

Abstract
Polycystic ovary syndrome (PCOS) is a heterogeneous and complex disorder that has both adverse reproductive and metabolic implications for affected women. However, there is generally poor understanding of its etiology. Varying expert-based diagnostic criteria utilize some combination of oligo-ovulation, hyperandrogenism, and the presence of polycystic ovaries. Criteria that require hyperandrogenism tend to identify a more severe reproductive and metabolic phenotype. The phenotype can vary by race and ethnicity, is difficult to define in the perimenarchal and perimenopausal period, and is exacerbated by obesity. The pathophysiology involves abnormal gonadotropin secretion from a reduced hypothalamic feedback response to circulating sex steroids, altered ovarian morphology and functional changes, and disordered insulin action in a variety of target tissues. PCOS clusters in families and both female and male relatives can show stigmata of the syndrome, including metabolic abnormalities. Genome-wide association studies have identified a number of candidate regions, although their role in contributing to PCOS is still largely unknown.
-
Definitions and Differential Diagnosis
NIH/Rotterdam/AE-PCOS Society diagnostic criteria
Exclusion of other endocrinopathies
Evaluation of features
PCOS in puberty and reproductive aging
Summary
-
Epidemiology
Obesity
Nutrition/exercise
Ethnicity/race
Endocrine-disrupting chemicals
Cardiovascular disease risk
Cancer risk
Psychosocial issues
Long-term outcome of children born to mothers with polycystic ovary syndrome
Summary
-
Pathophysiology
Introduction
Androgen excess
Effects of hyperandrogenemia on the hypothalamus-pituitary axis
Ovarian dysfunction and follicle development
Insulin sensitivity and secretion
Obesity, fat distribution, and adipose tissue function and morphology
Increased sympathetic nerve activity
Summary
-
Molecular Genetics
Genetic tools
Evidence for a genetic background
Candidate gene studies
Genome-wide association studies
Other approaches
Pharmacogenomic studies
Epigenetic influences
Summary
-
Future Research Directions
Diagnosis and epidemiology
Pathophysiology
Molecular genetics
I. Definitions and Differential Diagnosis
A. NIH/Rotterdam/AE-PCOS Society diagnostic criteria
In 1990, a group of investigators who attended a National Institutes of Health (NIH) sponsored conference defined polycystic ovary syndrome (PCOS) as hyperandrogenism and/or hyperandrogenemia (HA) with oligo-anovulation, excluding other endocrinopathies (on the basis of a consensus questionnaire) (1). In 2003, however, the Rotterdam consensus (based on closed session consensus among primarily European and American investigators) expanded the diagnostic criteria to include at least two of the following features: 1) clinical or biochemical hyperandrogenism; 2) oligo-anovulation; and 3) polycystic ovaries (PCO), excluding the same endocrinopathies (2). An Expert Panel from the 2012 NIH Evidence-based Methodology Workshop on PCOS recommended that clinicians use the more recent Rotterdam criteria for diagnosis (3). Consequently, the 6–10% prevalence of PCOS (as defined by 1990 NIH criteria) has doubled under the broader Rotterdam or Androgen Excess-PCOS Society criteria (4), with 1990 NIH-defined PCOS being the most common phenotype (4–6). The increased prevalence of PCOS with the Rotterdam criteria is due to the expansion of the syndrome to include women without documented ovulatory dysfunction or hyperandrogenism, but who have PCO (4).
Women with 1990 NIH-defined PCOS (with hyperandrogenism and oligo-ovulation) are at increased risk of developing reproductive and metabolic abnormalities (Table 1), including infertility and type 2 diabetes mellitus (T2DM), respectively. Although insulin resistance and obesity are commonly found in women with PCOS, they are not part of the diagnostic criteria. Ovulatory women with PCOS have a lower body mass index (BMI) and lesser degrees of hyperinsulinemia and hyperandrogenism than women with 1990 NIH-defined PCOS (7, 8). Women with PCO and oligo-anovulation (without androgen excess) are least affected and do not fulfill the diagnosis of PCOS by the Androgen Excess-PCOS Society (again based on expert consensus within the society), which, like the 1990 NIH criteria, emphasizes hyperandrogenism (7, 9).
Table 1.
Current Interpretation of the Relative Effects of Dyads of PCOS Stigmata on Common Abnormalities
| Hyperandrogenism and Anovulation | Hyperandrogenism and PCO | Anovulation and PCO | |
|---|---|---|---|
| Hirsutism | ++ | ++ | + |
| Infertility | ++ | + | ++ |
| Obesity | ++ | ++ | + |
| Glucose intolerance | ++ | ++ | + |
| Dyslipidemia | ++ | ++ | + |
| Mood disorders | ++ | ++ | + |
B. Exclusion of other endocrinopathies
To properly diagnose PCOS, clinicians need to exclude other endocrinopathies that mimic PCOS (1). These disorders include nonclassic adrenal hyperplasia, Cushing's syndrome, androgen-producing tumors, and drug-induced androgen excess. In addition, clinicians should rule out ovulatory dysfunction from other causes, including thyroid dysfunction and hyperprolactinemia, as well as pregnancy in reproductive-aged women.
C. Evaluation of features
1. Androgen excess
a. Measurement of hirsutism.
Hirsutism is excessive terminal (coarse) hair that appears in a male (midline) pattern (10) and should be distinguished from hypertrichosis, which is a diffuse increase in hair follicles. It is exacerbated by elevated circulating androgen levels and the conversion of T to DHT by 5α-reductase in the pilosebaceous unit (11). This, in turn, leads to the transformation of thin less visible vellus hair to terminal hair through androgen receptor activation in the pilosebaceous unit. Other growth factors and cytokines also contribute to pilosebaceous unit differentiation (12). Racial factors affecting the initial density of pilosebaceous units and their responsiveness to androgens and other hormones may result in differences in hair distribution and density in women with PCOS. For example, women of European ancestry with PCOS tend to have more marked midline hirsutism than women of East Asian ancestry (13) but a similar amount as African Americans (14). Consequently, scales of hirsutism assessment, such as the modified Ferriman-Gallwey scale (15), will have varying cutoffs depending on race (16).
b. Measurement of biochemical hyperandrogenism.
Circulating androgen levels can also help to identify those hirsute women with PCOS, particularly when hirsutism is severe, sudden in onset, and rapidly progressive, or when it is associated with menstrual dysfunction, obesity, acanthosis nigricans, or clitoromegaly (11). The accurate measurement of T requires sensitive methods that include extracting interfering phospholipids by liquid chromatography and tandem mass spectrometry, immunochemiluminescence, or RIA (17–20). High-quality T and SHBG assays (performed ideally in the follicular phase with reference values derived by comparison with normal levels in women with regular menses) are often used to determine total T and calculated free T (11, 17, 19, 21). Serum dehydroepiandrosterone sulfate (DHEAS) levels may be increased in women with androgen excess who have normal T levels (22) and are used as a marker of adrenal hyperandrogenism, despite the fact the DHEAS is a precursor of a very weak androgen (dehydroepiandrosterone [DHEA]). There are no clear cutoffs for abnormal levels, and levels decline with age. The clinical meaning of isolated elevation in DHEAS is uncertain (23). DHEAS levels are associated with insulin resistance in women with PCOS (24) because they are lowered with insulin-sensitizing treatment (25). Levels are also elevated in brothers of women with PCOS, suggesting familial clustering (26). Circulating total and free T and serum DHEAS levels are elevated in 75% of women with 1990 NIH-defined PCOS. This is because free T, the single most predictive assay, is increased in 60% of women with 1990 NIH-defined PCOS (27). There has been some evidence that androstenedione is also a sensitive and specific marker of hyperandrogenism in women with PCOS (28); however, convention and the greater availability of T assays has led to wider utilization of T in the diagnosis of PCOS.
2. Polycystic ovaries
a. Ultrasound methods: morphology and size.
Clinicians, largely on the basis of transabdominal ultrasound, originally defined polycystic ovarian morphology by the presence of 10 or more follicles measuring 2–8 mm in diameter, arranged peripherally around a dense core of stroma or scattered throughout an increased amount of stroma (29). In 2004, Rotterdam PCOS criteria updated the definition of PCO as the presence of 12 or more follicles in each ovary measuring 2–9 mm in diameter and/or an increased ovarian volume (>10 mL) in at least one ovary (2, 30, 31). Using these criteria for PCO, the number of 2- to 5-mm follicles positively correlates with serum androgen levels, whereas the number of 6- to 9-mm follicles negatively correlates with fasting serum insulin and T levels, as well as BMI (30). As a result of improved ovarian imaging, many believe this follicle number threshold value should be increased (32, 33), and an expert panel recommended using a cutoff of ≥ 25 follicles per ovary to diagnose a polycystic ovary (34). Some authors have argued, using principle component analysis, that follicle number per ovary correlates well with HA and thus is a good surrogate marker for ovarian hyperandrogenism (35). Multifollicular ovaries can be a normal stage of development in adolescence and early adulthood (36). Therefore, enlarged ovarian size (>10 mL) may be a simpler indicator of adult PCOS than follicular number in adolescent girls who have hyperandrogenism and oligomenorrhea for at least 2 years postmenarche (37). The prevalence of polycystic ovary morphology is three to four times that of PCOS in epidemiological studies because polycystic ovary morphology is commonly found in normal women (36, 38). Although some women with polycystic ovary morphology may display a relative HA when challenged with GnRH agonist (39), isolated polycystic ovary morphology does not indicate an endocrinopathy. Medication may affect ovarian morphology and size, most clearly by hormonal contraception, which can lower ovarian volume over time (39). However, it does not appear to result in complete resolution of polycystic ovary morphology in women with PCOS (40, 41).
b. Other imaging/screening modalities.
Magnetic resonance imaging (MRI) has the potential for greater resolution than current ultrasound technology because it is able to image antral follicles of 1 mm in diameter. Thus, MRI of women with PCO note higher antral follicle counts (42) and may result in more overlap with the morphology of ovaries of normal women (43). MRI is currently used only on a research basis.
Serum anti-Müllerian hormone (AMH), originating mostly from granulosa cells of large preantral/small antral follicles (44, 45), has emerged as a possible surrogate marker of PCO (31, 33). Elevated serum AMH levels in women with PCOS are related to both increased follicle numbers and granulosa cell hypersecretion (46–49). They also are positively correlated with LH and T levels and negatively predicted by BMI (50, 51). Although differences in circulating AMH levels by PCOS phenotype reflect the heterogeneity of the syndrome, threshold values to discriminate the various PCOS phenotypes remain unclear (33, 50–52).
3. Determination of chronic anovulation
All major classifications of PCOS include ovulatory dysfunction as a component, and it represents a major clinical concern for most patients (53). Of note, there is incomplete understanding of the process of normal ovulation, it varies over lifetime, and it is often difficult to measure objectively (3, 54). According to the Androgen Excess/PCOS Society Task Force Report, as many as 85% of women with PCOS have clinical evidence of menstrual irregularities (55).
Clinicians diagnose oligomenorrhea when menstrual cycles last longer than 35 days or occur less than eight times a year, although women with regular menstrual cycles may nonetheless have chronic anovulation (53, 55). Chang et al (56) reported that 16% of 316 women with PCOS (diagnosed using the NIH 1990 criteria) had normal-appearing cycles, despite having oligo-anovulation. When compared to anovulation, oligo-anovulation generally has a less severe phenotype (57). To confirm anovulation, clinicians may obtain a serum progesterone level during the suspected midluteal phase of the cycle and presume that the cycle is oligo-anovulatory if the level is lower than 3–4 ng/mL (58).
D. PCOS in puberty and reproductive aging
1. Prepubertal
Early pubarche or adrenarche has been linked to the later development of PCOS, although mechanisms are not well defined (59). However, animal models and studies in prepubertal children suggest that early exposure to androgens (particularly from an adrenal source) may be a risk factor for developing PCOS (60–62). Therefore, prospective follow-up in young children with early pubarche is warranted because these girls (particularly if obese) may have an increased risk of developing metabolic syndrome (MetS) (using the less stringent adolescent cutoffs) (63) and PCOS-like symptoms (64). The MetS is accepted as a cardiovascular risk factor and consists of elevated waist circumference, systolic and/or diastolic blood pressure, fasting blood glucose, and fasting serum triglycerides and decreased serum high-density lipoprotein cholesterol levels.
2. Adolescence
The median age for onset of menarche in the United States is 12.4 years, with a cycle interval typically of 21–45 days in the first gynecological year (mean, 32.2 d). The flow length is usually about 7 days. Longer cycles most often occur in the first 3 years after menarche, and the overall trend is toward shorter, more regular cycles with increasing age. Persistent infrequent menstrual cycles merit further investigation (65). By the third year after menarche, 60–80% of menstrual cycles are 21–34 days long, as is typical of adults (65). The age for onset of menarche is related to the time of regular ovulatory cycles. Girls who are less than 12 years of age at menarche have 50% ovulatory cycles by 1 year after menarche, whereas girls with onset of menarche at 12–13 years of age or greater than 13 years need 3 or 4–5 years, respectively, to establish 50% ovulatory cycles (66). van Hooff et al (67) assessed the risk of developing oligomenorrhea in young women with irregular menstrual cycles, comparing those with an average cycle length of 22–34 days to those with an average cycle length of 35–41 days. At 4 years after menarche, only 10% of adolescents with shorter average cycles had oligomenorrhea, whereas more than 50% of those with longer average cycles remained oligomenorrheic (67). Conversely, van Hooff has shown that after 3 years of follow-up, 12% of women with regular cycles at a mean age of 15 years develop irregular cycles at a mean age of 18, whereas 48% of adolescents with irregular cycles at mean age of 15 achieve regular cycling by age 18. So the predictive value of initial menstrual cyclicity history may not fully determine who is at risk for developing PCOS. A study by Carmina et al (37) suggests that by the sixth to 10th menstrual year, 70–80% of young women have regular ovulatory cycles.
All agree that many asymptomatic adolescent girls (range, 26–54%) have PCO by ovarian sonographic appearance (68–71), suggesting that PCO-like ovaries in adolescents 1.3–3.8 years after menarche may be normal. In a 3-year study of Chilean girls with PCO-like ovaries, PCO morphology was common and did not correlate over visits (70). The inability of clinicians to perform a transvaginal sonogram in many adolescents further complicates the accurate diagnosis of PCOS at this age (67, 68, 70, 72, 73). Although the diagnostic criteria are uncertain in adolescent girls, the diagnosis may lead to greater recognition of metabolic risk factors for diabetes and cardiovascular disease (CVD), with earlier intervention preventing these sequelae (74, 75).
3. Menopause
Studies have shown that women with PCOS can develop regular menstrual cycles with age (76–79). Studies suggest that with aging there are smaller ovarian volumes, smaller follicle counts, greater FSH levels, and lower inhibin B and AMH levels (36, 80, 81). Circulating androgen levels also decline with age, although normative data for the perimenopause remain unclear (55). Currently, there are no diagnostic criteria for PCOS in perimenopausal or menopausal woman; however, clinicians often make a reasonable diagnosis based upon a history of prior oligomenorrhea and hyperandrogenism (9, 82). Several studies that used this modified PCOS diagnostic criteria have suggested increased cardiovascular risk in women with PCOS (83–85), whereas a prospective study of perimenopausal women with PCOS (based on the 1990 NIH definition of hyperandrogenic chronic anovulation) did not see a progressive increase in cardiovascular risk or events with aging (86).
E. Summary
In summary, the diagnostic criteria for PCOS are based on expert consensus, not evidence. The consensus opinion has generally agreed that the ovary is central to the disorder and that it is necessary to exclude other endocrinological disorders before making the diagnosis. Improved and standardized androgen assays, novel methods for documenting chronic anovulation that go beyond menstrual history, and imaging technology may refine the diagnostic criteria. The diagnosis of PCOS in perimenarchal girls remains problematic due to the overlap of stigmata of PCOS with normal pubertal maturation.
II. Epidemiology
A. Obesity
Many women with PCOS are overweight or obese (87–90). Obesity in the United States affects approximately 80% of women with PCOS (91, 92), whereas outside the United States it affects only 50% of women with PCOS (90, 93–96). This increased association in the United States appears to be similar to the increased prevalence of obesity in the country overall.
Obesity does appear to exacerbate many aspects of the PCOS phenotype, particularly those risk factors related to MetS (97). Studies have suggested that women with PCOS are at risk for nonalcoholic fatty liver disease (98). However, it is uncertain whether PCOS is associated with body composition changes, which (independent and/or additive to obesity) may further exacerbate reproductive and metabolic aspects of the PCOS phenotype (99). Some studies suggest that in women with PCOS, body weight alone or BMI may not be as important with respect to metabolic impact as fat distribution, especially the adverse effects of centripetal obesity (100, 101). A small study by Kirchengast and Huber (102) showed a significantly greater amount of body fat and lower amount of lean body mass in women with PCOS compared to controls matched for age, weight, and BMI. However, an older study by Good et al (103) did not demonstrate differences in fat distribution between women with PCOS and lean controls. Similarly, a study involving MRI and computed tomography scans of visceral adipose tissue in women with PCOS and BMI-matched controls found little evidence that fat distribution increased risk for PCOS (even in women with increased waist:hip ratios) (104, 105). It also appears that if a patient had oligomenorrhea and hyperandrogenism in adolescence, there is an increased risk of developing obesity (BMI > 40 kg/m2) and MetS by age 24, suggesting a temporal association of PCOS with obesity even if a primary predisposition does not exist (106). Nonetheless, evidence suggests that subcutaneous adipocyte size is increased in obese women with PCOS, along with functional abnormalities in their adipose tissue, including a decrease in the lipolytic effects of catecholamines and lower circulating levels of adiponectin (104, 107–109).
It is currently debated whether obesity per se can cause PCOS. Obesity is associated with suppressed levels of SHBG leading to higher free androgen levels and prolonged follicular phases (without anovulation) leading to a longer menstrual cycle (110), which could be confused with a PCOS diagnosis. Massive weight loss in obese women with PCOS, such as results from bariatric surgery, has been shown to improve multiple reproductive and metabolic abnormalities in the syndrome (111), although there is not consistent evidence that all aspects of the syndrome resolve (112).
B. Nutrition/exercise
Weight loss is likely an important element to reducing the severity of PCOS phenotypic expression. Dietary modification is an important part of any weight-loss program, with most studies suggesting that exercise alone is inadequate to improve symptoms relating to PCOS phenotype. Some studies have suggested using a form of the Diabetes Prevention Program exercise schedule, whereby participants try to lose one pound a week (113, 114). A clinical guideline, developed in Australia for the diagnosis and treatment of women with PCOS, recommended lifestyle changes. These included changes in diet, exercise, and/or behaviors that benefit general health, including weight loss and weight gain prevention (115). However, there exists only limited data supporting the efficacy of lifestyle change in regard to PCOS (116, 117). However, there is general consensus that weight and glycemic parameters are improved. Furthermore, there is no clear evidence that there are any additional defects related to energy expenditure beyond obesity that would predispose women with PCOS to weight gain or compromise their ability to lose weight (118, 119). It is commonly thought that lowering insulin levels will result in weight loss in women with PCOS. However, although pharmacological studies of metformin use in women with PCOS showed insulin-lowering effects, there was no consistent pattern of weight loss (120). Similarly, women with PCOS taking troglitazone (a thiazolidinedione) experienced a dose-response decrease in fasting and glucose stimulated serum insulin levels and a dose-response increase in weight (121).
C. Ethnicity/race
PCOS is a common endocrinopathy in many racial and ethnic groups. Two gene loci (first identified in a genome-wide association of Han Chinese women) were replicated in women of European ancestry; this commonality suggests an ancient evolutionary trait (122–124). An examination of the racial variability of these genetic variants among publically available genomic databases provides evidence that PCOS ethnic variations are strongly determined by the genetic background in humans (125, 126).
Ethnic variations in PCOS phenotypic expression occur in North and South American women, including Canadians, Latinas, African Americans, Caribbean Hispanics, Icelanders, Europeans, South East Asians, Chinese, New Zealanders, and women from the Middle East (13, 127–133). Women of African descent with PCOS are more likely to have hypertension and CVD risk factors, whereas Hispanic women are more at risk for MetS and T2DM (134, 135). Despite these phenotypic differences, there may not be significant differences in these reproductive or metabolic features in younger populations (128).
D. Endocrine-disrupting chemicals
Environmental agents can act as endocrine disrupters. Studies have associated common household objects (known as plasticizers) with obesity, alterations in puberty, and ovulatory dysfunction (136). Specifically, elevated levels of bisphenol A (BPA), an estrogen-mimicking compound, may contribute to the pathogenesis of PCOS. Researchers have reported elevated levels of BPA in women with ovulatory dysfunction (137). Bisphenol S exposure in neonatal rats appears to be linked to both PCOS-like syndrome and abnormal glucose metabolism. Rat ovarian theca-interstitial cells cultured with BPA had elevated T synthesis, possibly through the increased mRNA expression of enzymes involved in the steroid production pathways (138–141). A study of lean and overweight women with PCOS reported that both groups had higher levels of BPA as compared to lean and overweight controls. The study also positively correlated BPA with proxies of insulin resistance (142). These limited data, however, have not established any clear cause-and-effect relationship with stigmata of PCOS.
E. Cardiovascular disease risk
Differences exist in several CVD risk factors between women with 1990 NIH-defined PCOS and normal women, and these differences are more profound in obese individuals (143–145). Impaired glucose tolerance or T2DM from insulin resistance develops in about 40% of women with 1990 NIH-defined PCOS by the fourth decade of life, with glycemic control worsening with age and weight gain (91, 92, 146–148). Women with PCOS also have dyslipidemia, including low levels of high-density lipoprotein-cholesterol; increased values of triglycerides and total and low-density lipoprotein-cholesterol; and altered low-density lipoprotein quality (149, 150). Because 1990 NIH-defined PCOS is characterized by a preferential increase in abdominal adiposity with weight gain, MetS is also highly prevalent in PCOS women compared to BMI-matched controls; conversely, MetS is lower in prevalence in individuals with less abdominal adiposity (134, 151, 152), as is the case in countries where obesity is less common (93, 153). Nevertheless, genetic, environmental, and hormonal factors coexist to regulate lipid metabolism in women with PCOS, which is not fully explained by body weight alone (149, 154). For example, nonobese women with PCOS exhibit elevated levels of lipoprotein-a (155). Lipoprotein-a is a stable, genetically and racially determined, lipid-rich, low-density lipoprotein that is distinct from lipoprotein-c. It can coexist with increased small low-density lipoprotein particles and an otherwise normal lipid profile (156). A study reported that cholesterol efflux capacity from macrophages, a measure of high-density lipoprotein-cholesterol function and an independent predictor of subclinical CVD, is abnormally suppressed in women with PCOS (157).
