smooth muscle (SM) cells (ASMCs) (51).
Gene targeting strategy and generation of α2SM-DN mice.
To generate α2SM-DN mice, the α2(1–120)flag transgene was engineered with a HindIII site and Kozak sequence at the 5′-end and an EcoRI site at the 3′-end (Fig. 2A). The PCR product was digested with HindIII and EcoRI and then subcloned into pBluescript PA plasmid upstream of two polyA sites (Fig. 2B). XhoI digestion of the plasmid yielded a 1-kb α2(1–120)flag-pA fragment, and the fragment was then introduced into the pGEX4T-SM-myosin heavy chain (MHC) plasmid at the SalI site immediately downstream of the SM-MHC promoter (Fig. 2C) (32). The transgene sequence was verified by sequencing the 3′- and 5′-ends of the cDNA. The successful recombinant plasmid was linearized by digestion with NotI, purified, and injected into the pronucleus of B6/D2F2 fertilized mouse eggs, which were then implanted into pseudopregnant female mice (at the Cornell Core Transgenic Mouse Facility, Cornell University, Ithaca, NY). Founder α2SM-DN mice generated by this procedure were identified by PCR and Southern blot analyses and then subsequently bred to WT C57Bl/6 mice. The presence of Na+ pump α2-DN was verified by genotyping of α2SM-DN progeny using PCR primer pairs (SM-MHC forward 5′-ATCTTCTAACTGGGTGGTGGTGGT-3′ and Flag reverse 5′-TGTAATCGGATCCTTCGTCCTCCA-3′) to yield a 500-bp product.
Fig. 2.

Diagram of the gene strategy used to generate mice that express the truncated NH2-terminal DN segment of the Na+ pump α2-subunit. Expression of the DN segment was under the control of the SM-specific myosin heavy chain (MHC) promoter. See text for details.
α2SM-DN mice were viable and exhibited no obvious behavioral or anatomic abnormalities. They bred normally and were inbred to generate mice with a maximum number of copies of the transgene. On two back crosses with WT C57Bl/6 mice, all offspring expressed the transgene (as verified by PCR).
Transgenic α2SM-Tg mice.
α2SM-Tg mice (on a FVB/n background) were generated at the University of Cincinnati (43). Heterozygote breeders (α2SM-Tg/−) were used by the University of Maryland investigators to generate two lines: one designated WT FVB/n, which had no transgene, and an inbred α2SM-Tg line to maximize transgene expression. On two back crosses with WT FVB/n mice, >90% of the offspring expressed the transgene (as identified by PCR).
BP Measurements
Telemetry.
In most experiments, BP was monitored continuously in awake, free-moving mice using implanted BP transmitters (TA11PA-C10, Data Science, St. Paul, MN). WT and transgenic mice (16–18 wk old) were anesthetized with 2–2.5% isoflurane supplemented with 100% O2. The right carotid artery was isolated through a midline neck incision and was ligated near its bifurcation onto internal and external branches. A small hole was made with a 25-gauge needle, and the transmitter catheter was inserted and threaded forward to the aortic arch. The transmitter body was passed subcutaneously to a subcutaneous pocket in the left abdominal flank. Seven to ten days after surgery, 24-h BPs were recorded by telemetry.
Tail cuff.
In a preliminary experiment, systolic arterial BP and heart rate were screened in conscious, restrained α2SM-DN mice with a commercial tail-cuff pulse BP system (model MC4000, Hatteras Instruments, Cary, NC). Many of these “first generation” α2SM-DN mice were then used as breeders; others were euthanized for tissue harvesting and immunoblot analysis. Ten BP readings were obtained, and the measurements were repeated on 3 consecutive days. These data were averaged to obtain a single basal systolic BP value for each mouse. The tail-cuff technique was validated by simultaneously recording BP by telemetry in three mice implanted with BP transmitters. The correlation power coefficient (r2) from the linear regression was 0.87, indicating good agreement between the two methods.
The BP data collection was performed “double blind”: the individual measuring the BP did not know the genotype. Animal code numbers were matched with genotype and BP after the data were collected. Male mice were used for telemetric BP data collection to simplify comparison with a previous study of α2SM-Tg male mice (43).
ANG II and Dietary Protocols
Once basal BPs were obtained, mice were anesthetized with 1.5% isofluorane inhalation and an ANG II-filled (acetate salt, Bachem Americas, Torrance, CA) or vehicle-filled osmotic minipump (Alzet model 2004, Durect, Cupertino, CA) was then implanted. It was inserted into a subcutaneous pocket in the right ventral abdominal wall via a small right groin incision.
The standard diet contained 0.4% NaCl; high-salt (HS) diets contained either 2% or 6% NaCl, as indicated in the results. All diets were purchased from Dyets (Bethlehem, PA).
Myogenic Tone and Reactivity
Details of the methods have been previously published (58). In brief, segments of fourth-order arteries (mean passive diameter ≈ 140 μm) were dissected from the mesenteric artery arcade on the external surface of the small intestine. Segments were cannulated at both ends, intraluminal pressurize was raised to 70 mmHg (no internal flow), and segments were superfused with physiological salt solution (PSS) at 35–37°C. When myogenic tone (MT) developed, intraluminal pressure was lowered to 10 mmHg and raised stepwise in 20 mmHg increments to 130 mmHg to determine myogenic reactivity (MR). Segments were then equilibrated in Ca2+-free (0Ca) PSS, and the sequence was repeated to determine the passive diameter at each pressure level. During this process, external diameter was continuously monitored with a custom online edge detector based on a Nikon TMS microscope (Nikon, Melville, NY). Artery segments that developed significant leaks were discarded.
Data are presented as graphs of artery diameter versus intraluminal pressure in normal Krebs solution and 0Ca PSS (passive diameter). The difference between the two curves is the MR.
Immunoblot Analysis
Preparation of tissue extracts.
The aorta, urinary bladder, heart, and brain from α2SM-DN mice and the aorta from α2SM-Tg mice were minced in homogenization buffer (HB1) at 4°C with a Polytron homogenizer (Brinkmann Instruments, Westbury, NY). The suspension was then centrifuged for 20 min at 16,000 g and 4°C. The pellet (cell membranes) from this centrifugation step was resuspended in HB2 buffer [similar to HB1 but with the addition of the nonionic detergent IGEPAL CA-630 (1%), Sigma-Aldrich] and incubated for 1 h on ice. The lysate was then centrifuged for 20 min at 16,000 g and 4°C. The supernatant was collected and stored at −80°C until use. The protein concentration was determined with a bicinchoninic acid assay (Bio-Rad, Hercules, CA) with BSA as a standard.
Immunoblot analysis.
One volume of protein preparation was mixed with an equal volume of 2× Laemmli Sample Buffer (Bio-Rad) containing 5% 2-mercaptoethanol, and proteins were separated on SDS-PAGE minigels (Bio-Rad). The protein on the gel was then transferred to a polyvinylidene difluoride membrane with an iBlot Dry Blotting System (Invitrogen, Carlsbad, CA). The membrane was blocked with 5% nonfat dry milk in Tris-buffered saline with Tween 20 for 2 h and subjected to immunoblot analysis using isoform-specific monoclonal or polyclonal antibodies. Details of our methods have been previously published (26). Immunoblot band densities were normalized to the respective loading controls (α-actin in the heart or β-actin in the aorta, bladder, and brain) and then compared with expression in the respective WT tissues (equal to 100%).
