

Abstract
Myelodysplastic syndromes (MDS) represent a variety of clonal abnormalities, possibly preleukemic and display numerous phenotypic manifestations. Specific mutations carry high morbidity and mortality rates due to cell line dysplasia. MDS commonly presents with symptoms related to anemia, and approximately two-thirds will develop thrombocytopenia, a rare, but potentially lethal complication that increases complexity in treatment and morbidity, and may be due to unique genetic mutations leading to refractory thrombocytopenia, ultimately leading to an overall reduction in survival. Careful identification and monitoring of this patient subdivision can significantly reduce morbidity and mortality, and potential identification of specific gene mutations and advances in treatment options will hopefully provide guidance on detecting at-risk patients in the future. We present a case of a man with MDS-U (karyotype 46, XY, del (20) (q11.2q13.3) (20) with no detected JAK2 V617F mutation), who in despite of appropriate evidenced based treatment, continued to exhibit refractory thrombocytopenia.
Key words: Thrombocytopenia, myelodysplasia, refractory, MDS, malignancy
Introduction
The myelodysplastic syndrome (MDS) are a heterogeneous assembly of hematologic disorders illustrated commonly by impaired hematopoiesis resulting in varied cytopenias and an increased threat of converting into acute myeloid leukemia (AML). High-throughput genomic methods currently uncover a diverse collection of mutated genes located within the genetic profile of many MDS patients.1 However, the role of cytogenic analysis and its corresponding benefit, diagnosis, prognosis, and therapeutic strategies, have not been fully established to date. MDS is frequently diagnosed in older males and those with previous exposure to benzene, chemotherapy or radiotherapy. 4000-50,000 new cases are diagnosed each year in the US alone,2 with and estimated incidence rate of 4.3 per 100,000.3 Up to half of all patients with MDS present asymptomatic with diagnosis being discovered after routine laboratory blood testing. Impaired hematopoietic stem cell proliferation and differentiation leads to abnormal cell morphology and physiology with subsequent increase in morbidity. Common symptoms vary and can include fatigue and weakness from anemia, infections from neutropenia, or bleeding from thrombocytopenia.
Studies have demonstrated that 40-65% of patients with MDS experience hemorrhagic complications due to thrombocytopenia, with these complications being cited as the cause of death in 10-20% of individuals with MDS.4 Patients who exhibit persistent refractory thrombocytopenia require physicians to more closely monitor bleeding complications and amplify the complexity of establishing correct treatment options. Identifying the patients who exhibit this rare subset type of MDS is critical in reducing morbidity and mortality in this patient population.
Case Report
A 71 year-old Caucasian male ex-smoker without any significant past medical history presented to the emergency department complaining of worsening redness and swelling in his left lower leg. He presented five days earlier and was given antibiotic treatment which did not resolve the problem. On physical exam BP 119/68 mHg, Pulse 61/min, Resp 17/min, Temp: 96.6°F, O2 saturation 96% on Room air. Laboratory testing showed hemoglobin 12.2 g/dL, WBC 7.3 K/uL, Platelet count 26 K/uL, Neutrophils 29, Lymphocytes 20%, MCV 73.2 fL, glucose 120 mg/dL, BUN 14 mg/dL, Creatinine 0.8 mg/dL, Calcium 8.7 mg/dL, albumin 4.3 g/dL, Sodium 137 mmol/L, Potassium 4 mmol/L, Chloride 100 mmol/L, and CO2 25 mmol/L. Liver enzymes were within normal limits. X-ray ruled out osseous contribution to his problem and venous Doppler was negative for DVT. Subsequently a diagnosis of cellulitis was made. CBC demonstrated multiple cytopenias, including thrombocytopenia (Tables 1 and 2). A review of hospital records noted previous findings of thrombocytopenia as far as four years earlier. Considering pancytopenia an oncology consult was called who reviewed the peripheral smear which showed slight anisocytosis, monocytosis with marked thrombocytopenia with some giant platelets, No pseudo-Pelger-Huet cells or circulating blasts were noted (Figure 1A). Given the laboratory findings, leukemia was considered as a differential and therefore a bone marrow biopsy was performed which showed the differential (Table 3) granulopoiesis, hypercellular marrow with increased myelomonocytic cells and megakaryocytogenesis (Figure 1B,C). Immunohistochemistry showed CD117: about 5-10% positive cells, consistent with immature cells/blasts, CD34: No increase in CD34 positive cells, CD163: Highlights predominantly monocytic component, CD123: Negative. Reticulin staining showed mild diffuse reticulin fibrosis. Immunophenotyping by flow cytometry analysis FLG13-859 shows mild myelomonocytic abnormalities. PCR study for JAK2 is negative. FISH was performed for BCR/ABL1 to detect the (9:22) translocation associated with CML and less commonly ALL or AML were normal. Cytogenic testing demonstrated an abnormal karyotype 46, XY, del(20)(q11.2q13.3)(20) (Figure 2). Other 15 metaphase spreads examined showed a normal male karyotype, 46, XY. Patient was diagnosed with myelodysplastic syndrome, Unclassified (MDS-U) with refractory thrombocytopenia.