There has been a plethora of studies linking PCOS to newer surrogate markers of CVD including increased left ventricular mass (158), endothelial dysfunction (159, 160), and arterial stiffness (161). Adjusting for age and BMI, women with PCOS also may be more susceptible to subclinical vascular disease than normal women, including increased carotid-intima media thickness (162, 163) and coronary artery calcification (164, 165), although studies have not always noted this (166).
Nevertheless, there have been limited data suggesting that women with PCOS experience increased CVD event rates. However, this is likely due to the relatively late onset of CVD events in women in the seventh to eighth decades of life and the paucity of studies that have included women with PCOS in this age range. The current evidence for increased CVD morbidity and mortality comes from studies that are cross-sectional with small numbers (167, 168), that are prospective with larger numbers but based primarily on ovarian morphology (169, 170), or that used cohorts where the diagnosis of PCOS was uncertain and extrapolated from existing data (83). Such studies of postmenopausal women have suggested a link between CVD and features of PCOS (84, 171). One of the most cited articles linking CVD events to a history of PCOS in postmenopausal women (85) has since been withdrawn by the authors due to their inability to replicate the original results (172). There are data from a meta-analysis that suggest a higher rate of nonfatal stroke (but not nonfatal coronary heart disease) in women with PCOS (Figure 1) (173). There are also data to suggest that the increased CVD risk noted in younger women with PCOS may plateau in women with PCOS as they age, and that women without PCOS may develop a more severe risk factor profile with age (78, 86, 174). Thus, the relationship between PCOS and cardiovascular events remains murky.
Figure 1.

Meta-analysis of stroke and coronary heart disease (CHD) in women with PCOS. This figure includes a forest plot comparing the risk of nonfatal stroke in women with PCOS compared to controls in the older age group (mean > 45 y) (top) and a forest plot comparing risk of nonfatal CHD in women with PCOS compared to controls in the older age group (bottom) (mean > 45 y). CI, confidence interval; M-H, Mantel-Haenszel. [Adapted from S. A. Anderson et al: Risk of coronary heart disease and risk of stroke in women with polycystic ovary syndrome: a systematic review and meta-analysis. Int J Cardiol. 2014;176:486–487 (173), with permission. © Elsevier.]
F. Cancer risk
Women with PCOS have multiple risk factors for endometrial cancer that include obesity, metabolic abnormalities (such as diabetes and hypertension), and a history of oligomenorrhea with prolonged exposure to unopposed estrogen. Studies have noted a 2.7-fold increased risk for developing endometrial cancer vs the general population (175, 176). This increased endometrial cancer risk in PCOS likely applies to a subgroup of PCOS women with obesity, because the risk is reduced but not eliminated when adjusted for BMI (177, 178). The increased risk for this malignancy in PCOS is largely from prolonged endometrial exposure to unopposed estrogen due to chronic anovulation (179), although secretory endometrium in women with PCOS shows progesterone resistance with dysregulated gene expression controlling steroid action and cell proliferation (180, 181).
Studies regarding PCOS and ovarian cancer risk are contradictory. In a long-term study of women diagnosed with PCOS through hospital records, mortality from ovarian cancer was not increased over the general population (170). Conversely, a case-control study of women with histologically confirmed epithelial ovarian cancer showed an increased, age-adjusted, 2.5-fold risk of developing ovarian cancer in women with self-reported PCOS (182). There is no apparent association of PCOS with breast cancer, and insufficient data exist to evaluate the relationships between PCOS and uterine leiomyosarcoma or vaginal, vulvar, or cervical cancers (6, 175).
G. Psychosocial issues
The prevalence of depression and anxiety is higher in women with PCOS than in the general population (183). Such mood disorders, capable of impairing quality of life, can be prominent in adolescents faced with issues of self-presentation, in young adult women concerned with fertility, and in women of all ages with respect to eating, overweight, and clinical manifestations of androgen excess (184, 185). A study associated PCOS with bipolar disorder (186), although the association may be with both the disorder and specific treatments of the disorder (187). Specifically, studies have associated valproate with weight gain and the development of oligomenorrhea, relative HA (188), and PCO (when used to treat epilepsy) (189). Administering valproate to normal human thecal cells in vitro has increased thecal androgen production similar to what is seen in polycystic ovary thecal cells (190).
H. Long-term outcome of children born to mothers with polycystic ovary syndrome
The long-term reproductive and metabolic risks for offspring of women with PCOS are an area of ongoing interest, but current data are limited. Offspring, like other first-degree relatives, are at increased risk for developing PCOS (191, 192) and can display varying degrees of reproductive and metabolic abnormalities (193, 194). However, not all offspring are affected, and there may be variable onset of symptoms that are puberty dependent (39). Currently there is no screening or genetic test to predict girls who will develop PCOS.
I. Summary
In summary, multiple factors are associated with phenotypic heterogeneity, including obesity, race, and ethnicity. The role of other factors such as endocrine disruptors and fetal origins remains uncertain. The best diet to prevent and control symptoms of PCOS lacks definitive scientific validation. Relevant long-term outcomes that require further epidemiological investigation include premature cardiovascular events, ovarian and breast cancers, and major psychiatric morbidity. Finally, the developmental issues and long-term health outcomes of children of women with PCOS need further study.
III. Pathophysiology
A. Introduction
The pathophysiology of PCOS is complex and reflects the interactions between genetic, metabolic, fetal, and environmental factors. The relative importance of these factors may vary in individual affected women (Figure 2). Among these factors, disordered gonadotropin secretion, HA, insulin resistance and hyperinsulinemia, ovarian dysfunction, and follicular arrest are prominent. The potential role of these factors and their actions are summarized in this section.
Figure 2.

Pathophysiology of PCOS—a vicious circle. Several theories have been proposed to explain the pathogenesis of PCOS. One of these is that neuroendocrine defects lead to increased pulse frequency and amplitude of LH and relatively low FSH. This causes intrinsic defects in ovarian androgen production. Also, there may be an alteration in cortisol metabolism and excessive adrenal androgen production. Insulin resistance with compensatory hyperinsulinemia further increases ovarian androgen production both directly and indirectly via the inhibition of hepatic SHBG production. Obesity, insulin resistance, and high circulating androgens are associated with increased sympathetic nerve activity. E, estradiol.
In normal women, the adrenal glands and the ovaries secrete androgens in response to ACTH and LH, respectively (195). Approximately half of the androgen production stems from direct secretion and half from enzymes peripherally converting 17-ketosteroids into androstenedione (predominantly) in skin, liver, and adipose tissue (196). The hypothalamic-pituitary axis does not directly regulate androgen production in the adrenal glands, and intraglandular autocrine and paracrine factors also influence androgen production throughout the body (197, 198). In women with PCOS, the ovary is the main source of androgen, but the adrenal contributes in some 30–50% of individuals who show enhanced 17-ketosteroid responses to ACTH (199, 200).
A consistent feature of PCOS is disordered gonadotropin secretion with elevated mean LH, low or low normal FSH, and a persistently rapid frequency of GnRH pulse secretion (201, 202). Below, we explore the role of HA in disrupting the normal steroidal regulation of GnRH and gonadotropin secretion.
B. Androgen excess
1. In utero
Experimentally, excess fetal T induces PCOS-like reproductive and metabolic traits in female mammals, from rodents to primates; however, not all features are reproduced in all models. Rodent models are convenient and inexpensive and show various features of PCOS after intrauterine exposure to T, DHT, or DHEA. These features include irregular or lengthened estrus cycles, increased LH pulsatility, HA, and varied metabolic changes (203). Rodent models with green fluorescent protein-labeled GnRH neurons show impaired progesterone inhibition of GnRH pulse firing in the presence of DHT and increased excitatory inputs to the GnRH neuron (204). A prenatal androgenization mouse study provided further data identifying markedly decreased progesterone receptor expression in the arcuate nucleus of the hypothalamus combined with increased GABAergic innervation of GnRH neurons, suggesting this as a neuroendocrine origin for PCOS (205). Prenatal T exposure alters placental steroidogenesis in rat models (as suggested by the increased protein expression of estrogen receptors α and β and androgen receptor and 17β-hydroxysteroid dehydrogenase-2) and results in the dysregulation of lipid metabolism in adult female offspring (206).
Sheep models of PCOS have provided useful information for a better understanding of hormonal regulation. Sheep exposed to prenatal T had increased LH pulsatility and impaired estrogen/progesterone feedback mechanisms, which resulted in altered ovulatory and follicular dynamics and the progressive loss of estrus cycles (207). Sheep models of PCOS also manifest insulin resistance and PCOS ovarian phenotype.
The most comprehensive and best PCOS mimic is found in monkeys, given their similar hypothalamic-pituitary-ovarian and hypothalamic-pituitary-adrenal axes to humans. In this PCOS-like animal model, excess fetal T (given exogenously prenatally) produces hormonal, reproductive, and metabolic abnormalities. After puberty, the offspring of T-exposed monkey mothers exhibit LH hypersecretion, ovulatory dysfunction, hyperandrogenism, and insulin resistance; in addition, roughly 50% of the offspring have enlarged ovaries with increased follicle counts (208). Some of these changes appear developmentally programmed in utero because second-generation female offspring also manifest elevated LH pulsatility from reduced hypothalamic steroid-negative feedback, exaggerated T responses to chorionic gonadotropin, diminished ovarian reserve, excess adrenal androgen production, and altered abdominal adipose characteristics (208–212). The monkey model has also shown that prenatal androgenization is associated with marked infant changes in pancreatic islet morphology and function (believed to be due to aberrant islet development), which leads to subtle adult differences in islet function, supporting postnatal islet plasticity in adapting to changing physiological demands of aging (208, 213).
Evidence for the in utero effects of excess androgen exposure in humans is less convincing. Earlier work had documented the occurrence of cystic ovaries and PCOS-like symptoms in girls with congenital adrenal virilizing disorders (214). The congenital virilizing adrenal hyperplasias, particularly 21-hydroxylase deficiency, provide the human experiment of nature regarding prenatal androgen exposure. Based on their observation of increased LH secretion in women with congenital adrenal hyperplasia, Barnes et al (214) hypothesized that prenatal androgen exposure altered hypothalamic-pituitary programming, resulting in an increased tendency of LH hypersecretion at puberty, which may contribute to the ovarian hyperandrogenism noted in adolescent and adult women with congenital adrenal hyperplasia.
Maternal T during pregnancy is elevated in women with PCOS (215), but whether this results in increased fetal exposure is unclear, given the markedly increased levels of SHBG and abundant placental aromatase activity during pregnancy (216). The origins of blood (fetal vs maternal) in umbilical vein samples are uncertain, but cord blood may provide a better estimate of androgen values at birth. There have been mixed reports about whether umbilical vein T levels are elevated in female babies born to women with PCOS (217, 218). There is also at least one study that has shown elevated amniotic fluid T levels in women with PCOS with female fetuses (not males) (219). However, amniotic fluid T is higher in boys than girls, and T levels do not alter during gestation in either sex (219, 220). Additionally, a prospective study did not demonstrate a relationship between maternal androgen levels during pregnancy and the subsequent development of PCOS symptoms in adolescence (69).
The effect of maternal androgen levels on placental steroidogenesis has not been well researched. A study demonstrated that higher maternal androgen levels increased 3β-hydroxysteroid dehydrogenase-1 and decreased cytochrome P450 placental protein expression, although the study did not directly measure placental androgen production (221).
Thus, whereas the availability of maternal androgens to the fetus is not well established, it remains possible that excess secretion from placental and fetal steroidogenic tissue may expose the developing hypothalamus to excess androgen before the final programming of steroid feedback and other regulatory mechanisms (206). The intrauterine environment, which leads to fetal growth restriction, may also contribute to an increased prevalence of hyperandrogenism and insulin resistance in girls (222), although the association of these stigmata with growth restriction is not universal (223).
2. Pre-/peripubertal girls
Adrenarche is the gradual increase in female humans (between ages 6–8 y) of adrenal androgens, DHEA, and DHEAS, which stimulate growth and may play a role in the activation of the hypothalamic-pituitary-ovarian axis. Premature adrenarche has been associated with subsequent ovarian HA, PCOS, and insulin resistance (59). The disorder may be idiopathic or may reflect increased insulin and IGF-1, which enhances androgen responses to ACTH. Obesity is associated with hyperinsulinemia, and HA is commonly present. Sixty-five percent of obese prepubertal and peripubertal girls (BMI > 95 percentile of ideal body weight for age) had elevated free T levels (224, 225). During puberty, girls with a history of premature adrenarche showed exaggerated androgen precursor responses to a GnRH agonist (226), which was consistent with increased cytochrome P450C17 α activity. By mid puberty, the rise in LH secretion stimulates ovarian androgen production. In insulin-resistant states, hyperinsulinemia enhances LH action. In 2012, approximately 17% of girls aged 2–19 years were at > 95 percentile ideal body weight (227), and this marked increase in obesity during the past 40 years has likely contributed to increased adrenal and ovarian androgen secretion and increased predisposition to the development of the PCOS. The incidence of premature adrenarche or pubarche among daughters of women with PCOS does not appear to be common, although metabolic and reproductive abnormalities may not be evident until the onset of puberty (39).
There is a surge of interest in defining early metabolic abnormalities in daughters born to women with PCOS. Sir-Petermann et al (193) demonstrated that 30 prepubertal and 69 pubertal daughters of women with PCOS had significantly higher ovarian volume and 2-hour insulin when compared to controls matched for age, Tanner stage, and BMI. In Tanner stages IV and V, basal T and post-GnRH-stimulated LH, T, and 17 hydroxyprogesterone levels were significantly higher in PCOS daughters compared to controls, suggesting that the metabolic abnormalities appear during late puberty (193). This group had earlier demonstrated lower levels of adiponectin in prepubertal daughters and pubertal daughters of women with PCOS compared to controls (228). Of equal interest is a similar study that demonstrated increased basal and stimulated DHEA levels during the onset of puberty and a modest advancement in bone age during both childhood and peripubertal period (ages 9–13 y) (193). These studies all suggest that reproductive abnormalities may exist in daughters of women with PCOS in early stages of sexual development. Sir-Petermann et al (194) showed higher levels of AMH in daughters of women with PCOS at all Tanner stages and suggested that the girls with the highest AMH levels had the most marked metabolic derangements.
3. Adults
In late adolescence and adulthood, the ovary is the main source of excess T production in response to LH and hyperinsulinemia. LH is the primary factor (225), with insulin augmenting ovarian theca cell androgen secretion (229, 230). The reduction of hyperinsulinemia through dieting, metformin, or troglitazone improves ovulatory rates and reduces plasma T by approximately 20% (121, 231, 232). A greater reduction in T is noted when a GnRH agonist desensitizes LH and suppresses its secretion (233). The adrenal glands continue to contribute to HA in adults with PCOS, and both basal and ACTH-stimulated androgen responses remain elevated (compared to controls) until menopause (234).
Thus, data suggest that HA is commonly present before puberty in some patients who later develop PCOS, and it is often associated with coincident obesity. Elevated androgens may also increase the risk of metabolic abnormalities (235), as well as the risk of anovulation and the altered steroidal regulation of LH secretion at the hypothalamus.
C. Effects of hyperandrogenemia on the hypothalamus-pituitary axis
Adult women with PCOS often have upper normal or elevated serum LH levels with elevated ovarian steroids. This is associated with a rapid LH pulse secretion frequency (and by inference, a rapid GnRH pulse secretion frequency) (236), although LH pulses can be suppressed after the infrequent ovulations. In rats, the frequency of GnRH pulse secretion modulates gonadotropin synthesis with rapid frequency pulses favoring α-subunit and LH β-subunit synthesis and slower frequency pulses favoring FSH β-subunit synthesis (237). In women with PCOS, the frequency of LH pulses is persistently at the highest level, approximately one pulse per hour before the midcycle surge. These data suggest impaired negative regulation of the GnRH pulse generator, and studies have shown reduced sensitivity to the inhibitory action of progesterone in the presence of estradiol (238, 239). Blocking the androgen receptor with flutamide reverses the insensitivity to progesterone, implicating T as a causative factor in altered steroid hormone feedback (240).
Hyperandrogenemic adolescent girls have a similar pattern of rapid frequency LH pulse secretion (241), which is present before menarche (242). Normal early pubertal girls are highly sensitive to the inhibitory effects of progesterone on GnRH-LH pulse secretion, and sensitivity declines as puberty progresses, similar to what is seen in adult women at Tanner stage III. By mid to late puberty, some girls with HA have impaired progesterone feedback, similar to adults with PCOS, and approximately half of HA adolescents are insensitive to progesterone inhibition. The mechanisms involved in the development of pubertal progesterone insensitivity remain unclear. The degree of free T elevation and the time since menarche were similar in progesterone-sensitive vs -insensitive girls. The only difference detected in progesterone-insensitive HA adolescents was elevated fasting insulin levels, suggesting that hyperinsulinemia may act on the central nervous system (243).
The apparent acceleration of pubertal maturation in girls with HA appears related to the advancement of day/night GnRH-LH secretory patterns (244). In early puberty (Tanner stages I and II), daytime LH pulses are few and of low amplitude, while sleep-related secretion shows increased frequency and amplitude. Interestingly, the frequency of sleep-related LH secretion remains constant from early through late puberty, approximately one pulse every 2 hours. However, with advancing puberty, daytime GnRH-LH secretion increases. By Tanner III, daytime GnRH-LH secretion equals pulse frequency during sleep, and by Tanner IV to V, daytime GnRH-LH secretion actually exceeds pulse frequency during sleep. Thus, in late puberty day/night patterns of LH secretion are similar to those in adult women where pulse frequency slows during sleep in the follicular phase. In normal puberty, there is usually a gradual extension of GnRH pulse secretion from adolescent sleep-related-only to an adult 24-hour pattern.
This normal pattern is accelerated in obese HA girls. After Tanner stage III, daytime LH pulse frequency exceeds that during sleep, and by late puberty pulse frequencies are higher than in normal girls throughout 24 hours (Figure 3) (243).
Figure 3.
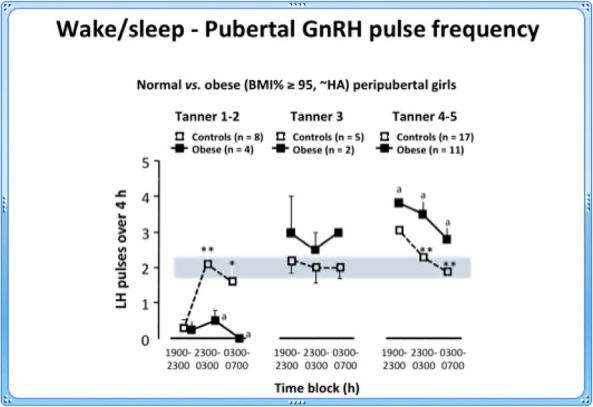
Day/night changes in GnRH pulse frequency in normal (open) and obese hyperandrogenemic (closed) girls through pubertal maturation. The shaded area indicates the range of pulse frequency during sleep and is unchanged throughout puberty. *, P < .05; **, P < .001, obese (a) vs controls. [Adapted from C.R. McCartney et al: Maturation of luteinizing hormone (gonadotropin-releasing hormone) secretion across puberty: evidence for altered regulation in obese peripubertal girls. J Clin Endocrinol Metab. 2009;94:56–66 (224), with permission. © The Endocrine Society.]
The mechanisms controlling normal wake/sleep-related GnRH secretion remain uncertain, but could include steroid hormones acting on the central nervous system. In normal early pubertal girls (Tanner stages I–III), both progesterone and T increase 2-fold overnight (224) and are suppressed by dexamethasone, suggesting an adrenal source in response to the overnight increase in ACTH.
Of interest, Collins et al (245) reported that progesterone exerts differential inhibitory feedback on GnRH, suppressing daytime but not sleep-related secretion. T gradually increases during normal puberty (224, 246) and may impair progesterone's inhibitory effect on daytime secretion. As T rises, reduced progesterone inhibition during the daytime leads to extended daytime secretion of GnRH and LH. The mechanisms involved are unclear, but T might inhibit hypothalamic progesterone receptor expression (247). The importance of progesterone and T in regulating the advancement of normal puberty remains to be proven. However, the LH secretory patterns in obese hyperandrogenemic girls support a role for T in pubertal maturation (248, 249). Although final proof awaits further experimentation in humans, data from nonhuman primates provides strong support. One study exposed prepubertal female monkeys to elevated exogenous T (3-fold) from 1 year of age. Three years later, LH pulse frequency increased 2.7-fold compared to cholesterol-implanted controls (250). These data provide a strong basis for implicating a gradual increase in T as a regulatory factor in normal pubertal maturation. It follows that elevating T may accelerate the normal evolutionary pattern of GnRH pulse secretion.
D. Ovarian dysfunction and follicle development
1. Changes in folliculogenesis and follicle recruitment and oocyte developmental competence
Ovulation results from synchronized signaling by the hypothalamus, pituitary, ovarian theca cells, ovarian granulosa cells, and the developing follicle. Growth of the primordial follicles is gonadotropin-independent. In the preantral stage, LH receptors are expressed, leading to LH-stimulated theca cell androgen secretion, which provides a substrate for granulosa cell estradiol production. Coordination and interaction of LH, FSH, insulin, IGF-1, AMH, steroidogenic enzyme function, and other factors culminates in ovulation. This process goes awry in women with PCOS where abnormal follicular development and apparent failure to select a dominant follicle results in anovulation (251). Studies have suggested an early defect in the folliculogenesis in PCO preceding follicular recruitment (252, 253). The ovulatory dysfunction in PCOS is characterized by increased follicular activation, but the growth of these follicles is arrested before they mature (254). Thus, women with PCOS have an increased proportion of primordial follicles and a corresponding increase in activated growing (primary) follicles (253, 255). The follicles in women with PCOS also have lower rates of atresia, which may explain why the ovaries in these women do not undergo a premature depletion of their follicular pools (256). The arrested follicle development can possibly be explained by the normal but relatively low circulating FSH levels (in reference to LH levels) in women with PCOS (257), levels that are not high enough to stimulate normal maturation processes (251). LH hypersecretion is also detrimental for follicular growth and ovulation in women with PCOS and may cause the premature luteinization of granulosa cells by decreasing FSH sensitivity (258–260).