Solutions and Antibodies
PSS contained (in mM) 112 NaCl, 25.7 NaHCO3, 4.9 KCl, 2.0 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 11.5 d-glucose, and 10 HEPES (pH 7.4; gas composition: 5% O2-5% CO2-90% N2). In 0Ca PSS, CaCl2 was omitted and 0.5 mM EGTA was added.
HB1 contained 140 mM NaCl, 10 mM NaH2PO4, 2 mM EDTA, 10 mM NaN3, and protease inhibitor cocktail tablets (2 tablets/50 ml, Roche Diagnostics, Mannheim, Germany). Tris-buffered saline with Tween 20 solution contained 50 mM Tris, 150 mM NaCl, and 0.05% Tween 20 at pH 7.6 (adjusted with HCl).
The following antibodies were used for immunoblot analysis: 1) polyclonal anti-Na+ pump α1-subunit raised against the NASE epitope (gift from T. Pressley, Texas Tech University, Lubbock, TX; see Fig. 1A); 2) polyclonal anti-Na+ pump α2-subunit raised against the HERED epitope (Millipore, Billerica, MA; see Fig. 1A); 3) monoclonal anti-NCX1 antibody (R3F1, gift from K. D. Philipson, University of California-Los Angeles, Los Angeles, CA); 4) monoclonal anti-SERCA2 (Affinity BioReagents, Golden, CO); 5) polyclonal anti-PMCA1 (Thermo Scientific, Rockford, IL); 6) monoclonal anti-Flag (Sigma-Aldrich); 7) polyclonal anti-transient receptor potential C-type channel protein 6 (TRPC6; Sigma-Aldrich); 8) monoclonal anti-cardiac α-actin (Sigma-Aldrich); 9) monoclonal anti-SM β-actin (Sigma-Aldrich); and 10) monoclonal anti-GAPDH (Abcam, Cambridge, MA).
Statistical Analyses
Data are expressed as means ± SE; n values denote numbers of mice. Comparisons of BP data were made using ANOVA or Student's paired or unpaired t-tests, as appropriate. Differences were considered significant at P < 0.05.
RESULTS
Genotype Verification of α2-Na+ Pump Expression and Basal BP in Genetically Engineered Mice
α2SM-DN mice.
Immunoblot data (Fig. 3) demonstrated that α2SM-DN mice expressed the α2(1–120)flag DN construct (identified with anti-Flag antibodies) in the aorta and urinary bladder, in which SM is prevalent. The DN construct was not detected in the heart or brain. Native α2 was identified with antibodies to the HERED epitope because this epitope is present in the native protein (41) but not in the DN construct (Fig. 1A). Native α2 expression was reduced by ∼50% in the urinary bladder and by ∼70% in the aorta of α2SM-DN mice, but normal levels were expressed in the heart and brain (Fig. 3). Thus, expression of the DN construct was SM specific. Although the heart and brain are highly vascularized, Na+ pumps (and NCX1) are ∼5- to 15-fold more prevalent in cardiac myocytes, glia, and neurons than in arteries (see, e.g., Refs. 16, 17, 35, and 57)1. Therefore, downregulation of α2 in cardiac and cerebral blood vessels would not have been detected in these heart and brain whole tissue immunoblots.
Fig. 3.

Several Ca2+ transporters are markedly downregulated in SM of α2SM-DN mice. A: representative immunoblots of aorta, urinary bladder, brain, and heart membrane proteins from wild-type (WT) and α2SM-DN mice. Na+ pump α1- and α2-isoforms were detected with antibodies raised against the amino acid sequences (NASE and HERED, respectively); see Fig. 1A and Ref. 41. Expressed DN peptide was detected with anti-Flag antibodies (see Fig. 1B). Expression of α2-Na+ pumps but not α1-Na+ pumps, Na+/Ca2+ exchanger 1 (NCX1), sarco(endo)plasmic Ca2+-ATPase 2 (SERCA2), PM Ca2+-ATPase 1 (PMCA1) and transient receptor potential C-type channel protein 6 (TRPC6) proteins were all reduced in the aorta and urinary bladder of α2SM-DN mice. Expression of all these proteins in the brain and heart was comparable in WT and α2SM-DN mice. Loading controls were α-actin (heart) or β-actin (aorta, bladder, and brain). B: summarized aorta and urinary bladder data from 3 to 8 mice. Values were normalized to the loading control (α-actin in the heart and β-actin in the aorta, bladder and brain) and then compared with expression in the respective WT tissues (equal to 100%). Note that α1 in the α2SM-DN mouse aorta and bladder and PMCA1 in the bladder were not markedly different from the WT group. Bar graphs for the brain and heart are not shown because expression levels for all tested proteins in α2SM-DN mice were within 100 ± 10% of WT mice (A).
Note that the DN construct has only one transmembrane segment and is missing the cation binding and translocation sites as well as the α2 catalytic machinery (Fig. 1) and is thus nonfunctional. Nevertheless, this construct contains the α2 sorting signal and is expressed in the PM (51). The reduced expression of native α2 in the aorta and bladder indicates that in vivo, as well as in vitro (51), expression of the DN construct downregulates native α2 in SMs.
Basal BP was modestly but significantly higher in α2SM-DN mice than in WT (C57Bl/6) control mice. Figure 4A shows systolic BPs (tail cuff) from the first male and female offspring of the founder mice. Figure 4B shows mean BP (MBP) data (telemetry) from a subsequent α2SM-DN generation of male mice.
Fig. 4.
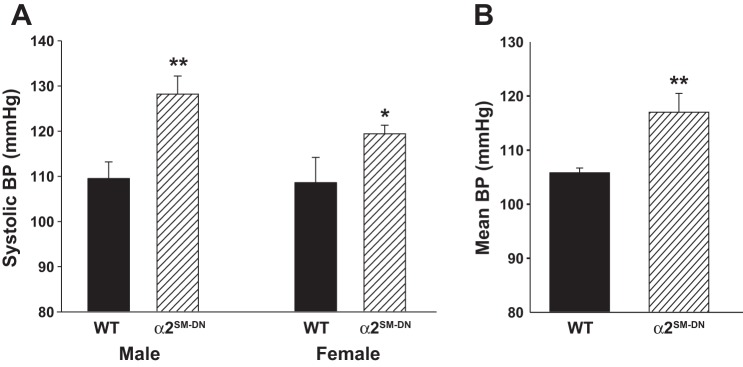
Basal blood pressure (BP) is elevated in α2SM-DN mice. A: systolic BP (tail cuff) data from the first male and female offspring of the α2SM-DN colony founder mice and their WT C57Bl/6 controls. BPs determined on 3 consecutive days were averaged to obtain a single value for each mouse. Male mice: n = 10 WT mice and 11 DN mice, **P < 0.01; female mice: n = 4 WT mice and 14 DN mice, *P < 0.03. B: basal 24-h mean BPs (MBPs) of male WT mice and a later generation of male α2SM-DN mice recorded by telemetry. MBPs from 3 consecutive days were averaged to obtain a single value for each mouse. n = 7 WT mice and 7 DN mice. **P < 0.01.
α2SM-Tg mice.
