

This study demonstrates for the first time that ANG II type 2 receptor (AT2) activation enhances ANG I-converting enzyme 2 (ACE2) expression and activity and prevents TNF-α-stimulated ICAM-1 expression via inhibition of NF-κB signaling, providing evidence that drugs that mimic or enhance the function of AT2 and/or ACE2 may be beneficial for the treatment of cardiovascular diseases.
Keywords: ANG II type 2 receptor, ANG I-converting enzyme 2, TNF-α, ICAM-1
Abstract
ANG II type 2 receptor (AT2) and ANG I-converting enzyme 2 (ACE2) are important components of the renin-ANG system. Activation of AT2 and ACE2 reportedly counteracts proinflammatory effects of ANG II. However, the possible interaction between AT2 and ACE2 has never been established. We hypothesized that activation of AT2 increases ACE2 activity, thereby preventing TNF-α-stimulated ICAM-1 expression via inhibition of NF-κB signaling. Human coronary artery endothelial cells were pretreated with AT2 antagonist PD123319 (PD) or ACE2 inhibitor DX600 and then stimulated with TNF-α in the presence or absence of AT2 agonist CGP42112 (CGP). We found that AT2 agonist CGP increased both ACE2 protein expression and activity. This effect was blunted by AT2 antagonist PD. ICAM-1 expression was very low in untreated cells but greatly increased by TNF-α. Activation of AT2 with agonist CGP or with ANG II under concomitant AT1 antagonist reduced TNF-α-induced ICAM-1 expression, which was reversed by AT2 antagonist PD or ACE2 inhibitor DX600 or knockdown of ACE2 with small interfering RNA. AT2 activation also suppressed TNF-α-stimulated phosphorylation of inhibitory κB (p-IκB) and NF-κB activity. Inhibition of ACE2 reversed the inhibitory effect of AT2 on TNF-α-stimulated p-IκB and NF-κB activity. Our findings suggest that stimulation of AT2 reduces TNF-α-stimulated ICAM-1 expression, which is partly through ACE2-mediated inhibition of NF-κB signaling.
NEW & NOTEWORTHY
This study demonstrates for the first time that ANG II type 2 receptor (AT2) activation enhances ANG I-converting enzyme 2 (ACE2) expression and activity and prevents TNF-α-stimulated ICAM-1 expression via inhibition of NF-κB signaling, providing evidence that drugs that mimic or enhance the function of AT2 and/or ACE2 may be beneficial for the treatment of cardiovascular diseases.
inflammation in the vascular wall is an important contributor to the pathophysiology of hypertension (10), atherosclerosis, and the development of cardiovascular disease (CVD) (36, 37). It is considered one of main mechanisms contributing to the progression of CVD (35) and may be involved in triggering myocardial and cerebral ischemia (19, 33). Patients with CVD have increased expression and plasma concentration of inflammatory markers and mediators (3, 39). In particular, plasma levels of TNF-α and IL-6, as well as ICAM-1, VCAM-1, and E-selectin have been reportedly increased (35) in hypertensive subjects.
TNF-α is one of the major inflammatory cytokines produced by a number of cells, including vascular endothelial cells (29). Stimulation of endothelial cells with TNF-α leads to activation of NF-kB and subsequently increases the expression of adhesion molecules, such as ICAM-1 and proinflammatory cytokines (8). The signaling pathway of NF-кB activation by TNF-α involves the activation of the IKK complex, which phosphorylates inhibitory κB (IκB), resulting in its ubiquitination and subsequent proteasomal degradation (14).
Accumulating evidence shows that the renin-ANG system (RAS) is a regulator of the inflammatory response in the vascular wall (32). ANG II, the main effector peptide of the RAS, binds to two major subtypes of receptors, namely, ANG II types 1 and 2 receptors (AT1 and AT2, respectively) (5). ANG II exerts proinflammatory effects on the vasculature via AT1, whereas activation of AT2 reportedly opposes the effects of AT1 and reduces inflammation. It has been demonstrated that ANG II, via AT1, modulates the secretion of TNF-α from many cell types, including endothelial cells (2). Whereas the proinflammatory features of AT1 are well established, less is known about the anti-inflammatory effects of AT2 (32). AT2 is believed to antagonize not only AT1-mediated proinflammatory actions but also other proinflammatory mediators, such as cytokines, via inhibitory effects on NF-кB signaling (32).
ANG I-converting enzyme 2 (ACE2) is an important component of the RAS and reportedly regulates endothelial cell responses to inflammation (21). It is highly expressed in coronary endothelial cells compared with vascular smooth muscle cells or cardiomyocytes (9, 27). Studies have shown that endothelial overexpression of ACE2 inhibited TNF-α-induced monocyte adhesion, whereas silencing of ACE2 increased monocyte-endothelial adhesion. Furthermore, ANG II-induced increases in cellular adhesion molecules were attenuated in endothelial cells overexpressing ACE2 (21). These data suggest that ACE2 plays a role in limiting endothelial cell inflammation, both under inflammatory and ANG II-stimulated conditions.
Both AT2 and ACE2 serve as negative regulators of the RAS and reportedly counteract ANG II or inflammatory cytokine-induced by proinflammatory actions. However, there are only a few reports implicating the possible association between AT2 and ACE2. Ali et al. (1) recently demonstrated that activation of AT2 with a specific peptide agonist CGP42112 (CGP) increased ACE2 activity in diabetic rats and human kidney-2 cells. Qi et al. (28) and Chang et al. (6) reported that overexpression of AT2 in the heart increased the ratios of ACE2/ACE and ANG 1-7 receptor (Mas)/AT1, and deletion of AT2 decreased ACE2 expression and accelerated diabetic nephropathy. Our preliminary study in cultured human coronary artery endothelial cells (HCAECs) also showed that activation of AT2 increases ACE2 expression. Based on these findings, we further tested the novel hypothesis that activation of AT2 negatively modulates TNF-α-stimulated NF-κB activation and ICAM-1 expression via an ACE2-mediated signaling cascade.
MATERIALS AND METHODS
Cell Culture and Reagents
HCAECs were purchased from Lonza (Basel, Switzerland) and maintained in endothelial cell growth medium-2 (EGM-2; Lonza). The cells were incubated at 37°C in 5% CO2 and 95% air. Recombinant human TNF-α was purchased from R&D Systems (Minneapolis, MN). AT2 agonist CGP was purchased from Bachem (Bubendorf, Switzerland); AT2 antagonist PD123319 (PD) was a gift from Pfizer (Groton, CT); and ACE2-specific peptide inhibitor DX600 was purchased from Phoenix Pharmaceuticals (Mannheim, Germany).