The most consistent biochemical abnormality in women with PCOS is the hypersecretion of androgens, which reflects an intrinsic dysfunction in the theca cells (261, 262) (Figure 4). Androgens play a critical role in impaired follicular growth by stimulating the initiation of primordial follicles and increasing the number of small antral follicles in the early gonadotropin-independent stage (263, 264). Furthermore, the LH hypersecretion seen in women with PCOS appears to amplify androgen production by the theca cells, whereas FSH levels are decreased, which inhibits follicular growth. This is further supported by the hyperresponsiveness of plasma 17-hydroxyprogesterone to a GnRH agonist challenge (265). Of note, women with PCOS and normal androgen levels may still have hyperandrogenic responses to a GnRH agonist challenge (266, 267). Insulin may also contribute to the inhibition of follicular maturation, especially at later stages when insulin, together with LH, inhibits granulosa cell proliferation and disrupts estrogen and progesterone synthesis (263, 268).
Figure 4.
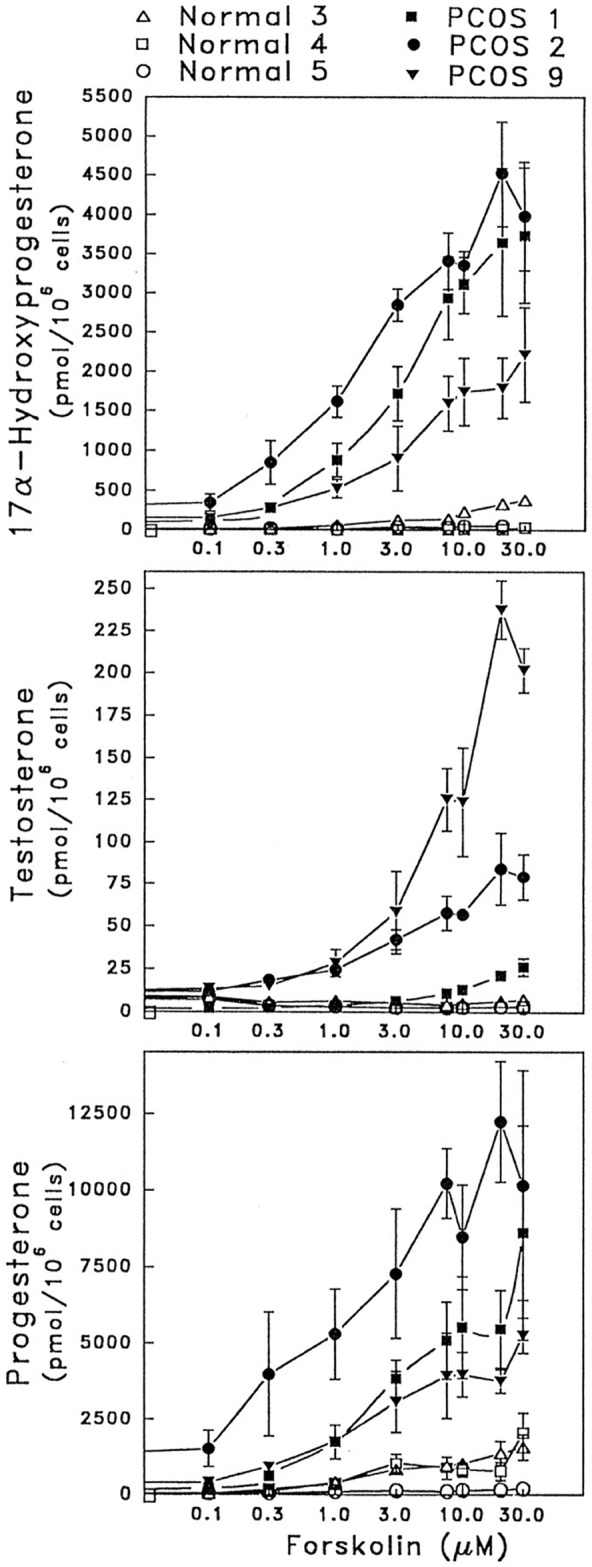
Excessive production of sex steroids by human thecal PCOS cells from women with PCOS in response to forskolin stimulation (mimicking gonadotropin action). [Adapted from V. L. Nelson et al: Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol. 1999;13(6):946–957 (262), with permission. © The Endocrine Society.]
Although androgen excess is detrimental to FSH-stimulated folliculogenesis, there is evidence that lower levels of androgens have a beneficial impact on folliculogenesis (269). These findings demonstrate that androgens attenuate follicular atresia through nuclear and extranuclear signaling pathways by enhancing the expression of the microRNA (miR) miR-125b, which suppresses proapoptotic protein expression. The study also indicates that, independent of transcription, androgens enhance FSH receptor expression, which then augments FSH-mediated follicle growth and development. These data suggest that it is a delicate balance between androgen excess and diminished circulating androgens, and alternate mechanisms might contribute to the unregulated follicle growth seen in PCOS.
Intraovarian factors involving follicular recruitment and growth—such as members of the TGF-β family (ie, AMH, inhibins, activins, bone morphogenic proteins, and growth differentiation factors [GDFs]), other growth factors, and cytokines (270, 271)—may also contribute to the abnormal follicle development and function seen in PCOS (260). The oocyte and its surrounding granulosa cells produce many of these factors because there is a close interaction between the signaling systems of the granulosa cells and oocytes (272, 273).
Gene expression of GDF9, an oocyte-derived growth factor affecting theca cell layer formation (274), is reduced in ovaries of anovulatory PCOS women (275), linking dysregulated oocyte GDF9 gene expression with altered folliculogenesis. AMH, a potential PCOS marker (35) produced by increased numbers of preantral and small antral follicles (276), may enhance theca cell androgen activity by inhibiting FSH and follicular development (272, 277). Reduced AMH protein levels in small primordial follicles and transitional follicles of anovulatory PCOS women may initially promote the recruitment of growing follicles and their oocytes (278), whereas increased granulosa cell AMH hypersecretion in small antral PCOS follicles could later impair further follicle growth (46, 47). In addition, decreased inhibin A and B levels occur in some small PCOS follicles despite normal amounts of activin and follistatin unbound to activin (279, 280). The roles of these TGF-β superfamily members in PCOS follicles remain to be clearly defined, as does a possible permissive effect of increased IGF-1 bioavailability on granulosa cell proliferation and steroidogenesis (54).
Intrafollicular cytokines and growth factors also affect oocyte development. Improved oocyte quality accompanies increased levels of granulocyte colony-stimulating factor; IL-12, IL-6, IL-8, and IL-18; brain-derived neurotropic factor; bone morphogenic protein 2; and amphiregulin, as well as decreased levels of IL-1 and IL-12 and vascular endothelial growth factor isoform (54). It is unclear, however, which cytokines and growth factors in follicular fluid are relevant for determining oocyte quality in PCOS (54).
E. Insulin sensitivity and secretion
Insulin resistance and its compensatory hyperinsulinemia are hallmarks of PCOS, and this puts women with PCOS at an increased risk of impaired glucose tolerance and T2DM (99, 281).
Research has shown that up to 30–40% of women with PCOS have impaired glucose tolerance and as many as 10% develop T2DM by the age of 40 (91, 92, 97, 282). Of note, there might be ethnic differences in the risk for glucose intolerance in PCOS (Italians, for example, display a lower risk) (282). And whereas obesity increases insulin resistance, lean women with PCOS have the same level of insulin sensitivity as obese controls (99, 104, 283), or in some cases lean controls (284, 285). The reason for not detecting differences in insulin sensitivity in lean women with PCOS might be explained by racial/ethnic differences, and this warrants further investigation (286, 287). Women with 1990 NIH-defined PCOS, which includes HA, have greater insulin resistance compared to anovulatory women with PCOS who have normal androgen levels and are relatively healthier metabolically (288–290), whereas a relatively normal weight (BMI < 27 kg/m2) significantly ameliorates this metabolic risk (134, 291). This highlights the importance of stratifying for phenotype. Compensatory hyperinsulinemia from insulin resistance can act at the dermis to induce acanthosis nigricans and skin tags (292–294).
Furthermore, the varying effects of obesity and ethnicity on insulin action might be due, in part, to defects in insulin secretion and reduced hepatic insulin clearance (295–297). Decompensation in insulin secretion in women with PCOS likely plays an important etiological role in the development of T2DM (296), and studies have shown that defects in insulin secretion are inherited in PCOS families (298). Studies have also shown that insulin sensitivity and insulin secretion have a hyperbolic and interdependent relationship (299), and thus must be viewed one in the context of the other, using measures such as the disposition index (an integrated measure of pancreatic insulin secretion and peripheral insulin sensitivity) (300, 301).
Importantly, insulin may play both direct and indirect roles in the pathogenesis of androgen excess in PCOS. Insulin stimulates ovarian androgen production and reduces hepatic SHBG synthesis, thereby increasing the levels of total and bioavailable androgens (302, 303). Insulin acts synergistically with LH to enhance theca cell androgen production in women with PCOS by activating a specific signaling pathway via its own receptor (304–306). In addition, insulin can stimulate human theca cell proliferation (307) and can enhance ovarian growth and follicular cyst formation in rats (308). However, evidence suggests that T levels are similar among the hyperandrogenic PCOS phenotypes (191, 192), which argues against a primary role for insulin in the pathogenesis of androgen excess.
When measuring insulin action and secretion, it is important to use methods that have both high sensitivity and high specificity. Both the euglycemic hyperinsulinemic clamp (309) and the frequent-sampling, intravenous glucose tolerance test (with minimal model analysis) are “gold standard” methods for measuring insulin sensitivity and secretion (310), whereas methods like homeostasis model of assessment for insulin resistance and oral glucose stimulated insulin sensitivity (eg, the Matsuda model) are less sensitive (311–313). There is no simple validated clinical test of insulin sensitivity.
1. Insulin resistance in muscle and adipose tissue
Insulin-mediated glucose uptake is limited to tissues that express the insulin-responsive glucose transporter GLUT4, such as cardiac and skeletal muscle and adipose tissue. Interestingly, studies have also reported GLUT4 expression in granulosa cells (314). Insulin also stimulates amino acid uptake and suppresses hepatic glucose production and lipolysis. A central question in PCOS is why there is resistance to some actions of insulin (eg, metabolic), whereas insulin action on steroidogenesis seems to be preserved.
Insulin resistance in skeletal muscle is defined as impaired glucose transport and muscle glycogen synthesis in response to insulin. And we define insulin resistance in adipose tissue as impaired glucose transport and the suppressed inhibition of lipolysis in response to insulin (315). Studies have found that free fatty acids induce insulin resistance and impair insulin action in obesity, T2DM, and PCOS (316, 317). Some research suggests that there are fat depot-specific differences in adipocyte lipolysis, and that visceral adipocytes are a main source of increased portal vein free fatty acids that can contribute further to hepatic-mediated insulin resistance. Other published works have extensively reviewed the molecular defects responsible for aberrant activities in skeletal muscle and adipose tissue and provide a more comprehensive review of this aspect of the pathophysiology of PCOS (318).
a. Muscle.
One study reported intrinsic abnormalities in glucose transport and insulin signaling in myotubes from affected women, and that these abnormalities contribute to insulin resistance (319). Another study reported impaired insulin responsiveness but not impaired sensitivity, and no change in GLUT4 protein expression (320). Another study found no evidence for defects in insulin-stimulated glycogen synthase activity in myotubes from women with PCOS (321). Research has demonstrated that one mechanism for the postbinding defect in skeletal muscle insulin signaling appears to be constitutive serine phosphorylation of the insulin receptor and downstream signaling molecules, such as insulin receptor substrate-1 (IRS-1) (319, 322, 323). There is an increased serine phosphorylation of insulin receptors in the skeletal muscle of women with PCOS (322). The decreased insulin-mediated (euglycemic hyperinsulinemic clamp) activation of skeletal muscle phosphatidylinositol 3 in vivo in PCOS is consistent with the serine phosphorylation-mediated impairment of insulin signaling (324). Decreases in protein kinase B (Akt) activation and Akt substrate of 160 kDa (238), which are downstream of phosphatidylinositol 3-kinase (AS160), are also consistent with a primary defect in insulin receptor-mediated signaling. The constitutive activation of kinases in the extracellular signal-regulated kinases 1/2 contributes to the increased serine phosphorylation of IRS-1 (323). These findings provide evidence of selective insulin resistance in skeletal muscle.
In addition to insulin signaling defects in the skeletal muscle of women with PCOS, research has also suggested that mitochondrial dysfunction is involved in PCOS (325). One study demonstrated mitochondrial dysfunction by reporting a decreased expression of genes involved in mitochondrial oxidative metabolism, such as peroxisome proliferator-activated receptor γ coactivator-1 α. However, the study found no differences in mitochondrial number or function in cultured myotubes (321), indicating that changes in mitochondrial gene expression in PCOS skeletal muscle are not a primary defect.
One study demonstrated that T exposure in female rats impairs insulin signal transduction (326), and a second study found similar signaling impairment in skeletal muscle from women with PCOS (319). Moreover, studies showed that T and DHT exposure in female rats reduces whole-body insulin sensitivity by modifying skeletal morphology, including reducing the number of insulin-sensitive fibers, increasing the number of less insulin-sensitive muscle fibers, reducing capillary density, impairing glycogen synthase activity, and decreasing GLUT4 protein expression (327–332). Importantly, studies have shown persistent defects in insulin action and/or signaling in cultured PCOS myotubes and a decreased responsiveness to insulin-stimulated glucose uptake (320, 321).
b. Adipocytes and adipose tissue.
In vitro studies indicate that androgens can directly induce selective insulin resistance in the adipocytes of women by acting via the androgen receptor (333, 334). Adipocytes from women with PCOS display decreased insulin sensitivity (319, 335–337). Studies have shown that the number of insulin receptors and their affinity for insulin appear to be normal, suggesting a postbinding defect in insulin signaling in adipocytes (108, 335, 336). Furthermore, reduced insulin responsiveness indicates postreceptor alterations that are probably related to the reduced levels of GLUT4 in the adipocytes of women with PCOS (336, 338). A study also noted increased miRNA93 expression in adipose tissue from women with PCOS, which inhibits GLUT4 gene expression (339). No studies have found major differences in the expression or activity of proteins in the insulin-signaling pathway downstream from insulin receptors in isolated adipocytes. There have been reports, however, of increased levels of phosphatidylinositol 3-kinase together with impaired phosphorylation pattern of IRS-1 in adipose tissue (333, 334).
Although skeletal muscle accounts for 85% of insulin-stimulated glucose uptake in the body (340), it is important to note that modest changes in adipose tissue glucose uptake may have substantial secondary effects on whole-body glucose metabolism as demonstrated in mice (341). Therefore, it is possible that the molecular defects observed in the insulin-signaling pathway of adipocytes from women with PCOS may have a significant effect on whole-body insulin resistance (342). Further research is needed to elucidate the altered molecular pathways in adipose tissue of women with PCOS.
F. Obesity, fat distribution, and adipose tissue function and morphology
It is well established that women with PCOS are often overweight or obese, and it is possible that the increasing global prevalence of obesity may play a key role in promoting the development of PCOS in susceptible individuals (343). A prevalence study investigated whether obesity increases the risk of PCOS in the general population. It demonstrated that the prevalence rates of PCOS in underweight, normal-weight, overweight, and obese women were 8.2, 9.8, 9.9, and 9.0%, respectively, similar to that observed in the general population (96). These results suggest that the risk of PCOS is only minimally increased with obesity. On the other hand, a Spanish prevalence study among overweight and obese subjects demonstrated a 28.3% prevalence of PCOS, which is markedly higher than the 5.5% prevalence of PCOS in lean women (93). Thus, the effect of obesity may vary by race and ethnicity. Obesity can also exacerbate pre-existing clinical, hormonal, and metabolic features in women with PCOS (343). In addition, studies have associated HA with abdominal fat accumulation in women and lower levels of circulating SHBG (344, 345). Iatrogenic HA increases visceral fat accumulation in female-to-male transsexuals (346). Furthermore, administering androgen receptor antagonists to women with PCOS decreased visceral/subcutaneous fat mass together with circulating androgens (347). Studies have also associated androgen exposure with increased fat accumulation in animals (332, 348). Studies exploring the effects of weight loss in obese women with PCOS support the impact of obesity on the severity of PCOS. Weight loss through diet, exercise, and lifestyle management reduces circulating androgens and increases SHBG levels (95), reduces ovarian volume and follicle count (349), improves insulin sensitivity, reduces hyperinsulinemia (95, 350, 351), and improves menstrual cyclicity and fertility (95, 349). The degree of weight loss (and the requisite time frame) necessary to induce significant improvement in the reproductive phenotype in women with PCOS remains poorly understood (343, 352). Obesity and other adipose tissue-related factors may, therefore, play a critical role in the promotion and/or the maintenance of PCOS.
Whether women with PCOS have increased amounts of abdominal/visceral fat accumulation remains uncertain. Clinicians can measure fat distribution using various methodologies, ranging from less-sensitive measures (such as waist circumference), to more accurate dual-energy x-ray absorptiometry-derived measures, to gold standard measures using computerized tomography or MRI. Two MRI studies demonstrated that although increased waist:hip ratio measurements indicated abdominal/visceral fat accumulation, MRI did not show this accumulation (104, 105). Subsequently, one study demonstrated lower visceral fat among lean women with PCOS (353), and another found larger accumulations of visceral fat among both normal-weight and overweight women with PCOS (354). However, these studies used different imaging protocols (ie, single slice vs multislice) and differing PCOS inclusion criteria, which may explain the conflicting results. Future studies should include cohorts of adequate size to control for confounders, use consistent diagnostic criteria, and utilize standardized MRI-based morphology. Such studies would further increase our understanding of the impact of obesity on abdominal/visceral fat accumulation.
1. Impaired adipocyte function
Studies have reported abnormal adipocyte function in women with PCOS. Examples include the disrupted secretion and release of adipokines from adipose tissue (eg, low circulating adiponectin levels) (104, 355); increased serum, adipocyte, and adipose tissue retinol-binding protein 4 (356); and increased adipose tissue and circulating visfatin (357). Several studies have associated PCOS with low-grade inflammation based on increased levels of several inflammatory mediators (358). Importantly, many of these inflammatory markers are produced in adipose tissue. The formation of clusters of macrophages around dead adipocytes in crown-like structures is a primary feature of chronic low-grade inflammation (359). A study demonstrated an increased expression of CD11c (a marker of inflammatory macrophages) and increased crown-like structure density in subcutaneous adipose tissue in normal-weight and overweight women with PCOS (354). These results indicate an increased inflammatory state in the adipose tissues of women with PCOS. However, whether the altered secretion of various adipokines and inflammatory markers is a unique feature of PCOS, a consequence of reduced insensitivity, or even a cause of the decreased insulin sensitivity warrants further investigation.
Studies have shown enlarged abdominal subcutaneous adipocytes in women with PCOS, independent of BMI (104, 108, 360, 361). Importantly, enlarged subcutaneous adipocytes are an independent predictor of risk for T2DM, and adipocyte size is a major contributing factor for the proper function of adipocytes and adipose tissue. Research has shown that enlarged fat cells and reduced serum adiponectin, together with a large waistline, are the strongest factors predicting insulin resistance in women with PCOS, and these appear to be central factors in the maintenance of insulin resistance in PCOS (104).
G. Increased sympathetic nerve activity
Metabolic and cardiovascular disorders are related to autonomic dysfunction (362, 363), and many of the PCOS-related clinical characteristics—including HA, hyperinsulinemia/insulin resistance, central obesity, hypertension, obstructive sleep apnea, and depression—are associated with increased activity in the sympathetic nervous system. Insulin increases muscle sympathetic nerve activity by increasing glucose metabolism in hypothalamic neurons. This suppresses inhibitory pathways between the hypothalamus and brainstem sympathetic nerve centers (364, 365). However, the relationship between hyperinsulinemia and sympathetic nervous system activation is complex and is affected by obesity. Obesity is associated with high sympathetic nerve activity, and visceral activity is more closely related to increased sympathetic activity than total and subcutaneous fat (366, 367). Thus, altered activity in the sympathetic nervous system may play a significant role in the progression of PCOS (368).
Research has shown that general muscle sympathetic nerve activity, as measured with the direct microneurographic technique (369), is increased in normal-weight women with PCOS compared to age- and BMI-matched controls (370). Research has also shown that acupuncture, when combined with low-frequency electrical stimulation and exercise, decreases the high levels of sympathetic nerve activity seen in women with PCOS (371); however, in another study, acupuncture did not differ from a sham control in regard to how it affected ovulation frequency (372). The latter study did not measure sympathetic nerve activity or heart rate variability (HRV) (373).
In addition to studies that have directly measured sympathetic nerve activity, there are a number of studies that have assessed autonomic activity through indirect measurements, such as HRV and heart rate recovery (HRR) (374–377). The HRV studies demonstrated increased sympathetic and decreased parasympathetic components of HRV in women with PCOS (376, 378). The HRR studies indicated an attenuated autonomic response as demonstrated by an exaggerated systolic blood pressure response to exercise and delayed recovery, both of which indicate sympathetic activation and increased peripheral vessel resistance (377). Because HRR after exercise is thought to be an indirect marker for parasympathetic activity, this result suggests that women with PCOS have lower vagal activity (377).
The hypothesis that the sympathetic nervous system plays a role in the etiology of PCOS is further strengthened by data showing a greater density of catecholaminergic nerve fibers in the ovaries of women with PCOS (379, 380) and altered catecholamine metabolism in adolescents with PCOS (381). Increased ovarian sympathetic nerve activity might exaggerate PCOS symptoms by stimulating androgen secretion (382). Research has identified nerve growth factor (NGF), a marker of sympathetic nerve activity, in both rodent and human ovaries; and its receptors are localized to the theca cells of the developing follicles (383). The concept that ovarian sympathetic nerves are involved in the development of ovulatory dysfunction and disturbed ovarian steroidogenesis arises from research showing that follicular cyst development in estradiol valerate-treated rats is associated with anovulation and increased ovarian NGF production (384). Researchers found that blocking the biological actions of NGF (by injecting NGF antibodies and antisense oligonucleotides [for the low-affinity NGF receptor] into the ovaries [385] or by injecting NGF antibodies into the peritoneal cavity) partially restored estrus cyclicity and ovulatory capacity in estradiol valerate-treated rats (386). The best results came from the surgical denervation of the superior ovarian nerve (the nerves associated with the secretory cells of the ovary) (382). However, whether overproduction of NGF increases ovarian androgen production or vice versa is presently unknown.
