

Chronic infusion of low-dose angiotensin II in the mouse induces diastolic dysfunction and HFpEF in the absence of pressure overload, LV systolic dysfunction, LV dilatation or hypertrophy, and metabolic abnormalities. This model may be considered a novel tool for mechanistic preclinical studies in HFpEF with translational potential.
Keywords: heart failure with preserved ejection fraction, diastolic dysfunction, animal model, angiotensin II, end-diastolic pressure, isovolumetric relaxation time
Abstract
Heart failure (HF) with preserved ejection fraction (HFpEF) is a clinical syndrome of HF symptoms associated with impaired diastolic function. Although it represents ∼50% of patients with HF, the mechanisms of disease are poorly understood, and therapies are generally ineffective in reducing HF progression. Animal models of HFpEF not due to pressure or volume overload are lacking, therefore limiting in-depth understanding of the pathophysiological mechanisms and the development of novel therapies. We hypothesize that a continuous infusion of low-dose angiotensin II (ATII) is sufficient to induce left ventricular (LV) diastolic dysfunction and HFpEF, without increasing blood pressure or inducing LV hypertrophy or dilatation. Osmotic pumps were implanted subcutaneously in 8-wk-old male mice assigned to the ATII (0.2 mg·kg−1·day−1) or volume-matched vehicle (N = 8/group) for 4 wk. We measured systolic and diastolic arterial blood pressures through a tail-cuff transducer, LV dimensions and ejection fraction through echocardiography, and LV relaxation through pulsed-wave Doppler and LV catheterization. Myocardial fibrosis and cardiomyocyte cross-sectional area were measured. ATII infusion had no effects on systemic arterial blood pressure. ATII induced significant impairment in LV diastolic function, as measured by an increase (worsening) in LV isovolumetric relaxation time, myocardial performance index, isovolumetric relaxation time constant, and LV end-diastolic pressure without altering LV dimensions, mass, or ejection fraction. Chronic infusion of low-dose ATII recapitulates the HFpEF phenotype in the mouse, without increasing systemic arterial blood pressure. This mouse model may provide insight into the mechanisms of HFpEF.
NEW & NOTEWORTHY
Chronic infusion of low-dose angiotensin II in the mouse induces diastolic dysfunction and HFpEF in the absence of pressure overload, LV systolic dysfunction, LV dilatation or hypertrophy, and metabolic abnormalities. This model may be considered a novel tool for mechanistic preclinical studies in HFpEF with translational potential.
heart failure (hf) with preserved ejection fraction (HFpEF) is a clinical syndrome of symptoms of HF, such as breathlessness and exercise intolerance, associated with impaired left ventricular (LV) diastolic function in the presence of a normal LV ejection fraction (LVEF > 50%) (2). HFpEF carries significant morbidity and mortality burdens, and the prevalence of the disease has increased over the past 30 yr (2, 17). To date, the mechanisms underlying diastolic dysfunction and the progression of HFpEF are poorly understood. The pathophysiology involves impaired LV relaxation and contractile reserve, increased LV stiffness, as well as abnormal renal sodium handling, arterial stiffness, and aberrant ventriculo-arterial coupling (2).
The limited availability of animal models of HFpEF has potentially represented a major limitation in conducting mechanistic studies in the field (11). The animal models of HFpEF have hitherto involved mostly studies of increased afterload and LV hypertrophy (i.e., aortic banding or systemic arterial hypertension), models of increased preload (i.e., aorto-caval fistulas), or models of altered metabolism (i.e., obesity, diabetes, hyperlipidemia), making it difficult to distinguish between the intrinsic mechanism(s) of diastolic dysfunction vs. the mechanism(s) causing dysfunction (11). Moreover, aortic banding and aorto-caval fistulas do not reflect clinically existing conditions, and the majority of patients with HFpEF continue to have HF symptoms, even when they have controlled blood pressure (BP). In fact, HFpEF patients have LV hypertrophy in <50% of cases and often show no evidence of increased preload or LV dilatation, thus making the value of the current preclinical models questionable (1, 2, 11).
Angiotensin II (ATII) occupies a central role in homeostasis, hypertension, and HF, regulating afterload, preload, cardiac hypertrophy, and fibrosis (4). Chronic infusion of ATII has been used as a model of chronic hypertension, while low-dose infusion of ATII affects the cardiovascular system without inducing systemic arterial hypertension (4).
We hypothesized that low-dose ATII would reproduce the cardiac phenotype of HFpEF in the mouse in the absence of elevations in systemic arterial BP or LV hypertrophy, and thus serve as a preclinical model of HFpEF free of confounding factors.
METHODS
Ethical aspects.
