

Antihypertensive use was previously associated with lower arterial stiffness in patients with elastin insufficiency. Here we show that biomechanical properties of Eln+/− large arteries are unchanged by antihypertensive treatment. Reduced stiffness is related to lower pulse pressure and a shift to a more compliant region of the pressure-diameter curve.
Keywords: elastin, arterial stiffness, hypertension, pulse pressure, Williams-Beuren syndrome
Abstract
Increased arterial stiffness is a common characteristic of humans with Williams-Beuren syndrome and mouse models of elastin insufficiency. Arterial stiffness is associated with multiple negative cardiovascular outcomes, including myocardial infarction, stroke, and sudden death. Therefore, identifying therapeutic interventions that improve arterial stiffness in response to changes in elastin levels is of vital importance. The goal of this study was to determine the effect of chronic pharmacologic therapy with different classes of antihypertensive medications on arterial stiffness in elastin insufficiency. Elastin-insufficient mice 4–6 wk of age and wild-type littermates were subcutaneously implanted with osmotic micropumps delivering a continuous dose of one of the following: vehicle, losartan, nicardipine, or propranolol for 8 wk. At the end of treatment period, arterial blood pressure and large artery compliance and remodeling were assessed. Our results show that losartan and nicardipine treatment lowered blood pressure and pulse pressure in elastin-insufficient mice. Elastin and collagen content of abdominal aortas as well as ascending aorta and carotid artery biomechanics were not affected by any of the drug treatments in either genotype. By reducing pulse pressure and shifting the working pressure range of an artery to a more compliant region of the pressure-diameter curve, antihypertensive medications may mitigate the consequences of arterial stiffness, an effect that is drug class independent. These data emphasize the importance of early recognition and long-term management of hypertension in Williams-Beuren syndrome and elastin insufficiency.
NEW & NOTEWORTHY
Antihypertensive use was previously associated with lower arterial stiffness in patients with elastin insufficiency. Here we show that biomechanical properties of Eln+/− large arteries are unchanged by antihypertensive treatment. Reduced stiffness is related to lower pulse pressure and a shift to a more compliant region of the pressure-diameter curve.
williams-beuren syndrome [WBS, also known as Williams syndrome, Online Mendelian Inheritance in Man (OMIM) No. 194050] is a rare multisystem disorder caused by a microdeletion at chromosome 7q11.23 (4, 43). Focal stenoses of medium- and large-sized elastic arteries are classic for the condition and are present to varying severity in ∼65% of individuals (22, 31). Hypertension is common, with a prevalence of ∼50%, and is often present independent of focal aortic or renal artery stenosis (8, 10, 11, 13, 31). Recently, children and adults with WBS were found to have increased arterial stiffness, even in the absence of hypertension (2, 22). This same study also reported increased risk of stroke (22) and previous studies cite a 25- to-100 fold increased risk of sudden death (5, 42) in those with WBS compared with unaffected individuals.
The vascular disease in WBS is caused by haploinsufficiency for an extracellular matrix protein, elastin (ELN). Elastin is initially produced by the cell as a soluble monomer, tropoelastin, that is cross linked in the extracellular space (41). As a fully cross-linked multimer, elastin provides recoil to elastic vessels. Arteries with reduced elastin are less compliant and develop increased number of smooth muscle lamellae in late gestation/early postnatal period (26, 40). Consequently, individuals with elastin insufficiency have developmental rather than induced vascular stiffness.
Arterial stiffness, as measured by pulse wave velocity (PWV) (37), is recognized as an important risk factor for cardiovascular events, including myocardial infarction and stroke. In the Rotterdam study, increasing aortic PWV was associated with increased coronary heart disease and stroke in apparently healthy individuals (27). In patients with chronic kidney disease stages 3–5, PWV was shown to be an independent predictor of cardiovascular events and all-cause mortality (6, 21), while in patients with type 1 diabetes, PWV was associated with all investigated complications, including cardiovascular, renal, retinal, and autonomic disease (36).
While there is an abundance of evidence implicating arterial stiffness in the development and/or progression of cardiovascular disease, the optimal intervention(s) necessary to attenuate arterial stiffness remain unclear. To that end, antihypertensive agents are among the most widely studied group of medications for their effect on arterial stiffness. In a recent meta-analysis of double-blind, randomized controlled trials, treatment with antihypertensive medications reduced arterial stiffness, as assessed by PWV, independently of blood pressure reduction (29). Interestingly, angiotensin-converting enzyme inhibitors were found to be more effective than calcium channel blockers in short-term trials while angiotensin-converting enzyme inhibitors, calcium channel blockers, and beta-blockers significantly reduced PWV compared with placebo in long-term trials (29). In the recent WBS arterial stiffness study, lower PWV was seen in individuals treated with antihypertensives but the study was underpowered to determine class superiority (22). Beta-blockers, angiotensin receptor blockers, and calcium channel blockers were commonly used in this and other studies (9, 22).
Humans and mice with elastin insufficiency have similar features. Hemizygous Eln mice (Eln+/−) are hypertensive, have increased large artery stiffness, and have increased number of lamellar units (12); all characteristics of humans with elastin insufficiency (26). Given the association of arterial stiffness with cardiovascular events and the potential beneficial effects of different classes of antihypertensive medications discussed above, we sought to determine whether elastin insufficiency-mediated arterial stiffness may be attenuated by chronic treatment with commonly used antihypertensive agents. The low prevalence of WBS complicates large population-based studies or randomized controlled clinical trials. Therefore, we utilized the elastin-haploinsufficient mouse (Eln+/−), a model with longstanding increased arterial stiffness that is noted as early as postnatal day 7, even preceding blood pressure elevation (24), to address the following questions. Can elastin insufficiency-mediated arterial stiffness be ameliorated by chronic treatment with antihypertensive medications? If so, is this effect specific to a particular class of antihypertensive medications? Also, what are the implications to the management of WBS patients with hypertension and/or arterial stiffness?