Overexpression of α2-Na+ pumps in the aorta and gastric antrum, but not in the heart, was described in the initial study (43) of α2SM-Tg male mice, which were modestly hypotensive (tail cuff). We confirmed that α2SM-Tg mice overexpress the α2-Na+ pump in the aorta (Fig. 5A) and have lower BP (telemetry) than WT-FVB/n control mice (Fig. 5B).
Fig. 5.
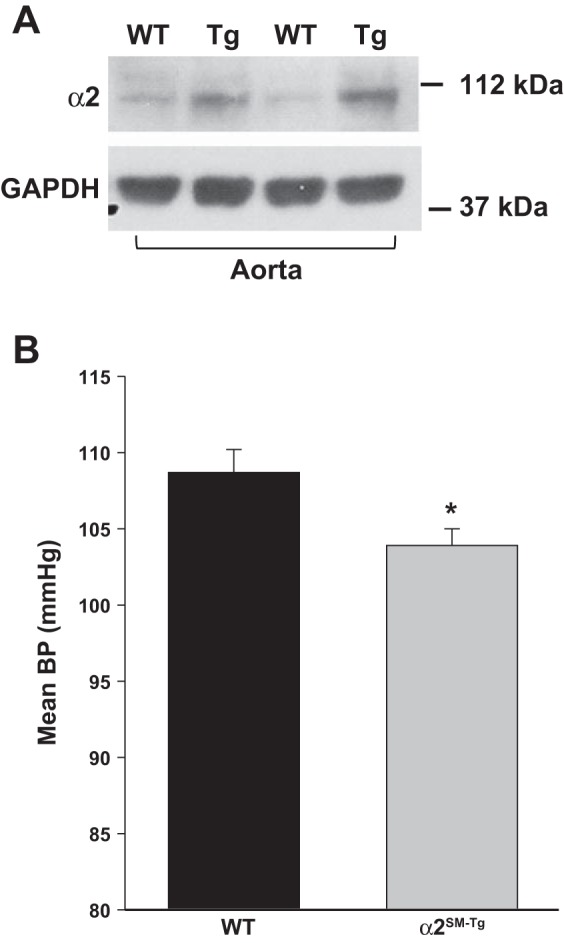
Expression of α2-Na+ pumps is increased and basal MBP is reduced in mice with a SM-specific α2-transgene (α2SM-Tg mice). A: immunoblots of aorta membrane proteins from WT FVB/n control and α2SM-Tg mice. B: basal 24-h MBPs of male WT and α2SM-Tg mice were recorded by telemetry. MBPs from 3 consecutive days were averaged to obtain a single value for each mouse. n = 9 WT mice and 15 Tg mice. *P < 0.02.
Ca2+ Transporter Expression Correlates With α2-Na+ Pump Expression
Prior immunoblot studies (42, 43) of the aorta and gastric antrum in α2SM-Tg mice revealed that not only the α2-Na+ pumps but also other Na+ and Ca2+ transporters were affected in these SM-containing tissues. α1-Na+ pump, NCX1, SERCA2, and PMCA proteins were all upregulated in both the aorta and gastric antrum. Contraction-related proteins (actin and 20-kDa myosin light chain) were, however, unaffected (42, 43).
Expression of α1-Na+ pumps is negligibly affected in α2SM-DN mice. However, a number of Ca2+ transporter proteins are markedly downregulated in both the aorta and urinary bladder (Fig. 3): NCX1, SERCA2, PMCA1, and TRPC6 (a component of receptor-operated Ca2+ channels). Of these, PMCA1 appears to be least affected, especially in the urinary bladder. Again, expression of all these proteins in the heart and brain was normal (Fig. 3A). These α2SM-DN and α2SM-Tg mouse data indicate that the expression of the several Ca2+ transporters in SM is directly correlated with SM-specific α2-Na+ pump expression. The fact that similar changes in Ca2+ transporter expression are seen in multiple SMs in the two models suggests a cause-effect relationship between α2-Na+ pump expression and these Ca2+ transporter changes. We attribute this to a compensatory effect on Ca2+ transport (see discussion).
Effects of ANG II and a HS Diet on BP in α2SM-DN and α2SM-Tg Mice
α2SM-DN mice.
The high basal BP in α2SM-DN mice (Fig. 4) raised the possibility that these mice might be “salt sensitive” and that their arteries might be hypercontractile, despite the downregulation of Ca2+ transporter proteins in the arteries. To explore this possibility, we measured the changes in BP during a 10-day period on a 6% NaCl (HS) diet (Fig. 6A) and during an 11-day subcutaneous infusion of low-dose ANG II (240 ng·kg−1·min−1; Fig. 6B). Both treatments increased BP more rapidly in α2SM-DN mice than in their WT counterparts (Fig. 6). Moreover, both treatments caused a sustained increase in BP in α2SM-DN mice but not in WT control mice (Fig. 6). At days 8–10 of treatment, the HS diet increased MBP by 6.1 mmHg in DN mice versus only 0.4 mmHg (i.e., no change from baseline) in WT mice, and ANG II increased MBP by 9.3 mmHg in DN mice vs 6.1 mmHg in WT mice. Thus, expression of the DN construct in SM augmented the responses to both HS and ANG II.
Fig. 6.
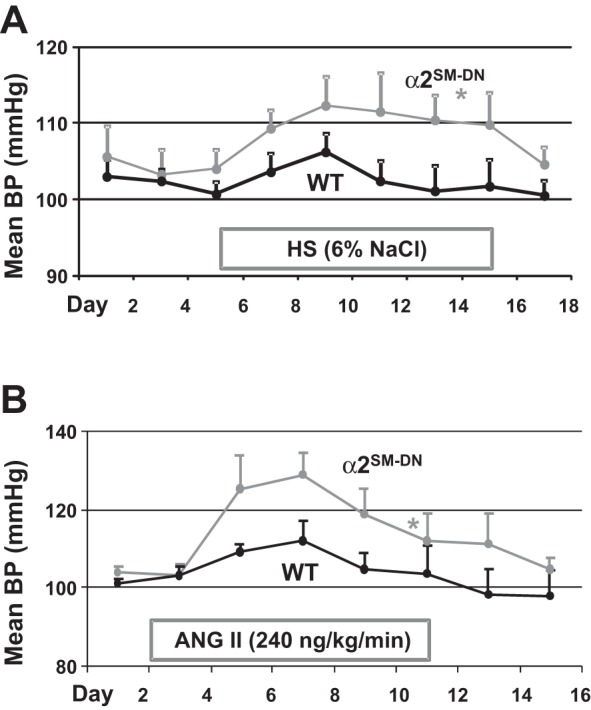
α2SM-DN mice are more sensitive to high dietary salt (HS) and low-dose ANG II than are WT mice. A: effect of a high-salt (HS; 6% NaCl) diet on 24-h MBP in WT and α2SM-DN mice. B: effect of 240 ng·kg−1·min−1 ANG II (subcutaneous infusion by osmotic minipump) on 24-h MBP in WT and α2SM-DN mice. n = 5 mice/group for both A and B. *P < 0.05 by ANOVA.
α2SM-Tg mice.