H. Summary
In summary, the pathophysiology of PCOS clearly involves both reproductive and metabolic manifestations; however, there are still large gaps in identifying a common molecular mechanism that would explain both aspects of the phenotype or the significant heterogeneity of the syndrome. There is a lack of animal models that spontaneously develop a PCOS phenotype because most are androgen induced. Similarly, the development of three-dimensional culture methods of human follicles that could recapitulate the in vivo PCOS ovary environment would be a significant advance over current models that use isolated human thecal or granulosa cell lines in two-dimensional culture. Impaired folliculogenesis and steroidogenesis in PCOS is multifaceted and is controlled by extra- and intraovarian factors as well as genetics. The abnormal intrafollicular environment that develops in PCOS can disturb the maturation of the oocyte and alter the oocyte gene expression pattern that is important for oocyte development. Future research should aim to increase our understanding of how different molecular mechanisms interact with each other to control ovarian functions. Such studies would significantly improve our understanding of how to correct the dysfunctional follicle growth and abnormal oocyte development that occurs in PCOS.
IV. Molecular Genetics
A. Genetic tools
PCOS is a complex polygenic disease that may involve the subtle interaction of environmental factors, susceptibility, and protective genomic variants. Earlier studies of the genetics of PCOS suggested an autosomal dominant mode of inheritance (387), but the recruitment of large families with multiple affected women likely biased these studies (388). Subsequent prospective studies have revealed smaller kindreds and a more complex inheritance pattern. In addition to the challenges inherent to gene discovery for any common disease, there are several notable factors that further complicate research into the genetic basis of PCOS. These include the multiple diagnostic criteria and heterogeneity of PCOS and the fact that PCOS can be diagnosed only in women of reproductive age. As noted above, diagnosing premenarchal girls, adolescent girls, and menopausal women is problematic. Furthermore, there is no accepted male phenotype, although males do appear to have androgen-related and metabolic dysfunction (26, 389, 390). Also, PCOS impairs fertility or delays fertility, which reduces family size (388, 391). Therefore, the availability of relatives for family-based genetic studies is limited (392).
Cytogenetic approaches have not identified major chromosomal aberrations (393). Indeed, aneuploidy rates of unfertilized oocytes in women suffering from PCOS did not differ from women with tubal factor infertility (394). Although researchers have used both linkage analysis and association to study the genetics of complex traits, they have not been particularly useful (395). The poor yield of such studies in dissecting the genetic origins of PCOS similarly suggests that it is a complex genetic trait. Most published studies of genes in PCOS have been genetic association studies of poor quality that would not meet the current, more stringent, guidelines outlined in the Strengthening the Reporting of Observational Studies in Epidemiology–Molecular Epidemiology statement (396). Limitations include small sample size and diagnostic heterogeneity, population stratification, failure to examine the entire candidate gene (usually only one variant is examined), not correcting for multiple testing (within the same study or separate reports from the same cohort), confounding phenotypes (eg, obesity), and the lack of replication by an independent cohort in the same study. Alternatively, researchers could examine founder populations that are genetically more homogeneous or use family-based association studies that only require a proband and usually both parents. Several whole genome approaches are now available that do assess the role of positional or functional candidate genes in PCOS. Initially, researchers used microarrays with equally distant microsatellites, but in the late 1990s, single nucleotide polymorphism (SNP) arrays spanning the whole genome became available. Next-generation techniques sequencing either all exons or the whole genome have become an affordable option for studying the genetic background of PCOS (397). Of course, identifying a significant variant is only the first step. Confirming the biological relevance of PCOS-associated variants by molecular genetic analyses (eg, expression analysis or targeted genetic disruption in cell culture or another organism) is critical. Studies rarely perform this next step, and this a real problem with all significant findings from genome-wide association studies (GWAS).
B. Evidence for a genetic background
1. Twin studies
There are two studies that assessed the heritability of PCOS in twins by comparing the degree of concordance between mono- and dizygotic twin pairs (398, 399). One small study suggests that PCOS is not the result of a single autosomal genetic defect, but that environmental factors, perhaps both intrauterine and extrauterine, are involved in the pathogenesis of this disorder, or that PCOS may be an X-linked disorder, or the result of polygenic factors. However, fasting insulin level, androstanediol glucuronide, and BMI did appear to be significantly influenced by genetics (398).
The second larger study revealed a strong contribution of genetic factors to PCOS and indicates that a model including additive genetic factors and unique environmental factors is the most parsimonious. In this study the variance in the pathogenesis of PCOS is 72% due to genetic influences (399). The same group published a second paper in which they tried to evaluate the prevalence of PCOS in women from opposite-sex twin pairs compared to women from same-sex twin pairs, sisters, and female spouses of twins. They did not report any difference in the prevalence of PCOS between these groups, indicating that possible androgen exposure of the female fetus (caused by a shared intrauterine environment with a male fetus) does not result in PCOS-like traits (400).
2. Family-based studies
Evidence for the role of genetics in PCOS includes a well-documented familial clustering of PCOS, with sisters more likely to be affected with signs and symptoms of the disorder, and first-degree relatives having higher rates of metabolic abnormalities including insulin resistance, decreased β-cell function, dyslipidemia, and MetS. In one prospective family study, 22% of the sisters of women with PCOS fulfilled diagnostic criteria for PCOS. In addition, another 24% of the sisters had HA combined with regular menstrual cycles (191). Circulating T levels in both of these groups of sisters were significantly increased compared with unaffected sisters and control women with normal menstrual cycles who did not have PCOS. Probands, sisters with PCOS, and hyperandrogenemic sisters also had elevated DHEAS and serum LH levels compared with control women. Studies have noted this familial aggregation of HA (with or without oligomenorrhea) in other PCOS kindreds, which suggests that it is a genetic trait (191, 192, 401, 402).
PCOS and its associated metabolic abnormalities cluster in families, suggesting that there is a genetic susceptibility to these defects (191, 403). Compared to unaffected sisters and women of similar age, weight, and ethnicity, sisters and mothers of women with PCOS have higher rates of HA, insulin resistance, and MetS (404, 405). Male relatives of women with PCOS had increased prevalence rates of MetS and obesity compared to the general US male population. In contrast to women with PCOS and their female relatives, the higher prevalence of MetS in male relatives was attributed to elevated BMI. These findings suggest that the high rates of MetS in male relatives of women with PCOS are related to higher rates of obesity compared with the general population (406, 407). Some studies have also suggested increased rates of CVD events in parents of women with PCOS. Other studies confirm that both race and family history of diabetes have a substantial impact on the metabolic and glycemic status of women with PCOS. A history of T2DM in a first-degree relative appears to be an important factor in predicting the risks of metabolic abnormalities, impaired glucose tolerance, and T2DM in women with PCOS (132, 298, 408–410).
3. Population-based studies
Population-based (case control) studies have been more popular than family-based studies due to easier subject recruitment (no need to obtain parental information and samples), reduced costs (no need to genotype parents), and lower levels of identity by descent (397). And whereas population-based studies have tested a large number of functional candidate genes for association or linkage with PCOS phenotypes, the findings are more negative than positive. However, a lack of universally accepted diagnostic criteria makes comparing such studies problematic. Furthermore, the particularly small sample size of the study populations in case-control and family-based studies compared to GWAS appear to be major limitations for population-based genetic studies of PCOS (411).
C. Candidate gene studies
Previously, PCOS genetic research predominantly used candidate gene analyses. Although this method has identified several very promising PCOS susceptibility genes or loci (eg, fibrillin-3, genes involved in insulin metabolism [insulin (INS), INS receptor, and INS receptor substrate 1], transcription factor 7 like 2, calpain 10, the fat mass and obesity-associated gene [FTO], SHBG, and the FSH receptor gene), no single gene has been successfully replicated and identified as truly causative across all studies (397). A number of substantially robust family-based studies identified and replicated a marker within the fibrillin-3 gene (412, 413), which was partially replicated by case-control studies (414, 415). The associated variant is within a dinucleotide repeat marker, and no SNP within fibrillin-3 is as strongly associated. The latter may account for the failure of fibrillin-3 to be significantly associated with PCOS in GWAS (122, 416).
One case-control study replicated the insulin receptor (414), and several independent studies have reported evidence for an association with IRS-1 (417). Studies have not significantly associated the diabetes susceptibility region of the TCF7L2 gene with the PCOS reproductive phenotype, although studies have associated a novel region of the gene with PCOS-affected status (418). However, these findings have not been replicated. Studies have associated the obesity susceptibility variants of the FTO gene with body weight rather than reproductive features (419, 420), whereas other regions of the gene may be associated with PCOS (421). The FTO allele associated with obesity represses mitochondrial thermogenesis in adipocyte precursor cells in a tissue-autonomous manner (422). The FSH receptor is of interest because it appears to be a GWAS locus; the case-control analysis had the usual constraints of limited sample size, lack of replication, and high chance of a type 1 error (423).
Among the reasons for lack of replication are inadequate sample size (which could lead to false-positive or false-negative findings) and incomplete coverage of the candidate gene (ie, many studies testing associations with just a few variants in each gene). Lastly, the candidate gene approach can only be used if there is an a priori hypothesis about the involvement of a given gene in the etiology of PCOS because of its proposed role in the pathogenesis. Hence, it falls short of identifying novel genes or genes whose functions are less well known (397).
D. Genome-wide association studies
In the first GWAS that targeted PCOS, Chinese researchers identified causative genes in Han Chinese women who were diagnosed with PCOS using the Rotterdam criteria (122). This study only examined women who fulfilled all three criteria (ie, ovulatory dysfunction, hyperandrogenism, and polycystic ovarian morphology). Their discovery set included 744 PCOS cases and 895 controls. Replications involved two independent cohorts of 2840 PCOS cases and 5012 controls from northern Han Chinese, and 498 cases and 780 controls from southern and central Han Chinese, respectively. However, it should be mentioned that apart from the other diagnostic characteristics, T levels were only marginally elevated in the replication cohorts. The authors of this paper identified strong evidence of associations between PCOS and three loci (eg, 2p16.3, 2p21, and 9q33.3) that were significantly associated with PCOS. Candidates within these loci were the FSH receptor, the LH receptor, and the DENND1A genes on chromosome 2, and the thyroid adenoma-associated (THADA) gene on chromosome 9 (122). Several studies tried to replicate these findings in other cohorts, mostly of European descent. One study failed to replicate the identified SNPs (424), whereas two others did replicate at least some of the identified susceptibility loci (123, 124). Hence, the DENND1A and THADA genes seem to be associated with PCOS in European populations and are likely to be important in the etiology of PCOS, regardless of ethnicity. Evidence supporting a role for the DENND1A.V2 isoform in the pathophysiology of PCOS includes the finding of increased amounts of this variant in theca cells derived from women with PCOS and studies in which targeted overexpression of the DENND1A.V2 isoform in theca cells obtained from normal women recapitulated hyperandrogenic theca cell function (Figure 5) (425). The finding of the DENND1A.V2 variant in urinary exosomes of women with PCOS may prove to be a useful diagnostic tool in the future (425). These findings illustrate how results from GWAS can be utilized to understand the pathophysiology of PCOS. It has been suggested that DENND1A and other proteins such as LHCGR and INSR form a signaling cascade that influences theca cell androgen biosynthesis (426). Further research is needed to test this hypothesis.
Figure 5.

Overexpression of DENND1A isoforms leading to increased androgen production. Forced expression of DENND1A.V2 in normal theca cells results in augmented androgen and progestin production. A, DHEA production after infection of normal theca cells, with 0.3, 1.0, 3.0, and 10 pfu per cell of either empty (Null) or DENND1A.V2 (DENN.V2) adenovirus, treated in the absence (C) or presence (F) of 20 μm forskolin for 72 hours. B, Quantitative Western analysis after the infection of normal theca cells with 3 pfu Null or DENND1A.V2 adenovirus to confirm DENND1A.V2 protein expression. C—F, DHEA (C), 17OHP4 (D), T (E), and progesterone (F) biosynthesis in normal theca cells infected with either 3 pfu per cell of DENND1A.V2 or control (Null) adenovirus and treated in the absence (C) or presence (F) of 20 μm forskolin for 72 hours. DENND1A.V2 infection increased basal 17OHP4 (*, P < .01), T (*, P < .05), and P4 (*, P < .05) accumulation compared with control (Null) adenovirus. DENND1A.V2 infection also increased forskolin-stimulated DHEA (*, P < .001), 17OHP4 (**, P < .001), and P4 (**, P < .001) compared with control (Null) adenovirus. 17OHP4, 17-hydroxyprogesterone; P4, vaginal progesterone. [Adapted from J. M. McAllister et al: Overexpression of a DENND1A isoform produces a polycystic ovary syndrome theca phenotype. Proc Natl Acad Sci USA. 2014;111(15):E1519–E1527 (425), with permission. © National Academy of Sciences, USA.]
The analysis of the LHCG receptor in the Chinese GWAS was probably not sufficiently powered to detect modest effects (427). Subsequently, this Chinese group published another even larger GWAS in Han Chinese women with PCOS (also diagnosed using the Rotterdam criteria), which includes the original cohort discussed above (416) (Figure 6). This larger study identified eight new risk loci for PCOS. For this study, the researchers analyzed data from the previous cohort together with a new cohort of Han Chinese—a discovery cohort of 1510 cases and 2016 controls, and a replication cohort of 8226 cases and 7578 controls. A subsequent meta-analysis in this second study confirmed the previously identified loci and another eight new genome-wide, significant, PCOS-associated signals at 9q22.32, 11q22.1, 12q13.2, 12q14.3, 16q12.1, 19p13.3, and 20q13.2; and a second independent signal at 2p16.3 within the FSHR gene. These newly identified loci contain genes with evidence for involvement of insulin signaling, sexual hormone function, and T2DM. Other candidate genes were related to calcium signaling and endocytosis (416). Previous studies had already reported the involvement of the FSHR gene. In this study, the Ser/Ser FSHR polymorphism (located at position 608 within the intracellular portion of the FSHR) was associated with higher serum FSH levels and seemed to represent a risk allele in normo-gonadotropic anovulatory patients as well as in women suffering from PCOS (423). An independent study assessing a cohort of European ancestry reported similar findings (428). These authors found strong evidence for an association of PCOS with previously identified SNPs in the 2p16.3 locus. Important to note, the marker with the strongest association with PCOS in the Chinese-cohort studies was not informative in the European-cohort studies. However, the European studies did identify and genotype three other markers within this SNP, and one of those seemed to be nominally associated with PCOS. With regard to SNP mapping, the strongest evidence for association in the European studies was to the formerly identified FSHR. Therefore, it seems that products of the LH-choriogonadotropin receptor and FSHR genes could be involved in the etiology of PCOS, regardless of ethnicity (Figure 7) (428).
Figure 6.
Genome-wide Manhattan plot for the GWAS meta-analysis. Shown are the −log10 P values for the SNPs that passed quality control. The solid horizontal line indicates P < 1 × 10−5. Markers with 50 kb of a SNP associated with PCOS are marked in red for those identified in a previous GWAS and replicated here and in blue for those first identified in the current study. Associations at THADA, LHCGR, and DENND1A were also reported in a previous GWAS. [Adapted from Y. Shi et al: Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat Genet. 2012;44(9):1020–1025 (416), with permission. © Nature Publishing Group.]
Figure 7.

Median FSH levels in women with PCOS, stratified according to the number of allelic variants in the FSH receptor [FSHR (Ser680)] and LH receptor [LHR (Asn312)], ie, carriers of zero to four polymorphic alleles. The total number of variant alleles was significantly associated with increasing FSH levels. [Adapted from O. Valkenburg et al: Genetic polymorphisms of GnRH and gonadotrophic hormone receptors affect the phenotype of polycystic ovary syndrome. Hum Reprod. 2009;24(8):2014–2022 (432), with permission. © European Society of Human Reproduction and Embryology.]
Another GWAS identified a gene involved in the genetic predisposition to obesity in Korean women with PCOS, linking the GYS2 gene with PCOS and BMI. Mutations in this gene are associated with the autosomal recessive disorder glycogen storage disease type 0 (GSD0) and, interestingly, PCO have been noted in girls and women with glycogen storage diseases (429). The Korean GWAS also reported some pleiotropic effects of GYS2 on childhood obesity and gestational diabetes (430). However, there was no replication cohort in this study.
One of the two latest large GWAS expanded on these findings and examined common genetic susceptibility loci in European ancestry women diagnosed with PCOS based on the National Institutes of Health criteria (hyperandrogenism and chronic anovulation) (431). This GWAS also included well phenotyped normal reproductive controls in the replication phases, instead of population-based controls. Three loci reached genome-wide significance—two novel loci, chr 8p32.1 in the region of GATA4 and NEIL2 and chr 11p14.1 in the region of the FSH B polypeptide gene; and one previously found in Chinese PCOS (416), chr 9q22.32 in the region of c9orf3/FANCC. The same chr 11p14.1 SNP, rs11031006, in the region of the FSH B polypeptide gene, also reached genome-wide significance in the meta-analysis of the quantitative LH levels within this study. The study also found significance in the case-control meta-analysis, with two novel loci mapping to chr 8p32.1 and chr11p14.1, and a chr 9q22.32 locus previously found in Chinese PCOS. This same chr 11p14.1 SNP, rs11031006, also strongly associates with PCOS diagnosis and LH levels (Figure 8).
Figure 8.
Manhattan plots for LH levels in women with PCOS. Alternating blue and red colors indicate genotyped SNPs, and accompanying black and grey colors indicate imputed variants, on odd and even chromosomes, respectively. The red horizontal red line indicates genomewide significance. QQ plots and lGC/ lGC1000 are inset in the upper right corner of the plot. For LH levels, P values are from sample-size weighted two-strata meta-analysis of strata-specific linear regression P values. (Stage 1: 645 PCOS cases; Stage 2: 399 PCOS cases). (Adapted from Hayes MG, et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun. 2015;6:7502.) (431)
E. Other approaches
1. Functional genomic studies
Functional genomic technologies, such as cDNA microarray, have been applied in various biological studies to identify differentially expressed genes and obtain a global view of those genes that might be involved in the development of PCOS. Samples of 119 known genes taken from ovaries of women with PCOS showed differential expressions compared to samples from ovaries of normal controls (433). These differentially expressed genes are involved in various biological functions—such as cell division/apoptosis, regulation of gene expression, and metabolism—reflecting the complexity of clinical manifestations of PCOS (262, 434–436). Most of this research studied long-term cultures of theca cells from either PCOS or normal ovaries, which might have influenced their expression profile. However, DNA expression profiles from whole PCOS ovaries were very similar to profiles from PCOS theca cells in long-term cultures (433).
Another study demonstrated that morphologically indistinguishable, high-quality oocytes from women with and without PCOS exhibit different gene expression profiles (437). Promoter analysis suggests that androgens and other activators of nuclear receptors may play a role in differential gene expression in the PCOS oocyte (437). Likewise, the annotation of the differentially expressed genes suggests that defects in meiosis or early embryonic development may contribute to reduced developmental competency of PCOS oocytes (437).
F. Pharmacogenomic studies
Several studies have examined candidate genes known to be involved in drug response and tried to identify predictive alleles. Such studies have identified genetic regions associated with a response to metformin, including a SNP in the serine-threonine kinase 11 gene associated with ovulation (438), and one in the OCT-1 transporter gene associated with a favorable reduction in cholesterol (439). Studies have associated FSHR gene polymorphisms with varying responses to gonadotropin ovulation induction, which may prove useful in treating women with PCOS (440). However, like other candidate gene studies in women with PCOS, replication is required from larger cohorts in multicenter clinical trials.
G. Epigenetic influences
The androgen receptor is located on the X chromosome, and it contains a polymorphic CAG repeat region in exon 1. The length of this CAG repeat region shows an inverse relationship with androgen sensitivity. Skewed X-inactivation could give rise to an over-representation of paternally or maternally derived androgen receptors with different androgen sensitivity. Hence, skewing might lead to differences in androgen sensitivity. Indeed, some patients with high serum androgen levels had a longer repeat stretch than patients with lower serum androgen levels (441), which contradicts the expectation of longer repeats associated with decreased androgen sensitivity. Another study compared the frequency distributions of CAG repeat alleles and their pattern of expression via X-inactivation analysis among 83 fertile women and 122 infertile women with PCOS (442). The infertile women with PCOS exhibited a greater frequency of CAG alleles (>22 repeats) compared to both the fertile control group and the general population. The findings warrant a closer inspection of X-linked genes in PCOS that considers both genotype and epigenotype because the meta-analysis of genetic association studies of CAG repeats in the androgen receptors have shown no differences between those of controls (443).
It may be necessary to examine the epigenetic modifications to the genetic code to unravel some of the confounding evidence. A pilot study found no significant difference in the global methylation of peripheral leukocyte DNA between patients with PCOS and matched controls. Global methylation arrays might not be as sensitive as approaches that target specific genes of interest. Moreover, studying methylation profiles in leukocytes might not detect important differences that could exist between different tissues. Therefore, methylation in specific target genes or regions as well as key tissues other than peripheral leukocytes (such as human ovaries, adipose tissue, or adrenals) should be investigated. Aberrant DNA methylation patterns could serve as epigenetic biomarkers for early PCOS detection, and understanding the epigenetic mechanisms involved in PCOS may provide novel avenues for the diagnosis and treatment of this common disorder (444).
H. Summary
Early data supporting a single gene etiology to PCOS has been refuted by multiple GWAS showing PCOS to be consistent with a complex genetic disorder with multiple alleles associated with a small degree of risk. There has been heterogeneity in the diagnostic criteria used to identify PCOS, as well as variability in the design of the studies—including the choice for controls and replication strategies. Furthermore, many GWAS remain to be published, and an eventual meta-analysis of existing GWAS in PCOS is in the planning phase. There has been a lack of functional genomic studies to explain the possible pathophysiological significance of identified genetic variants, although clearly the gonadotropin and gonadotropin receptor variants are consistent with phenotypic abnormalities in PCOS.
V. Future Research Directions
As illustrated in this statement, there are multiple areas of uncertainty in the diagnosis, epidemiology, pathophysiology, and genetics of PCOS. One of the lingering controversies is the name “polycystic ovary syndrome,” which incompletely reflects the full reproductive and metabolic aspects of the syndrome (445). The Expert Panel of the Scientific Statement Workshop on PCOS recommended renaming the syndrome. The Expert Panel also generated a list of several research priorities based on the presentations and recommendations of leading experts from around the world (Table 2).