MATERIALS AND METHODS
Animals.
Eln+/− mice carrying a hemizygous deletion of 4 kb of the promoter and exon 1 of the elastin gene were generated by Li et al. (25, 26) in R1 embryonic stem cells and backcrossed into the C57Bl/6J background for over five generations as previously described. Additional back-crossing of more than five additional generations to C57Bl/6J mice occurred in our laboratory to reduce background contamination with 129/Sv from the embryonic stem line. Mice were genotyped for the Eln locus as previously described (17). Male Eln+/− and wild-type (WT) littermates were used for experiments in this study. Mice were housed together under standard conditions with free access to food and water. All surgical procedures were performed in accordance with protocols approved by the Animal Studies Committee of Washington University School of Medicine.
Drug selection.
We chose three classes of antihypertensive agents to treat Eln+/− mice and WT littermates: angiotensin II type 1 receptor blocker (losartan), beta-blocker (propranolol), and calcium channel blocker (nicardipine). The doses used in this study were based on previous studies showing a hemodynamic effect from administering these agents via osmotic micropumps (14, 20, 30, 39). Of the beta-blockers, propranolol was selected as it is commonly used clinically and in mouse models of extracellular matrix-associated vasculopathy (16, 18). For the calcium channel blocker nicardipine, a dihydropyridine was chosen, as dihydorpyridines have greater selectivity for vascular sites than myocardial sites, hence exhibiting greater vasodilatory effects with minimal electrophysiologic or negative ionotropic effects compared with nondihydropyridine calcium channel blockers, such as diltiazem and verapamil.
Drugs and osmotic micropump implantation.
At 4–6 wk of age, male WT and Eln+/− mice were subcutaneously implanted with ALZET microosmotic pumps (model 1004; DURECT, Cupertino, CA), delivering a continuous dose of one of the following reagents: water, 50% dimethylsulfoxide (DMSO) in water, losartan (30 mg·kg−1·day−1) (20), propranolol (10 mg·kg−1·day−1) (7, 28, 30), or nicardipine (5 mg·kg−1·day−1) (14, 39) for a total of 8 wk in a volume of 200 μl. Losartan and propranolol were dissolved in water while nicardipine was dissolved in 50% DMSO. All reagents were purchased from Sigma-Aldrich (St. Louis, MO). Five to eight mice were used for each experimental group. To implant the osmotic micropumps, mice were anesthetized with 1.5% isofluorane and placed in prone position. The interscapular region was shaved to remove hair and skin was wiped with betadine. A 1-cm mid-scapular incision was made; forceps were used to spread the subcutaneous tissue creating a pocket where the filled pump was inserted delivery portal first. The incision was closed with two to three sutures (6-0 nylon; Ethicon). Since each osmotic pump is designed to deliver the reagent for 28 days, on day 28, the first pump was removed and replaced by a new pump using the same surgical technique. Physiological studies were done on day 56, when mice were killed. At replacement and death, pumps were removed and delivery of the drug was confirmed by assessing the volume of solution remaining in each pump.
Blood pressure and heart rate measurements.
Due to the smaller and more tortuous carotid arteries in Eln+/− mice compared with WT littermates, measurement of blood pressure in conscious animals via radiotelemetry was not technically feasible. Therefore, at the end of the treatment period (8 wk), mice were anesthetized with 1.5% inhaled isofluorane and restrained on a heated pad to maintain body temperature at 37°C. A Millar pressure transducer (model SPR-671; Houston, TX) was introduced into the right common carotid artery and advanced to the ascending aorta where heart rate and systolic (SBP) and diastolic blood pressure (DBP) were recorded via the PowerLab data acquisition system (ADInstruments, Colorado Springs, CO). Data were analyzed using LabChart 7 for MAC software (ADInstruments).
Arterial pressure-diameter curve measurements.
After arterial blood pressure measurement, mice were killed under isofluorane anesthesia. As previously described (12), ascending aorta and left common carotid artery were excised and placed in a physiologic saline solution (PSS) of the following composition: 130 mM NaCl, 4.7 mM KCl, 1.6 mM CaCl2, 1.18 mM MgSO4-7H2O, 1.17 mM KH2PO4, 14.8 mM NaHCO3, 5.5 mM dextrose, and 0.026 mM EDTA (pH 7.4). Vessels were then cleaned of surrounding fat and connective tissue and mounted onto a pressure arteriograph (Danish Myotechnology, Aarhus, Denmark). Experiments were done at 37°C in an organ bath containing PSS. Mounted vessels were visualized using an inverted microscope connected to a charged-coupled device camera and a computerized system allowing continuous recording of vessel diameters. Vessel outer diameters were recorded while increasing intravascular pressure from 0 to 175 mmHg by increments of 25 mmHg (12 s per step). The average of three outer diameter measurements was taken at each pressure.
Quantitative real-time reverse transcription PCR.
At the end of treatment period, descending thoracic aortas were isolated and stored in RNAlater at −80°C until mRNA was extracted using Trizol per manufacturer's protocol (Life Technologies, Grand Island, NY). Given the anticipated small amount of mRNA per aorta, 10 μg of glycogen were added to each sample as carrier at the time of precipitation. One microgram of mRNA was used for first-strand cDNA synthesis using High-Capacity RNA-to-cDNA Kit per manufacturer's protocol (20-μl reaction). One-microliter cDNA was then used for real time PCR using TaqMan Fast Universal PCR Master Mix and TaqMan assays (primers/probes) purchased from Life Technologies. Reactions were done in duplicate using ViiA 7 real-time PCR system and experimental genes were normalized to Gapdh and Ppia. TaqMan assays used were Mm00514670_m1 (Eln), Mm00801666_g1 (Col1a1), Mm99999915_g1 (Gapdh), and Mm03302254_g (Ppia).