Table 1.
Complete blood count.
| Patient’s values | Normal range | |
|---|---|---|
| White blood cells | 7.3 | 3.6-11.0 K/uL |
| Red blood cells | 5.17 | 3.80-5.60 M/uL |
| Hemoglobin | 12.2 | 11.6-16.8 g/dL |
| Hematocrit | 37.8 | 35.1-50.0% |
| Mean corpuscular volume | 73.2 | 73.5-96.5 fL |
| Mean corpuscular hemoglobin | 23.7 | 23.9-33.6 pg |
| Mean corpuscular hemoglobin concentration | 32.3 | 32-35 g/dL |
| Red cell distribution width | 20.0 | 12.1-16.5% |
| Platelets | 37.0 | 150-375 K/uL |
Table 2.
Peripheral blood differential count.
| Patient’s value | Normal range, % | |
|---|---|---|
| Neutrophils | 29 | 43-76 |
| Bands | 1 | 0-10 |
| Lymphocytes | 20 | 17-45 |
| Atypical lymphocytes | 7 | 0-6 |
| Monocytes | 18 | 5-12 |
| Eosinophils | 0 | 0-8 |
Figure 1.

A) Peripheral smear showing slight anisocytosis, monocytosis with marked thrombocytopenia with some giant platelets; B, C) Bone marrow biopsy showing granulopoiesis, hypercellular marrow with increased myelomonocytic cells and megakaryocytogenesis at lower (B) and higher (C) magnification.
Table 3.
Bone marrow differential count.
| Patient’s value, % | |
|---|---|
| Erythroblasts | 22 |
| Blasts | 5 |
| Neutrophils/precursors | 16 |
| Eosinophils | 0 |
| Basophils | 1 |
| Lymphocytes | 4 |
| Monocytes | 50 |
| Plasma Cells | 2 |
| Promonocytes | 1 |
Figure 2.
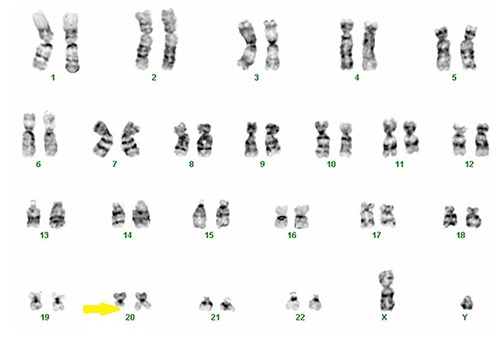
Karyotype 46, XY, del(20)(q11.2q13.3)(20).