Table 2.
Research Recommendations by the Expert Panel of the NIH-Sponsored Evidence-Based Methodology Workshop on PCOS
| Recommendations |
| Conduct adequately powered, carefully phenotyped, multiethnic cohort studies to establish the genetic or epigenetic cause(s) of the syndrome. |
| Establish the prevalence of abnormal glucose tolerance in women wishing to conceive, and determine whether treating abnormal glucose tolerance before or early post-conception alters maternal-fetal outcomes. |
| Conduct translational research to determine the mechanisms by which the syndrome alters ovarian, hypothalamic-pituitary-adrenal, and metabolic function to establish model systems that can be used to identify novel therapeutic approaches. |
| Conduct sufficiently large, well-controlled epidemiological studies determining the prevalence, phenotypes, and morbidities of PCOS in multiethnic longitudinal studies to determine: |
| a. Whether the syndrome is associated with increased cardiovascular and diabetic complications. |
| b. Whether the risk of these cardiovascular and diabetic complications (or the lack thereof) is associated with specific phenotypes. |
| c. Whether treatment of metabolic abnormalities reduces the risk of cardiovascular and diabetic complications. |
| Conduct suitably powered studies to determine whether the syndrome is associated with endometrial, breast, and ovarian cancers, and, if so, determine optimal prevention, detection, and treatment. |
| Identify optimal therapies to treat the most common symptoms and patient complaints of the syndrome, such as hirsutism and obesity. |
| Identify optimal therapies and best practices to achieve successful pregnancy. |
A. Diagnosis and epidemiology
The long-term consequences of PCOS are unclear. Therefore, it is crucial to establish the diagnostic criteria for PCOS early in adolescence. In this way, longitudinal studies can explore the natural progression of the PCOS throughout childhood. This will help us better determine the value of interventions that target PCOS and the various reproductive, metabolic, and psychological diseases associated with PCOS. Genetic and environmental factors (eg, socioeconomic status, lifestyle) that contribute to ethnic variances in PCOS are also important to consider in developing individualized strategies for treating PCOS. Better and more convincing studies are needed to show a clear link between PCOS and environmental disruptors. Similarly, we need to better understand how the maternal-fetal environment affects the developmental origins of PCOS. Because experimental constraints exist on human studies, more animal models and in vitro cell culture studies are needed to help us explore how developmentally relevant endocrine/paracrine factors and genes interact to influence reproduction and metabolism. With such information, new clinical strategies targeting the prevention of reproductive and metabolic dysfunction are likely to improve fertility, reduce CVD risk, and enhance quality of life in women with PCOS.
B. Pathophysiology
PCOS is likely a heterogeneous disorder with multiple perturbations leading to a variety of final phenotypes. A significant breakthrough in the pathophysiology of PCOS is to discover a pathway that links hyperandrogenism, decreased insulin sensitivity, and follicle arrest, all in one phenotype. Further examining how this pathway relates to dysfunction in affected tissues (eg, ovary, adipose, muscle, skin) is a top research priority. The disturbed function of the hypothalamic-pituitary-ovarian axis in relation to reproductive development, insulin-sensitizing agents, hormonal contraceptives, pregnancy, and lactation remains poorly understood. Human mechanistic studies that examine the various ways these pathways are disrupted will guide new treatments for women with PCOS. Similarly, larger studies that further examine proposed dysfunctional pathways, such as acupuncture to treat sympathetic overactivity in women with PCOS (446), may expand the use of such treatment options. Furthermore, in light of the letrozole study (which broadened a new avenue of infertility treatment in women with PCOS) (447), the development of newer agents that specifically target disrupted hypothalamic-pituitary-ovarian pathways in women with PCOS appears to be promising.
C. Molecular genetics
Identifying genetic variants that would improve the diagnosis of PCOS or variants that would decrease the heterogeneity of the syndrome remains an unfulfilled promise of genetic studies to date. GWAS from other PCOS populations in the world (to date they have been focused on East Asian and Caucasian populations) are anticipated, and the subsequent meta-analyses of these GWAS findings should yield more definitive targets for functional genomic studies. We need further research that identifies the role of the most promising GWAS regions in creating stigmata of the PCOS phenotype. Researchers are just now beginning to explore the functional role of some of the genetic variants linked to PCOS. These genomic findings may lead to a better understanding of the pathophysiology of PCOS. There should be more extensive pharmacogenomics research in women with PCOS. Prospective randomized trials that identify response genes to therapy and their validation will help us better individualize patient care. In addition, future epigenetic studies may provide insight into the relationship between the intrauterine environment and the parental genetic contribution to the development of PCOS.
Acknowledgments
This work was supported by National Institutes of Health Grants 1R01HD056510, 2 U54 HD034449, and 2 U10 HD 38992 and by University of Virginia Grant U54 HD 28934.
Disclosure Summary: D.A.D., S.E.O., and E.S.-V. report no conflicts. J.S.L. reports fees and grant support from Ferring, Genovum, Merck-Serono, Organon, Schering-Plough, and Serono. J.C.M. reports consulting for Astra Zeneca. R.S.L. reports consulting for Astra Zeneca, Euroscreen, Clarus Therapeutics, Takeda, Kindex, and Sprout, with grant support from Ferring and Astra Zeneca.
Footnotes
- AMH
- anti-Müllerian hormone
- BMI
- body mass index
- BPA
- bisphenol A
- CVD
- cardiovascular disease
- DHEA
- dehydroepiandrosterone
- DHEAS
- DHEA sulfate
- FTO
- fat mass and obesity-associated gene
- GDF
- growth differentiation factor
- GLUT
- glucose transporter
- GWAS
- genome-wide association studies
- HA
- hyperandrogenemia
- HRR
- heart rate recovery
- HRV
- heart rate variability
- INS
- insulin
- IRS-1
- insulin receptor substrate-1
- MetS
- metabolic syndrome
- MRI
- magnetic resonance imaging
- NGF
- nerve growth factor
- PCO
- polycystic ovaries
- PCOS
- polycystic ovary syndrome
- SNP
- single nucleotide polymorphism
- T2DM
- type 2 diabetes mellitus
- THADA
- thyroid adenoma-associated.
Reference
- 1. Zawadzki J, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif A, Givens HR, Haseltine FP, Merriam GR, ed. Polycystic Ovary Syndrome. Boston, MA: Blackwell Scientific; 1992:377–384. [Google Scholar]
- 2. Atkins D, Best D, Briss PA, et al. Grading quality of evidence and strength of recommendations. BMJ. 2004;328(7454):1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. NIH Office of Disease Prevention. Evidence-based Methodology Workshop on Polycystic Ovary Syndrome. 2012 Expert Panel Guidelines on PCOS. https://prevention.nih.gov/docs/programs/pcos/FinalReport.pdf Accessed December 3–5, 2012.
- 4. March WA, Moore VM, Willson KJ, Phillips DI, Norman RJ, Davies MJ. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum Reprod. 2010;25(2):544–551. [DOI] [PubMed] [Google Scholar]
- 5. Azziz R, Woods KS, Reyna R, Key TJ, Knochenhauer ES, Yildiz BO. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab. 2004;89(6):2745–2749. [DOI] [PubMed] [Google Scholar]
- 6. Fauser BC, Tarlatzis BC, Rebar RW, et al. Consensus on women's health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil Steril. 2012;97(1):28–38.e25. [DOI] [PubMed] [Google Scholar]
- 7. Welt CK, Gudmundsson JA, Arason G, et al. Characterizing discrete subsets of polycystic ovary syndrome as defined by the Rotterdam criteria: the impact of weight on phenotype and metabolic features. J Clin Endocrinol Metab. 2006;91(12):4842–4848. [DOI] [PubMed] [Google Scholar]
- 8. Carmina E, Chu MC, Longo RA, Rini GB, Lobo RA. Phenotypic variation in hyperandrogenic women influences the findings of abnormal metabolic and cardiovascular risk parameters. J Clin Endocrinol Metab. 2005;90(5):2545–2549. [DOI] [PubMed] [Google Scholar]
- 9. Azziz R, Carmina E, Dewailly D, et al. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab. 2006;91(11):4237–4245. [DOI] [PubMed] [Google Scholar]
- 10. Escobar-Morreale HF, Carmina E, Dewailly D, et al. Epidemiology, diagnosis and management of hirsutism: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update. 2012;18(2):146–170. [DOI] [PubMed] [Google Scholar]
- 11. Martin KA, Chang RJ, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93(4):1105–1120. [DOI] [PubMed] [Google Scholar]
- 12. Deplewski D, Rosenfield RL. Role of hormones in pilosebaceous unit development. Endocr Rev. 2000;21(4):363–392. [DOI] [PubMed] [Google Scholar]
- 13. Carmina E, Koyama T, Chang L, Stanczyk FZ, Lobo RA. Does ethnicity influence the prevalence of adrenal hyperandrogenism and insulin resistance in polycystic ovary syndrome? Am J Obstet Gynecol. 1992;167(6):1807–1812. [DOI] [PubMed] [Google Scholar]
- 14. DeUgarte CM, Woods KS, Bartolucci AA, Azziz R. Degree of facial and body terminal hair growth in unselected black and white women: toward a populational definition of hirsutism. J Clin Endocrinol Metab. 2006;91(4):1345–1350. [DOI] [PubMed] [Google Scholar]
- 15. Hatch R, Rosenfield RL, Kim MH, Tredway D. Hirsutism: implications, etiology, and management. Am J Obstet Gynecol. 1981;140(7):815–830. [DOI] [PubMed] [Google Scholar]
- 16. Zhao X, Ni R, Li L, et al. Defining hirsutism in Chinese women: a cross-sectional study. Fertil Steril. 2011;96(3):792–796. [DOI] [PubMed] [Google Scholar]
- 17. Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab. 2007;92(2):405–413. [DOI] [PubMed] [Google Scholar]
- 18. Kushnir MM, Blamires T, Rockwood AL, et al. Liquid chromatography-tandem mass spectrometry assay for androstenedione, dehydroepiandrosterone, and testosterone with pediatric and adult reference intervals. Clin Chem. 2010;56(7):1138–1147. [DOI] [PubMed] [Google Scholar]
- 19. Legro RS, Schlaff WD, Diamond MP, et al. Total testosterone assays in women with polycystic ovary syndrome: precision and correlation with hirsutism. J Clin Endocrinol Metab. 2010;95(12):5305–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ognibene A, Drake CJ, Jeng KY, et al. A new modular chemiluminescence immunoassay analyser evaluated. Clin Chem Lab Med. 2000;38(3):251–260. [DOI] [PubMed] [Google Scholar]
- 21. Moghetti P, Tosi F, Bonin C, et al. Divergences in insulin resistance between the different phenotypes of the polycystic ovary syndrome. J Clin Endocrinol Metab. 2013;98(4):E628–E637. [DOI] [PubMed] [Google Scholar]
- 22. Azziz R, Sanchez LA, Knochenhauer ES, et al. Androgen excess in women: experience with over 1000 consecutive patients. J Clin Endocrinol Metab. 2004;89(2):453–462. [DOI] [PubMed] [Google Scholar]
- 23. Goodarzi MO, Carmina E, Azziz R. DHEA, DHEAS and PCOS. J Steroid Biochem Mol Biol. 2015;145:213–225. [DOI] [PubMed] [Google Scholar]
- 24. Brennan K, Huang A, Azziz R. Dehydroepiandrosterone sulfate and insulin resistance in patients with polycystic ovary syndrome. Fertil Steril. 2009;91(5):1848–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Azziz R, Ehrmann DA, Legro RS, Fereshetian AG, O'Keefe M, Ghazzi MN. Troglitazone decreases adrenal androgen levels in women with polycystic ovary syndrome. Fertil Steril. 2003;79(4):932–937. [DOI] [PubMed] [Google Scholar]
- 26. Legro RS, Kunselman AR, Demers L, Wang SC, Bentley-Lewis R, Dunaif A. Elevated dehydroepiandrosterone sulfate levels as the reproductive phenotype in the brothers of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2002;87(5):2134–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang A, Brennan K, Azziz R. Prevalence of hyperandrogenemia in the polycystic ovary syndrome diagnosed by the National Institutes of Health 1990 criteria. Fertil Steril. 2010;93(6):1938–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Reilly MW, Taylor AE, Crabtree NJ, et al. Hyperandrogenemia predicts metabolic phenotype in polycystic ovary syndrome: the utility of serum androstenedione. J Clin Endocrinol Metab. 2014;99(3):1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adams J, Polson DW, Franks S. Prevalence of polycystic ovaries in women with anovulation and idiopathic hirsutism. Br Med J (Clin Res Ed). 1986;293(6543):355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jonard S, Robert Y, Cortet-Rudelli C, Pigny P, Decanter C, Dewailly D. Ultrasound examination of polycystic ovaries: is it worth counting the follicles? Hum Reprod. 2003;18(3):598–603. [DOI] [PubMed] [Google Scholar]
- 31. Balen AH, Laven JS, Tan SL, Dewailly D. Ultrasound assessment of the polycystic ovary: international consensus definitions. Hum Reprod Update. 2003;9(6):505–514. [DOI] [PubMed] [Google Scholar]
- 32. Allemand MC, Tummon IS, Phy JL, Foong SC, Dumesic DA, Session DR. Diagnosis of polycystic ovaries by three-dimensional transvaginal ultrasound. Fertil Steril. 2006;85(1):214–219. [DOI] [PubMed] [Google Scholar]
- 33. Dewailly D, Gronier H, Poncelet E, et al. Diagnosis of polycystic ovary syndrome (PCOS): revisiting the threshold values of follicle count on ultrasound and of the serum AMH level for the definition of polycystic ovaries. Hum Reprod. 2011;26(11):3123–3129. [DOI] [PubMed] [Google Scholar]
- 34. Dewailly D, Lujan ME, Carmina E, et al. Definition and significance of polycystic ovarian morphology: a task force report from the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update. 2014;20(3):334–352. [DOI] [PubMed] [Google Scholar]
- 35. Dewailly D, Pigny P, Soudan B, et al. Reconciling the definitions of polycystic ovary syndrome: the ovarian follicle number and serum anti-Müllerian hormone concentrations aggregate with the markers of hyperandrogenism. J Clin Endocrinol Metab. 2010;95(9):4399–4405. [DOI] [PubMed] [Google Scholar]
- 36. Johnstone EB, Rosen MP, Neril R, et al. The polycystic ovary post-Rotterdam: a common, age-dependent finding in ovulatory women without metabolic significance. J Clin Endocrinol Metab. 2010;95(11):4965–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carmina E, Oberfield SE, Lobo RA. The diagnosis of polycystic ovary syndrome in adolescents. Am J Obstet Gynecol. 2010;203(3):201.e1–5. [DOI] [PubMed] [Google Scholar]
- 38. Polson DW, Adams J, Wadsworth J, Franks S. Polycystic ovaries–a common finding in normal women. Lancet. 1988;1(8590):870–872. [DOI] [PubMed] [Google Scholar]
- 39. Kent SC, Gnatuk CL, Kunselman AR, Demers LM, Lee PA, Legro RS. Hyperandrogenism and hyperinsulinism in children of women with polycystic ovary syndrome: a controlled study. J Clin Endocrinol Metab. 2008;93(5):1662–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mes-Krowinkel MG, Louwers YV, Mulders AG, de Jong FH, Fauser BC, Laven JS. Influence of oral contraceptives on anthropomorphometric, endocrine, and metabolic profiles of anovulatory polycystic ovary syndrome patients. Fertil Steril. 2014;101(6):1757–1765.e1. [DOI] [PubMed] [Google Scholar]
- 41. Mulders AG, ten Kate-Booij M, Pal R, et al. Influence of oral contraceptive pills on phenotype expression in women with polycystic ovary syndrome. Reprod Biomed Online. 2005;11(6):690–696. [DOI] [PubMed] [Google Scholar]
- 42. Barber TM, Alvey C, Greenslade T, et al. Patterns of ovarian morphology in polycystic ovary syndrome: a study utilising magnetic resonance imaging. Eur Radiol. 2010;20(5):1207–1213. [DOI] [PubMed] [Google Scholar]
- 43. Leonhardt H, Hellström M, Gull B, et al. Ovarian morphology assessed by magnetic resonance imaging in women with and without polycystic ovary syndrome and associations with antimüllerian hormone, free testosterone, and glucose disposal rate. Fertil Steril. 2014;101(6):1747–1756.e1–e3. [DOI] [PubMed] [Google Scholar]
- 44. Knight PG, Glister C. Local roles of TGF-β superfamily members in the control of ovarian follicle development. Anim Reprod Sci. 2003;78:165–183. [DOI] [PubMed] [Google Scholar]
- 45. Weenen C, Laven JS, Von Bergh AR, et al. Anti-Müllerian hormone expression pattern in the human ovary: potential implications for initial and cyclic follicle recruitment. Mol Hum Reprod. 2004;10(2):77–83. [DOI] [PubMed] [Google Scholar]
- 46. Pellatt L, Hanna L, Brincat M, et al. Granulosa cell production of anti-Müllerian hormone is increased in polycystic ovaries. J Clin Endocrinol Metab. 2007;92(1):240–245. [DOI] [PubMed] [Google Scholar]
- 47. Fallat ME, Siow Y, Marra M, Cook C, Carrillo A. Müllerian-inhibiting substance in follicular fluid and serum: a comparison of patients with tubal factor infertility, polycystic ovary syndrome, and endometriosis. Fertil Steril. 1997;67(5):962–965. [DOI] [PubMed] [Google Scholar]
- 48. La Marca A, Orvieto R, Giulini S, Jasonni VM, Volpe A, De Leo V. Mullerian-inhibiting substance in women with polycystic ovary syndrome: relationship with hormonal and metabolic characteristics. Fertil Steril. 2004;82(4):970–972. [DOI] [PubMed] [Google Scholar]
- 49. Cook CL, Siow Y, Brenner AG, Fallat ME. Relationship between serum Müllerian-inhibiting substance and other reproductive hormones in untreated women with polycystic ovary syndrome and normal women. Fertil Steril. 2002;77(1):141–146. [DOI] [PubMed] [Google Scholar]
- 50. Piouka A, Farmakiotis D, Katsikis I, Macut D, Gerou S, Panidis D. Anti-Mullerian hormone levels reflect severity of PCOS but are negatively influenced by obesity: relationship with increased luteinizing hormone levels. Am J Physiol Endocrinol Metab. 2009;296(2):E238–E243. [DOI] [PubMed] [Google Scholar]
- 51. Homburg R, Ray A, Bhide P, et al. The relationship of serum anti-Mullerian hormone with polycystic ovarian morphology and polycystic ovary syndrome: a prospective cohort study. Hum Reprod. 2013;28(4):1077–1083. [DOI] [PubMed] [Google Scholar]
- 52. Rosenfield RL, Wroblewski K, Padmanabhan V, Littlejohn E, Mortensen M, Ehrmann DA. Antimüllerian hormone levels are independently related to ovarian hyperandrogenism and polycystic ovaries. Fertil Steril. 2012;98(1):242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Broekmans FJ, Knauff EA, Valkenburg O, Laven JS, Eijkemans MJ, Fauser BC. PCOS according to the Rotterdam consensus criteria: Change in prevalence among WHO-II anovulation and association with metabolic factors. BJOG. 2006;113(10):1210–1217. [DOI] [PubMed] [Google Scholar]
- 54. Dumesic DA, Richards JS. Ontogeny of the ovary in polycystic ovary syndrome. Fertil Steril. 2013;100(1):23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Azziz R, Carmina E, Dewailly D, et al. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report. Fertil Steril. 2009;91(2):456–488. [DOI] [PubMed] [Google Scholar]
- 56. Chang WY, Knochenhauer ES, Bartolucci AA, Azziz R. Phenotypic spectrum of polycystic ovary syndrome: clinical and biochemical characterization of the three major clinical subgroups. Fertil Steril. 2005;83(6):1717–1723. [DOI] [PubMed] [Google Scholar]
- 57. Burgers JA, Fong SL, Louwers YV, et al. Oligoovulatory and anovulatory cycles in women with polycystic ovary syndrome (PCOS): what's the difference? J Clin Endocrinol Metab. 2010;95(12):E485–E489. [DOI] [PubMed] [Google Scholar]
- 58. Legro RS, Barnhart HX, Schlaff WD, et al. Clomiphene, metformin, or both for infertility in the polycystic ovary syndrome. N Engl J Med. 2007;356(6):551–566. [DOI] [PubMed] [Google Scholar]
- 59. Ibáñez L, Dimartino-Nardi J, Potau N, Saenger P. Premature adrenarche–normal variant or forerunner of adult disease? Endocr Rev. 2000;21(6):671–696. [DOI] [PubMed] [Google Scholar]
- 60. Abbott DH, Tarantal AF, Dumesic DA. Fetal, infant, adolescent and adult phenotypes of polycystic ovary syndrome in prenatally androgenized female rhesus monkeys. Am J Primatol. 2009;71(9):776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ibáñez L, López-Bermejo A, Díaz M, Marcos MV, de Zegher F. Early metformin therapy (age 8–12 years) in girls with precocious pubarche to reduce hirsutism, androgen excess, and oligomenorrhea in adolescence. J Clin Endocrinol Metab. 2011;96(8):E1262–E1267. [DOI] [PubMed] [Google Scholar]
- 62. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. 2011;96(6):1610–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zimmet P, Alberti G, Kaufman F, et al. The metabolic syndrome in children and adolescents. Lancet. 2007;369(9579):2059–2061. [DOI] [PubMed] [Google Scholar]