Since α2SM-Tg mice have reduced basal BP (Fig. 5) (43), we hypothesized that these mice, in contrast to α2SM-DN mice, might be “salt resistant” and relatively insensitive to the BP elevating effect of ANG II. Indeed, subcutaneous infusion of a modest dose of ANG II (400 ng·kg−1·min−1) for 12 days elevated MBP in WT control mice by ∼30 mmHg but by only ∼20 mmHg in α2SM-Tg mice (Fig. 7A). In other words, the ANG II-induced MBP elevation from the respective baselines was significantly attenuated in Tg mice. Furthermore, the ANG II-induced rise in MBP was slow in these mice. MBP did not peak until 96 h after the ANG II infusion was started in α2SM-Tg mice, whereas MBP peaked by 48 h in WT mice.
Fig. 7.
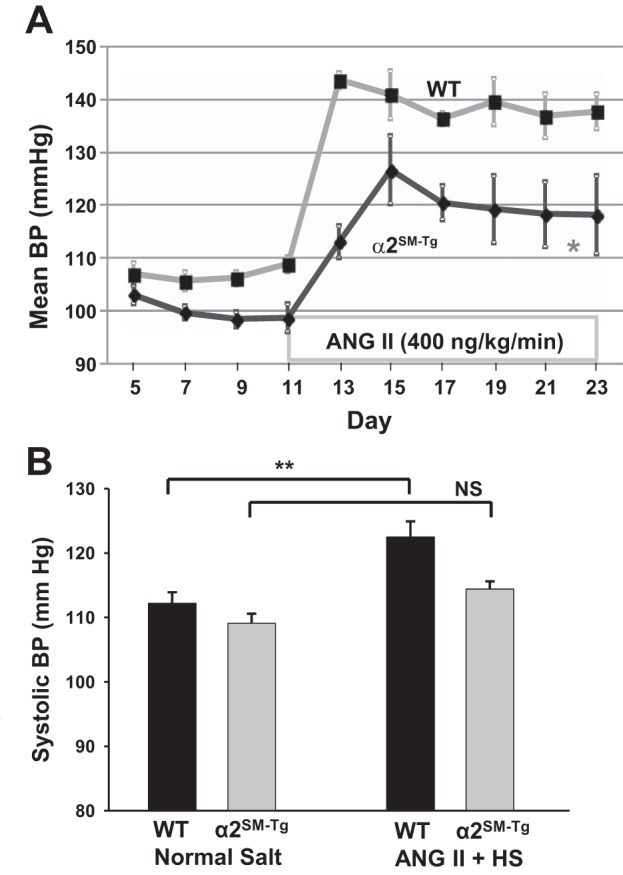
α2SM-Tg mice have reduced sensitivity to ANG II and ANG II + HS. A: effects of a moderate dose of ANG II (400 ng·kg−1·min−1 sc) on 24-h MBP in 5 WT mice and 5 α2SM-Tg mice. *P < 0.05 by ANOVA. B: effects of 12 days of a low dose of ANG II (300 ng·kg−1·min−1 sc) plus HS (2% NaCl diet) on systolic BP in 4 WT mice and 9 α2SM-Tg mice. Systolic BPs in each mouse were the means of three 12-h light period recordings. NS, not significant. **P < 0.01 by ANOVA.
ANG II and HS are “convergent signals” that act on the brain to elevate BP (9, 37); low-dose ANG II was therefore used to sensitize the mice to HS (9), as shown in Fig. 7B. WT and α2SM-Tg mice were fed a 2% NaCl (HS) diet during subcutaneous infusion with ANG II (300 ng·kg−1·min−1) for 12 days. This treatment elevated BP modestly in both groups of mice, but the increase in systolic BP in α2SM-Tg mice was only ∼60% of that in WT mice. Hence, the responses of α2SM-Tg mice to ANG II and to ANG II + HS diet were attenuated, in contrast to the augmented responses in α2SM-DN mice. This was reflected in both the rates of the response to ANG II and the magnitudes of the changes in MBP.
MR in Isolated Arteries From α2SM-DN and α2SM-Tg Mice
Davis and Hill (8) noted that the level of tone in isolated arteries “is often comparable to that observed in the same vessels in vivo.” With this concept in mind, we measured MT as a function of the intralumenal pressure (i.e., MR) in mesenteric small arteries from α2SM-DN and α2SM-Tg mice. As shown in Fig. 8A, the mean passive diameter of α2SM-Tg mouse arteries was slightly smaller than that of their WT FVB/n counterparts. When this was taken into account, MR was virtually identical in α2SM-Tg and WT FVB/n mouse arteries (Fig. 8B).
Fig. 8.

Expression of a Na+ pump α2-transgene in SMs does not affect myogenic reactivity (MR) in mouse mesenteric small arteries. A: pressure-diameter curves for WT (FVB/n) and α2SM-Tg mouse arteries. B: MR of WT and α2SM-Tg mouse arteries. MR is the difference between the passive diameter (PD) and the diameter measured in normal physiological salt solution (PSS). MR data are presented as percent decreases in arterial diameter relative to the PD at 130 mmHg intraluminal pressure (PD130), graphed as a function of intraluminal pressure.
The mean passive diameter of arteries from α2SM-DN mice was identical to that of their WT C57Bl/6 control counterparts (Fig. 9A). Interestingly, not only was the MR of α2SM-DN arteries significantly less than that of control arteries (Fig. 9B), but α2SM-DN arteries developed MT much more slowly than did WT arteries. All WT C57Bl/6 arteries developed steady tone ∼25–35 min after arteries were pressurized to 70 mmHg and warmed to ∼36°C, similar to both α2SM-Tg and WT FVB/n arteries. In contrast, it took at least 60 min for all six α2SM-DN arteries to develop tone, and several arteries required priming with one to two doses of 1 μM phenylephrine for 5 min before MT developed. As discussed below, we attribute this slow tone generation to the greatly reduced NCX1 and SERCA2 expression (Fig. 3).
Fig. 9.

Expression of a Na+ pump α2 DN construct in SMs reduces MR in mouse mesenteric small arteries. A: pressure-diameter curves for WT (C57Bl/6) and α2SM-DN mouse arteries. B: MR of WT and α2SM-DN mouse arteries (see Fig. 8 for further information). *P < 0.05 by ANOVA.
DISCUSSSION
Basal BP and BP Sensitivity to ANG II and Salt Correlate Inversely With α2-Na+ Pump Expression
Previously, we reported that basal BP was elevated in α2+/− mice, in which α2-Na+ pump expression in ASMCs was reduced by ∼50% (57). α2+/− mice are global heterozygotes; global homozygous null mutants (α2−/−) are born, but the neonates do not survive (20). To investigate further the role of arterial α2-Na+ pumps in BP modulation, we generated mice with a SM-specific reduction of expressed α2 based on a DN strategy (51). As described in the results, in vivo SM-specific expression of the NH2-terminal 120-amino acid segment of α2, a DN construct (51), reduced functional, full-length α2 expression in arteries by >50% (Fig. 3) and modestly elevated basal BP (Fig. 4). This is consistent with α2+/− mouse data (57), albeit seemingly not with the evidence that mice with cardiovascular (CV)-specific α2 knockout (KO; α2CV-KO mice) have normal basal BP (48) (but see How Can We Reconcile the Elevated Basal BP in α2SM-DN and α2+/− Mice With the Normal Basal BP in α2CV-KO Mice? below).