Experimental Protocols
HCAECs were incubated overnight in six-well plates with EGM-2 medium containing 0.5% FBS and then switched to serum-free endothelial basal medium-2 (Lonza) before the experiments.
To determine the effect of AT2 activation on ACE2 protein expression and activity.
Cells were either untreated or pretreated with the AT2 antagonist PD (10 μM) for 30 min and then treated with AT2 agonist CGP (0.1 μM) for 6 h. Afterwards, cells were lysed to determine ACE2 expression by Western blot or activity.
To determine the effect of AT2 activation with agonist CGP or ANG II in the presence of the AT1 antagonist on TNF-α-mediated ICAM-1 expression.
Cells were untreated or pretreated with the AT2 antagonist PD (10 μM) for 30 min and then treated with AT2 agonist CGP (0.1 μM) for an additional 30 min. To determine the effect of AT2 activation with ANG II in the presence of AT1 antagonist valsartan (Val; 1 μM) on TNF-α-mediated ICAM-1 expression, cells were untreated or pretreated with Val, with or without the AT2 antagonist PD for 30 min, and then treated with ANG II (0.1 μM) for an additional 30 min. Afterwards, cells were stimulated with TNF-α (0.5 ng/ml) for 6 h and lysed to determine ICAM-1 expression by Western blot.
To determine whether ACE2 inhibition diminishes the effect of AT2 on TNF-α-mediated ICAM-1 expression.
Cells were pretreated with the ACE2 inhibitor DX600 (1 μM) for 30 min and then treated with AT2 agonist CGP (0.1 μM) for an additional 30 min. Afterwards, cells were stimulated with TNF-α (0.5 ng/ml) for 6 h and lysed to determine ICAM-1 expression by Western blot. To ensure that the inhibitory effect of DX600 is specific for ACE2, we performed an additional experiment using ACE2 small interfering RNA (siRNA) to determine whether knockdown of ACE2 diminishes the inhibitory effects of AT2 on TNF-α-mediated ICAM-1 expression. Cells were transfected with either control-scrambled siRNA or ACE2 siRNA for 48 h and then treated with the AT2 agonist CGP (0.1 μM) for 30 min. Afterwards, cells were stimulated with TNF-α (0.5 ng/ml) for 6 h and then lysed to determine ICAM-1 expression by Western blot. ACE2 expression was also determined to check the transfection efficacy.
To determine whether ACE2 inhibition blocks the effect of AT2 on TNF-α-induced IκB phosphorylation and NF-κB activity.
Cells were pretreated with DX600 (1 μM) for 30 min and then treated with AT2 agonist CGP (0.1 μM) for an additional 30 min. For IκB phosphorylation (p-IκB), cells were stimulated with TNF-α (0.5 ng/ml) for 15 min and then lysed. p-IκB was determined by Western blot. For NF-κB activity, cells were stimulated with TNF-α (0.5 ng/ml) for 1 h and harvested to extract nuclear proteins. NF-κB activity was determined by EMSA.
Western Blot for ACE2 and ICAM-1 Expression and p-IкB
Cells were harvested in cell lysis buffer with protease inhibitors and phosphatase inhibitors, as described previously (45). Lysates were centrifuged at 14,000 g for 10 min to remove insoluble material. Following SDS-PAGE, proteins were transferred onto a polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA), which was blocked with 5% nonfat powdered milk in Tris-buffered saline solution and probed with primary polyclonal antibody to ICAM-1 (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA), ACE2 (1 μg/ml; R&D Systems), p-IкB (1:1,000; Cell Signaling Technology, Danvers, MA), IкB (1:1,000; Cell Signaling Technology), or GAPDH (1:1,000; Cell Signaling Technology). Horseradish peroxidase (HRP)-conjugated anti-rabbit IgG or anti-mouse IgG (both from Cell Signaling Technology) or anti-goat IgG (Thermo Scientific, Waltham, MA) was used to visualize proteins by a chemiluminescence reaction (ECL kit; GE Healthcare, Waukesha, WI). ACE2 or ICAM-1 expression was normalized to GAPDH, and p-IкB was normalized to total IкB.
ACE2 Activity Assay
ACE2 activity was measured by SensoLyte 390 ACE2 activity assay kit (AnaSpec, Fremont, CA), following the manufacturer's instructions. HCAECs were lysed in assay buffer containing 0.1% Triton X-100, incubated for 10 min at 4°C, and centrifuged at 20,000 g at 4°C, and the supernatant was collected for ACE2 activity assay. Samples (50 μl) were incubated with ACE2 substrate (Mca/Dnp), 10 μM captopril (to inhibit ACE activity), or both 10 μM captopril and 10 μM DX600 and assay buffer in a final reaction volume of 100 μl. Fluorescence intensity was measured at an excitation wavelength of 330 nm and an emission wavelength of 390 nm. Specific ACE2 activity was determined by subtracting the activity in the presence of both 10 μM captopril and 10 μM DX600 from the activity in the presence of only captopril (16). The protein content of the samples was determined by bicinchoninic acid (BCA) protein assay (Bio-Rad Laboratories, Hercules, CA). The specific ACE2 activity was calculated by comparing the known amounts of recombinant human ACE2 (Calbiochem, Merck Millipore, Billerica, MA) and normalized to total protein.
ANG 1-7 Measurement
ANG 1-7 in the culture medium was assessed using an ELISA kit (Peninsula Laboratories International, San Carlos, CA), following the manufacturer's instructions. In brief, HCAECs were untreated or pretreated with AT1 antagonist Val (1 μM), with or without the AT2 antagonist PD (10 μM), for 30 min and then treated with ANG II (0.1 μM) for 6 h. Afterwards, cell culture medium was collected for ANG 1-7 measurement. For normalization, cells were harvested and total protein content determined by BCA protein assay (Bio-Rad Laboratories). ANG 1-7 concentration was normalized to total protein and expressed as picogram·milliliter−1·microgram−1 protein.