The experiments were conducted under the guidelines of the “Guide for the Care and Use of Laboratory Animals”, published by the National Institutes of Health (revised 2011). The study protocol was approved by the Virginia Commonwealth Institutional Animal Care and Use Committee.
Experimental model.
Eight-week-old outbred male CD1 mice were supplied by Harlan (Indianapolis, IN). Mice were sedated with pentobarbital (50–70 mg/kg), and osmotic mini-pumps (DURECT, Cupertino, CA) were sterilely implanted in the intrascapular space to allow subcutaneous infusion of low-dose ATII (0.2 mg·kg−1·day−1) or vehicle sterile water (at the same flow rate), for 28 days. Mice were randomly assigned to different groups: sham-operated mice (no implantation of any pump; N = 8); mice implanted with an infusion pump that infused vehicle for 28 days (N = 8); and mice implanted with an infusion pump with ATII for 28 days (N = 8). The infusion of vehicle had no significant effects on any of the measured parameters and was indistinguishable from the sham-operated mice, and, therefore, throughout the rest of the paper, only data of vehicle are shown compared with ATII pump, while data on the sham-operated mice are not presented.
Noninvasive arterial BP measurement.
We measured systolic and diastolic arterial BP at baseline and at 28 days using a noninvasive tail-cuff BP analyzer (CODA System, Kent Scientific, Torrington, CT). Ten cycle measurements were collected through a dedicated software, and the mean was calculated for both the systolic and the diastolic BP (7).
Echocardiography.
All mice underwent transthoracic echocardiography at baseline (before surgery) and at 28 days, under light anesthesia (50 mg/kg pentobarbital sodium). Echocardiography was performed with the Vevo770 imaging system (VisualSonics, Toronto, Ontario, Canada) and a 30-MHz probe (3). The heart was visualized in B-mode from parasternal short-axis and apical views. We measured the LV end-diastolic diameter, LV end-systolic diameter, LV anterior wall diastolic thickness, and LV posterior wall diastolic thickness at M-mode, as previously described and according to the American Society of Echocardiography recommendations (3). LV fractional shortening, LVEF, and LV mass were calculated from the measurements of wall thickness and chamber diameters. Right ventricular (RV) systolic function was estimated using M-mode and measuring the tricuspidal annular plane systolic excursion (24). The transmitral LV outflow tract Doppler spectra (E, A, ET) was recorded from an apical four-chamber view, and the myocardial performance index (MPI) was calculated as the ratio of isovolumetric contraction time and isovolumetric relaxation time (IRT) divided by the ejection time (21). Tissue Doppler was used to measure the lateral E′ spectra, and calculate the E-to-E′ ratio (E/E′). Two-dimensional video loops were reviewed for abnormalities in the pericardial structure (effusion or thickening), RV dilatation, or interventricular dependence. The investigators performing and reading the echocardiograms were blinded to the treatment allocation.
LV catheterization.
Mice were anesthetized (70 mg/kg pentobarbital sodium), and a pressure probe catheter (AD Instruments, Colorado Springs, CO) was retrogradely inserted in the LV from the right carotid artery. LV end-diastolic pressures (LVEDP), and the pressure-volume loops were recorded and measured using LabChart Pro 5 (AD Instruments). The end-diastolic pressure-volume relationship (EDPVR) and end-systolic pressure-volume relationship (ESPVR) were calculated by pressure-volume measurements by concomitant Millar catheter and transthoracic echocardiography (29). Changes in preload were studied in a closed chest system, applying a resistance to preload by inflating the lungs artificially using a standard tidal volume (7.5 μm/g body wt) and rate (300 breaths/min). To determine changes in preload, the ventilator was turned off, allowing for a progressive increase of venous return and recording several consecutive beats. The EDVPR and ESPVR were then calculated according to the changes in pressure-volume loops by interpolating the points of end-diastolic pressure and end-systolic pressure with and without preload. The IRT constant, τ, was measured using the formula P = P0e−t/τ, where P represents the diastolic pressure, P0 is the pressure at the moment of maximum −dP/dT, and t is the IRT.
Sample collection.
Immediately following LV catheterization, hearts were rapidly excised and placed in 10% formalin and fixed for a minimum of 48 h and then embedded in paraffin. The lungs and the pericardium were inspected for gross anatomical abnormalities. Embedded heart tissues were sectioned into 5-μm slides and stained with Masson's trichrome (Sigma-Aldrich) to detect collagen fibers. The area of myocardial fibrosis was measured as percentage of collagen area on total myocardial tissue area. The cross-sectional area (CSA) of cardiomyocytes was measured to determine cardiomyocyte hypertrophy. Computer morphometry was performed using Image J software.
Statistical analysis.