BP may be directly correlated with Ca2+ levels and Ca2+ signaling in the arteries (13, 57, 58). If this is the case, we reasoned that mice with higher basal BP (α2SM-DN) and, presumably, higher arterial Ca2+ levels, should exhibit increased sensitivity to ANG II and HS, factors that foster vasoconstriction and hypertension (9, 37). Our observations demonstrated that the α2SM-DN mice were, indeed, more sensitive to ANG II and HS than WT mice (Fig. 6).
In contrast, α2-Na+ pump transgenic mice (α2SM-Tg), which overexpress the α2-subunit specifically in SM, including ASMCs, have low basal BP, as measured by tail cuff (43) and by telemetry (Fig. 5). Based on the aforementioned reasoning, these mice may be presumed to have reduced arterial Ca2+ levels and, therefore, may be expected to exhibit attenuated responses to ANG II and HS, as observed (Fig. 7). Thus, SM-specific α2 expression correlated inversely with both basal BP and sensitivity to ANG II and HS in these mouse lines.
This speculation about Ca2+-dependent mechanisms in both α2SM-DN and α2SM-Tg mice obviously requires verification by direct measurements of cytosolic Ca2+ concentration. Nevertheless, the results, as shown in Figs. 6 and 7, are consistent with these expectations.
MR Does Not Correlate With SM-Specific Expression of α2-Na+ Pumps
In earlier studies, we observed that MT and MR were correlated directly with basal BP in α2+/− mice (57) and in mice with SM-specific KO (53, 58) and SM-specific overexpression (18) of NCX1 (NCX1SM-KO and NCX1SM-Tg, respectively). This, plus the aforementioned reasoning about Ca2+ homeostasis and the reported association between arterial tone in vivo and in isolated arteries (8), led us to anticipate that MR might be inversely correlated with arterial α2-Na+ pump expression. This was not the case, however. MR was virtually identical in arteries from α2SM-Tg mice and their WT control counterparts (Fig. 8B). Furthermore, contrary to expectations, despite the higher basal BP, isolated small arteries from α2SM-DN mice had less MR than did control arteries (Fig. 9B).
These findings indicate that the relationship between arterial tone in vivo and in isolated arteries in vitro is more complicated than previously anticipated (8). One factor is that the in vitro tone is “purely” myogenic, whereas in vivo tone normally includes neural, humoral, and endothelial (shear stress) contributions. The myogenic component likely depends directly on myocyte Ca2+ homeostasis. MT is generated when an isolated arterial segment is pressurized and warmed to 36°C, and the cytosolic Ca2+ concentration rises (57). Because the expression of multiple Ca2+ transporters is altered in engineered mice (e.g., Fig. 3) (43), it is difficult to predict the net effect on Ca2+ homeostasis of these changes (and, likely, others that have not yet been identified). Also, membrane potential plays an important role in the generation of MT (23), and transporters that affect membrane potential might also be altered in engineered mice. Some of these factors, for example, the greatly reduced expression of NCX1, which mainly mediates Ca2+ entry in ASMCs (18, 53, 58), and SERCA2 (Fig. 3), may contribute to the delayed generation of MT (22, 46) in α2SM-DN mouse arteries.
Arterial Ca2+ Transporter Expression and Function in Mice With Genetically Altered SM-Specific Expression of α2-Na+ Pumps
It is instructive to compare the phenotypes in these mice with primary (targeted) genetic manipulation of α2-Na+ pumps and secondary (untargeted) changes in NCX1 and in mice with primary genetic alteration of NCX1. In the latter, basal BP correlated directly with arterial NCX1 expression (18, 53, 58): engineered downregulation of arterial NCX1 in NCX1SM-KO mice lowered cytosolic Ca2+ concentration and reduced basal MBP by 8–10 mmHg (53, 58). Conversely, SM-specific upregulation of NCX1 in NCX1SM-Tg mice raised cytosolic Ca2+ concentration and increased basal systolic BP by ∼10 mmHg (18). This was attributed to the higher cytosoli Ca2+ concentration and enhanced signaling in mice expressing more NCX1, thereby favoring Ca2+ entry in ASMCs (18, 58).
In mice with engineered α2-Na+ pumps (α2SM-Tg, α2SM-DN, and α2−/−), however, the expression and function of arterial NCX1 and other Ca2+ transporters (e.g., SERCA2, TRPC6, and PMCA1) are also greatly altered, i.e., there are secondary, compensatory changes [Fig. 3 (31, 43)]. In α2SM-Tg and α2SM-DN models, basal BP correlates inversely with NCX1 expression: in α2SM-DN mice, despite the reduced NCX1 expression, basal BP is elevated (Figs. 3 and 4), whereas basal BP is reduced in α2SM-Tg mice even though NCX1 is overexpressed (Fig. 5) (42). Also, in α2+/− mice, which have elevated basal BP, the fractional SEA0400 (NCX1 blocker)-induced decrease in MT was ∼35% less than in WT control mice (Fig. 9C in Ref. 57), which implies reduced NCX1 activity. Ca2+ homeostasis and signaling and, thus, arterial tone and BP are obviously influenced by the expression, in arterial myocytes, of α2-Na+ pumps as well as NCX1 and other Ca2+ transporters. These relationships may be complex, but the monogenic mutations of SM α2 and NCX1 suggest a common thread based on an understanding of the distribution and function of these transporters.
As a result of the functional coupling of α2 and NCX1 (see Introduction), decreased α2-Na+ pump-mediated Na+ extrusion, either because of reduced expression or pump inhibition, raises cytosolic Na+ concentration and, via NCX1, cytosolic Ca2+ concentration and enhances Ca2+ signals and contraction (1, 57). Therefore, NCX1 (and other Ca2+ transporters) may be downregulated to compensate for the reduced α2 expression and tendency to Ca2+ overload in α2SM-DN mice. Although protein expression was not measured, reduced NCX1 activity in α2−/− mouse ASMCs (31) and in α2+/− arteries (57) is consistent with this concept. Conversely, NCX1 (and other Ca2+ transporters) may be upregulated to compensate for the enhanced α2 expression and, presumably, reduce local cytosolic Na+ concentration and the tendency, therefore, to deplete Ca2+ in α2SM-Tg mice.
Primary Versus Secondary (Compensatory) Mechanisms That Influence BP in Mice With Genetically Engineered SM-α2 and SM-NCX1
What is the explanation for this apparently paradoxical relationship between NCX1 expression and basal BP? In NCX1SM-Tg and NCX1SM-KO mice, the primary defects are in one Ca2+ transport mechanism. Other compensatory mechanisms are therefore required to attenuate Ca2+ overload or Ca2+ depletion, respectively. In α2SM-DN and α2SM-Tg mice, the changes in α2-Na+ pump expression/activity alter the local Na+ electrochemical gradient driving force on NCX1 (1), thereby favoring NCX1-mediated Ca2+ entry and overload or Ca2+ extrusion and depletion, respectively. In both α2 engineered mouse lines, the modifications in NCX1 expression reflect, in part, the myocyte compensatory mechanisms needed to minimize the Ca2+ overload or depletion.