Small Interfering RNA
The ACE2 siRNAs were purchased from Ambion, Life Technologies (Grand Island, NY; Cat. #4392420), and the control scrambled siRNA was purchased from Qiagen (Valencia, CA). The siRNA was transfected into HCAECs in six-well plates using Lipofectamine 2000 (Life Technologies). It was diluted in Opti-MEM I, and the mixture was incubated for 5 min at room temperature. After the incubation, it was mixed with diluted siRNA in Opti-MEM I and incubated for 20 min at room temperature to allow a complex to form, after which, the siRNA-Lipofectamine mixture containing 100 pmol siRNA was added to each well.
EMSA for NF-κB Activity
NF-κB binding activity in nuclear extracts was assessed by EMSA using a kit from Panomics (Fremont, CA), according to the manufacturer's instructions. HCAECs were harvested following the Nuclear Extract Kit protocol (Active Motif, Carlsbad, CA) to extract nuclear proteins. EMSA was then conducted to measure NF-κB binding activity. Nuclear extract (2 μg) was incubated with polydeoxyinosinic-polydeoxycytidylic acid at room temperature for 5 min and then incubated with biotin-labeled probes at 15°C for 30 min. After electrophoresis on a 6% polyacrylamide gel, the samples on the gel were transferred onto a presoaked Pall Life Sciences Biodyne B nylon membrane (VWR International, Radnor, PA), which was crosslinked using a UV crosslinker for 3 min and then developed by adding the blocking buffer and streptavidin-HRP conjugate. Optical density of the bands was compared, as described previously (30).
Statistical Analysis
Results are expressed as means ± SE. Contrast statements in an ANOVA routine were used to examine all pairwise comparisons. A Hochberg's method was used to adjust for multiple comparisons. A difference was considered significant if adjusted P < 0.05.
RESULTS
Effect of AT2 Activation on ACE2 Protein Expression and Activity
Figure 1 shows that activation of AT2 with agonist CGP increased ACE2 protein expression by 1.79 ± 0.04-fold (n = 5, P < 0.01) compared with control. This effect was reduced by the AT2 antagonist PD, which alone, did not affect ACE2 protein expression. We examined further whether activation of AT2 regulates ACE2 activity. As shown in Fig. 2, AT2 activation increased ACE2 activity from 0.61 ± 0.05 to 0.95 ± 0.03 (pg·μl−1·h−1·μg−1 protein; n = 8, P < 0.01) compared with control. This increase was blocked by the AT2 antagonist PD. Treatment with PD alone did not affect ACE2 activity. These data suggest that activation of AT2 enhances both ACE2 protein expression and activity. In addition, activation of AT2 by ANG II (in the presence of the AT1 antagonist Val) increased ANG 1-7 in the medium from 0.1 ± 0.05 to 15.6 ± 0.5 (pg·ml−1·μg−1 protein; n = 6, P < 0.01) compared with control. This effect was blocked significantly by the AT2 antagonist PD (P < 0.01; Fig. 3).
Fig. 1.
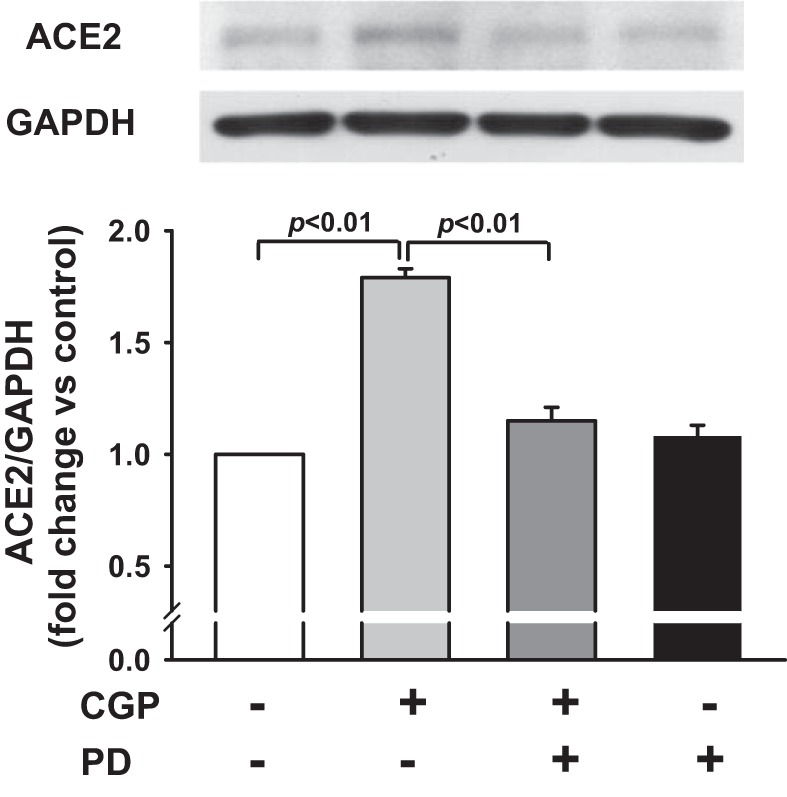
Effect of ANG II type 2 receptor (AT2) agonist CGP42112 (CGP) on ANG I-converting enzyme 2 (ACE2) protein expression. Human coronary artery endothelial cells (HCAECs) were pretreated with or without AT2 antagonist PD123319 (PD; 10 μM) and stimulated with AT2 agonist CGP (0.1 μM). Top: representative Western blots. Bottom: quantitative analysis of ACE2 protein expression presented as fold increase in relation to untreated controls (set at a value of 1.0 arbitrary unit); n = 5 separate experiments.
Fig. 2.

Effect of AT2 agonist CGP on ACE2 activity. HCAECs were pretreated with or without AT2 antagonist PD (10 μM) and then stimulated with CGP (0.1 μM). ACE2 activity was assessed with a SensoLyte 390 ACE2 activity assay kit using Mca/Dnp fluorescence resonance energy transfer peptide to detect ACE2 activity; n = 7–8 separate experiments.
Fig. 3.