All statistical analyses were performed using SPSS 21.0 package for Windows (Chicago, IL). Continuous variables are expressed as mean and SE, and one-way ANOVA to compare between three or more groups at any individual time point, followed by Bonferroni-corrected T-test for unpaired data was used. For interval changes over time, comparing ATII and vehicle infusion, an ANOVA for repeated measures was used, assessing the time × group interaction. P values < 0.05 were considered statistically significant.
RESULTS
Procedural data.
Sixteen mice underwent infusion pump implantation without any acute or late mortality (100% survival). Morphometric data are reported in Table 1.
Table 1.
Morphological parameters
| Group | BW, g | LVM, mg | LVEDD, mm | LVESD, mm |
|---|---|---|---|---|
| ATII | 38.4 ± 0.8 | 108 ± 7 | 3.96 ± 0.10 | 2.58 ± 0.13 |
| Vehicle | 39.0 ± 0.9 | 96 ± 5 | 4.19 ± 0.06 | 2.63 ± 0.13 |
Values are group means ± SE; N = 8 animals/group. ATII, angiotensin II; BW, body weight; LVM, left ventricular mass; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end systolic diameter. All P > 0.15.
Systemic arterial BP measurement.
Systolic and diastolic arterial BP values, measured noninvasively with a tail cuff, were within the normal range before implantation of the pump and remained unchanged after 28-day infusion of ATII (N = 4, all P > 0.81, Fig. 1).
Fig. 1.
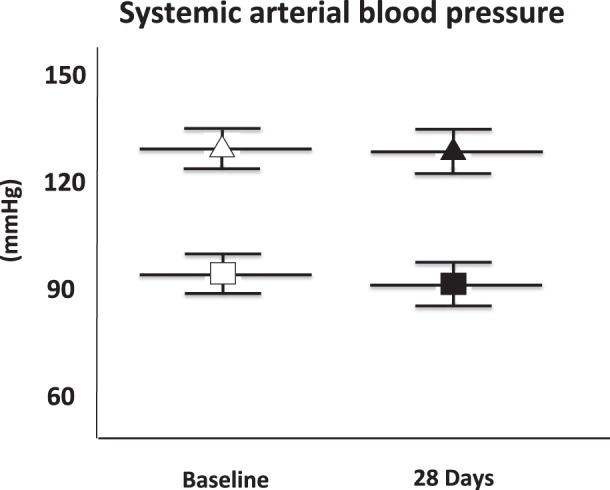
Systemic arterial blood pressure. The figure shows the lack of effect of a low dose of angiotensin II (ATII) infusion on systemic arterial blood pressure. Systolic and diastolic blood pressure values measured with a tail cuff at baseline and after 28 days of infusion of ATII are shown. Triangles, mean arterial systolic blood pressure; squares, mean arterial diastolic blood pressure. Values are group means ± SE; N = 4 animals/time point.
Echocardiography.
LV mass, LV end-diastolic diameter, LVEF, and tricuspidal annular plane systolic excursion values were unchanged between baseline and after 28 days of infusion of ATII or vehicle (all P > 0.22, Fig. 2). In the ATII group, the RV and the pericardium did not show abnormalities.
Fig. 2.
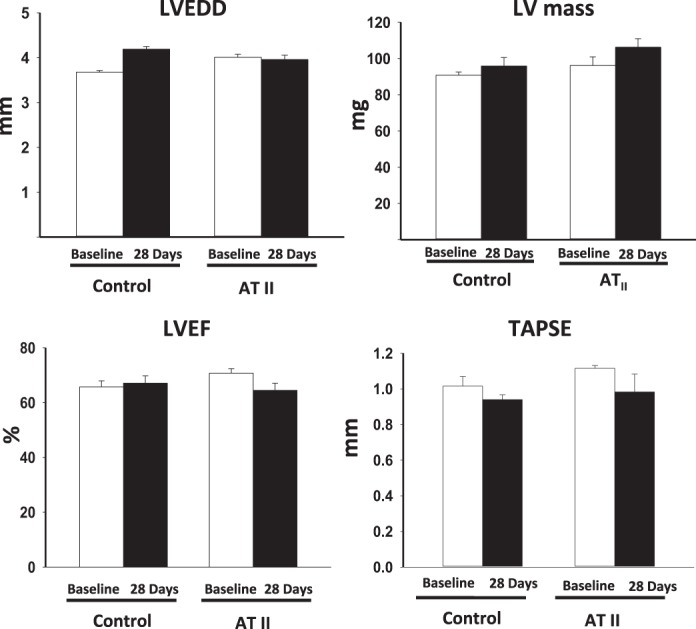
Echocardiographic measurement of left ventricular (LV) dimensions and systolic function. The figure shows lack of effect of a low dose of ATII infusion on LV end-diastolic diameter (LVEDD, expressed in mm), the LV mass (expressed in mg), LV ejection fraction (LVEF, expressed as percentage), and tricuspid annulus plane systolic excursion (TAPSE, expressed in mm). Values are means ± SE; N = 8 animals/group.