Discussion and Conclusions
MDS is a hematologic disorder that typically presents with one or more cytopenias. Anemia is ordinarily part of a bi- or pancytopenia, with independent neutropenia and thrombocytopenia cell lines being unusual. Refractory thrombocytopenia is a rare, but potentially lethal complication that increases complexity in treatment for the physician and increased morbidity for the patient. Genetic abnormalities are playing an expanded role in classifying prognosis and treatment, as well as the potential for MDS to transform into CML. This is especially true where there is a high proportion of cells in the G1 stage of the cell cycle.5 One potential cytogenic marker for clonal evolution is del(20), which was present in our patient. The del(20) is commonly found in patients with CML after tyrosine-kinase inhibitor therapy with and without remission.6 This marker does not necessarily indicate a worse case scenario of CML clonal divergence or disease progression and only 1.7% of people with MDS exhibit this alteration. From a prognostic standpoint, this clonal abnormality is considered good with a median survival of 4.8 years and a progression to AML median time at 9.4 years.7
Our patient had a del(20) with break points at 11.2 and 13.3. The 11.2 break point is the locus for the ASXL1 gene mutation. This particular mutation is found in approximately 21% of MDS cases and has been shown to be a non-favorable prognostic mutation. The ASXL1 (Additional Sex Combs Like 1) is implicated in chromatin remodeling and gene repression. It is believed to be responsible for repression of multiple transcription genes. The 13.3 break point correlates to the GNAS locus and is found in less than 1% of MDS diagnoses.8 This GNAS complex (Guanine Nucleotide Regulatory Protein) activated G-proteins, modulates various hormonal controls, and even stimulates the RAS pathway. Its role in MDS is less understood and hasn’t been as well studied in hematologic cancers as much as in solid tumors.9
Treatment options continue to be reasonably general in patients with MDS. Cytotoxic agents, antibodies or immunosuppression treatments, allogeneic stem cell transplantation, and blood products to correct hypocellularity have been the mainstay of most regimens. Multiple therapies are introduced as a result of the theory that different cell lines are arrested and/or deranged in various stages of the cell cycle.5 Because of the preleukemic and potential myeloblastic nature of MDS, dozens of cytotoxic agents have been utilized to combat cell abnormal cell differentiation, but have had varied success to date. The outcomes of theses treatments are very dependent on which somatic gene mutation(s) are present and whether or not they are responsive to the chemotherapeutic agent used. Allogeneic stem cell transplantation is usually limited to younger high-risk patients and rarely in very healthy older patients. As of date, this is the only curative treatment of higher-risk myelodysplastic syndromes, with prolonged disease-free survival of 35-50%.1 Immune mediated hematopoietic apoptosis caused by cytotoxic T-Cells, IFN-γ producing CD4+ cells, and Th17 cells is reduced by administering immune modulators. This has shown to not only help increase the reduced cell lines, but gives patients a more robust immune response which is crucial to reducing their morbidity and mortality. Specific and standard therapeutic approaches are made to increasing single or multiple cell line deficiencies. The goal in these treatments is to correct the cytopenias(s) involved as with any related reduction of the hematopoietic cell lines.
A reduction in any cell line can lead to potential adverse outcomes in patients. After an extensive review of the current literature, it is evident that thrombocytopenia is one of the most serious problems found in many patients with MDS, yet normally successfully treated. In few cases, due to potentially unique genetic mutations, platelet counts remain low despite evidenced based treatment. Careful identification and monitoring of this patient subdivision can significantly reduce morbidity and mortality. Identification of specific gene mutations and advances in treatment options will hopefully provide forthcoming guidance on detecting at-risk patients in the future.
References
- 1.Ades L, Itzykson PF, Fenaux P. Myelodysplastic syndromes. Lancet 2014;383:2239-52. [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian H, Giles F, List A, et al. The incidence and impact of thrombocytopenia in myelodysplastic syndromes. Cancer 2007;109:1705-14. [DOI] [PubMed] [Google Scholar]
- 3.Visconte V, Selleri C, Maciejewski J, Tiu M. Molecular pathogenesis of myelodysplastic syndromes. Transl Med UniSa 2014;8:19-30. [PMC free article] [PubMed] [Google Scholar]
- 4.Boruchov A. Thrombocytopenia in myelodysplastic syndromes and myelofibrosis. Sem Hematol 2009;46:S37-S43 [DOI] [PubMed] [Google Scholar]
- 5.Jacobs A. Myelodysplastic syndromes: pathogenesis, functional abnormalities, and clinical implications. J Clin Pathol 1985;38:1201-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serio B, Risitano AM, Giudince V, et al. Immunological derangement in hypocellular myelodysplastic syndromes. Transl Med UniSa 2014;8:31-42. [PMC free article] [PubMed] [Google Scholar]
- 7.Nybakken G, Bagg A. The genetic basis and expanding role of molecular analysis in the diagnosis, prognosis, and therapeutic design of myelodysplastic syndromes. J Mol Diagn 2014;16:145-58. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Manero G. Myelodysplastic syndromes: 2012 update on diagnosis, risk-stratification and management. Am J Hematol 2012;87:692-701. [DOI] [PubMed] [Google Scholar]
- 9.Bejar R, Stephenson K, Abdel-Wahah O, et al. Clinical effect of point mutations in myelodysplastic syndromes. New Engl J Med 2011;364:2496-506. [DOI] [PMC free article] [PubMed] [Google Scholar]