Desmosine and hydroxyproline content measurement.
Abdominal aortas (from the left renal artery to the iliac bifurcation) were isolated, washed in water, and frozen at −70°C for 10 min followed by drying for 2 h using a lyophilizer. After samples were weighed, 20 μl of 6 N hydrochloric acid (Thermo Scientific, Rockford, IL) were added to each sample, which was hydrolyzed for 48 h at 105°C. After drying for 1.5 h at 65°C using a vacuum concentrator (Speedvac), samples were dissolved in 400 μl water and filtered using a 0.45-μm filter.
Total protein content in each sample was determined using a ninhydrin-based assay (35). Briefly, an aliquot of the hydrolyzed sample was reacted at 85°C for 10 min with 100 μl ninhydrin (100 mM) dissolved in 75% ethylene glycol containing 2.5 mg/ml SnCL2 and 1 M sodium acetate pH 5.5. Absorbance at 575 nm was determined using a Synergy H4 Multi-Mode Reader (BioTek, Winooski, VT). Sample protein concentration was determined by comparison with a standard curve generated using a calibration standard for protein hydrolysis (Pickering Laboratories, Mountain View CA).
Hydroxyproline was determined using a chloramine-T colorimetric assay as described previously (32). Briefly, duplicate samples were oxidized with 1.5 mg chloramine-T for 20 min at room temperature then reacted for 20 min at 65°C with 20 mg p-dimethylaminobenzaldehyde dissolved in propanol containing 20% perchoric acid. The absorbance was determined at 550 nm using Synergy H4 Multi-Mode plate reader and the amount of hydroxyproline was determined by comparison to a hydroxyproline standard curve. All reagents used in this assay were obtained from Sigma-Aldrich.
Desmosine levels in tissue hydrolyzates were quantified using a nonequilibrium competitive enzyme-linked immunosorbent assay (15). An aliquot of the sample was added to a desmosine-ovalbumin-coated well in a microtiter plate followed by a 1:4,000 dilution of rabbit antidesmosine antiserum. After 1 h of incubation, nonbound antibody was removed by washing. A secondary goat anti-rabbit IgG antibody conjugated to horseradish peroxidase was added for 1 h to detect primary antibody bound to the plate, nonbound secondary antibody-peroxidase was removed by washing, and peroxidase activity was determined using SureBlue TMB peroxidase substrate. Absorbance at 650 nm was determined using a Synergy H4 Multi-Mode Reader (BioTek) and desmosine content was determined by extrapolation from a desmosine standard curve run on the same plate. The antisera, desmosine standards, and desmosine-ovalbumin conjugate were obtained from Elastin products (Owensville, MO). Blocking buffer, dilution buffer, wash solution and the SureBlue TMB peroxidase substrate came from KPL (Gathersburg, MD). Secondary antibody-peroxidase conjugate (NA934V) was a product of GE Health Sciences.
Statistical analysis.
One-way ANOVA with Dunnett's multiple comparisons test was used to determine the effect of each treatment on SBP, DBP, pulse pressure, heart rate, and desmosine and hydroxyproline content. Two-tailed unpaired t-test was used when only two groups were compared. Outer vessel diameters at different pressures were compared using two-way ANOVA with Dunnett's multiple comparisons test with the two variables being treatment group and pressure. The software Prism 6 for Mac OS X (GraphPad Software, La Jolla, CA) was used to run statistical analyses. Data are presented as means ± SD. P ≤ 0.05 was considered statistically significant differences. Table 1 lists the P values for each ANOVA performed in this study.
Table 1.
P values for all ANOVA tests performed in the study
| Figure | Each Group Compared with | ANOVA P Value |
|---|---|---|
| Figure 1 | ||
| A (systolic pressure) | H2O-Het | 0.0018 |
| DMSO-Het | 0.0015 | |
| B (systolic pressure) | H2O-WT | 0.0314 |
| C (diastolic pressure) | H2O-Het | 0.0142 |
| DMSO-Het | 0.0446 | |
| D (diastolic pressure) | H2O-WT | 0.0189 |
| E (pulse pressure) | H2O-Het | <0.0001 |
| DMSO-Het | <0.0001 | |
| F (pulse pressure) | H2O-WT | 0.7466 |
| Figure 2 | ||
| A (heart rate) | H2O-Het | <0.0001 |
| DMSO-Het | 0.5760 | |
| B (heart rate) | H2O-WT | <0.0001 |
| Figure 3 | ||
| A (aorta pressure-diameter) | H2O-Het | <0.0001 for treatment |
| <0.0001 for pressure | ||
| DMSO-Het | <0.0001 for treatment | |
| <0.0001 for pressure | ||
| B (carotid pressure-diameter) | H2O-Het | <0.0001 for treatment |
| <0.0001 for pressure | ||
| DMSO-Het | <0.0001 for treatment | |
| <0.0001 for pressure | ||
| C (aorta pressure-diameter) | H2O-WT | <0.0001 for pressure |
| 0.0177 for treatment | ||
| D (carotid pressure-diameter) | H2O-WT | <0.0001 for pressure |
| 0.0199 for treatment | ||
| E (slope) | H2O-Het | <0.0001 for pressure range |
| H2O-Het | 0.0012 for treatment group | |
| F (slope) | DMSO-Het | <0.001 for pressure range |
| DMSO-Het | 0.035 for treatment group | |
| Figure 4 | ||
| A (mEln qRT-PCR) | H2O-Het | 0.0074 |
| DMSO-Het | 0.0065 | |
| B (mCol1a1 qRT-PCR) | H2O-Het | 0.8918 |
| DMSO-Het | 0.9345 | |
| C (desmosine) | H2O-Het | <0.0001 |
| DMSO-Het | 0.013 | |
| D (hydroxyproline) | H2O-Het | 0.2553 |
| DMSO-Het | 0.6176 | |
| E (desmosine) | H2O-WT | 0.1521 |
| F (hydroxyproline) | H2O-WT | 0.7715 |
Het, elastin-insufficient mice; WT, wild-type mice.