Representative images of the pulsed wave Doppler spectra are shown in Fig. 3. Table 2 reports Doppler data for each group. ATII infusion induced a significant impairment in LV diastolic function, as measured by a significant increase of the IRT and of the MPI vs. baseline values (P = 0.018 and P < 0.001, respectively), whereas vehicle infusion had no significant effects on IRT or MPI (all P > 0.23) (Fig. 3). No changes in E/E′ were observed between the groups.
Fig. 3.
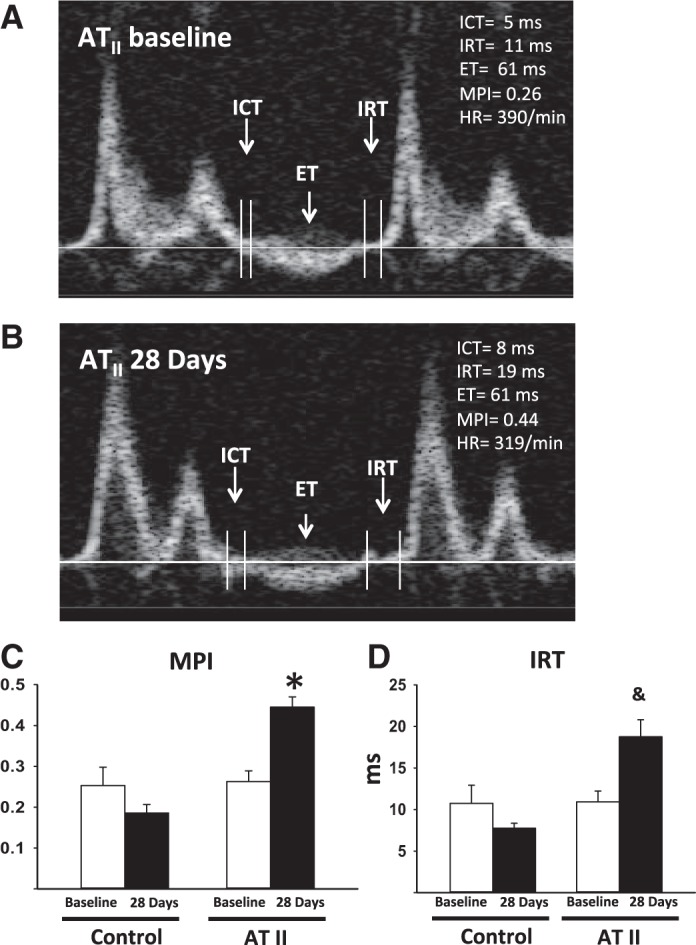
Pulse-wave Doppler spectra. A and B: representative images of pulse-wave Doppler recordings at baseline before ATII infusion and after 28 days, respectively. The isovolumetric contraction time (ICT), ejection time (ET), and isovolumetric relaxation time (IRT) are indicated. Myocardial performance index (MPI; C) and IRT (expressed in ms; D) of the mice infused with ATII or the control vehicle at baseline and after 28 days of infusion are shown. Values are means ± SE; N = 8 animals/group. *P < 0.001. &P = 0.018.
Table 2.
Waive Doppler and tissue Doppler parameters
| Group | E | A | E/A | S′ | E′ | A′ | E/E′ |
|---|---|---|---|---|---|---|---|
| ATII | 703 ± 45 | 266 ± 35 | 2.52 ± 0.27 | 15.9 ± 0.7 | 15.0 ± 1.4 | 11.6 ± 0.8 | 47 ± 5 |
| Vehicle | 668 ± 13 | 300 ± 51 | 2.42 ± 0.39 | 15.5 ± 0.6 | 12.4 ± 1.2 | 9.3 ± 1.4 | 57 ± 5 |
Values are group mean ± SE; N = 8 animals/group. All P > 0.13.
LVEDP.
Representative images of the pressure changes over time are reported in Fig. 4. ATII-treated mice showed significantly increased LVEDP, τ, and EDPVR compared with vehicle-treated mice (P = 0.002, P < 0.001, and P = 0.040, respectively, Fig. 4), reflecting increased LV elastance. The ESPVR was unchanged, reflecting similar systolic performance between the control vehicle and the ATII-treated mice.
Fig. 4.