Consider, for example, the behavior of small arteries (e.g., passive diameter = 140 μm, with MT at 70 mmHg = 15% of passive diameter; see Figs. 8 and 9). When α2-Na+ pumps are acutely inhibited by ∼50%, e.g., by low-dose ouabain (equivalent to a 50% decrease in α2 expression), local cytosolic Na+ concentration will rise and promote NCX1-mediated Ca2+ gain. MT at 70 mmHg will increase by ∼10% (Fig. 3D in Ref. 57) as the diameter declines from 119 to 114 μm. According to Poiseuille's law, at constant cardiac output, this corresponds to an 18% increase in systemic vascular resistance and, thus, an increase in MBP from 105 to 125 mmHg–a large increase! In vivo, of course, rapidly recruited baroreflex mechanisms would be expected to attenuate the rise in MBP, but this is not likely to be sustained. When α2-Na+ pump-mediated transport is congenitally reduced by ∼50%, however, as in α2SM-DN mice, slower compensatory mechanisms such as a decrease in NCX1 expression may (should) come into play, thereby chronically minimizing the MBP elevation–as we observed. This scenario illustrates how potentially large changes in Ca2+ homeostasis and, thus, arterial constriction, induced here by monogenic changes in ASMC Na+ or Ca2+ transporters may be masked by compensatory “Ca2+ transporter reprogramming.”
How Can We Reconcile the Elevated Basal BP in α2SM-DN and α2+/− Mice With the Normal Basal BP in α2CV-KO Mice?
The fact that α2SM-DN and α2+/− mice have modestly elevated basal BP (Fig. 4) (57), whereas α2CV-KO mice do not (48), appears to conflict with the general idea proposed above. Consideration of model-specific differences, however, suggests an explanation. First, α2SM-DN and α2+/− mouse arteries express 30–50% of the normal α2 protein (Fig. 3) (56), whereas the level in α2CV-KO mice, 11 ± 7% (48), is not significantly greater than 0%. [A very low level of α2 expression is expected, however, because endothelial cells also express α2-Na+ pumps (54) and this should not be knocked out in α2CV-KO mice.] Second, NCX1 is not downregulated in α2CV-KO mice (48), which indicates that the normal basal BP cannot be explained by the type of compensatory Ca2+ transporter regulation that apparently attenuates BP elevation in α2SM-DN mice. The implication is that the PM-sarco(endo)thelial reticulum junctional complexes, where NCX1 normally functionally couples to α2-Na+ pumps (25, 51, 57), are evidently disrupted by the virtually complete KO of α2 in α2CV-KO mice. As a result, NCX1-mediated Ca2+ transport becomes linked to the Na+ concentration gradient established by α1-Na+ pumps. Because α1-Na+ pumps have higher affinity for cytosolic Na+ concentration than do α2-Na+ pumps [α2 apparent Km = 12 mM and α2 Km = 22 mM cytosolic Na+ concentration (55)], they help maintain a steady, low cytosolic Ca2+ concentration in quiescent myocytes when α2 is absent.
Furthermore, α2CV-KO mice are resistant to adrenocorticotropic hormone-induced hypertension, an EO-dependent form of hypertension (10), as expected for mice with no arterial ouabain-sensitive α2-Na+ pumps (10, 30). In contrast, the BP in mice with a reduced, but finite, number of these pumps (vs. WT control mice) should be elevated under conditions that raise plasma EO, such as ANG II and HS (5, 6, 15), as observed in α2SM-DN mice (Fig. 6). Thus, in summary, the contrast between α2SM-DN and α2CV-KO mice, rather than undermining our view about the relationship between α2-Na+ pumps and BP, actually supports it.
Implications for the Pathogenesis of Hypertension
Our findings provide insights that appear relevant to the pathogenesis of hypertension. In numerous rodent hypertension models, including the Dahl, spontaneously hypertensive rat, ANG II, DOCA-salt, Milan, and ouabain-induced models, these same Ca2+ transporters (e.g., NCX1, SERCA2, and/or TRPC6) are markedly upregulated (2, 15, 44, 45, 52, 59, 60). In both the α2SM-Tg and α2SM-DN models, the Ca2+ transporter reprogramming appears to be “protective”: it either minimizes Ca2+ depletion and attenuates BP decline (α2SM-Tg mice) or minimizes Ca2+ overload and attenuates BP elevation (α2SM-DN mice). In the aforementioned hypertension models, however, the Ca2+ transporter protein reprogramming can be expected to enhance Ca2+ gain and signaling by myocytes (29, 45, 59) and thereby promote the rise in BP. The implication is that this protein reprogramming, which we attribute to a centrally regulated, sustained increase in circulating EO (7, 15) that acts directly on myocytes (28, 45, 60), contributes significantly to the elevated BP in all those hypertension models.
Conclusions
This report reinforces the long-standing idea (4, 47) that Na+ pumps and NCX are important factors in the regulation of arterial Ca2+ homeostasis and constriction. Although SM-specific monogenic alteration of expression of either protein might be expected to modify, profoundly, Ca2+ homeostasis and, thus, BP, compensatory changes in other proteins may markedly attenuate the responses and confound the interpretation. The resulting modest net changes in basal BP may be misleading and, therefore, overlooked, unless the functional significance of all these factors is taken into account.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01-HL-107555 and P01-HL-78870 (to M. P. Blaustein) and R01-HL-107654 (to J. Zhang).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.C., M.I.K., R.J.P., J.Z., and M.P.B. conception and design of research; L.C., H.S., Y.W., J.C.L., and T.J.P. performed experiments; L.C., H.S., Y.W., M.I.K., T.J.P., R.J.P., J.Z., and M.P.B. analyzed data; L.C., H.S., M.I.K., T.J.P., R.J.P., J.Z., and M.P.B. interpreted results of experiments; L.C., H.S., J.Z., and M.P.B. prepared figures; L.C., H.S., M.I.K., T.J.P., R.J.P., J.Z., and M.P.B. edited and revised manuscript; L.C., H.S., Y.W., J.C.L., M.I.K., T.J.P., R.J.P., J.Z., and M.P.B. approved final version of manuscript; M.P.B. drafted manuscript.
ACKNOWLEDGMENTS
The authors thank Meng Li for management of the mouse colonies.