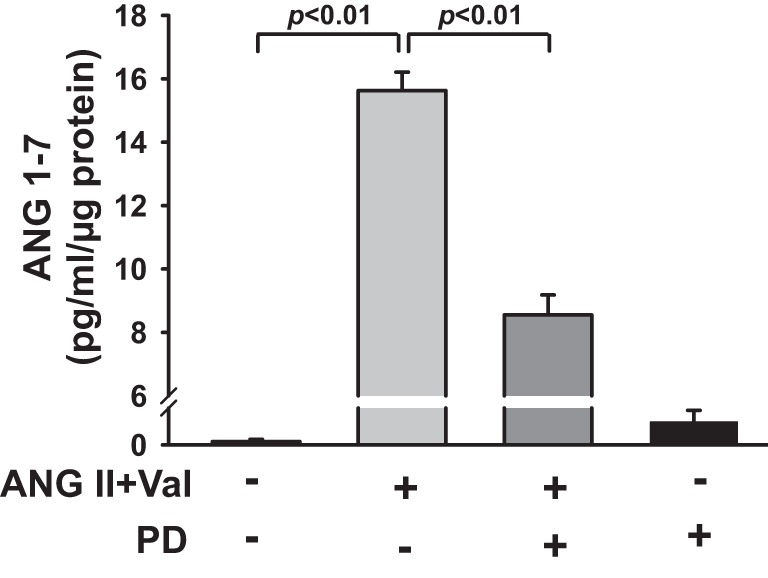
Effect of AT2 activation on ANG 1-7 in culture medium. HCAECs were pretreated with valsartan (Val; 1 μM) in the presence or absence of AT2 antagonist PD (10 μM) and then stimulated with ANG II (0.1 μM). ANG 1-7 in culture medium was assessed with an ELISA kit. ANG 1-7 concentration was normalized to total protein and expressed as picogram·milliliter−1·microgram−1 protein; n = 6 separate experiments.
Effect of AT2 Activation on TNF-α-Mediated ICAM-1 Expression
ICAM-1 expression was very low in untreated cells but greatly increased by TNF-α (Fig. 4A). AT2 agonist CGP significantly reduced the TNF-α-mediated ICAM-1 expression from 1.0 arbitrary unit to 0.47 ± 0.02 (n = 10, P < 0.01). This effect was reversed by the AT2 antagonist PD, demonstrating the specific effects of AT2. PD, per se, had no effect on TNF-α-mediated ICAM-1 expression. In addition, activation of AT2 by ANG II (in the presence of AT1 antagonist Val) reduced the TNF-α-mediated ICAM-1 expression from 1.0 arbitrary unit to 0.50 ± 0.05 (n = 5, P < 0.01), and this effect was reversed by the AT2 antagonist PD (Fig. 4B). These results suggest that activation of AT2 is effective in blocking TNF-α stimulation of ICAM-1 in HCAECs.
Fig. 4.

A: effect of AT2 agonist CGP on TNF-α-mediated ICAM-1 protein expression. HCAECs were pretreated with CGP (0.1 μM) in the presence or absence of AT2 antagonist PD (10 μM) and then stimulated with TNF-α (0.5 ng/ml). Top: representative Western blots. Bottom: quantitative analysis of ICAM-1 protein expression presented as fold change in relation to TNF-α-alone group (set at a value of 1.0 arbitrary unit); n = 5–10 separate experiments. B: effect of ANG II plus AT1 antagonist Val on TNF-α-mediated ICAM-1 protein expression. HCAECs were pretreated with ANG II (0.1 μM) plus Val (1 μM) in the presence or absence of AT2 antagonist PD (10 μM) and then stimulated with TNF-α (0.5 ng/ml). Top: representative Western blots. Bottom: quantitative evaluation of ICAM-1 protein expression presented as fold changes in relation to TNF-α-alone group (set at a value of 1.0 arbitrary unit); n = 5 separate experiments.
ACE2 Inhibition Diminished the Effect of AT2 on TNF-α-Mediated ICAM-1 Expression
DX600 is a potent inhibitor of ACE2, which has been widely used for in vitro studies (43). We tested whether inhibition of ACE2 with DX600 blocks the effects of AT2 on TNF-α-mediated ICAM-1 expression. Similar to data shown in Fig. 4A, AT2 agonist CGP significantly reduced the TNF-α-mediated ICAM-1 expression (n = 10, P < 0.01), and this effect was partially reversed by ACE2 inhibitor DX600 (from 0.47 ± 0.04 to 0.84 ± 0.02; n = 5–10, P < 0.01; Fig. 5). DX600, per se, did not affect TNF-α-mediated ICAM-1 expression. We examined further whether knockdown of ACE2 with a siRNA blocks the inhibitory effect of AT2 on TNF-α-mediated ICAM-1 expression. First, we tested the efficacy of ACE2 siRNA and found that it reduced ACE2 protein expression by 74% compared with scrambled siRNA (Fig. 6A). Our data showed further that the inhibitory effect of the AT2 agonist CGP on TNF-α-mediated ICAM-1 expression was blunted significantly by the ACE2 siRNA compared with the scrambled control (Fig. 6B). These data indicated that the inhibitory effect of AT2 activation on TNF-α-mediated ICAM-1 expression is partially mediated via ACE2.
Fig. 5.
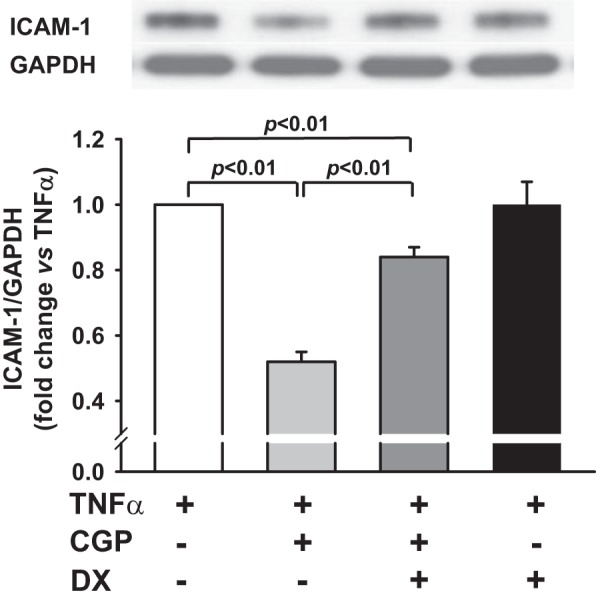
Effect of ACE2 blockade on AT2-induced inhibition of TNF-α-stimulated ICAM-1 protein expression. HCAECs were pretreated with CGP (0.1 μM) in the presence or absence of the ACE2 inhibitor DX600 (DX; 1 μM) and then stimulated with TNF-α (0.5 ng/ml). Top: representative Western blots. Bottom: quantitative evaluation of ICAM-1 protein expression presented as fold changes in relation to TNF-α-alone group (set at a value of 1.0 arbitrary unit); n = 5–10 separate experiments.