- 64. Ibáñez L, Potau N, Chacon P, Pascual C, Carrascosa A. Hyperinsulinaemia, dyslipaemia and cardiovascular risk in girls with a history of premature pubarche. Diabetologia. 1998;41(9):1057–1063. [DOI] [PubMed] [Google Scholar]
- 65. ACOG Committee on Adolescent Health Care. ACOG Committee Opinion no. 349, November 2006: menstruation in girls and adolescents: using the menstrual cycle as a vital sign. Obstet Gynecol. 2006;108(5):1323–1328. [DOI] [PubMed] [Google Scholar]
- 66. Apter D, Vihko R. Early menarche, a risk factor for breast cancer, indicates early onset of ovulatory cycles. J Clin Endocrinol Metab. 1983;57(1):82–86. [DOI] [PubMed] [Google Scholar]
- 67. van Hooff MH, Voorhorst FJ, Kaptein MB, Hirasing RA, Koppenaal C, Schoemaker J. Predictive value of menstrual cycle pattern, body mass index, hormone levels and polycystic ovaries at age 15 years for oligo-amenorrhoea at age 18 years. Hum Reprod. 2004;19(2):383–392. [DOI] [PubMed] [Google Scholar]
- 68. Mortensen M, Rosenfield RL, Littlejohn E. Functional significance of polycystic-size ovaries in healthy adolescents. J Clin Endocrinol Metab. 2006;91(10):3786–3790. [DOI] [PubMed] [Google Scholar]
- 69. Hickey M, Sloboda DM, Atkinson HC, et al. The relationship between maternal and umbilical cord androgen levels and polycystic ovary syndrome in adolescence: a prospective cohort study. J Clin Endocrinol Metab. 2009;94(10):3714–3720. [DOI] [PubMed] [Google Scholar]
- 70. Merino PM, Codner E, Cassorla F. A rational approach to the diagnosis of polycystic ovarian syndrome during adolescence. Arq Bras Endocrinol Metabol. 2011;55(8):590–598. [DOI] [PubMed] [Google Scholar]
- 71. Villarroel C, Merino PM, López P, et al. Polycystic ovarian morphology in adolescents with regular menstrual cycles is associated with elevated anti-Mullerian hormone. Hum Reprod. 2011;26(10):2861–2868. [DOI] [PubMed] [Google Scholar]
- 72. Shah B, Parnell L, Milla S, Kessler M, David R. Endometrial thickness, uterine, and ovarian ultrasonographic features in adolescents with polycystic ovarian syndrome. J Pediatr Adolesc Gynecol. 2010;23(3):146–152. [DOI] [PubMed] [Google Scholar]
- 73. Villa P, Rossodivita A, Sagnella F, et al. Ovarian volume and gluco-insulinaemic markers in the diagnosis of PCOS during adolescence. Clin Endocrinol (Oxf). 2013;78(2):285–290. [DOI] [PubMed] [Google Scholar]
- 74. Carreau AM, Baillargeon JP. PCOS in adolescence and type 2 diabetes. Curr Diab Rep. 2015;15(1):564. [DOI] [PubMed] [Google Scholar]
- 75. Roe AH, Prochaska E, Smith M, Sammel M, Dokras A. Using the androgen excess-PCOS society criteria to diagnose polycystic ovary syndrome and the risk of metabolic syndrome in adolescents. J Pediatr. 2013;162(5):937–941. [DOI] [PubMed] [Google Scholar]
- 76. Elting MW, Korsen TJ, Rekers-Mombarg LT, Schoemaker J. Women with polycystic ovary syndrome gain regular menstrual cycles when ageing. Hum Reprod. 2000;15(1):24–28. [DOI] [PubMed] [Google Scholar]
- 77. Elting MW, Kwee J, Korsen TJ, Rekers-Mombarg LT, Schoemaker J. Aging women with polycystic ovary syndrome who achieve regular menstrual cycles have a smaller follicle cohort than those who continue to have irregular cycles. Fertil Steril. 2003;79(5):1154–1160. [DOI] [PubMed] [Google Scholar]
- 78. Schmidt J, Brännström M, Landin-Wilhelmsen K, Dahlgren E. Reproductive hormone levels and anthropometry in postmenopausal women with polycystic ovary syndrome (PCOS): a 21-year follow-up study of women diagnosed with PCOS around 50 years ago and their age-matched controls. J Clin Endocrinol Metab. 2011;96(7):2178–2185. [DOI] [PubMed] [Google Scholar]
- 79. Carmina E, Campagna AM, Lobo RA. Emergence of ovulatory cycles with aging in women with polycystic ovary syndrome (PCOS) alters the trajectory of cardiovascular and metabolic risk factors. Hum Reprod. 2013;28(8):2245–2252. [DOI] [PubMed] [Google Scholar]
- 80. Alsamarai S, Adams JM, Murphy MK, et al. Criteria for polycystic ovarian morphology in polycystic ovary syndrome as a function of age. J Clin Endocrinol Metab. 2009;94(12):4961–4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sowers MR, Eyvazzadeh AD, McConnell D, et al. Anti-mullerian hormone and inhibin B in the definition of ovarian aging and the menopause transition. J Clin Endocrinol Metab. 2008;93(9):3478–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Puurunen J, Piltonen T, Morin-Papunen L, et al. Unfavorable hormonal, metabolic, and inflammatory alterations persist after menopause in women with PCOS. J Clin Endocrinol Metab. 2011;96(6):1827–1834. [DOI] [PubMed] [Google Scholar]
- 83. Solomon CG, Hu FB, Dunaif A, et al. Menstrual cycle irregularity and risk for future cardiovascular disease. J Clin Endocrinol Metab. 2002;87(5):2013–2017. [DOI] [PubMed] [Google Scholar]
- 84. Krentz AJ, von Mühlen D, Barrett-Connor E. Searching for polycystic ovary syndrome in postmenopausal women: evidence of a dose-effect association with prevalent cardiovascular disease. Menopause. 2007;14(2):284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shaw LJ, Bairey Merz CN, Azziz R, et al. Postmenopausal women with a history of irregular menses and elevated androgen measurements at high risk for worsening cardiovascular event-free survival: results from the National Institutes of Health–National Heart, Lung, and Blood Institute sponsored Women's Ischemia Syndrome Evaluation. J Clin Endocrinol Metab. 2008;93(4):1276–1284. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 86. Polotsky AJ, Allshouse AA, Crawford SL, et al. Hyperandrogenic oligomenorrhea and metabolic risks across menopausal transition. J Clin Endocrinol Metab. 2014;99(6):2120–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Diamanti-Kandarakis E. Role of obesity and adiposity in polycystic ovary syndrome. Int J Obes (Lond). 2007;31(suppl 2):S8–S13; discussion S31–S32. [DOI] [PubMed] [Google Scholar]
- 88. Hoeger KM, Oberfield SE. Do women with PCOS have a unique predisposition to obesity? Fertil Steril. 2012;97(1):13–17. [DOI] [PubMed] [Google Scholar]
- 89. Legro RS. The genetics of obesity. Lessons for polycystic ovary syndrome. Ann NY Acad Sci. 2000;900:193–202. [DOI] [PubMed] [Google Scholar]
- 90. Legro RS. Obesity and PCOS: implications for diagnosis and treatment. Semin Reprod Med. 2012;30(6):496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ehrmann DA, Barnes RB, Rosenfield RL, Cavaghan MK, Imperial J. Prevalence of impaired glucose tolerance and diabetes in women with polycystic ovary syndrome. Diabetes Care. 1999;22(1):141–146. [DOI] [PubMed] [Google Scholar]
- 92. Legro RS, Kunselman AR, Dodson WC, Dunaif A. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999;84(1):165–169. [DOI] [PubMed] [Google Scholar]
- 93. Alvarez-Blasco F, Botella-Carretero JI, San Millán JL, Escobar-Morreale HF. Prevalence and characteristics of the polycystic ovary syndrome in overweight and obese women. Arch Intern Med. 2006;166(19):2081–2086. [DOI] [PubMed] [Google Scholar]
- 94. Balen AH, Conway GS, Kaltsas G, et al. Polycystic ovary syndrome: the spectrum of the disorder in 1741 patients. Hum Reprod. 1995;10(8):2107–2111. [DOI] [PubMed] [Google Scholar]
- 95. Kiddy DS, Hamilton-Fairley D, Bush A, et al. Improvement in endocrine and ovarian function during dietary treatment of obese women with polycystic ovary syndrome. Clin Endocrinol (Oxf). 1992;36(1):105–111. [DOI] [PubMed] [Google Scholar]
- 96. Yildiz BO, Knochenhauer ES, Azziz R. Impact of obesity on the risk for polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93(1):162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Moran LJ, Misso ML, Wild RA, Norman RJ. Impaired glucose tolerance, type 2 diabetes and metabolic syndrome in polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod Update. 2010;16(4):347–363. [DOI] [PubMed] [Google Scholar]
- 98. Baranova A, Tran TP, Birerdinc A, Younossi ZM. Systematic review: association of polycystic ovary syndrome with metabolic syndrome and non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2011;33(7):801–814. [DOI] [PubMed] [Google Scholar]
- 99. Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes. 1989;38(9):1165–1174. [DOI] [PubMed] [Google Scholar]
- 100. Pasquali R, Casimirri F, Venturoli S, et al. Body fat distribution has weight-independent effects on clinical, hormonal, and metabolic features of women with polycystic ovary syndrome. Metabolism. 1994;43(6):706–713. [DOI] [PubMed] [Google Scholar]
- 101. Pasquali R, Gambineri A, Biscotti D, et al. Effect of long-term treatment with metformin added to hypocaloric diet on body composition, fat distribution, and androgen and insulin levels in abdominally obese women with and without the polycystic ovary syndrome. J Clin Endocrinol Metab. 2000;85(8):2767–2774. [DOI] [PubMed] [Google Scholar]
- 102. Kirchengast S, Huber J. Body composition characteristics and body fat distribution in lean women with polycystic ovary syndrome. Hum Reprod. 2001;16(6):1255–1260. [DOI] [PubMed] [Google Scholar]
- 103. Good C, Tulchinsky M, Mauger D, Demers LM, Legro RS. Bone mineral density and body composition in lean women with polycystic ovary syndrome. Fertil Steril. 1999;72(1):21–25. [DOI] [PubMed] [Google Scholar]
- 104. Mannerås-Holm L, Leonhardt H, Kullberg J, et al. Adipose tissue has aberrant morphology and function in PCOS: enlarged adipocytes and low serum adiponectin, but not circulating sex steroids, are strongly associated with insulin resistance. J Clin Endocrinol Metab. 2011;96(2):E304–E311. [DOI] [PubMed] [Google Scholar]
- 105. Barber TM, Golding SJ, Alvey C, et al. Global adiposity rather than abnormal regional fat distribution characterizes women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93(3):999–1004. [DOI] [PubMed] [Google Scholar]
- 106. Glueck CJ, Morrison JA, Daniels S, Wang P, Stroop D. Sex hormone-binding globulin, oligomenorrhea, polycystic ovary syndrome, and childhood insulin at age 14 years predict metabolic syndrome and class III obesity at age 24 years. J Pediatr. 2011;159(2):308–313.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med. 2001;7(8):947–953. [DOI] [PubMed] [Google Scholar]
- 108. Dunaif A, Segal KR, Shelley DR, Green G, Dobrjansky A, Licholai T. Evidence for distinctive and intrinsic defects in insulin action in polycystic ovary syndrome. Diabetes. 1992;41(10):1257–1266. [DOI] [PubMed] [Google Scholar]
- 109. Ek I, Arner P, Bergqvist A, Carlström K, Wahrenberg H. Impaired adipocyte lipolysis in nonobese women with the polycystic ovary syndrome: a possible link to insulin resistance? J Clin Endocrinol Metab. 1997;82(4):1147–1153. [DOI] [PubMed] [Google Scholar]
- 110. Legro RS, Dodson WC, Gnatuk CL, et al. Effects of gastric bypass surgery on female reproductive function. J Clin Endocrinol Metab. 2012;97(12):4540–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Escobar-Morreale HF, Botella-Carretero JI, Alvarez-Blasco F, Sancho J, San Millán JL. The polycystic ovary syndrome associated with morbid obesity may resolve after weight loss induced by bariatric surgery. J Clin Endocrinol Metab. 2005;90(12):6364–6369. [DOI] [PubMed] [Google Scholar]
- 112. Eid GM, Cottam DR, Velcu LM, et al. Effective treatment of polycystic ovarian syndrome with Roux-en-Y gastric bypass. Surg Obes Relat Dis. 2005;1(2):77–80. [DOI] [PubMed] [Google Scholar]
- 113. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Harrison CL, Lombard CB, Moran LJ, Teede HJ. Exercise therapy in polycystic ovary syndrome: a systematic review. Hum Reprod Update. 2011;17(2):171–183. [DOI] [PubMed] [Google Scholar]
- 115. Teede HJ, Misso ML, Deeks AA, et al. Assessment and management of polycystic ovary syndrome: summary of an evidence-based guideline. Med J Aust. 2011;195(6):S65–S112. [DOI] [PubMed] [Google Scholar]
- 116. Moran LJ, Hutchison SK, Norman RJ, Teede HJ. Lifestyle changes in women with polycystic ovary syndrome. Cochrane Database Syst Rev. 2011(7):CD007506. [DOI] [PubMed] [Google Scholar]
- 117. Domecq JP, Prutsky G, Mullan RJ, et al. Lifestyle modification programs in polycystic ovary syndrome: systematic review and meta-analysis. J Clin Endocrinol Metab. 2013;98(12):4655–4663. [DOI] [PubMed] [Google Scholar]
- 118. Segal KR, Dunaif A. Resting metabolic rate and postprandial thermogenesis in polycystic ovarian syndrome. Int J Obes. 1990;14(7):559–567. [PubMed] [Google Scholar]
- 119. Robinson S, Chan SP, Spacey S, Anyaoku V, Johnston DG, Franks S. Postprandial thermogenesis is reduced in polycystic ovary syndrome and is associated with increased insulin resistance. Clin Endocrinol (Oxf). 1992;36(6):537–543. [DOI] [PubMed] [Google Scholar]
- 120. Tang T, Lord JM, Norman RJ, Yasmin E, Balen AH. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst Rev. 2012;5:CD003053. [DOI] [PubMed] [Google Scholar]
- 121. Azziz R, Ehrmann D, Legro RS. Troglitazone improves ovulation and hirsutism in the polycystic ovary syndrome: a multicenter, double blind, placebo-controlled trial. J Clin Endocrinol Metab. 2001;86(4):1626–1632. [DOI] [PubMed] [Google Scholar]
- 122. Chen ZJ, Zhao H, He L. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat Genet. 2011;43(1):55–59. [DOI] [PubMed] [Google Scholar]
- 123. Goodarzi MO, Jones MR, Li X, et al. Replication of association of DENND1A and THADA variants with polycystic ovary syndrome in European cohorts. J Med Genet. 2012;49(2):90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Welt CK, Styrkarsdottir U, Ehrmann DA, et al. Variants in DENND1A are associated with polycystic ovary syndrome in women of European ancestry. J Clin Endocrinol Metab. 2012;97(7):E1342–E1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Casarini L, Brigante G. The polycystic ovary syndrome evolutionary paradox: a genome-wide association studies-based, in silico, evolutionary explanation. J Clin Endocrinol Metab. 2014;99(11):E2412–E2420. [DOI] [PubMed] [Google Scholar]
- 126. Corbett S, Morin-Papunen L. The polycystic ovary syndrome and recent human evolution. Mol Cell Endocrinol. 2013;373(1–2):39–50. [DOI] [PubMed] [Google Scholar]
- 127. Goodarzi MO, Quiñones MJ, Azziz R, Rotter JI, Hsueh WA, Yang H. Polycystic ovary syndrome in Mexican-Americans: prevalence and association with the severity of insulin resistance. Fertil Steril. 2005;84(3):766–769. [DOI] [PubMed] [Google Scholar]
- 128. Ladson G, Dodson WC, Sweet SD, et al. Racial influence on the polycystic ovary syndrome phenotype: a black and white case-control study. Fertil Steril. 2011;96(1):224–229.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Park HR, Choi Y, Lee HJ, Oh JY, Hong YS, Sung YA. Phenotypic characteristics according to insulin sensitivity in non-obese Korean women with polycystic ovary syndrome. Diabetes Res Clin Pract. 2007;77(suppl 1):S233–S237. [DOI] [PubMed] [Google Scholar]
- 130. Sorbara LR, Tang Z, Cama A, et al. Absence of insulin receptor gene mutations in three insulin-resistant women with the polycystic ovary syndrome. Metabolism. 1994;43(12):1568–1574. [DOI] [PubMed] [Google Scholar]
- 131. Welt CK, Arason G, Gudmundsson JA, et al. Defining constant versus variable phenotypic features of women with polycystic ovary syndrome using different ethnic groups and populations. J Clin Endocrinol Metab. 2006;91(11):4361–4368. [DOI] [PubMed] [Google Scholar]
- 132. Dunaif A, Sorbara L, Delson R, Green G. Ethnicity and polycystic ovary syndrome are associated with independent and additive decreases in insulin action in Caribbean-Hispanic women. Diabetes. 1993;42(10):1462–1468. [DOI] [PubMed] [Google Scholar]
- 133. Wijeyaratne CN, Balen AH, Barth JH, Belchetz PE. Clinical manifestations and insulin resistance (IR) in polycystic ovary syndrome (PCOS) among South Asians and Caucasians: is there a difference? Clin Endocrinol (Oxf). 2002;57(3):343–350. [DOI] [PubMed] [Google Scholar]
- 134. Ehrmann DA, Liljenquist DR, Kasza K, Azziz R, Legro RS, Ghazzi MN. Prevalence and predictors of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91(1):48–53. [DOI] [PubMed] [Google Scholar]
- 135. Lo JC, Feigenbaum SL, Yang J, Pressman AR, Selby JV, Go AS. Epidemiology and adverse cardiovascular risk profile of diagnosed polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91(4):1357–1363. [DOI] [PubMed] [Google Scholar]
- 136. Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30(4):293–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Takeuchi T, Tsutsumi O, Ikezuki Y, Takai Y, Taketani Y. Positive relationship between androgen and the endocrine disruptor, bisphenol A, in normal women and women with ovarian dysfunction. Endocr J. 2004;51(2):165–169. [DOI] [PubMed] [Google Scholar]
- 138. Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A. The estrogenic effect of bisphenol A disrupts pancreatic β-cell function in vivo and induces insulin resistance. Environ Health Perspect. 2006;114(1):106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Newbold RR, Jefferson WN, Padilla-Banks E. Prenatal exposure to bisphenol A at environmentally relevant doses adversely affects the murine female reproductive tract later in life. Environ Health Perspect. 2009;117(6):879–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zhou W, Liu J, Liao L, Han S. Effect of bisphenol A on steroid hormone production in rat ovarian theca-interstitial and granulosa cells. Mol Cell Endocrinol. 2008;283:12–18. [DOI] [PubMed] [Google Scholar]
- 141. Fernández M, Bourguignon N, Lux-Lantos V, Libertun C. Neonatal exposure to bisphenol A and reproductive and endocrine alterations resembling the polycystic ovarian syndrome in adult rats. Environ Health Perspect. 2010;118(9):1217–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kandaraki E, Chatzigeorgiou A, Livadas S, et al. Endocrine disruptors and polycystic ovary syndrome (PCOS): elevated serum levels of bisphenol A in women with PCOS. J Clin Endocrinol Metab. 2011;96(3):E480–E484. [DOI] [PubMed] [Google Scholar]
- 143. Legro RS, Kunselman AR, Dunaif A. Prevalence and predictors of dyslipidemia in women with polycystic ovary syndrome. Am J Med. 2001;111(8):607–613. [DOI] [PubMed] [Google Scholar]
- 144. Macut D, Damjanovic S, Panidis D, et al. Oxidised low-density lipoprotein concentration - early marker of an altered lipid metabolism in young women with PCOS. Eur J Endocrinol. 2006;155(1):131–136. [DOI] [PubMed] [Google Scholar]
- 145. Taponen S, Martikainen H, Järvelin MR, et al. Metabolic cardiovascular disease risk factors in women with self-reported symptoms of oligomenorrhea and/or hirsutism: Northern Finland Birth Cohort 1966 Study. J Clin Endocrinol Metab. 2004;89(5):2114–2118. [DOI] [PubMed] [Google Scholar]
- 146. Legro RS, Gnatuk CL, Kunselman AR, Dunaif A. Changes in glucose tolerance over time in women with polycystic ovary syndrome: a controlled study. J Clin Endocrinol Metab. 2005;90(6):3236–3242. [DOI] [PubMed] [Google Scholar]
- 147. Norman RJ, Masters L, Milner CR, Wang JX, Davies MJ. Relative risk of conversion from normoglycaemia to impaired glucose tolerance or non-insulin dependent diabetes mellitus in polycystic ovarian syndrome. Hum Reprod. 2001;16(9):1995–1998. [DOI] [PubMed] [Google Scholar]
- 148. Boudreaux MY, Talbott EO, Kip KE, Brooks MM, Witchel SF. Risk of T2DM and impaired fasting glucose among PCOS subjects: results of an 8-year follow-up. Curr Diab Rep. 2006;6(1):77–83. [DOI] [PubMed] [Google Scholar]
- 149. Valkenburg O, Steegers-Theunissen RP, Smedts HP, et al. A more atherogenic serum lipoprotein profile is present in women with polycystic ovary syndrome: a case-control study. J Clin Endocrinol Metab. 2008;93(2):470–476. [DOI] [PubMed] [Google Scholar]
- 150. Wild RA, Carmina E, Diamanti-Kandarakis E, et al. Assessment of cardiovascular risk and prevention of cardiovascular disease in women with the polycystic ovary syndrome: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) Society. J Clin Endocrinol Metab. 2010;95(5):2038–2049. [DOI] [PubMed] [Google Scholar]
- 151. Apridonidze T, Essah PA, Iuorno MJ, Nestler JE. Prevalence and characteristics of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90(4):1929–1935. [DOI] [PubMed] [Google Scholar]
- 152. Dokras A, Bochner M, Hollinrake E, Markham S, Vanvoorhis B, Jagasia DH. Screening women with polycystic ovary syndrome for metabolic syndrome. Obstet Gynecol. 2005;106(1):131–137. [DOI] [PubMed] [Google Scholar]
- 153. Carmina E, Napoli N, Longo RA, Rini GB, Lobo RA. Metabolic syndrome in polycystic ovary syndrome (PCOS): lower prevalence in southern Italy than in the USA and the influence of criteria for the diagnosis of PCOS. Eur J Endocrinol. 2006;154(1):141–145. [DOI] [PubMed] [Google Scholar]
- 154. Essah PA, Nestler JE, Carmina E. Differences in dyslipidemia between American and Italian women with polycystic ovary syndrome. J Endocrinol Invest. 2008;31(1):35–41. [DOI] [PubMed] [Google Scholar]