LV catheterization. A and B: representative images of pressure-volume loop recordings of vehicle or ATII infusion for 28 days, respectively. The LV end-diastolic pressure (LVEDP) values were used to calculate the end-diastolic pressure-volume relationship (EDPVR) originated by pressure-volume loops with preload impediment by positive pressure ventilation and on spontaneous respiration. C: the LVDEP (expressed in mmHg) measured after 28 days of ATII or control vehicle infusion. D: the EDPVR slope values (expressed as mmHg/μl) of ATII or control vehicle infusion. E: the ESPVR slope values (expressed as mmHg/μl) of ATII or control vehicle infusion. F: the IRT time constant τ. Values are group means ± SE; N = 6–8 animals/group. *P = 0.002. &P = 0.04. #P = 0.001.
Cardiomyocyte hypertrophy and interstitial myocardial fibrosis.
Histological analysis revealed that ATII led to a significantly increased cardiomyocyte CSA vs. vehicle (P < 0.01, Fig. 5), reflecting cardiomyocyte hypertrophy. ATII treatment also led to increased interstitial myocardial fibrosis compared with the vehicle-treated group (P = 0.036, Fig. 5). The associations between the cardiomyocyte CSA or the interstitial fibrosis and IRT, MPI, and LVEDP are shown in Fig. 6. We found no statistically significant correlation between cardiomyocytes hypertrophy and fibrosis (R = 0.38, P = 0.28), or between these parameters with IRT, MPI, and LVEDP within the individual animals (Fig. 5).
Fig. 5.

Measurement of cardiomyocyte hypertrophy and quantification of the interstitial fibrosis. A and B: Masson's Trichrome stain on heart sections were used to visualize the cross-sectional areas of cardiomyocytes (×20 magnification) of mice infused with control vehicle or ATII, respectively. C and D: areas of magnification of A and B, respectively. E: quantification of the cardiomyocyte cross-sectional areas of the control vehicle and ATII infused mice. F and G: representative pictures of Masson's Trichrome staining to quantify the fibrosis in tissue sections of hearts of mice infused with control vehicle or ATII, respectively (×20 magnification). H and I: areas of magnification of F and G, respectively. J: quantification of the interstitial fibrosis reported as percentage (%) of the area of fibrotic tissue on the total area. Values are group means ± SE; N = 8 animals/group. *P < 0.05.
Fig. 6.

Relationship between hypertrophy and fibrosis with diastolic dysfunction. The graphs report the distribution of the values of fibrosis (top) or cross-sectional area (bottom) in relation to the MPI, end-diastolic pressure (EDP), and IRT in the control vehicle group (open circles) and in the low-dose ATII group (solid squares). Differences between groups are statistically significant. No significant correlations were observed in the distribution of the values. Values are group means ± SE.
DISCUSSION
Chronic infusion of low-dose ATII recapitulates the phenotype of HFpEF in the mouse, independent of systemic arterial hypertension and/or LV hypertrophy. HFpEF is a clinical syndrome of breathlessness, fatigue, and exercise intolerance, despite preserved LV systolic function (LVEF >50%), and direct or indirect evidence of diastolic dysfunction and/or elevated LV filling pressures, in the absence of other structural or functional abnormalities that may explain such symptoms (2, 19). HFpEF is a clinically heterogeneous syndrome, and, unlike other cardiac diseases, the incidence of HFpEF is increasing (2). Therapeutic strategies that benefit patients with HF with reduced ejection fraction (EF) (or systolic HF) have proven to be marginally effective or ineffective in patients with HFpEF (2). Despite the disease burden of HFpEF and associated morbidity and mortality, the pathophysiology remains unclear (2). Understanding the mechanisms involved in the progression of HFpEF is further complicated by the many comorbidities and risk factors affecting HFpEF patients, including age, obesity, hypertension, and diabetes. Therefore, there is a need for additional and more accurate preclinical models to understand the pathogenesis of HFpEF and potentially develop targeted therapies.
In this regard, an animal model of HFpEF that explores diastolic dysfunction independent of LV hypertrophy and/or pressure overload and of metabolic changes is lacking, although several animal models have been proposed to study HFpEF (11). The mouse model of transaortic constriction is a pressure-overload model where a band is placed around the aorta, and, as the mouse grows, the band becomes obstructive by preventing sufficient enlargement of the aortic CSA, thus increasing afterload (5, 15, 23). This model is subject to procedural variability based on the size of the banding, the age of mice at time of surgery, and the length of follow up, but most importantly the diastolic dysfunction is secondary to pressure overload and dependent on the severity and rapidity of onset of the constriction. As such, this model represents more closely a model of progressive aortic valve stenosis rather than a model of HFpEF. This model also develops decompensated eccentric LV hypertrophy associated with LV systolic dysfunction, which is an uncommon feature in HFpEF, as the evolution to HF with reduced EF is uncommon in patients with HFpEF (2). Of note, in some experimental settings, the banding was applied to induce a relatively rapid pressure overload (18). However, the clinical translational value of these models is difficult to understand, since, with the exception of acute prosthetic aortic valve thrombosis, or perhaps an acute hypertensive crisis in a patient without hypertension, a clinical scenario of acutely increased afterload is not observed.