Fig. 6.
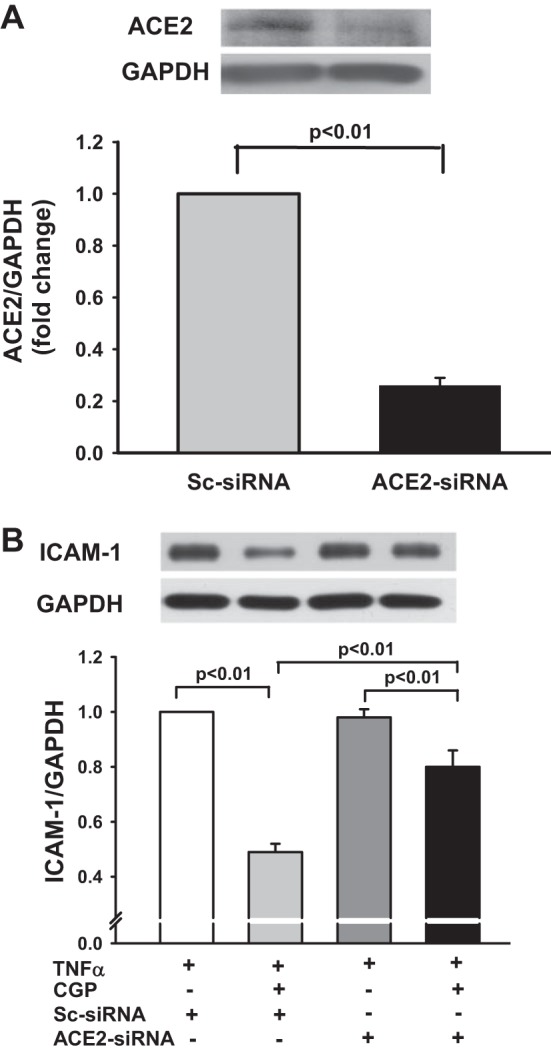
A: effect of ACE2 small interfering RNA (siRNA) on ACE2 protein expression. HCAECs were transfected with scrambled siRNA (Sc-siRNA) or ACE2-siRNA, and ACE2 protein expression was detected by Western blots. Top: representative Western blots. Bottom: quantitative evaluation of ACE2 protein expression presented as fold changes in relation to Sc-siRNA (set at a value of 1.0 arbitrary unit); n = 4 separate experiments. B: effect of ACE2 knockdown on AT2-induced inhibition of TNF-α-stimulated ICAM-1 protein expression. HCAECs were transfected with Sc-siRNA or ACE2-siRNA and then stimulated with TNF-α (0.5 ng/ml) in the presence or absence of AT2 agonist CGP (0.1 μM). Top: representative Western blots. Bottom: quantitative evaluation of ICAM-1 protein expression presented as fold changes in relation to TNF-α + Sc-siRNA group (set at a value of 1.0 arbitrary unit); n = 4 separate experiments.
ACE2 Inhibition Blocked the Effect of AT2 on TNF-α-Induced p-IκB
TNF-α is a classical activator of NF-κB (4). The activation of NF-κB is preceded by p-IκB. Analysis of cell extracts using p-IκB-specific antibodies showed that stimulation of the cells with TNF-α caused rapid p-IκB compared with untreated cells. AT2 agonist CGP significantly decreased p-IκB (n = 6, P < 0.01; Fig. 7). These data indicated that activation of AT2 inhibits TNF-α-induced NF-κB activation.
Fig. 7.
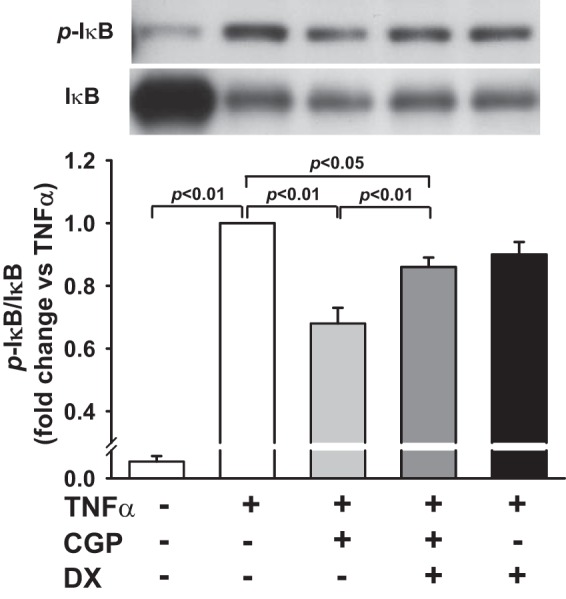
Effect of ACE2 blockade on AT2-induced inhibition of TNF-α-stimulated inhibitory κB phosphorylation (p-IκB). HCAECs were pretreated with AT2 agonist CGP (0.1 μM) in the presence or absence of the ACE2 inhibitor DX (1 μM) and then stimulated with TNF-α (0.5 ng/ml). Top: representative Western blots. Bottom: quantitative evaluation of p-IκB expression presented as fold changes in relation to TNF-α-alone group (set at a value of 1.0 arbitrary unit); n = 5–6 separate experiments.
We examined further whether the inhibitory effect of AT2 on TNF-α-induced p-IκB is mediated, in part, via ACE2 pathways. We found that pretreatment with ACE2 inhibitor DX600 partially reversed the effect of AT2 on TNF-α-induced p-IκB from 0.68 ± 0.05 (n = 6) to 0.86 ± 0.03 (n = 5, P < 0.01; Fig. 7). DX600, per se, did not affect TNF-α-induced p-IκB. These data suggested that ACE2 contributes to the inhibitory effect of AT2 on TNF-α stimulated NF-κB activation.
ACE2 Inhibition Suppressed the Effect of AT2 on TNF-α-Mediated NF-κB Activity
To confirm the involvement of ACE2 in the inhibitory actions of AT2 on TNF-α-induced NF-κB activation, we further measured NF-κB activity using the EMSA. As seen in Fig. 8A, TNF-α significantly enhanced NF-κB DNA binding activity compared with vehicle control. This induction was blocked by the AT2 agonist CGP. The intensity of DNA-protein complexes obtained by densitometric scanning was 0.67 ± 0.04-fold vs. TNF-α-treated cells (n = 5, P < 0.01; Fig. 8B). Pretreatment with ACE2 inhibitor DX600 partially reversed the effect of AT2 on TNF-α-induced NF-κB DNA binding activity (0.85 ± 0.04-fold vs. TNF-α-treated cells; n = 5, P < 0.05; Fig. 8B). These data further supported the contributions of ACE2 in the inhibitory effect of AT2 on TNF-α-stimulated activation of NF-κB.