RESULTS
Effect of chronic antihypertensive therapy on blood pressure in elastin insufficiency.
After 8 wk of continuous treatment of Eln+/− (Het) mice and WT littermates postweaning with vehicle (water or 50% DMSO in water), losartan (angiotensin II type 1 receptor blocker), nicardipine (calcium channel blocker), or propranolol (beta-blocker), arterial blood pressure was measured and recorded. Similar to untreated Eln+/− mice (12), vehicle-exposed Eln+/− mice had significantly higher SBP compared with WT littermates (Fig. 1A), while the DBP was not different (Fig. 1C). Treatment with losartan significantly decreased SBP and DBP in Eln+/− mice (Fig. 1, A and C). While SBP was significantly lowered by nicardipine treatment in Eln+/− mice (Fig. 1A), its effect on lowering DBP neared statistical significance (Fig. 1C). Interestingly, in WT mice, losartan significantly reduced SBP and DBP, but nicardipine did not significantly lower either (Fig. 1, B and D). Although propranolol treatment significantly reduced heart rate (Fig. 2) and resulted in a trend toward lower SBP and DBP in WT and Eln+/− mice, it did not reach statistical significance (Fig. 1). Losartan and nicardipine did not alter heart rate in either genotype (Fig. 2). No sudden death or other negative cardiovascular events were noted in animals treated with any of the above medications.
Fig. 1.

Effect of chronic antihypertensive therapy on systolic (SBP; A and B), diastolic blood pressure (DBP; C and D), and pulse pressure (E and F) in elastin-insufficient (Het) and littermate control [wild-type (WT)] mice. Pulse pressure was calculated as the difference between measured SBP and DBP. Each bar represents a different treatment group and is labeled below. H2O was the vehicle for losartan (LOS) and propranolol (PRO), while 50% DMSO was used to dissolve nicardipine (NIC). The dotted line separates the treatment groups using different vehicles, which were analyzed independently. Each treatment group was compared with the vehicle-treated Het group in A and B or to the vehicle-treated WT group in C and D. Data are mean values ± SD; n = 5–8 mice per treatment group. Significant difference in the post hoc Dunnett's multiple comparisons test: *P < 0.05, **P < 0.005, ***P < 0.001, ****P < 0.0001, between indicated groups.
Fig. 2.

Treatment with PRO but not LOS or NIC lowers heart rate. Effect of chronic antihypertensive therapy on heart rate in elastin-insufficient-Het mice (A) and littermates-WT (B). Each bar represents a different treatment group and is labeled below. H2O (water) was the vehicle for LOS and PRO, while 50% DMSO was used to dissolve NIC. The dotted line separates the treatment groups using different vehicles, which were analyzed independently. Each treatment group was compared with the vehicle-treated Het group in A or vehicle-treated WT group in B. Data are mean values ± SD; n = 5–8 mice per treatment group. Significant difference: ****P < 0.0001, between indicated groups.
Effect of chronic antihypertensive therapy on large artery mechanics in elastin insufficiency.
We examined the effect of chronic antihypertensive therapy on large artery stiffness by generating pressure-diameter curves of ascending aortas and left common carotid arteries obtained from Eln+/− mice and WT littermates treated with antihypertensive agents as described above. In a typical arterial pressure-diameter relationship, the curve initially has a steep slope (increased compliance), owing to the contribution of elastin [reviewed in Ref. 34]. At higher pressures where elastin is already fully extended, the curve becomes flat and the biomechanical properties of the vessel are controlled largely by collagen [reviewed in Ref. 34]. The slope of the pressure-diameter curve, and therefore compliance, is similar in WT and Eln+/− ascending aorta in the range of 75–100 mmHg (Fig. 3, E and F). At higher pressures, >100 mmHg, the slope is reduced in Eln+/− compared with WT (Fig. 3, E and F). None of the tested antihypertensives changed the large artery pressure-diameter curves for aorta or carotid arteries in either Eln+/− or WT mice (Fig. 3, A–D), and no differences in the slope were noted either (Fig. 3, E and F).
Fig. 3.

Chronic antihypertensive therapy has no effect on large artery mechanics. Pressure-diameter relationships of ascending aortas (A and C) and left common carotid arteries (B and D) from Eln+/− mice (Het) and littermates (WT) treated with various reagents: vehicle (H2O or 50% DMSO), LOS, PRO, and NIC. For A–D, solid lines represent vehicle-treated groups while dotted lines represent drug-treated groups; black represents WT while grey represents Eln+/− mice. In A and B, the black lines representing H2O-WT and DMSO-WT appear separately above all grey curves representing the 5 Het groups. Each treatment group was compared with the vehicle-treated Het group in A and B or to the vehicle-treated WT group in C and D. For E and F, the slope of the ascending aorta pressure-diameter curve between designated pressures (75–100 or 100–125 mmHg) was calculated by taking the difference in vessel outer diameter (OD) at chosen pressures and dividing it by the pressure difference (25 mmHg in this case) for the indicated treatment groups. Within each pressure range, each treatment group was compared with the vehicle-treated Het group. Data are mean values ± SD; n = 5–8 mice per treatment group. Significant difference in the post hoc Dunnett's multiple comparisons test: *P < 0.05, **P < 0.005.