- 155. Rizzo M, Berneis K, Hersberger M, et al. Milder forms of atherogenic dyslipidemia in ovulatory versus anovulatory polycystic ovary syndrome phenotype. Hum Reprod. 2009;24(9):2286–2292. [DOI] [PubMed] [Google Scholar]
- 156. Berneis K, Rizzo M, Hersberger M, et al. Atherogenic forms of dyslipidaemia in women with polycystic ovary syndrome. Int J Clin Pract. 2009;63(1):56–62. [DOI] [PubMed] [Google Scholar]
- 157. Roe A, Hillman J, Butts S, et al. Decreased cholesterol efflux capacity and atherogenic lipid profile in young women with PCOS. J Clin Endocrinol Metab. 2014;99(5):E841–E847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Wang ET, Ku IA, Shah SJ, et al. Polycystic ovary syndrome is associated with higher left ventricular mass index: the CARDIA women's study. J Clin Endocrinol Metab. 2012;97(12):4656–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Paradisi G, Steinberg HO, Hempfling A, et al. Polycystic ovary syndrome is associated with endothelial dysfunction. Circulation. 2001;103(10):1410–1415. [DOI] [PubMed] [Google Scholar]
- 160. Kravariti M, Naka KK, Kalantaridou SN, et al. Predictors of endothelial dysfunction in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90(9):5088–5095. [DOI] [PubMed] [Google Scholar]
- 161. Soares GM, Vieira CS, Martins WP, et al. Increased arterial stiffness in nonobese women with polycystic ovary syndrome (PCOS) without comorbidities: one more characteristic inherent to the syndrome? Clin Endocrinol (Oxf). 2009;71(3):406–411. [DOI] [PubMed] [Google Scholar]
- 162. Talbott EO, Guzick DS, Sutton-Tyrrell K, et al. Evidence for association between polycystic ovary syndrome and premature carotid atherosclerosis in middle-aged women. Arterioscler Thromb Vasc Biol. 2000;20(11):2414–2421. [DOI] [PubMed] [Google Scholar]
- 163. Luque-Ramírez M, Mendieta-Azcona C, Alvarez-Blasco F, Escobar-Morreale HF. Androgen excess is associated with the increased carotid intima-media thickness observed in young women with polycystic ovary syndrome. Hum Reprod. 2007;22(12):3197–3203. [DOI] [PubMed] [Google Scholar]
- 164. Christian RC, Dumesic DA, Behrenbeck T, Oberg AL, Sheedy PF, 2nd, Fitzpatrick LA. Prevalence and predictors of coronary artery calcification in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88(6):2562–2568. [DOI] [PubMed] [Google Scholar]
- 165. Talbott EO, Zborowski JV, Rager JR, Boudreaux MY, Edmundowicz DA, Guzick DS. Evidence for an association between metabolic cardiovascular syndrome and coronary and aortic calcification among women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89(11):5454–5461. [DOI] [PubMed] [Google Scholar]
- 166. Chang AY, Ayers C, Minhajuddin A, et al. Polycystic ovarian syndrome and subclinical atherosclerosis among women of reproductive age in the Dallas Heart Study. Clin Endocrinol (Oxf). 2011;74(1):89–96. [DOI] [PubMed] [Google Scholar]
- 167. Cibula D, Cífková R, Fanta M, Poledne R, Zivny J, Skibová J. Increased risk of non-insulin dependent diabetes mellitus, arterial hypertension and coronary artery disease in perimenopausal women with a history of the polycystic ovary syndrome. Hum Reprod. 2000;15(4):785–789. [DOI] [PubMed] [Google Scholar]
- 168. Elting MW, Korsen TJ, Bezemer PD, Schoemaker J. Prevalence of diabetes mellitus, hypertension and cardiac complaints in a follow-up study of a Dutch PCOS population. Hum Reprod. 2001;16(3):556–560. [DOI] [PubMed] [Google Scholar]
- 169. Wild S, Pierpoint T, McKeigue P, Jacobs H. Cardiovascular disease in women with polycystic ovary syndrome at long-term follow-up: a retrospective cohort study. Clin Endocrinol (Oxf). 2000;52(5):595–600. [DOI] [PubMed] [Google Scholar]
- 170. Pierpoint T, McKeigue PM, Isaacs AJ, Wild SH, Jacobs HS. Mortality of women with polycystic ovary syndrome at long-term follow-up. J Clin Epidemiol. 1998;51(7):581–586. [DOI] [PubMed] [Google Scholar]
- 171. Azevedo GD, Duarte JM, Souza MO, Costa-E-Silva TD, Soares EM, Maranhão TM. Menstrual cycle irregularity as a marker of cardiovascular risk factors at postmenopausal years [in Portuguese]. Arq Bras Endocrinol Metabol. 2006;50(5):876–883. [DOI] [PubMed] [Google Scholar]
- 172. Withdrawn by author: retraction of postmenopausal women with a history of irregular menses and elevated androgen measurements at high risk for worsening cardiovascular event-free survival: results from the National Institutes of Health–National Heart, Lung, and Blood Institute sponsored by Women's Ischemia Syndrome Evaluation. J Clin Endocrinol Metab. 2015;100(3):1206. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 173. Anderson SA, Barry JA, Hardiman PJ. Risk of coronary heart disease and risk of stroke in women with polycystic ovary syndrome: a systematic review and meta-analysis. Int J Cardiol. 2014;176(2):486–487. [DOI] [PubMed] [Google Scholar]
- 174. Talbott E, Clerici A, Berga SL, et al. Adverse lipid and coronary heart disease risk profiles in young women with polycystic ovary syndrome: results of a case-control study. J Clin Epidemiol. 1998;51(5):415–422. [DOI] [PubMed] [Google Scholar]
- 175. Chittenden BG, Fullerton G, Maheshwari A, Bhattacharya S. Polycystic ovary syndrome and the risk of gynaecological cancer: a systematic review. Reprod Biomed Online. 2009;19(3):398–405. [DOI] [PubMed] [Google Scholar]
- 176. Haoula Z, Salman M, Atiomo W. Evaluating the association between endometrial cancer and polycystic ovary syndrome. Hum Reprod. 2012;27(5):1327–1331. [DOI] [PubMed] [Google Scholar]
- 177. Hardiman P, Pillay OC, Atiomo W. Polycystic ovary syndrome and endometrial carcinoma. Lancet. 2003;361(9371):1810–1812. [DOI] [PubMed] [Google Scholar]
- 178. Fader AN, Arriba LN, Frasure HE, von Gruenigen VE. Endometrial cancer and obesity: epidemiology, biomarkers, prevention and survivorship. Gynecol Oncol. 2009;114(1):121–127. [DOI] [PubMed] [Google Scholar]
- 179. Coulam CB, Annegers JF, Kranz JS. Chronic anovulation syndrome and associated neoplasia. Obstet Gynecol. 1983;61(4):403–407. [PubMed] [Google Scholar]
- 180. Schindler AE. Progestogen deficiency and endometrial cancer risk. Maturitas. 2009;62(4):334–337. [DOI] [PubMed] [Google Scholar]
- 181. Savaris RF, Groll JM, Young SL, et al. Progesterone resistance in PCOS endometrium: a microarray analysis in clomiphene citrate-treated and artificial menstrual cycles. J Clin Endocrinol Metab. 2011;96(6):1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Schildkraut JM, Schwingl PJ, Bastos E, Evanoff A, Hughes C. Epithelial ovarian cancer risk among women with polycystic ovary syndrome. Obstet Gynecol. 1996;88:554–559. [DOI] [PubMed] [Google Scholar]
- 183. Dokras A. Mood and anxiety disorders in women with PCOS. Steroids. 2012;77(4):338–341. [DOI] [PubMed] [Google Scholar]
- 184. Elsenbruch S, Hahn S, Kowalsky D, et al. Quality of life, psychosocial well-being, and sexual satisfaction in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88(12):5801–5807. [DOI] [PubMed] [Google Scholar]
- 185. de Niet JE, de Koning CM, Pastoor H, et al. Psychological well-being and sexarche in women with polycystic ovary syndrome. Hum Reprod. 2010;25(6):1497–1503. [DOI] [PubMed] [Google Scholar]
- 186. Matsunaga H, Sarai M. Elevated serum LH and androgens in affective disorder related to the menstrual cycle: with reference to polycystic ovary syndrome. Jpn J Psychiatry Neurol. 1993;47(4):825–842. [DOI] [PubMed] [Google Scholar]
- 187. Rasgon NL, Altshuler LL, Fairbanks L, et al. Reproductive function and risk for PCOS in women treated for bipolar disorder. Bipolar Disord. 2005;7(3):246–259. [DOI] [PubMed] [Google Scholar]
- 188. Joffe H, Cohen LS, Suppes T, et al. Valproate is associated with new-onset oligoamenorrhea with hyperandrogenism in women with bipolar disorder. Biol Psychiatry. 2006;59(11):1078–1086. [DOI] [PubMed] [Google Scholar]
- 189. Isojärvi JI, Laatikainen TJ, Pakarinen AJ, Juntunen KT, Myllylä VV. Polycystic ovaries and hyperandrogenism in women taking valproate for epilepsy. N Engl J Med. 1993;329(19):1383–1388. [DOI] [PubMed] [Google Scholar]
- 190. Nelson-DeGrave VL, Wickenheisser JK, Cockrell JE, et al. Valproate potentiates androgen biosynthesis in human ovarian theca cells. Endocrinology. 2004;145(2):799–808. [DOI] [PubMed] [Google Scholar]
- 191. Legro RS, Driscoll D, Strauss JF, 3rd, Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci USA. 1998;95(25):14956–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Franks S, Webber LJ, Goh M, et al. Ovarian morphology is a marker of heritable biochemical traits in sisters with polycystic ovaries. J Clin Endocrinol Metab. 2008;93(9):3396–3402. [DOI] [PubMed] [Google Scholar]
- 193. Sir-Petermann T, Codner E, Pérez V, et al. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94(6):1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Sir-Petermann T, Ladrónde Guevara A, Codner E, et al. Relationship between anti-Müllerian hormone (AMH) and insulin levels during different Tanner stages in daughters of women with polycystic ovary syndrome. Reprod Sci. 2012;19(4):383–390. [DOI] [PubMed] [Google Scholar]
- 195. Rosenfield RL. Ovarian and adrenal function in polycystic ovary syndrome. Endocrinol Metab Clin North Am. 1999;28(2):265–293. [DOI] [PubMed] [Google Scholar]
- 196. Burger HG. Androgen production in women. Fertil Steril. 2002;77(suppl 4):S3–S5. [DOI] [PubMed] [Google Scholar]
- 197. Ehrmann DA, Barnes RB, Rosenfield RL. Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocr Rev. 1995;16(3):322–353. [DOI] [PubMed] [Google Scholar]
- 198. McGee AE, Strauss JF., III Ovarian hormone synthesis. In: Jameson JL, De Groot LJ, eds.: Endocrinology: Adult and Pediatric (7th ed). Philadelphia: Saunders/Elsevier; 2016:2192–2206. [Google Scholar]
- 199. Azziz R, Fox LM, Zacur HA, Parker CR, Jr, Boots LR. Adrenocortical secretion of dehydroepiandrosterone in healthy women: highly variable response to adrenocorticotropin. J Clin Endocrinol Metab. 2001;86(6):2513–2517. [DOI] [PubMed] [Google Scholar]
- 200. Moran C, Reyna R, Boots LS, Azziz R. Adrenocortical hyperresponsiveness to corticotropin in polycystic ovary syndrome patients with adrenal androgen excess. Fertil Steril. 2004;81(1):126–131. [DOI] [PubMed] [Google Scholar]
- 201. Zumoff B, Freeman R, Coupey S, Saenger P, Markowitz M, Kream J. A chronobiologic abnormality in luteinizing hormone secretion in teenage girls with the polycystic-ovary syndrome. N Engl J Med. 1983;309(20):1206–1209. [DOI] [PubMed] [Google Scholar]
- 202. Kazer RR, Kessel B, Yen SS. Circulating luteinizing hormone pulse frequency in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1987;65(2):233–236. [DOI] [PubMed] [Google Scholar]
- 203. Walters KA, Allan CM, Handelsman DJ. Rodent models for human polycystic ovary syndrome. Biol Reprod. 2012;86(5):149, 1–12. [DOI] [PubMed] [Google Scholar]
- 204. Sullivan SD, Moenter SM. Prenatal androgens alter GABAergic drive to gonadotropin-releasing hormone neurons: implications for a common fertility disorder. Proc Natl Acad Sci USA. 2004;101(18):7129–7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Moore AM, Prescott M, Marshall CJ, Yip SH, Campbell RE. Enhancement of a robust arcuate GABAergic input to gonadotropin-releasing hormone neurons in a model of polycystic ovarian syndrome. Proc Natl Acad Sci USA. 2015;112(2):596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Sun M, Maliqueo M, Benrick A, et al. Maternal androgen excess reduces placental and fetal weights, increases placental steroidogenesis, and leads to long-term health effects in their female offspring. Am J Physiol Endocrinol Metab. 2012;303(11):E1373–E1385. [DOI] [PubMed] [Google Scholar]
- 207. Padmanabhan V, Veiga-Lopez A. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol. 2013;373:8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Abbott DH, Barnett DK, Bruns CM, Dumesic DA. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update. 2005;11(4):357–374. [DOI] [PubMed] [Google Scholar]
- 209. Abbott DH, Nicol LE, Levine JE, Xu N, Goodarzi MO, Dumesic DA. Nonhuman primate models of polycystic ovary syndrome. Mol Cell Endocrinol. 2013;373:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Keller E, Chazenbalk GD, Aguilera P, et al. Impaired preadipocyte differentiation into adipocytes in subcutaneous abdominal adipose of PCOS-like female rhesus monkeys. Endocrinology. 2014;155(7):2696–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Dumesic DA, Patankar MS, Barnett DK, Lesnick TG, Hutcherson BA, Abbott DH. Early prenatal androgenization results in diminished ovarian reserve in adult female rhesus monkeys. Hum Reprod. 2009;24(12):3188–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Xu N, Kwon S, Abbott DH, et al. Epigenetic mechanism underlying the development of polycystic ovary syndrome (PCOS)-like phenotypes in prenatally androgenized rhesus monkeys. PLoS One. 2011;6(11):e27286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Nicol LE, O'Brien TD, Dumesic DA, Grogan T, Tarantal AF, Abbott DH. Abnormal infant islet morphology precedes insulin resistance in PCOS-like monkeys. PLoS One. 2014;9(9):e106527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Barnes RB, Rosenfield RL, Ehrmann DA, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab. 1994;79(5):1328–1333. [DOI] [PubMed] [Google Scholar]
- 215. Sir-Petermann T, Maliqueo M, Angel B, Lara HE, Pérez-Bravo F, Recabarren SE. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod. 2002;17(10):2573–2579. [DOI] [PubMed] [Google Scholar]
- 216. Dmitrovic R, Katcher HI, Kunselman AR, Legro RS. Continuous glucose monitoring during pregnancy in women with polycystic ovary syndrome. Obstet Gynecol. 2011;118(4):878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Barry JA, Kay AR, Navaratnarajah R, et al. Umbilical vein testosterone in female infants born to mothers with polycystic ovary syndrome is elevated to male levels. J Obstet Gynaecol. 2010;30(5):444–446. [DOI] [PubMed] [Google Scholar]
- 218. Anderson H, Fogel N, Grebe SK, Singh RJ, Taylor RL, Dunaif A. Infants of women with polycystic ovary syndrome have lower cord blood androstenedione and estradiol levels. J Clin Endocrinol Metab. 2010;95(5):2180–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Palomba S, Marotta R, Di Cello A, et al. Pervasive developmental disorders in children of hyperandrogenic women with polycystic ovary syndrome: a longitudinal case-control study. Clin Endocrinol (Oxf). 2012;77(6):898–904. [DOI] [PubMed] [Google Scholar]
- 220. Sarkar P, Bergman K, Fisk NM, O'Connor TG, Glover V. Amniotic fluid testosterone: relationship with cortisol and gestational age. Clin Endocrinol (Oxf). 2007;67(5):743–747. [DOI] [PubMed] [Google Scholar]
- 221. Maliqueo M, Lara HE, Sánchez F, Echiburú B, Crisosto N, Sir-Petermann T. Placental steroidogenesis in pregnant women with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol. 2013;166(2):151–155. [DOI] [PubMed] [Google Scholar]
- 222. Ibáñez L, Potau N, Francois I, de Zegher F. Precocious pubarche, hyperinsulinism, and ovarian hyperandrogenism in girls: relation to reduced fetal growth. J Clin Endocrinol Metab. 1998;83(10):3558–3562. [DOI] [PubMed] [Google Scholar]
- 223. Legro RS, Roller RL, Dodson WC, Stetter CM, Kunselman AR, Dunaif A. Associations of birthweight and gestational age with reproductive and metabolic phenotypes in women with polycystic ovarian syndrome and their first-degree relatives. J Clin Endocrinol Metab. 2010;95(2):789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. McCartney CR, Blank SK, Prendergast KA, et al. Obesity and sex steroid changes across puberty: evidence for marked hyperandrogenemia in pre- and early pubertal obese girls. J Clin Endocrinol Metab. 2007;92(2):430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Knudsen KL, Blank SK, Burt Solorzano C, et al. Hyperandrogenemia in obese peripubertal girls: correlates and potential etiological determinants. Obesity. 2010;18(11):2118–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Ibáñez L, Potau N, Zampolli M, Street ME, Carrascosa A. Girls diagnosed with premature pubarche show an exaggerated ovarian androgen synthesis from the early stages of puberty: evidence from gonadotropin-releasing hormone agonist testing. Fertil Steril. 1997;67(5):849–855. [DOI] [PubMed] [Google Scholar]
- 227. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA. 2014;311(8):806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Maliqueo M, Galgani JE, Pérez-Bravo F, et al. Relationship of serum adipocyte-derived proteins with insulin sensitivity and reproductive features in pre-pubertal and pubertal daughters of polycystic ovary syndrome women. Eur J Obstet Gynecol Reprod Biol. 2012;161(1):56–61. [DOI] [PubMed] [Google Scholar]
- 229. Franks S, Gilling-Smith C, Watson H, Willis D. Insulin action in the normal and polycystic ovary. Endocrinol Metab Clin North Am. 1999;28(2):361–378. [DOI] [PubMed] [Google Scholar]
- 230. Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab. 1998;83(6):2001–2005. [DOI] [PubMed] [Google Scholar]
- 231. Creanga AA, Bradley HM, McCormick C, Witkop CT. Use of metformin in polycystic ovary syndrome: a meta-analysis. Obstet Gynecol. 2008;111(4):959–968. [DOI] [PubMed] [Google Scholar]
- 232. Norman RJ, Davies MJ, Lord J, Moran LJ. The role of lifestyle modification in polycystic ovary syndrome. Trends Endocrinol Metab. 2002;13(6):251–257. [DOI] [PubMed] [Google Scholar]
- 233. Steingold K, De Ziegler D, Cedars M, et al. Clinical and hormonal effects of chronic gonadotropin-releasing hormone agonist treatment in polycystic ovarian disease. J Clin Endocrinol Metab. 1987;65(4):773–778. [DOI] [PubMed] [Google Scholar]
- 234. Puurunen J, Piltonen T, Jaakkola P, Ruokonen A, Morin-Papunen L, Tapanainen JS. Adrenal androgen production capacity remains high up to menopause in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94(6):1973–1978. [DOI] [PubMed] [Google Scholar]
- 235. Coviello AD, Legro RS, Dunaif A. Adolescent girls with polycystic ovary syndrome have an increased risk of the metabolic syndrome associated with increasing androgen levels independent of obesity and insulin resistance. J Clin Endocrinol Metab. 2006;91(2):492–497. [DOI] [PubMed] [Google Scholar]
- 236. Waldstreicher J, Santoro NF, Hall JE, Filicori M, Crowley WF., Jr Hyperfunction of the hypothalamic-pituitary axis in women with polycystic ovarian disease: indirect evidence for partial gonadotroph desensitization. J Clin Endocrinol Metab. 1988;66(1):165–172. [DOI] [PubMed] [Google Scholar]
- 237. Dalkin AC, Haisenleder DJ, Ortolano GA, Ellis TR, Marshall JC. The frequency of gonadotropin-releasing-hormone stimulation differentially regulates gonadotropin subunit messenger ribonucleic acid expression. Endocrinology. 1989;125(2):917–924. [DOI] [PubMed] [Google Scholar]
- 238. Pastor CL, Griffin-Korf ML, Aloi JA, Evans WS, Marshall JC. Polycystic ovary syndrome: evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 1998;83(2):582–590. [DOI] [PubMed] [Google Scholar]
- 239. Daniels TL, Berga SL. Resistance of gonadotropin releasing hormone drive to sex steroid-induced suppression in hyperandrogenic anovulation. J Clin Endocrinol Metab. 1997;82(12):4179–4183. [DOI] [PubMed] [Google Scholar]
- 240. Eagleson CA, Gingrich MB, Pastor CL, et al. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 2000;85(11):4047–4052. [DOI] [PubMed] [Google Scholar]
- 241. Porcu E, Venturoli S, Longhi M, Fabbri R, Paradisi R, Flamigni C. Chronobiologic evolution of luteinizing hormone secretion in adolescence: developmental patterns and speculations on the onset of the polycystic ovary syndrome. Fertil Steril. 1997;67(5):842–848. [DOI] [PubMed] [Google Scholar]
- 242. Apter D, Bützow T, Laughlin GA, Yen SS. Accelerated 24-hour luteinizing hormone pulsatile activity in adolescent girls with ovarian hyperandrogenism: relevance to the developmental phase of polycystic ovarian syndrome. J Clin Endocrinol Metab. 1994;79(1):119–125. [DOI] [PubMed] [Google Scholar]
- 243. Blank SK, McCartney CR, Chhabra S, et al. Modulation of gonadotropin-releasing hormone pulse generator sensitivity to progesterone inhibition in hyperandrogenic adolescent girls–implications for regulation of pubertal maturation. J Clin Endocrinol Metab. 2009;94(7):2360–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Apter D, Bützow TL, Laughlin GA, Yen SS. Gonadotropin-releasing hormone pulse generator activity during pubertal transition in girls: pulsatile and diurnal patterns of circulating gonadotropins. J Clin Endocrinol Metab. 1993;76(4):940–949. [DOI] [PubMed] [Google Scholar]