Fig. 8.
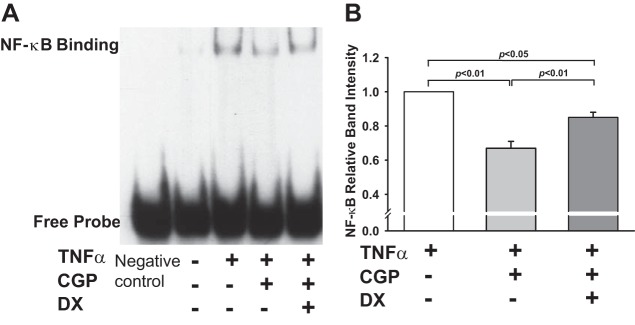
Effect of ACE2 blockade on AT2-induced inhibition of TNF-α-stimulated NF-κB activity. HCAECs were pretreated with AT2 agonist CGP (0.1 μM) in the presence or absence of the ACE2 inhibitor DX (1 μM) and then stimulated with TNF-α (0.5 ng/ml). NF-κB activity was measured by EMSA as DNA binding intensity. A: representative NF-κB DNA binding; Negative control, EMSA probe without nuclear extract. B: quantitative analysis of NF-κB DNA binding intensity presented as fold changes in relation to TNF-α-alone group (set at a value of 1.0 arbitrary unit); n = 4–5 separate experiments.
DISCUSSION
In the present study, we reported for the first time that activation of AT2 enhances ACE2 protein expression and ACE2 activity, which contributes to the inhibitory effects of AT2 on TNF-α-induced NF-κB activation and ICAM-1 expression.
The RAS represents one of the major players in vascular inflammation. The main effector peptide of the RAS, ANG II, exerts proinflammatory effects via AT1, whereas AT2 reportedly exerts anti-inflammatory actions (15, 42). Studies have shown that AT2 activation, using the specific agonist C21, suppressed inflammatory cytokines in vitro (31) and in vivo (11, 13, 17, 23, 31). The AT2 agonist CGP also has anti-inflammatory actions by attenuating LPS-induced cyclooxygenase-2 (COX-2) expression in rat intestinal epithelial cells (40) and by reducing plasma and kidney cortex inflammatory markers (TNF-α, IL-6) in obese rats (34). TNF-α is a proinflammatory cytokine that primarily targets vascular endothelial cells (22). It upregulates specific adhesion molecules, such as ICAM-1, an important regulator of vascular inflammatory processes (24). In the present study, we demonstrated that TNF-α increased ICAM-1 expression in HCAECs, which is in agreement with the previous reports (25). We then asked whether AT2 decreases TNF-α-stimulated ICAM-1 expression. Our data showed that activation of AT2 by the agonist CGP or ANG II (with AT1 blockade) indeed inhibited TNF-α-mediated ICAM-1 expression, and this action was AT2 specific, since AT2 agonist CGP- or ANG II-induced effects were blunted by the specific AT2 antagonist, PD.
ACE2 serves as a negative regulator of the RAS (21). It is highly expressed in vascular endothelial cells and regulates endothelial cell responses to inflammation (21). Lovren et al. (21) reported that endothelial overexpression of ACE2 inhibited TNF-α-induced monocyte adhesion, whereas the silencing of ACE2 increased monocyte-endothelial adhesion. Although a number of reports have described the role of ACE2 or AT2 in limiting endothelial cell inflammation (21, 23, 31, 32, 38, 44), only a few reports describe the possible association between AT2 and ACE2. Qi et al. (28) reported that overexpression of AT2 in the heart increased ACE2/ACE and Mas/AT1 ratios. Ali et al. (1) reported recently that chronic activation of AT2 with the agonist CGP increased renal ACE2 activity in diabetic rats, whereas deletion of AT2 decreased ACE2 expression and accelerated diabetic nephropathy (6). In the present study, we demonstrated for the first time that activation of AT2 enhances not only ACE2 protein expression but also ACE2 activity in HCAECs. We further demonstrated that the AT2-mediated increase in ACE2 contributes to the anti-inflammatory effects of AT2, since inhibition of ACE2 reversed the inhibitory effects of AT2 on TNF-α-stimulated ICAM-1 expression and p-IкB. Taken together, our data support a role of ACE2 in AT2-mediated anti-inflammatory actions.
The mechanisms responsible for the anti-inflammatory actions of AT2 and ACE2 are not well understood. Our data provide evidence that activation of AT2 leads to suppression of p-IkB and NF-kB activation. NF-кB is known as a key transcription factor implicated in the regulation of a variety of genes participating in inflammatory responses. It has been shown that activation of NF-кB by TNF-α is required for the transcriptional activation of endothelial cell adhesion molecules (8). A key step in NF-kB activation involves p-IkB. In the present study, we demonstrated that activation of AT2 inhibited TNF-α-induced p-IkB and NF-kB binding activity. These data are consistent with previous studies showing that stimulation of AT2 exerts an anti-inflammatory action through inhibition of NF-kB (31, 42).
Previously, we demonstrated that activation of AT2 increases Src homology region 2 domain-containing phosphatase-1 (SHP-1) activity in mouse coronary artery endothelial cells (46). Activation of ACE2 also reportedly suppresses NF-kB activation via stimulation of SHP-1 (12, 41). Since SHP-1 is capable of dephosphorylating and stabling IkB (32), it is possible that AT2-induced suppression of NF-kB is modulated, in part, by ACE2-mediated signaling via activation of SHP-1. On the other hand, it has been reported that AT2-induced suppression of NF-kB is achieved by activation of mothers against decapentaplegic homolog 7 (Smad7) (18, 26). Liu et al. (20) recently demonstrated that deletion of ACE2 downregulated Smad7 protein and enhanced ANG-II-stimulated NF-kB activation and renal inflammation. Accordingly, ACE2-mediated Smad7 signaling may also be involved in AT2-dependent attenuation of NF-kB activation. In addition to activating NF-κB pathway, TNF-α activates the MAPK pathway (7). These pathways involve signaling through ERK and p38 MAPKs. Activation of AT2 may interfere with other proinflammatory signaling cascades, such as reduction of COX-2 expression (40) and inhibition of ERK and STAT activation (15). Clarification of these mechanisms warrants further studies.