Other commonly used models of diastolic dysfunction are related to systemic hypertension in the rat (16). The spontaneous hypertensive rat and the salt-sensitive rat models are models of cardiac dysfunction in the setting of large increases in systemic arterial BP (27). These models are useful in studying hypertensive cardiomyopathy, which also evolves through concentric LV hypertrophy with preserved EF to eccentric LV hypertrophy with reduced EF (11). While many patients with HFpEF have arterial hypertension, less than one-half have LV hypertrophy, and eccentric LV hypertrophy with reduced EF is not a common feature of HFpEF (2).
The rodent model of aorto-caval fistula is a model of volume overload characterized by biventricular hypertrophy, LV enlargement, and increased systolic and diastolic pressure (1). This model is used to study congestive HF, and, therefore, the prominent systolic nature of ventricular dysfunction represents a limitation of the use of this model to study diastolic dysfunction (26, 28).
The db/db leptin receptor-deficient morbidly obese mouse has been used as a model of HFpEF; however, diastolic dysfunction is associated with morbid obesity and severe hyperglycemia secondary to diabetes (22). The db/db model has an inherent value in exploring the contribution of obesity and metabolic syndrome, often seen in patients with HFpEF, but may not be representative of all patients with HFpEF. The ZSF1 obese rat model is a model of metabolic dysfunction in which HFpEF is associated with obesity and diabetes (9). The rat develops HF in association with hyperglycemia and insulin resistance, and, therefore, while it may represent a good model for HFpEF with metabolic alterations, it is unlikely to be representative of all patients with HFpEF. Genetic modifications of titin and cardiac myosin binding protein C have also been proposed as models of HFpEF (12, 25). These genetic models allow for the intrinsic changes to develop progressively as the mouse grows and ages, but do not reflect the clinical HFpEF syndrome.
Unlike other models of ATII infusion at higher doses, which induce significant systemic hypertension, we used a subpressor dose of ATII that recapitulates the enhanced activation of the renin-angiotensin-aldosterone system (RAAS) in patients with HFpEF, devoid of systemic arterial hypertension (17, 28). The proposed model, therefore, differs from prior models by the absence of increased afterload, hypertrophy, dilatation, or metabolic abnormalities, and as such it is likely to provide information that is not available from the others, yet it is complementary to the already available models. This model shows that, in the mouse, ATII signaling is sufficient to induce diastolic dysfunction and HFpEF. This may appear at odds with clinical trials, failing to clearly show significant benefits in survival with blockers of the RAAS (2). It should be considered, however, that RAAS blockers in HFpEF have been mostly tested on top of other vasodilators and allowed for crossover to open-label treatments, thus biasing toward the null hypothesis (8, 13). Moreover, failure of RAAS blocker to improve outcomes in HFpEF should not be viewed as proof of a pathogenic role of ATII in HFpEF, as it may simply reflect the inability of RAAS blockers to reverse the disease process once fully established.
We describe that infusion of subpressor doses of ATII in the mouse leads to increased IRT, τ, MPI, EDPVR, and LVEDP, indicative of impaired myocardial relaxation, increased elastance, wall stress, and diastolic dysfunction. The inability to assess for symptoms of HF is an obvious limitation to any animal study. Nevertheless, the increase in LVEDP is considered a reliable sign of HF. The increase in IRT and τ and the upward shift in EDPVR in absence of changes in the pericardium or RV suggest that the LVEDP is indeed due to abnormal LV relaxation and/or elastance. We did not find a change in E/E′ in this study: while this appears to be at odds with the reported finding of increased E/E′ in patients with HFpEF, it should be considered that elevation in the E/E′ may only occur when LVEDP is markedly increase (i.e., >20 mmHg in patients with HFpEF) and, therefore, be a less sensitive marker of HFpEF (14). We measured changes in LVEDP and EDPVR in a closed chest/abdomen model, representing a potential way to reduce confounders related to surgery, bleeding, pain, and changes in temperature in the chest. Pentobarbital sodium has become the preferred sedative in our experiments, as it does not interfere with physiological or pharmacological preconditioning, and, although it likely reduced heart rate, contractility, and lusitropy in all groups, we expect the effects to be equal in all groups and thus not interfere with the comparisons between groups.
The histological analysis revealed also mild, but significant, increase in cardiomyocyte size (hypertrophy) and interstitial fibrosis, indicative of increased cardiac elastance. These changes developed in the absence of hypertension, systolic dysfunction, or LV remodeling. This model of chronic low-dose ATII infusion may, therefore, be a novel and useful tool to provide in-depth mechanistic insight for understanding the development and contribution of diastolic dysfunction in HFpEF, independent of genetic modifications, metabolic abnormalities, or pressure and volume overload.