Chronic antihypertensive therapy does not alter elastin or collagen content of abdominal aortas.
Inhibitors of the renin angiotensin system and calcium channel blockers exert direct antifibrotic effects on conduit vessels by downregulating expression of transforming growth factor-β, thereby reducing collagen deposition and vessel stiffness (3, 34, 38). To assess whether chronic antihypertensive treatment had an effect on elastin or collagen content of large arteries, we assessed elastin and collagen gene expression in descending thoracic aorta of Eln+/− mice and WT littermates treated with various antihypertensive agents as above. Eln+/− aortas have ∼50% less elastin mRNA than WT littermates, a finding that is not affected by any of the drug treatments (Fig. 4A). Collagen gene expression (Col1a1) is not different between aorta of Eln+/− mice and WT littermates, and it is not affected by any of the drug treatments (Fig. 4B). In addition, to assess mature protein content, we quantified desmosine (an amino acid unique to cross-linked elastin) and hydroxyproline (amino acid common in collagen) in the abdominal aorta of these animals. Consistent with previous report of untreated large arteries (12), abdominal aorta from vehicle exposed Eln+/− mice had 31% less desmosine than WT littermates (Fig. 4C) while hydroxyproline content (Fig. 4D) was not different. Treatment with losartan, nicardipine, or propranolol for 8 wk did not lead to statistically significant differences in desmosine or hydroxyproline content in the abdominal aorta of Eln+/− or WT mice (Fig. 4, C–F).
Fig. 4.

Chronic antihypertensive therapy does not alter elastin or collagen content in elastin-insufficient mice. A and B: relative gene expression of elastin (mEln; A) and collagen (mCol1a1; B) in thoracic descending aorta of Eln+/− (Het) mice compared with littermate (WT)-treated mice as measured by quantitative reverse transcription real time PCR. Desmosine (C and E) or hydroxyproline (D and F) content of abdominal aorta from Eln+/− (Het) or littermate (WT) mice. Each treatment group is labeled below. The dotted line separates the treatment groups using different vehicles, which were analyzed independently. Each treatment group was compared with the vehicle-treated Het group in A–D or to the vehicle-treated WT group in E and F. Data are mean values ± SD; n = 5–8 mice per treatment group. Significant difference in the post hoc Dunnett's multiple comparisons test: *P < 0.05, **P < 0.01, ****P < 0.0001, between indicated groups.
Blood pressure reduction improves pulse pressure in elastin insufficiency.
The elastic nature of large arteries facilitates dampening of the pulse pressure by storing energy during systole and releasing it during diastole, propelling blood toward the periphery. Therefore, arterial elasticity (or lack thereof, i.e., stiffness) is a major determinant of pulse pressure (9, 33). As shown in Fig. 1E, vehicle-treated Eln+/− mice have higher pulse pressure than WT littermates, a reflection of stiffer vessels. Chronic treatment with losartan or nicardipine significantly reduced pulse pressure in Eln+/− mice, while propranolol did not (Fig. 1E). Importantly, while losartan treatment significantly lowered SBP and DBP in WT mice (Fig. 1, B and D), it did not affect pulse pressure in those animals (Fig. 1F). Similarly, nicardipine and propranolol treatments also had no effect on pulse pressure in WT mice (Fig. 1F).
DISCUSSION
In human essential hypertension, eutrophic or hypertrophic remodeling occurs in small resistance arteries based on duration and severity of hypertension. In large elastic arteries, in contrast, structural changes occur that lead to increased stiffness of the artery (1, 9). These structural changes include collagen deposition and elastin fragmentation, producing a thicker and stiffer vessel wall. The changes that occur in large and small vessels as a part of essential hypertension are not mutually exclusive, as remodeling of arterioles increases vascular resistance and blood pressure, which in turn increases arterial stiffness leading to a vicious cycle (9). This is in contrast to the developmental changes in vascular wall structure seen in genetic conditions of elastin insufficiency. Here, large arteries of Eln+/− mice develop with a 35% increase in the number of elastic lamellae and smooth muscle cell layers. Their elastic laminas are continuous rather than fragmented; however, their overall elastin content is reduced compared with that of WT littermate controls, a consequence of having thinner elastic lamellae (26). Furthermore, aortic collagen content is comparable between Eln+/− mice and WT littermates (26). These structural differences are complete shortly after birth (40) and result in a distinct form of congenital vascular stiffness.
Data from the WBS SAVE study showed reduced PWV in individuals maintained on antihypertensives (22). However, there was not enough power in the study to determine drug class superiority for treatment. In a recent study describing presentation and management of hypertension in 41 children with WBS, calcium channel blockers, inhibitors of the renin-angiotensin-aldosterone system (angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor blockers), and beta-blockers were most commonly used (8). All were associated with good blood pressure control in most cases, although more than one agent was necessary in 36% of the treated patients (8). Similarly, angiotensin-converting enzyme inhibitors/angiotensin II type 1 receptor blockers, calcium channel blockers, and beta-blockers are considered first line agents for the management of primary hypertension by 47, 37, and 6.6% of pediatric nephrologists, respectively (19). Therefore, we tested the effect of long-term treatment with these three classes of antihypertensive medications on vascular stiffness in elastin-insufficient mice.