Limitation
We demonstrate in vitro that activation of AT2 increases ACE2 protein expression and activity and prevents TNF-α-stimulated ICAM-1 expression via inhibition of NF-κB signaling. However, the role of ACE2 in the anti-inflammatory actions of AT2 has not been studied in vivo, which warrants further investigation. Nevertheless, this limitation does not negate the evidence that ACE2 plays an important role in mediating the anti-inflammatory effects of AT2.
Summary and Perspective
The present study demonstrates for the first time the potential role of ACE2 in AT2-mediated anti-inflammatory actions in HCAECs. It is reported that patients with CVD have increased expression and/or plasma concentrations of inflammatory markers, such as TNF-α, which primarily target vascular endothelial cells and upregulate specific adhesion molecules, such as ICAM-1, contributing to the pathophysiology of CVD, whereas AT2 and ACE2 are highly expressed in vascular endothelial cells and may act as anti-inflammatory mediators in vascular inflammation. Indeed, our study demonstrated that activation of AT2 and ACE2 diminished a TNF-α-induced increase of ICAM-1 in human coronary endothelial cells; however, further studies are needed to elucidate the precise mechanisms by which AT2 and ACE2 act in cooperation to inhibit inflammatory responses both in vitro and in vivo. Nevertheless, our data, along with others, do support the possibility that drugs that mimic or enhance the function of AT2 and/or ACE2 may be beneficial for the treatment of CVDs with inflammatory and fibrotic components.
GRANTS
Support for this work was provided by Henry Ford Health System Institutional Fund (to X-P. Yang) and National Heart, Lung, and Blood Institute Grant HL-28982 (to O. A. Carretero).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: L.Z. and X-P.Y. conception and design of research; L.Z., J.X., N.R., and X.G. performed experiments; L.Z., P.H., E.P., and X-P.Y. analyzed data; L.Z. and X-P.Y. interpreted results of experiments; L.Z. and X-P.Y. prepared figures; L.Z. drafted manuscript; L.Z., O.A.C., P.H., and X-P.Y. edited and revised manuscript; L.Z., O.A.C., J.X., P.H., N.R., X.G., E.P., and X-P.Y. approved final version of manuscript.
REFERENCES
- 1.Ali Q, Wu Y, Hussain T. Chronic AT2 receptor activation increases renal ACE2 activity, attenuates AT1 receptor function and blood pressure in obese Zucker rats. Kidney Int 84: 931–939, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arenas IA, Xu Y, Lopez-Jaramillo P, Davidge ST. Angiotensin II-induced MMP-2 release from endothelial cells is mediated by TNF-alpha. Am J Physiol Cell Physiol 286: C779–C784, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Blake GJ, Ridker PM. Novel clinical markers of vascular wall inflammation. Circ Res 89: 763–771, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Borner C, Hollt V, Kraus J. Mechanisms of the inhibition of nuclear factor-kappaB by morphine in neuronal cells. Mol Pharmacol 81: 587–597, 2012. [DOI] [PubMed] [Google Scholar]
- 5.Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev 24: 261–271, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Chang SY, Chen YW, Chenier I, Tran Sle M, Zhang SL. Angiotensin II type II receptor deficiency accelerates the development of nephropathy in type I diabetes via oxidative stress and ACE2. Exp Diabetes Res 2011: 521076, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu WM. Tumor necrosis factor. Cancer Lett 328: 222–225, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J 9: 899–909, 1995. [PubMed] [Google Scholar]
- 9.Danilczyk U, Eriksson U, Oudit GY, Penninger JM. Physiological roles of angiotensin-converting enzyme 2. Cell Mol Life Sci 61: 2714–2719, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De CC, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol 25: 2106–2113, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Dhande I, Ali Q, Hussain T. Proximal tubule angiotensin AT2 receptors mediate an anti-inflammatory response via interleukin-10: role in renoprotection in obese rats. Hypertension 61: 1218–1226, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Hashim AZ, Renno WM, Raghupathy R, Abduo HT, Akhtar S, Benter IF. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-kappaB-dependent pathways. Br J Pharmacol 166: 1964–1976, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gelosa P, Pignieri A, Fandriks L, De GM, Hallberg A, Banfi C, Castiglioni L, Turolo L, Guerrini U, Tremoli E, Sironi L. Stimulation of AT2 receptor exerts beneficial effects in stroke-prone rats: focus on renal damage. J Hypertens 27: 2444–2451, 2009. [DOI] [PubMed] [Google Scholar]
- 14.Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 72: 1493–1505, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Horiuchi M, Hayashida W, Akishita M, Tamura K, Daviet L, Lehtonen JY, Dzau VJ. Stimulation of different subtypes of angiotensin II receptors, AT1 and AT2 receptors, regulates STAT activation by negative crosstalk. Circ Res 84: 876–882, 1999. [DOI] [PubMed] [Google Scholar]
- 16.Huentelman MJ, Zubcevic J, Katovich MJ, Raizada MK. Cloning and characterization of a secreted form of angiotensin-converting enzyme 2. Regul Pept 122: 61–67, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Kaschina E, Grzesiak A, Li J, Foryst-Ludwig A, Timm M, Rompe F, Sommerfeld M, Kemnitz UR, Curato C, Namsolleck P, Tschope C, Hallberg A, Alterman M, Hucko T, Paetsch I, Dietrich T, Schnackenburg B, Graf K, Dahlof B, Kintscher U, Unger T, Steckelings UM. Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation 118: 2523–2532, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Lan HY. Smad7 as a therapeutic agent for chronic kidney diseases. Front Biosci 13: 4984–4992, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation 104: 365–372, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Liu Z, Huang XR, Chen HY, Penninger JM, Lan HY. Loss of angiotensin-converting enzyme 2 enhances TGF-beta/Smad-mediated renal fibrosis and NF-kappaB-driven renal inflammation in a mouse model of obstructive nephropathy. Lab Invest 92: 650–661, 2012. [DOI] [PubMed] [Google Scholar]