Footnotes
REFERENCES
- 1.Arnon A, Hamlyn JM, Blaustein MP. Ouabain augments Ca2+ transients in arterial smooth muscle without raising cytosolic Na+. Am J Physiol Heart Circ Physiol 279: H679–H691, 2000. [DOI] [PubMed] [Google Scholar]
- 2.Bae YM, Kim A, Lee YJ, Lim W, Noh YH, Kim EJ, Kim J, Kim TK, Park SW, Kim B, Cho SI, Kim DK, Ho WK. Enhancement of receptor-operated cation current and TRPC6 expression in arterial smooth muscle cells of deoxycorticosterone acetate-salt hypertensive rats. J Hypertens 25: 809–817, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol Renal Physiol 275: F633–F650, 1998. [DOI] [PubMed] [Google Scholar]
- 4.Blaustein MP. Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am J Physiol Cell Physiol 232: C165–C173, 1977. [DOI] [PubMed] [Google Scholar]
- 5.Blaustein MP, Chen L, Song H, Leenen FHH, Hamlyn JM. How does the brain talk to the arteries and heart? (Abstract). FASEB J 29: 984.983, 2015. [Google Scholar]
- 6.Blaustein MP, Chen L, Li M, Gao J, Hamlyn JM. Angiotensin II triggers the same pressor mechanisms in salt-sensitive hypertension and during salt depletion. Hypertension 62: A400, 2013. [Google Scholar]
- 7.Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, Van Huysse JW, Zhang J, Wier WG. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol 302: H1031–H1049, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev 79: 387–423, 1999. [DOI] [PubMed] [Google Scholar]
- 9.DeClue JW, Guyton AC, Cowley AW Jr, Coleman TG, Norman RA Jr, McCaa RE. Subpressor angiotensin infusion, renal sodium handling, and salt-induced hypertension in the dog. Circ Res 43: 503–512, 1978. [DOI] [PubMed] [Google Scholar]
- 10.Dostanic-Larson I, Van Huysse JW, Lorenz JN, Lingrel JB. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc Natl Acad Sci USA 102: 15845–15850, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dostanic I, Paul RJ, Lorenz JN, Theriault S, Van Huysse JW, Lingrel JB. The α2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol 288: H477–H485, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Doursout MF, Chelly JE, Liang YY, Buckley JP. The ouabain-dependent Na+-K+ pump and the brain renin-angiotensin system. Clin Exp Hypertens A Theory Pract 14: 393–411, 1992. [DOI] [PubMed] [Google Scholar]
- 13.Fairfax ST, Mauban JR, Hao S, Rizzo MA, Zhang J, Wier WG. Ca2+ signaling in arterioles and small arteries of conscious, restrained, optical biosensor mice. Front Physiol 5: 387, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrandi M, Minotti E, Salardi S, Florio M, Bianchi G, Ferrari P. Ouabainlike factor in Milan hypertensive rats. Am J Physiol Renal Fluid Electrolyte Physiol 263: F739–F748, 1992. [DOI] [PubMed] [Google Scholar]
- 15.Hamlyn JM, Linde CI, Gao J, Huang BS, Golovina VA, Blaustein MP, Leenen FH. Neuroendocrine humoral and vascular components in the pressor pathway for brain angiotensin II: a new axis in long term blood pressure control. PLos One 9: e108916, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han F, Tucker AL, Lingrel JB, Despa S, Bers DM. Extracellular potassium dependence of the Na+-K+-ATPase in cardiac myocytes: isoform specificity and effect of phospholemman. Am J Physiol Cell Physiol 297: C699–C705, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hund TJ, Mohler PJ. Ankyrin-based targeting pathway regulates human sinoatrial node automaticity. Channels 2: 404–406, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Iwamoto T, Kita S, Zhang J, Blaustein MP, Arai Y, Yoshida S, Wakimoto K, Komuro I, Katsuragi T. Salt-sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type-1 in vascular smooth muscle. Nat Med 10: 1193–1199, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Jacobs BE, Liu Y, Pulina MV, Golovina VA, Hamlyn JM. Normal pregnancy: mechanisms underlying the paradox of a ouabain-resistant state with elevated endogenous ouabain, suppressed arterial sodium calcium exchange, and low blood pressure. Am J Physiol Heart Circ Physiol 302: H1317–H1329, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA, Lingrel JB. Identification of a specific role for the Na,K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol Cell 3: 555–563, 1999. [DOI] [PubMed] [Google Scholar]
- 21.Juhaszova M, Blaustein MP. Distinct distribution of different Na+ pump alpha subunit isoforms in plasmalemma. Physiological implications. Ann NY Acad Sci 834: 524–536, 1997. [DOI] [PubMed] [Google Scholar]
- 22.Kashihara T, Nakayama K, Matsuda T, Baba A, Ishikawa T. Role of Na+/Ca2+ exchanger-mediated Ca2+ entry in pressure-induced myogenic constriction in rat posterior cerebral arteries. J Pharmacol Sci 110: 218–222, 2009. [DOI] [PubMed] [Google Scholar]
- 23.Knot HJ, Nelson MT. Regulation of arterial diameter and wall Ca2+ in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508 199–209, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krep H, Price DA, Soszynski P, Tao QF, Graves SW, Hollenberg NK. Volume sensitive hypertension and the digoxin-like factor. Reversal by a Fab directed against digoxin in DOCA-salt hypertensive rats. Am J Hypertens 8: 921–927, 1995. [DOI] [PubMed] [Google Scholar]
- 25.Lee MY, Song H, Nakai J, Ohkura M, Kotlikoff MI, Kinsey SP, Golovina VA, Blaustein MP. Local subplasma membrane Ca2+ signals detected by a tethered Ca2+ sensor. Proc Natl Acad Sci USA 103: 13232–13237, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lencesova L, O'Neill A, Resneck WG, Bloch RJ, Blaustein MP. Plasma membrane-cytoskeleton-endoplasmic reticulum complexes in neurons and astrocytes. J Biol Chem 279: 2885–2893, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Lighthall G, Manunta P, Hamlyn J. Increased circulating ouabain in Dahl SS/JR rats. J Hypertens 12: S158, Abst 870, 1994. [Google Scholar]
- 28.Linde CI, Antos LK, Golovina VA, Blaustein MP. Nanomolar ouabain increases NCX1 expression and enhances Ca2+ signaling in human arterial myocytes: a mechanism that links salt to increased vascular resistance? Am J Physiol Heart Circ Physiol 303: H784–H794, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linde CI, Karashima E, Raina H, Zulian A, Wier WG, Hamlyn JM, Ferrari P, Blaustein MP, Golovina VA. Increased arterial smooth muscle Ca2+ signaling, vasoconstriction, and myogenic reactivity in Milan hypertensive rats. Am J Physiol Heart Circ Physiol 302: H611–H620, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lorenz JN, Loreaux EL, Dostanic-Larson I, Lasko V, Schnetzer JR, Paul RJ, Lingrel JB. ACTH-induced hypertension is dependent on the ouabain-binding site of the α2-Na+-K+-ATPase subunit. Am J Physiol Heart Circ Physiol 295: H273–H280, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lynch RM, Weber CS, Nullmeyer KD, Moore ED, Paul RJ. Clearance of store-released Ca2+ by the Na+-Ca2+ exchanger is diminished in aortic smooth muscle from Na+-K+-ATPase α2-isoform gene-ablated mice. Am J Physiol Heart Circ Physiol 294: H1407–H1416, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Manabe I, Owens GK. The smooth muscle myosin heavy chain gene exhibits smooth muscle subtype-selective modular regulation in vivo. J Biol Chem 276: 39076–39087, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Manunta P, Ferrandi M, Bianchi G, Hamlyn JM. Endogenous ouabain in cardiovascular function and disease. J Hypertens 27: 9–18, 2009. [DOI] [PubMed] [Google Scholar]