The data presented herein demonstrate several important observations. First, chronic antihypertensive therapy does not alter large artery biomechanics when administered postweaning in elastin-insufficient or WT mice. Treatment with any of the three classes of antihypertensives used in this study showed no direct effect on either collagen or elastin deposition in aortas as shown by unchanged Cola1 and Eln gene expression as well as hydroxyproline and desmosine content in descending thoracic and abdominal aorta, respectively. Like the ascending aorta, the descending thoracic and abdominal aortas are large elastic arteries that each show reduced elastin content and increased number of thinner continuous elastic lamellae in Eln+/− relative to WT mice (12). While an imperfect match for the ascending aorta, their use allows comparison of multiple markers (physiology, desmosine/hydroxyproline, and RNA) to be made from the same animals. The postweaning time for therapy initiation was chosen to reflect current clinical practice. It is possible that earlier treatment administration could generate other effects.
Second, the potential beneficial effects of antihypertensive treatment on arterial stiffness stem from pulse pressure reduction and from shifting the working pressure range to a more compliant segment (steeper slope) of the pressure-diameter curve where elastin rather than collagen largely controls arterial stiffness (Fig. 5); the effect of which is to reduce afterload on the heart. Pulse pressure, which results from intermittent ventricular ejection, is affected by two main factors, the dampening capacity of the proximal elastic conduit arteries and the timing and intensity of wave reflections (33). The cushioning capacity of conduit vessels is determined by arterial stiffness, assessed by compliance. Wave reflections are affected by number, structure, function, and geometry of small muscular arteries and arterioles (33). Antihypertensive therapy in our study did not affect the biomechanical properties of the large arteries; the pressure-diameter curves are nearly identical. Treatment with either losartan or nicardipine, however, significantly reduced pulse pressure in Eln+/− mice, suggesting an effect on pulse wave reflections, an effect likely mediated by changes at the small resistance artery/arteriole level. Interestingly, despite lowering SBP and DBP in Eln+/− and WT mice, losartan treatment reduced pulse pressure only in Eln+/− but not WT mice suggesting that the beneficial effect exerted by this treatment requires the presence of systolic hypertension and/or elastin insufficiency.
Fig. 5.
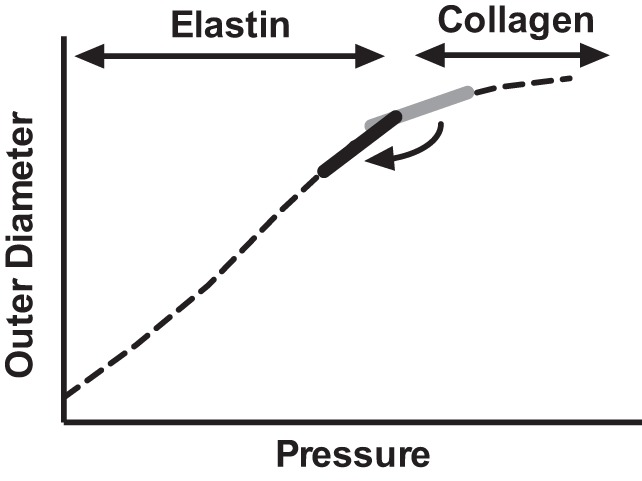
Model illustrating the functional effect of blood pressure reduction on large artery physiology in Eln+/− mice. Lowering blood pressure in Eln+/− mice results in a shift of the working pressure from the portion of the pressure-diameter curve depicted in grey (higher pressure, reduced slope, and reduced compliance) to the region underlying the black solid line (lower pressure, steeper slope, and increased compliance). Elastin controls the biomechanical properties of the vessel at lower pressures; but at higher pressures where the elastin is fully extended, collagen dominates. Eln+/− mice have less elastin. Consequently, the transition from the elastin mediated “compliant” portion of the curve to the flatter collagen controlled part of the curve occurs as lower pressures in Eln+/− relative to WT.
Third, this effect is not necessarily specific to a particular class of antihypertensive agents, rather any medication that sufficiently lowers blood pressure, particularly SBP, has the potential to improve functional stiffness. Consequently, we cannot conclude that any one class of antihypertensive agents is superior to another in elastin insufficiency. Rather, we propose that by lowering the blood pressure, particularly SBP, antihypertensive medications can shift the working pressure of a vessel to a more compliant/elastic (increased slope) part of the pressure-diameter curve, reducing afterload on the heart, potentially protecting the individual from the negative effects of chronic arterial stiffness. The observation of a medication class-independent effect of antihypertensives is in concert with a recent randomized clinical trial showing no difference in the rate of aortic root dilatation in individuals with Marfan's syndrome treated with either losartan or atenolol for 3 yr (23). In the current study, there was no mortality associated with treatment with any of these medications.
Of particular importance to the WBS/supravalvular aortic stenosis (SVAS) community is the implied effect of reduced arterial elastin content on vessel biomechanics. Functional arterial stiffness increases at higher blood pressures, where forces are shifted from elastic fibers to collagen. That transition occurs at a lower pressure in elastin-insufficient vessels than in ones with typical amounts of elastin (at ∼100 mmHg in Eln+/− vs. ∼125 mmHg in WT vessels) as evidenced by the more acute reduction in slope in Eln+/− relative to WT over these intervals. Consequently, individuals with elastin insufficiency, even children, for whom calcification of the vessels does not contribute to stiffness, may see increased stiffness and downstream negative cardiovascular outcomes even though they are not overtly hypertensive by current definition. This finding is also in line with findings from the WBS SAVE PWV study, which showed 58% of the WBS participants had stiffness without overt hypertension (22) and suggests the need to clinically evaluate and treat arterial stiffness in individuals with WBS and SVAS.
In conclusion, elastin insufficiency causes vascular stiffness, a phenotype known to be associated with negative cardiovascular outcomes. While chronic antihypertensive treatment does not induce large vessel remodeling in WT or Eln+/− mice, overall cardiovascular health is improved by antihypertensive treatment by lowering pulse pressure and by moving the arterial mechanics to a more compliant portion of the pressure diameter curve. For individuals with WBS, the optimal working pressure for the vessel may lie at a lower pressure than for individuals with normal elastin content and clinical assessment of vascular stiffness by PWV would allow that determination. The choice to treat is complicated by concerns about the effects of focal stenosis and end organ perfusion, and those questions require further study. However, given our knowledge of the impact of stiffness on phenotypes like heart attack, stroke, and sudden death, it is imperative to begin to consider the role of arterial stiffness as part of the treatment paradigm.