- 21.Lovren F, Pan Y, Quan A, Teoh H, Wang G, Shukla PC, Levitt KS, Oudit GY, Al-Omran M, Stewart DJ, Slutsky AS, Peterson MD, Backx PH, Penninger JM, Verma S. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am J Physiol Heart Circ Physiol 295: H1377–H1384, 2008. [DOI] [PubMed] [Google Scholar]
- 22.Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp Mol Pathol 70: 317–325, 2001. [DOI] [PubMed] [Google Scholar]
- 23.Matavelli LC, Huang J, Siragy HM. Angiotensin AT(2) receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension 57: 308–313, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mo SJ, Son EW, Lee SR, Lee SM, Shin DH, Pyo S. CML-1 inhibits TNF-alpha-induced NF-kappaB activation and adhesion molecule expression in endothelial cells through inhibition of IkBalpha kinase. J Ethnopharmacol 109: 78–86, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Myers CL, Wertheimer SJ, Schembri-King J, Parks T, Wallace RW. Induction of ICAM-1 by TNF-alpha, IL-1 beta, and LPS in human endothelial cells after downregulation of PKC. Am J Physiol Cell Physiol 263: C767–C772, 1992. [DOI] [PubMed] [Google Scholar]
- 26.Nagarajan RP, Chen F, Li W, Vig E, Harrington MA, Nakshatri H, Chen Y. Repression of transforming-growth-factor-beta-mediated transcription by nuclear factor kappaB. Biochem J 348: 591–596, 2000. [PMC free article] [PubMed] [Google Scholar]
- 27.Oudit GY, Crackower MA, Backx PH, Penninger JM. The role of ACE2 in cardiovascular physiology. Trends Cardiovasc Med 13: 93–101, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Qi Y, Li H, Shenoy V, Li Q, Wang F, Raizada M, Sumners C, Katovich M. Moderate cardiac-selective overexpression of angiotensin type 2 receptor protects cardiac functions from ischemic injury. Exp Physiol 97: 89–101, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ranta V, Orpana A, Carpen O, Turpeinen U, Ylikorkala O, Viinikka L. Human vascular endothelial cells produce tumor necrosis factor-alpha in response to proinflammatory cytokine stimulation. Crit Care Med 27: 2184–2187, 1999. [DOI] [PubMed] [Google Scholar]
- 30.Rhaleb NE, Pokharel S, Sharma UC, Peng H, Peterson E, Harding P, Yang XP, Carretero OA. N-Acetyl-Ser-Asp-Lys-Pro inhibits interleukin-1beta-mediated matrix metalloproteinase activation in cardiac fibroblasts. Pflugers Arch 465: 1487–1495, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rompe F, Artuc M, Hallberg A, Alterman M, Stroder K, Thone-Reineke C, Reichenbach A, Schacherl J, Dahlof B, Bader M, Alenina N, Schwaninger M, Zuberbier T, Funke-Kaiser H, Schmidt C, Schunck WH, Unger T, Steckelings UM. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor kappaB. Hypertension 55: 924–931, 2010. [DOI] [PubMed] [Google Scholar]
- 32.Rompe F, Unger T, Steckelings UM. The angiotensin AT2 receptor in inflammation. Drug News Perspect 23: 104–111, 2010. [DOI] [PubMed] [Google Scholar]
- 33.Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med 340: 115–126, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Sabuhi R, Ali Q, Asghar M, Al-Zamily NR, Hussain T. Role of the angiotensin II AT2 receptor in inflammation and oxidative stress: opposing effects in lean and obese Zucker rats. Am J Physiol Renal Physiol 300: F700–F706, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Savoia C, Sada L, Zezza L, Pucci L, Lauri FM, Befani A, Alonzo A, Volpe M. Vascular inflammation and endothelial dysfunction in experimental hypertension. Int J Hypertens 2011: 281240, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savoia C, Schiffrin EL. Inflammation in hypertension. Curr Opin Nephrol Hypertens 15: 152–158, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Savoia C, Schiffrin EL. Reduction of C-reactive protein and the use of anti-hypertensives. Vasc Health Risk Manag 3: 975–983, 2007. [PMC free article] [PubMed] [Google Scholar]
- 38.Simôes e Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol 169: 477–492, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suematsu M, Suzuki H, DeLano FA, Schmid-Schonbein GW. The inflammatory aspect of the microcirculation in hypertension: oxidative stress, leukocytes/endothelial interaction, apoptosis. Microcirculation 9: 259–276, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Tani T, Ayuzawa R, Takagi T, Kanehira T, Maurya DK, Tamura M. Angiotensin II bi-directionally regulates cyclooxygenase-2 expression in intestinal epithelial cells. Mol Cell Biochem 315: 185–193, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tao X, Fan J, Kao G, Zhang X, Su L, Yin Y, Zrenner B. Angiotensin-(1-7) attenuates angiotensin II-induced signalling associated with activation of a tyrosine phosphatase in Sprague-Dawley rats cardiac fibroblasts. Biol Cell 106: 182–192, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Wu L, Iwai M, Li Z, Shiuchi T, Min LJ, Cui TX, Li JM, Okumura M, Nahmias C, Horiuchi M. Regulation of inhibitory protein-kappaB and monocyte chemoattractant protein-1 by angiotensin II type 2 receptor-activated Src homology protein tyrosine phosphatase-1 in fetal vascular smooth muscle cells. Mol Endocrinol 18: 666–678, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Xu P, Sriramula S, Lazartigues E. ACE2/ANG-(1-7)/Mas pathway in the brain: the axis of good. Am J Physiol Regul Integr Comp Physiol 300: R804–R817, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhong JC, Yu XY, Lin QX, Li XH, Huang XZ, Xiao DZ, Lin SG. Enhanced angiotensin converting enzyme 2 regulates the insulin/Akt signalling pathway by blockade of macrophage migration inhibitory factor expression. Br J Pharmacol 153: 66–74, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu L, Carretero OA, Liao TD, Harding P, Li H, Sumners C, Yang XP. Role of prolylcarboxypeptidase in angiotensin II type 2 receptor-mediated bradykinin release in mouse coronary artery endothelial cells. Hypertension 56: 384–390, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu L, Carretero OA, Xu J, Wang L, Harding P, Rhaleb NE, Yang JJ, Sumners C, Yang XP. Angiotensin II type 2 receptor-stimulated activation of plasma prekallikrein and bradykinin release: role of SHP-1. Am J Physiol Heart Circ Physiol 302: H2553–H2559, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]