- 34.Manunta P, Rogowski AC, Hamilton BP, Hamlyn JM. Ouabain-induced hypertension in the rat: relationships among plasma and tissue ouabain and blood pressure. J Hypertens 12: 549–560, 1994. [PubMed] [Google Scholar]
- 35.Muller-Ehmsen J, McDonough AA, Farley RA, Schwinger RH. Sodium pump isoform expression in heart failure: implication for treatment. Basic Res Cardiol 97, Suppl 1: I25–I30, 2002. [DOI] [PubMed] [Google Scholar]
- 36.O'Brien WJ, Lingrel JB, Wallick ET. Ouabain binding kinetics of the rat alpha two and alpha three isoforms of the sodium-potassium adenosine triphosphate. Arch Biochem Biophys 310: 32–39, 1994. [DOI] [PubMed] [Google Scholar]
- 37.Osborn JW, Fink GD, Sved AF, Toney GM, Raizada MK. Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr Hypertens Rep 9: 228–235, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Oshiro N, Dostanic-Larson I, Neumann JC, Lingrel JB. The ouabain-binding site of the α2 isoform of Na,K-ATPase plays a role in blood pressure regulation during pregnancy. Am J Hypertens 23: 1279–1285, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pierdomenico SD, Bucci A, Manunta P, Rivera R, Ferrandi M, Hamlyn JM, Lapenna D, Cuccurullo F, Mezzetti A. Endogenous ouabain and hemodynamic and left ventricular geometric patterns in essential hypertension. Am J Hypertens 14: 44–50, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Poburko D, Liao CH, Lemos VS, Lin E, Maruyama Y, Cole WC, van Breemen C. Transient receptor potential channel 6-mediated, localized cytosolic Na+ transients drive Na+/Ca2+ exchanger-mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ Res 101: 1030–1038, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Pressley TA. Phylogenetic conservation of isoform-specific regions within α-subunit of Na+-K+-ATPase. Am J Physiol Cell Physiol 262: C743–C751, 1992. [DOI] [PubMed] [Google Scholar]
- 42.Pritchard TJ, Bowman PS, Jefferson A, Tosun M, Lynch RM, Paul RJ. Na+-K+-ATPase and Ca2+ clearance proteins in smooth muscle: a functional unit. Am J Physiol Heart Circ Physiol 299: H548–H556, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pritchard TJ, Bullard DP, Lynch RM, Lorenz JN, Paul RJ. Transgenic mice expressing Na+-K+ ATPase in smooth muscle decreases blood pressure. Am J Physiol Heart Circ Physiol 293: H1172–H1182, 2007. [DOI] [PubMed] [Google Scholar]
- 44.Pulina MV, Zulian A, Baryshnikov SG, Linde CI, Karashima E, Hamlyn JM, Ferrari P, Blaustein MP, Golovina VA. Cross talk between plasma membrane Na+/Ca2+ exchanger-1 and TRPC/Orai-containing channels: key players in arterial hypertension. Adv Exp Med Biol 961: 365–374, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pulina MV, Zulian A, Berra-Romani R, Beskina O, Mazzocco-Spezzia A, Baryshnikov SG, Papparella I, Hamlyn JM, Blaustein MP, Golovina VA. Upregulation of Na+ and Ca2+ transporters in arterial smooth muscle from ouabain-induced hypertensive rats. Am J Physiol Heart Circ Physiol 298: H263–H274, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raina H, Ella SR, Hill MA. Decreased activity of the smooth muscle Na+/Ca2+ exchanger impairs arteriolar myogenic reactivity. J Physiol 586: 1669–1681, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reuter H, Blaustein MP, Haeusler G. Na-Ca exchange and tension development in arterial smooth muscle. Philos Trans R Soc Lond B Biol Sci 265: 87–94, 1973. [DOI] [PubMed] [Google Scholar]
- 48.Rindler TN, Dostanic I, Lasko VM, Nieman ML, Neumann JC, Lorenz JN, Lingrel JB. Knockout of the Na,K-ATPase α2-isoform in the cardiovascular system does not alter basal blood pressure but prevents ACTH-induced hypertension. Am J Physiol Heart Circ Physiol 301: H1396–H1404, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rossi G, Manunta P, Hamlyn JM, Pavan E, De Toni R, Semplicini A, Pessina AC. Immunoreactive endogenous ouabain in primary aldosteronism and essential hypertension: relationship with plasma renin, aldosterone and blood pressure levels. J Hypertens 13: 1181–1191, 1995. [DOI] [PubMed] [Google Scholar]
- 50.Shelly DA, He S, Moseley A, Weber C, Stegemeyer M, Lynch RM, Lingrel J, Paul RJ. Na+ pump α2-isoform specifically couples to contractility in vascular smooth muscle: evidence from gene-targeted neonatal mice. Am J Physiol Cell Physiol 286: C813–C820, 2004. [DOI] [PubMed] [Google Scholar]
- 51.Song H, Lee MY, Kinsey SP, Weber DJ, Blaustein MP. An N-terminal sequence targets and tethers Na+ pump α2 subunits to specialized plasma membrane microdomains. J Biol Chem 281: 12929–12940, 2006. [DOI] [PubMed] [Google Scholar]
- 52.Taniguchi S, Furukawa K, Sasamura S, Ohizumi Y, Seya K, Motomura S. Gene expression and functional activity of sodium/calcium exchanger enhanced in vascular smooth muscle cells of spontaneously hypertensive rats. J Cardiovasc Pharmacol 43: 629–637, 2004. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, Chen L, Li M, Cha H, Iwamoto T, Zhang J. Conditional knockout of smooth muscle sodium calcium exchanger type-1 lowers blood pressure and attenuates angiotensin II-salt hypertension. Physiol Rep 3: e12273, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zahler R, Sun W, Ardito T, Kashgarian M. Na-K-ATPase α-isoform expression in heart and vascular endothelia: cellular and developmental regulation. Am J Physiol Cell Physiol 270: C361–C371, 1996. [DOI] [PubMed] [Google Scholar]
- 55.Zahler R, Zhang ZT, Manor M, Boron WF. Sodium kinetics of Na,K-ATPase alpha isoforms in intact transfected HeLa cells. J Gen Physiol 110: 201–213, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J, Chen L, Lingrel JB, Philipson KD, Blaustein MP. Arterial myocyte Na+ pumps and Na+/Ca2+ exchangers modulate Ca2+ signaling, contractility and long-term blood pressure. J Hypertens 24, Suppl 6: S61, 2006. [Google Scholar]
- 57.Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, Bianchi G, Ferrari P, Hamlyn JM, Iwamoto T, Lingrel JB, Matteson DR, Wier WG, Blaustein MP. Sodium pump α2 subunits control myogenic tone and blood pressure in mice. J Physiol 569: 243–256, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J, Ren C, Chen L, Navedo MF, Antos LK, Kinsey SP, Iwamoto T, Philipson KD, Kotlikoff MI, Santana LF, Wier WG, Matteson DR, Blaustein MP. Knockout of Na+/Ca2+ exchanger in smooth muscle attenuates vasoconstriction and L-type Ca2+ channel current, and lowers blood pressure. Am J Physiol Heart Circ Physiol 298: H1472–H1483, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zulian A, Baryshnikov SG, Linde CI, Hamlyn JM, Ferrari P, Golovina VA. Upregulation of Na+/Ca2+ exchanger and TRPC6 contributes to abnormal Ca2+ homeostasis in arterial smooth muscle cells from Milan hypertensive rats. Am J Physiol Heart Circ Physiol 299: H624–H633, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zulian A, Linde CI, Pulina MV, Baryshnikov SG, Papparella I, Hamlyn JM, Golovina VA. Activation of c-SRC underlies the differential effects of ouabain and digoxin on Ca2+ signaling in arterial smooth muscle cells. Am J Physiol Cell Physiol 304: C324–C333, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]