GRANTS
This work was funded by National Institutes of Health (NIH) Training Grants 5T32-HL-7873-15 and 2T32-HD-043010-11 and Physician-Scientist Training Program Fellow support by the Mallinckrodt Foundation (to C. M. Halabi), NIH Grants HL-53325, HL-105314, and H74138 (to R. P. Mecham), and NIH Grant K08-HL-109076 (to B. A. Kozel). B. A. Kozel also received funding through the Children's Discovery Institute at St. Louis Children's Hospital and Washington University School of Medicine.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: C.M.H. and B.A.K. conception and design of research; C.M.H., T.J.B., R.H.K., and L.Y. performed experiments; C.M.H., T.J.B., R.H.K., and B.A.K. analyzed data; C.M.H., T.J.B., R.P.M., and B.A.K. interpreted results of experiments; C.M.H. prepared figures; C.M.H. drafted manuscript; C.M.H., R.P.M., and B.A.K. edited and revised manuscript; C.M.H., T.J.B., R.H.K., L.Y., R.P.M., and B.A.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Terese Hall and Joshua Danback for administrative assistance.
REFERENCES
- 1.Agabiti-Rosei E, Heagerty AM, Rizzoni D. Effects of antihypertensive treatment on small artery remodelling. J Hypertens 27: 1107–1114, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Bassareo PP, Mercuro G. Increased arterial stiffness in children with Williams syndrome and normal blood pressure. Blood Press Monit 15: 257–261, 2010. [DOI] [PubMed] [Google Scholar]
- 3.Benetos A, Gautier S, Lafleche A, Topouchian J, Frangin G, Girerd X, Sissmann J, Safar ME. Blockade of angiotensin II type 1 receptors: effect on carotid and radial artery structure and function in hypertensive humans. J Vasc Res 37: 8–15; discussion 68–70, 2000. [DOI] [PubMed] [Google Scholar]
- 4.Beuren AJ, Apitz J, Harmjanz D. Supravalvular aortic stenosis in association with mental retardation and a certain facial appearance. Circulation 26: 1235–1240, 1962. [DOI] [PubMed] [Google Scholar]
- 5.Bird LM, Billman GF, Lacro RV, Spicer RL, Jariwala LK, Hoyme HE, Zamora-Salinas R, Morris C, Viskochil D, Frikke MJ, Jones MC. Sudden death in Williams syndrome: report of ten cases. J Pediatr 129: 926–931, 1996. [DOI] [PubMed] [Google Scholar]
- 6.Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness on survival in end-stage renal disease. Circulation 99: 2434–2439, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Boluyt MO, Long X, Eschenhagen T, Mende U, Schmitz W, Crow MT, Lakatta EG. Isoproterenol infusion induces alterations in expression of hypertrophy-associated genes in rat heart. Am J Physiol Heart Circ Physiol 269: H638–H647, 1995. [DOI] [PubMed] [Google Scholar]
- 8.Bouchireb K, Boyer O, Bonnet D, Brunelle F, Decramer S, Landthaler G, Liutkus A, Niaudet P, Salomon R. Clinical features and management of arterial hypertension in children with Williams-Beuren syndrome. Nephrol Dial Transplant 25: 434–438, 2010. [DOI] [PubMed] [Google Scholar]
- 9.Briet M, Schiffrin EL. Treatment of arterial remodeling in essential hypertension. Curr Hypertens Rep 15: 3–9, 2013. [DOI] [PubMed] [Google Scholar]
- 10.Broder K, Reinhardt E, Ahern J, Lifton R, Tamborlane W, Pober B. Elevated ambulatory blood pressure in 20 subjects with Williams syndrome. Am J Med Genet 83: 356–360, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Eronen M, Peippo M, Hiippala A, Raatikka M, Arvio M, Johansson R, Kahkonen M. Cardiovascular manifestations in 75 patients with Williams syndrome. J Med Genet 39: 554–558, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest 112: 1419–1428, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrero GB, Biamino E, Sorasio L, Banaudi E, Peruzzi L, Forzano S, di Cantogno LV, Silengo MC. Presenting phenotype and clinical evaluation in a cohort of 22 Williams-Beuren syndrome patients. Eur J Med Genet 50: 327–337, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Gao X, Iwai M, Inaba S, Tomono Y, Kanno H, Mogi M, Horiuchi M. Attenuation of monocyte chemoattractant protein-1 expression via inhibition of nuclear factor-kappaB activity in inflammatory vascular injury. Am J Hypertens 20: 1170–1175, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Gunja-Smith Z. An enzyme-linked immunosorbent assay to quantitate the elastin crosslink desmosine in tissue and urine samples. Anal Biochem 147: 258–264, 1985. [DOI] [PubMed] [Google Scholar]
- 16.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312: 117–121, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirano E, Knutsen RH, Sugitani H, Ciliberto CH, Mecham RP. Functional rescue of elastin insufficiency in mice by the human elastin gene: implications for mouse models of human disease. Circ Res 101: 523–531, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Huang J, Yamashiro Y, Papke CL, Ikeda Y, Lin Y, Patel M, Inagami T, Le VP, Wagenseil JE, Yanagisawa H. Angiotensin-converting enzyme-induced activation of local angiotensin signaling is required for ascending aortic aneurysms in fibulin-4-deficient mice. Sci Transl Med 5: 183ra158, 181-111, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ingelfinger JR. Clinical practice. The child or adolescent with elevated blood pressure. N Engl J Med 370: 2316–2325, 2014. [DOI] [PubMed] [Google Scholar]