This model, however, is not without limitations. First, we only studied young adult male CD1 mice. In the future, additional studies should initiate low-dose ATII infusion in aging mice to understand how the disease progression may be altered with age. HFpEF is more prevalent in women; therefore, it will be crucial to examine this model in female mice, at various stages of their life (i.e., pre- and post-ovarian senescence or ovariectomized mice). The characteristic hypertrophic response in females may be protective in some instances but deleterious in others, such as in HFpEF (20). The infusion may also be extended to determine whether disease severity increases with a longer infusion period, and to see if mice will eventually develop systemic arterial hypertension, LV hypertrophy, and/or LV systolic dysfunction. The mechanisms by which ATII induces diastolic dysfunction in this HFpEF model are also not explored in this study. Impaired relaxation may be a dynamic effect on calcium handling, or dependent on the structural changes seen in interstitial fibrosis and cardiomyocyte hypertrophy (10). An additional limitation is the lack of data on isolated cardiomyocyte stiffness, shown to be abnormal in HFpEF (6). We also did not measure changes in titin isoform expression or phosphorylation status, which appear to regulate the cardiomyocyte stiffness (9). The effects of ATII on cardiomyocyte stiffness and titin will require further focused investigations.
In conclusion, a 28-day infusion of subpressor dose of ATII recapitulates the HFpEF features of impaired LV relaxation and increased LV elastance in the absence of pressure overload, LV systolic dysfunction, LV dilatation or hypertrophy, and metabolic abnormalities and may, therefore, be considered as a novel tool for mechanistic preclinical studies in HFpEF with immense translational potential.
GRANTS
S. Toldo is supported by an American Heart Association Post-Doctoral Award.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.A.R., B.W.V.T., A.A., and S.T. conception and design of research; J.A.R., A.G.M., S.C., C.M., A.A., and S.T. performed experiments; J.A.R., A.G.M., S.C., C.M., R.G., E.M., B.W.V.T., A.A., and S.T. analyzed data; J.A.R., S.C., E.M., J.V.R., F.N.S., A.A., and S.T. interpreted results of experiments; J.A.R., A.G.M., A.A., and S.T. prepared figures; J.A.R., F.N.S., A.A., and S.T. drafted manuscript; J.A.R., A.G.M., S.C., C.M., R.G., J.V.R., F.N.S., B.W.V.T., A.A., and S.T. approved final version of manuscript; A.G.M., S.C., C.M., R.G., E.M., J.V.R., F.N.S., A.A., and S.T. edited and revised manuscript.
REFERENCES
- 1.Abassi Z, Goltsman I, Karram T, Winaver J, Hoffman A. Aortocaval fistula in rat: a unique model of volume-overload congestive heart failure and cardiac hypertrophy. J Biomed Biotechnol 2011: 729497, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbate A, Arena R, Abouzaki N, Van Tassell BW, Canada J, Shah K, Biondi-Zoccai G, Voelkel NF. Heart failure with preserved ejection fraction: refocusing on diastole. Int J Cardiol 179: 430–440, 2015. [DOI] [PubMed] [Google Scholar]
- 3.Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Biondi-Zoccai GG, Houser JE, Qureshi IZ, Ownby ED, Gustini E, Biasucci LM, Severino A, Capogrossi MC, Vetrovec GW, Crea F, Baldi A, Kukreja RC, Dobrina A. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 117: 2670–2683, 2008. [DOI] [PubMed] [Google Scholar]
- 4.Allen AM, Zhuo J, Mendelsohn FA. Localization and function of angiotensin AT1 receptors. Am J Hypertens 13: 31S–38S, 2000. [DOI] [PubMed] [Google Scholar]
- 5.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A 103: 10086–10091, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borbely A, van der Velden J, Papp Z, Bronzwaer JG, Edes I, Stienen GJ, Paulus WJ. Cardiomyocyte stiffness in diastolic heart failure. Circulation 111: 774–781, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Daugherty A, Rateri D, Hong L, Balakrishnan A. Measuring blood pressure in mice using volume pressure recording, a tail-cuff method. J Vis Exp 27: 1291, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang JC. Heart failure therapy: what should clinicians believe? J Am Med Assoc 308: 2144–2146, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Hamdani N, Franssen C, Lourenco A, Falcao-Pires I, Fontoura D, Leite S, Plettig L, Lopez B, Ottenheijm CA, Becher PM, Gonzalez A, Tschope C, Diez J, Linke WA, Leite-Moreira AF, Paulus WJ. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail 6: 1239–1249, 2013. [DOI] [PubMed] [Google Scholar]
- 10.Hillestad V, Kramer F, Golz S, Knorr A, Andersson KB, Christensen G. Long-term levosimendan treatment improves systolic function and myocardial relaxation in mice with cardiomyocyte-specific disruption of the Serca2 gene. J Appl Physiol 115: 1572–1580, 2013. [DOI] [PubMed] [Google Scholar]
- 11.Horgan S, Watson C, Glezeva N, Baugh J. Murine models of diastolic dysfunction and heart failure with preserved ejection fraction. J Card Fail 20: 984–995, 2014. [DOI] [PubMed] [Google Scholar]
- 12.LeWinter MM, Granzier H. Cardiac titin: a multifunctional giant. Circulation 121: 2137–2145, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lund LHBL, Dahlström U, Edner M. Association between use of renin-angiotensin system antagonists and mortality in patients with heart failure and preserved ejection fraction. J Am Med Assoc 20: 2108–2117, 2012. [DOI] [PubMed] [Google Scholar]
- 14.Oh JK, Park SJ, Nagueh SF. Established and novel clinical applications of diastolic function assessment by echocardiography. Circ Cardiovasc Imaging 4: 444–455, 2011. [DOI] [PubMed] [Google Scholar]
- 15.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 110: 705–712, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Okamoto K. Spontaneous hypertension in rats. Int Rev Exp Pathol 7: 227–270, 1969. [PubMed] [Google Scholar]
- 17.Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 355: 251–259, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Patten RD, Hall-Porter MR. Small animal models of heart failure: development of novel therapies, past and present. Circ Heart Fail 2: 138–144, 2009. [DOI] [PubMed] [Google Scholar]
- 19.Paulus WJ, Tschope C, Sanderson JE, Rusconi C, Flachskampf FA, Rademakers FE, Marino P, Smiseth OA, De Keulenaer G, Leite-Moreira AF, Borbely A, Edes I, Handoko ML, Heymans S, Pezzali N, Pieske B, Dickstein K, Fraser AG, Brutsaert DL. How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J 28: 2539–2550, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Piro M, Della Bona R, Abbate A, Biasucci LM, Crea F. Sex-related differences in myocardial remodeling. J Am Coll Cardiol 55: 1057–1065, 2010. [DOI] [PubMed] [Google Scholar]
- 21.Porter TR, Shillcutt SK, Adams MS, Desjardins G, Glas KE, Olson JJ, Troughton RW. Guidelines for the use of echocardiography as a monitor for therapeutic intervention in adults: a report from the American Society of Echocardiography. J Am Soc Echocardiogr 28: 40–56, 2015. [DOI] [PubMed] [Google Scholar]
- 22.Reil JC, Hohl M, Reil GH, Granzier HL, Kratz MT, Kazakov A, Fries P, Muller A, Lenski M, Custodis F, Graber S, Frohlig G, Steendijk P, Neuberger HR, Bohm M. Heart rate reduction by If-inhibition improves vascular stiffness and left ventricular systolic and diastolic function in a mouse model of heart failure with preserved ejection fraction. Eur Heart J 34: 2839–2849, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarnavski O, McMullen JR, Schinke M, Nie Q, Kong S, Izumo S. Mouse cardiac surgery: comprehensive techniques for the generation of mouse models of human diseases and their application for genomic studies. Physiol Genomics 16: 349–360, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Toldo S, Bogaard HJ, Van Tassell BW, Mezzaroma E, Seropian IM, Robati R, Salloum FN, Voelkel NF, Abbate A. Right ventricular dysfunction following acute myocardial infarction in the absence of pulmonary hypertension in the mouse. PloS One 6: e18102, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tong CW, Nair NA, Doersch KM, Liu Y, Rosas PC. Cardiac myosin-binding protein-C is a critical mediator of diastolic function. Pflügers Arch 466: 451–457, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Treskatsch S, Feldheiser A, Rosin AT, Sifringer M, Habazettl H, Mousa SA, Shakibaei M, Schafer M, Spies CD. A modified approach to induce predictable congestive heart failure by volume overload in rats. PloS One 9: e87531, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamori Y, Ooshima A, Okamoto K. Genetic factors involved in spontaneous hypertension in rats an analysis of F 2 segregate generation. Japn Circ J 36: 561–568, 1972. [DOI] [PubMed] [Google Scholar]
- 28.Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, Loibner H, Wang XH, Penninger JM, Kassiri Z, Oudit GY. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 122: 717–728, 2010. [DOI] [PubMed] [Google Scholar]
- 29.Zile MR, Baicu CF, Gaasch WH. Diastolic heart failure–abnormalities in active relaxation and passive stiffness of the left ventricle. N Engl J Med 350: 1953–1959, 2004. [DOI] [PubMed] [Google Scholar]