- 20.Jiang F, Jones GT, Dusting GJ. Failure of antioxidants to protect against angiotensin II-induced aortic rupture in aged apolipoprotein(E)-deficient mice. Br J Pharmacol 152: 880–890, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karras A, Haymann JP, Bozec E, Metzger M, Jacquot C, Maruani G, Houillier P, Froissart M, Stengel B, Guardiola P, Laurent S, Boutouyrie P, Briet M. Large artery stiffening and remodeling are independently associated with all-cause mortality and cardiovascular events in chronic kidney disease. Hypertension 60: 1451–1457, 2012. [DOI] [PubMed] [Google Scholar]
- 22.Kozel BA, Danback JR, Waxler JL, Knutsen RH, de Las Fuentes L, Reusz GS, Kis E, Bhatt AB, Pober BR. Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension 63: 74–79, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacro RV, Dietz HC, Sleeper LA, Yetman AT, Bradley TJ, Colan SD, Pearson GD, Tierney ES, Levine JC, Atz AM, Benson DW, Braverman AC, Chen S, De Backer J, Gelb BD, Grossfeld PD, Klein GL, Lai WW, Liou A, Loeys BL, Markham LW, Olson AK, Paridon SM, Pemberton VL, Pierpont ME, Pyeritz RE, Radojewski E, Roman MJ, Sharkey AM, Stylianou MP, Wechsler SB, Young LT, Mahony L. Atenolol vs. losartan in children and young adults with Marfan's syndrome. N Engl J Med 371: 2061–2071, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le VP, Knutsen RH, Mecham RP, Wagenseil JE. Decreased aortic diameter and compliance precedes blood pressure increases in postnatal development of elastin-insufficient mice. Am J Physiol Heart Circ Physiol 301: H221–H229, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature 393: 276–280, 1998. [DOI] [PubMed] [Google Scholar]
- 26.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest 102: 1783–1787, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattace-Raso FU, van der Cammen TJ, Hofman A, van Popele NM, Bos ML, Schalekamp MA, Asmar R, Reneman RS, Hoeks AP, Breteler MM, Witteman JC. Arterial stiffness and risk of coronary heart disease and stroke: the Rotterdam Study. Circulation 113: 657–663, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Naghshin J, McGaffin KR, Witham WG, Mathier MA, Romano LC, Smith SH, Janczewski AM, Kirk JA, Shroff SG, O'Donnell CP. Chronic intermittent hypoxia increases left ventricular contractility in C57BL/6J mice. J Appl Physiol (1985) 107: 787–793, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ong KT, Delerme S, Pannier B, Safar ME, Benetos A, Laurent S, Boutouyrie P. Aortic stiffness is reduced beyond blood pressure lowering by short-term and long-term antihypertensive treatment: a meta-analysis of individual data in 294 patients. J Hypertens 29: 1034–1042, 2011. [DOI] [PubMed] [Google Scholar]
- 30.Patrizio M, Musumeci M, Stati T, Fecchi K, Mattei E, Catalano L, Marano G. Propranolol promotes Egr1 gene expression in cardiomyocytes via beta-adrenoceptors. Eur J Pharmacol 587: 85–89, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Pober BR, Johnson M, Urban Z. Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. J Clin Invest 118: 1606–1615, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reddy GK, Enwemeka CS. A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem 29: 225–229, 1996. [DOI] [PubMed] [Google Scholar]
- 33.Safar ME, Levy BI, Struijker-Boudier H. Current perspectives on arterial stiffness and pulse pressure in hypertension and cardiovascular diseases. Circulation 107: 2864–2869, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Schiffrin EL. Circulatory therapeutics: use of antihypertensive agents and their effects on the vasculature. J Cell Mol Med 14: 1018–1029, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Starcher B. A ninhydrin-based assay to quantitate the total protein content of tissue samples. Anal Biochem 292: 125–129, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Theilade S, Lajer M, Persson F, Joergensen C, Rossing P. Arterial stiffness is associated with cardiovascular, renal, retinal, and autonomic disease in type 1 diabetes. Diabetes Care 36: 715–721, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomlinson LA. Methods for assessing arterial stiffness: technical considerations. Curr Opin Nephrol Hypertens 21: 655–660, 2012. [DOI] [PubMed] [Google Scholar]
- 38.Van Bortel LM, Kool MJ, Boudier HA, Struijker Boudier HA. Effects of antihypertensive agents on local arterial distensibility and compliance. Hypertension 26: 531–534, 1995. [DOI] [PubMed] [Google Scholar]
- 39.Vazquez E, Coronel I, Bautista R, Romo E, Villalon CM, Avila-Casado MC, Soto V, Escalante B. Angiotensin II-dependent induction of AT2 receptor expression after renal ablation. Am J Physiol Renal Physiol 288: F207–F213, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP. Reduced vessel elasticity alters cardiovascular structure and function in newborn mice. Circ Res 104: 1217–1224, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagenseil JE, Mecham RP. New insights into elastic fiber assembly. Birth Defects Res C Embryo Today 81: 229–240, 2007. [DOI] [PubMed] [Google Scholar]
- 42.Wessel A, Gravenhorst V, Buchhorn R, Gosch A, Partsch CJ, Pankau R. Risk of sudden death in the Williams-Beuren syndrome. Am J Med Genet A 127A: 234–237, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Williams JC, Barratt-Boyes BG, Lowe JB. Supravalvular aortic stenosis. Circulation 24: 1311–1318, 1961. [DOI] [PubMed] [Google Scholar]