- 245. Collins JS, Marshall JC, McCartney CR. Differential sleep-wake sensitivity of gonadotropin-releasing hormone secretion to progesterone inhibition in early pubertal girls. Neuroendocrinology. 2012;96(3):222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Mitamura R, Yano K, Suzuki N, Ito Y, Makita Y, Okuno A. Diurnal rhythms of luteinizing hormone, follicle-stimulating hormone, testosterone, and estradiol secretion before the onset of female puberty in short children. J Clin Endocrinol Metab. 2000;85(3):1074–1080. [DOI] [PubMed] [Google Scholar]
- 247. Foecking EM, Szabo M, Schwartz NB, Levine JE. Neuroendocrine consequences of prenatal androgen exposure in the female rat: absence of luteinizing hormone surges, suppression of progesterone receptor gene expression, and acceleration of the gonadotropin-releasing hormone pulse generator. Biol Reprod. 2005;72(6):1475–1483. [DOI] [PubMed] [Google Scholar]
- 248. Blank SK, McCartney CR, Marshall JC. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Hum Reprod Update. 2006;12(4):351–361. [DOI] [PubMed] [Google Scholar]
- 249. Burt Solorzano CM, Beller JP, Abshire MY, Collins JS, McCartney CR, Marshall JC. Neuroendocrine dysfunction in polycystic ovary syndrome. Steroids. 2012;77(4):332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. McGee WK, Bishop CV, Bahar A, et al. Elevated androgens during puberty in female rhesus monkeys lead to increased neuronal drive to the reproductive axis: a possible component of polycystic ovary syndrome. Hum Reprod. 2012;27(2):531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Franks S, Stark J, Hardy K. Follicle dynamics and anovulation in polycystic ovary syndrome. Hum Reprod Update. 2008;14(4):367–378. [DOI] [PubMed] [Google Scholar]
- 252. Hughesdon PE. Morphology and morphogenesis of the Stein-Leventhal ovary and of so-called “hyperthecosis”. Obstet Gynecol Surv. 1982;37(2):59–77. [DOI] [PubMed] [Google Scholar]
- 253. Webber LJ, Stubbs S, Stark J, et al. Formation and early development of follicles in the polycystic ovary. Lancet. 2003;362(9389):1017–1021. [DOI] [PubMed] [Google Scholar]
- 254. Jonard S, Dewailly D. The follicular excess in polycystic ovaries, due to intra-ovarian hyperandrogenism, may be the main culprit for the follicular arrest. Hum Reprod Update. 2004;10(2):107–117. [DOI] [PubMed] [Google Scholar]
- 255. Maciel GA, Baracat EC, Benda JA, et al. Stockpiling of transitional and classic primary follicles in ovaries of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89(11):5321–5327. [DOI] [PubMed] [Google Scholar]
- 256. Webber LJ, Stubbs SA, Stark J, et al. Prolonged survival in culture of preantral follicles from polycystic ovaries. J Clin Endocrinol Metab. 2007;92(5):1975–1978. [DOI] [PubMed] [Google Scholar]
- 257. Rebar R, Judd HL, Yen SS, Rakoff J, Vandenberg G, Naftolin F. Characterization of the inappropriate gonadotropin secretion in polycystic ovary syndrome. J Clin Invest. 1976;57(5):1320–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Richards JS, Jonassen JA, Kersey K. Evidence that changes in tonic luteinizing hormone secretion determine the growth of preovulatory follicles in the rat. Endocrinology. 1980;107(3):641–648. [DOI] [PubMed] [Google Scholar]
- 259. Gougeon A. Dynamics of follicular growth in the human: a model from preliminary results. Hum Reprod. 1986;1(2):81–87. [DOI] [PubMed] [Google Scholar]
- 260. Qiao J, Feng HL. Extra- and intra-ovarian factors in polycystic ovary syndrome: impact on oocyte maturation and embryo developmental competence. Hum Reprod Update. 2011;17(1):17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Gilling-Smith C, Story H, Rogers V, Franks S. Evidence for a primary abnormality of thecal cell steroidogenesis in the polycystic ovary syndrome. Clin Endocrinol (Oxf). 1997;47:93–99. [DOI] [PubMed] [Google Scholar]
- 262. Nelson VL, Legro RS, Strauss JF, 3rd, McAllister JM. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol. 1999;13(6):946–957. [DOI] [PubMed] [Google Scholar]
- 263. Nisenblat V, Norman RJ. Androgens and polycystic ovary syndrome. Curr Opin Endocrinol Diabetes Obes. 2009;16(3):224–231. [DOI] [PubMed] [Google Scholar]
- 264. Weil S, Vendola K, Zhou J, Bondy CA. Androgen and follicle-stimulating hormone interactions in primate ovarian follicle development. J Clin Endocrinol Metab. 1999;84(8):2951–2956. [DOI] [PubMed] [Google Scholar]
- 265. Ehrmann DA, Rosenfield RL, Barnes RB, Brigell DF, Sheikh Z. Detection of functional ovarian hyperandrogenism in women with androgen excess. N Engl J Med. 1992;327(3):157–162. [DOI] [PubMed] [Google Scholar]
- 266. Chang PL, Lindheim SR, Lowre C, et al. Normal ovulatory women with polycystic ovaries have hyperandrogenic pituitary-ovarian responses to gonadotropin-releasing hormone-agonist testing. J Clin Endocrinol Metab. 2000;85(3):995–1000. [DOI] [PubMed] [Google Scholar]
- 267. Mortensen M, Ehrmann DA, Littlejohn E, Rosenfield RL. Asymptomatic volunteers with a polycystic ovary are a functionally distinct but heterogeneous population. J Clin Endocrinol Metab. 2009;94(5):1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Franks S, Mason H, Willis D. Follicular dynamics in the polycystic ovary syndrome. Mol Cell Endocrinol. 2000;163:49–52. [DOI] [PubMed] [Google Scholar]
- 269. Sen A, Prizant H, Light A, et al. Androgens regulate ovarian follicular development by increasing follicle stimulating hormone receptor and microRNA-125b expression. Proc Natl Acad Sci USA. 2014;111(8):3008–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Diamanti-Kandarakis E. Polycystic ovarian syndrome: pathophysiology, molecular aspects and clinical implications. Expert Rev Mol Med. 2008;10:e3. [DOI] [PubMed] [Google Scholar]
- 271. Raja-Khan N, Urbanek M, Rodgers RJ, Legro RS. The role of TGF-β in polycystic ovary syndrome. Reprod Sci. 2014;21(1):20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Visser JA, Schipper I, Laven JS, Themmen AP. Anti-Müllerian hormone: an ovarian reserve marker in primary ovarian insufficiency. Nat Rev Endocrinol. 2012;8(6):331–341. [DOI] [PubMed] [Google Scholar]
- 273. Dumesic DA, Abbott DH. Implications of polycystic ovary syndrome on oocyte development. Semin Reprod Med. 2008;26(1):53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Young JM, McNeilly AS. Theca: the forgotten cell of the ovarian follicle. Reproduction. 2010;140(4):489–504. [DOI] [PubMed] [Google Scholar]
- 275. Teixeira Filho FL, Baracat EC, Lee TH, et al. Aberrant expression of growth differentiation factor-9 in oocytes of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2002;87(3):1337–1344. [DOI] [PubMed] [Google Scholar]
- 276. Pellatt L, Rice S, Mason HD. Anti-Müllerian hormone and polycystic ovary syndrome: a mountain too high? Reproduction. 2010;139(5):825–833. [DOI] [PubMed] [Google Scholar]
- 277. Pigny P, Merlen E, Robert Y, et al. Elevated serum level of anti-mullerian hormone in patients with polycystic ovary syndrome: relationship to the ovarian follicle excess and to the follicular arrest. J Clin Endocrinol Metab. 2003;88(12):5957–5962. [DOI] [PubMed] [Google Scholar]
- 278. Stubbs SA, Hardy K, Da Silva-Buttkus P, et al. Anti-müllerian hormone protein expression is reduced during the initial stages of follicle development in human polycystic ovaries. J Clin Endocrinol Metab. 2005;90(10):5536–5543. [DOI] [PubMed] [Google Scholar]
- 279. Magoffin DA, Jakimiuk AJ. Inhibin A, inhibin B and activin A concentrations in follicular fluid from women with polycystic ovary syndrome. Hum Reprod. 1998;13:2693–2698. [DOI] [PubMed] [Google Scholar]
- 280. Welt CK, Taylor AE, Fox J, Messerlian GM, Adams JM, Schneyer AL. Follicular arrest in polycystic ovary syndrome is associated with deficient inhibin A and B biosynthesis. J Clin Endocrinol Metab. 2005;90(10):5582–5587. [DOI] [PubMed] [Google Scholar]
- 281. Chang RJ, Nakamura RM, Judd HL, Kaplan SA. Insulin resistance in nonobese patients with polycystic ovarian disease. J Clin Endocrinol Metab. 1983;57(2):356–359. [DOI] [PubMed] [Google Scholar]
- 282. Gambineri A, Patton L, Altieri P, et al. Polycystic ovary syndrome is a risk factor for type 2 diabetes: results from a long-term prospective study. Diabetes. 2012;61(9):2369–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Stepto NK, Cassar S, Joham AE, et al. Women with polycystic ovary syndrome have intrinsic insulin resistance on euglycaemic-hyperinsulaemic clamp. Hum Reprod. 2013;28(3):777–784. [DOI] [PubMed] [Google Scholar]
- 284. Holte J, Bergh T, Berne C, Berglund L, Lithell H. Enhanced early insulin response to glucose in relation to insulin resistance in women with polycystic ovary syndrome and normal glucose tolerance. J Clin Endocrinol Metab. 1994;78(5):1052–1058. [DOI] [PubMed] [Google Scholar]
- 285. Gennarelli G, Rovei V, Novi RF, et al. Preserved insulin sensitivity and β-cell activity, but decreased glucose effectiveness in normal-weight women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90(6):3381–3386. [DOI] [PubMed] [Google Scholar]
- 286. Ovesen P, Moller J, Ingerslev HJ, et al. Normal basal and insulin-stimulated fuel metabolism in lean women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 1993;77(6):1636–1640. [DOI] [PubMed] [Google Scholar]
- 287. Vrbíková J, Cibula D, Dvoráková K, et al. Insulin sensitivity in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89(6):2942–2945. [DOI] [PubMed] [Google Scholar]
- 288. Yildiz BO, Bozdag G, Yapici Z, Esinler I, Yarali H. Prevalence, phenotype and cardiometabolic risk of polycystic ovary syndrome under different diagnostic criteria. Hum Reprod. 2012;27(10):3067–3073. [DOI] [PubMed] [Google Scholar]
- 289. Barber TM, Wass JA, McCarthy MI, Franks S. Metabolic characteristics of women with polycystic ovaries and oligo-amenorrhoea but normal androgen levels: implications for the management of polycystic ovary syndrome. Clin Endocrinol (Oxf). 2007;66(4):513–517. [DOI] [PubMed] [Google Scholar]
- 290. Diamanti-Kandarakis E, Panidis D. Unravelling the phenotypic map of polycystic ovary syndrome (PCOS): a prospective study of 634 women with PCOS. Clin Endocrinol (Oxf). 2007;67(5):735–742. [DOI] [PubMed] [Google Scholar]
- 291. Legro RS, Brzyski RG, Diamond MP, et al. The Pregnancy in Polycystic Ovary Syndrome II Study: baseline characteristics and effects of obesity from a multicenter randomized clinical trial. Fertil Steril. 2014;101(1):258–269.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Flier JS, Eastman RC, Minaker KL, Matteson D, Rowe JW. Acanthosis nigricans in obese women with hyperandrogenism. Characterization of an insulin-resistant state distinct from the type A and B syndromes. Diabetes. 1985;34(2):101–107. [DOI] [PubMed] [Google Scholar]
- 293. Dunaif A, Hoffman AR, Scully RE, et al. Clinical, biochemical, and ovarian morphologic features in women with acanthosis nigricans and masculinization. Obstet Gynecol. 1985;66(4):545–552. [PubMed] [Google Scholar]
- 294. Dunaif A, Graf M, Mandeli J, Laumas V, Dobrjansky A. Characterization of groups of hyperandrogenic women with acanthosis nigricans, impaired glucose tolerance, and/or hyperinsulinemia. J Clin Endocrinol Metab. 1987;65(3):499–507. [DOI] [PubMed] [Google Scholar]
- 295. O'Meara NM, Blackman JD, Ehrmann DA, et al. Defects in β-cell function in functional ovarian hyperandrogenism. J Clin Endocrinol Metab. 1993;76(5):1241–1247. [DOI] [PubMed] [Google Scholar]
- 296. Ehrmann DA, Sturis J, Byrne MM, Karrison T, Rosenfield RL, Polonsky KS. Insulin secretory defects in polycystic ovary syndrome. Relationship to insulin sensitivity and family history of non-insulin-dependent diabetes mellitus. J Clin Invest. 1995;96(1):520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Ciampelli M, Fulghesu AM, Cucinelli F, et al. Heterogeneity in β cell activity, hepatic insulin clearance and peripheral insulin sensitivity in women with polycystic ovary syndrome. Hum Reprod. 1997;12(9):1897–1901. [DOI] [PubMed] [Google Scholar]
- 298. Colilla S, Cox NJ, Ehrmann DA. Heritability of insulin secretion and insulin action in women with polycystic ovary syndrome and their first degree relatives. J Clin Endocrinol Metab. 2001;86(5):2027–2031. [DOI] [PubMed] [Google Scholar]
- 299. Kahn SE, Prigeon RL, McCulloch DK, et al. Quantification of the relationship between insulin sensitivity and β-cell function in human subjects. Evidence for a hyperbolic function. Diabetes. 1993;42(11):1663–1672. [DOI] [PubMed] [Google Scholar]
- 300. Bergman RN, Ader M, Huecking K, Van Citters G. Accurate assessment of β-cell function: the hyperbolic correction. Diabetes. 2002;51(suppl 1):S212–S220. [DOI] [PubMed] [Google Scholar]
- 301. Dunaif A, Finegood DT. β-Cell dysfunction independent of obesity and glucose intolerance in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1996;81(3):942–947. [DOI] [PubMed] [Google Scholar]
- 302. Baillargeon JP, Nestler JE. Commentary: polycystic ovary syndrome: a syndrome of ovarian hypersensitivity to insulin? J Clin Endocrinol Metab. 2006;91(1):22–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Nestler JE, Powers LP, Matt DW, et al. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 1991;72(1):83–89. [DOI] [PubMed] [Google Scholar]
- 304. Nestler JE. Insulin regulation of human ovarian androgens. Hum Reprod. 1997;12(suppl 1):53–62. [DOI] [PubMed] [Google Scholar]
- 305. Poretsky L, Cataldo NA, Rosenwaks Z, Giudice LC. The insulin-related ovarian regulatory system in health and disease. Endocr Rev. 1999;20(4):535–582. [DOI] [PubMed] [Google Scholar]
- 306. Diamanti-Kandarakis E, Argyrakopoulou G, Economou F, Kandaraki E, Koutsilieris M. Defects in insulin signaling pathways in ovarian steroidogenesis and other tissues in polycystic ovary syndrome (PCOS). J Steroid Biochem Mol Biol. 2008;109:242–246. [DOI] [PubMed] [Google Scholar]
- 307. Duleba AJ, Spaczynski RZ, Olive DL. Insulin and insulin-like growth factor I stimulate the proliferation of human ovarian theca-interstitial cells. Fertil Steril. 1998;69(2):335–340. [DOI] [PubMed] [Google Scholar]
- 308. Poretsky L, Clemons J, Bogovich K. Hyperinsulinemia and human chorionic gonadotropin synergistically promote the growth of ovarian follicular cysts in rats. Metabolism. 1992;41(8):903–910. [DOI] [PubMed] [Google Scholar]
- 309. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237(3):E214–E223. [DOI] [PubMed] [Google Scholar]
- 310. Bergman RN. Lilly lecture 1989. Toward physiological understanding of glucose tolerance. Minimal-model approach. Diabetes. 1989;38(12):1512–1527. [DOI] [PubMed] [Google Scholar]
- 311. Hücking K, Watanabe RM, Stefanovski D, Bergman RN. OGTT-derived measures of insulin sensitivity are confounded by factors other than insulin sensitivity itself. Obesity. 2008;16(8):1938–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Diamanti-Kandarakis E, Kouli C, Alexandraki K, Spina G. Failure of mathematical indices to accurately assess insulin resistance in lean, overweight, or obese women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89(3):1273–1276. [DOI] [PubMed] [Google Scholar]
- 313. Ader M, Stefanovski D, Richey JM, et al. Failure of homeostatic model assessment of insulin resistance to detect marked diet-induced insulin resistance in dogs. Diabetes. 2014;63(6):1914–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Roberts R, Stark J, Iatropoulou A, Becker DL, Franks S, Hardy K. Energy substrate metabolism of mouse cumulus-oocyte complexes: response to follicle-stimulating hormone is mediated by the phosphatidylinositol 3-kinase pathway and is associated with oocyte maturation. Biol Reprod. 2004;71(1):199–209. [DOI] [PubMed] [Google Scholar]
- 315. Hardy OT, Czech MP, Corvera S. What causes the insulin resistance underlying obesity? Curr Opin Endocrinol Diabetes Obes. 2012;19(2):81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Kashyap S, Belfort R, Gastaldelli A, et al. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes. 2003;52(10):2461–2474. [DOI] [PubMed] [Google Scholar]
- 317. Roden M, Price TB, Perseghin G, et al. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97(12):2859–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev. 2012;33(6):981–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Corbould A, Kim YB, Youngren JF, et al. Insulin resistance in the skeletal muscle of women with PCOS involves intrinsic and acquired defects in insulin signaling. Am J Physiol Endocrinol Metab. 2005;288(5):E1047–E1054. [DOI] [PubMed] [Google Scholar]
- 320. Ciaraldi TP, Aroda V, Mudaliar S, Chang RJ, Henry RR. Polycystic ovary syndrome is associated with tissue-specific differences in insulin resistance. J Clin Endocrinol Metab. 2009;94(1):157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Eriksen M, Pørneki AD, Skov V, et al. Insulin resistance is not conserved in myotubes established from women with PCOS. PLoS One. 2010;5(12):e14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Dunaif A, Xia J, Book CB, Schenker E, Tang Z. Excessive insulin receptor serine phosphorylation in cultured fibroblasts and in skeletal muscle. A potential mechanism for insulin resistance in the polycystic ovary syndrome. J Clin Invest. 1995;96(2):801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Corbould A, Zhao H, Mirzoeva S, Aird F, Dunaif A. Enhanced mitogenic signaling in skeletal muscle of women with polycystic ovary syndrome. Diabetes. 2006;55(3):751–759. [DOI] [PubMed] [Google Scholar]
- 324. Dunaif A, Wu X, Lee A, Diamanti-Kandarakis E. Defects in insulin receptor signaling in vivo in the polycystic ovary syndrome (PCOS). Am J Physiol Endocrinol Metab. 2001;281(2):E392–E399. [DOI] [PubMed] [Google Scholar]
- 325. Skov V, Glintborg D, Knudsen S, et al. Reduced expression of nuclear-encoded genes involved in mitochondrial oxidative metabolism in skeletal muscle of insulin-resistant women with polycystic ovary syndrome. Diabetes. 2007;56(9):2349–2355. [DOI] [PubMed] [Google Scholar]
- 326. Allemand MC, Irving BA, Asmann YW, et al. Effect of testosterone on insulin stimulated IRS1 Ser phosphorylation in primary rat myotubes–a potential model for PCOS-related insulin resistance. PLoS One. 2009;4(1):e4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Holmäng A, Larsson BM, Brzezinska Z, Björntorp P. Effects of short-term testosterone exposure on insulin sensitivity of muscles in female rats. Am J Physiol. 1992;262(6):E851–E855. [DOI] [PubMed] [Google Scholar]
- 328. Holmäng A, Svedberg J, Jennische E, Björntorp P. Effects of testosterone on muscle insulin sensitivity and morphology in female rats. Am J Physiol. 1990;259(4):E555–E560. [DOI] [PubMed] [Google Scholar]
- 329. Holmäng A, Niklasson M, Rippe B, Lönnroth P. Insulin insensitivity and delayed transcapillary delivery of insulin in oophorectomized rats treated with testosterone. Acta Physiol Scand. 2001;171(4):427–438. [DOI] [PubMed] [Google Scholar]
- 330. Rincon J, Holmäng A, Wahlström EO, et al. Mechanisms behind insulin resistance in rat skeletal muscle after oophorectomy and additional testosterone treatment. Diabetes. 1996;45(5):615–621. [DOI] [PubMed] [Google Scholar]
- 331. Johansson J, Feng Y, Shao R, Lönn M, Billig H, Stener-Victorin E. Intense electroacupuncture normalizes insulin sensitivity, increases muscle GLUT4 content, and improves lipid profile in a rat model of polycystic ovary syndrome. Am J Physiol Endocrinol Metab. 2010;299:E551–E559. [DOI] [PubMed] [Google Scholar]
- 332. Mannerås L, Cajander S, Holmäng A, et al. A new rat model exhibiting both ovarian and metabolic characteristics of polycystic ovary syndrome. Endocrinology. 2007;148(8):3781–3791. [DOI] [PubMed] [Google Scholar]
- 333. Corbould A. Chronic testosterone treatment induces selective insulin resistance in subcutaneous adipocytes of women. J Endocrinol. 2007;192(3):585–594. [DOI] [PubMed] [Google Scholar]
- 334. Capllonch-Amer G, Lladó I, Proenza AM, García-Palmer FJ, Gianotti M. Opposite effects of 17-β estradiol and testosterone on mitochondrial biogenesis and adiponectin synthesis in white adipocytes. J Mol Endocrinol. 2014;52(2):203–214. [DOI] [PubMed] [Google Scholar]
- 335. Ciaraldi TP, el-Roeiy A, Madar Z, Reichart D, Olefsky JM, Yen SS. Cellular mechanisms of insulin resistance in polycystic ovarian syndrome. J Clin Endocrinol Metab. 1992;75(2):577–583. [DOI] [PubMed] [Google Scholar]
- 336. Seow KM, Juan CC, Hsu YP, Hwang JL, Huang LW, Ho LT. Amelioration of insulin resistance in women with PCOS via reduced insulin receptor substrate-1 Ser312 phosphorylation following laparoscopic ovarian electrocautery. Hum Reprod. 2007;22(4):1003–1010. [DOI] [PubMed] [Google Scholar]
- 337. Chang W, Goodarzi MO, Williams H, Magoffin DA, Pall M, Azziz R. Adipocytes from women with polycystic ovary syndrome demonstrate altered phosphorylation and activity of glycogen synthase kinase 3. Fertil Steril. 2008;90(6):2291–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Rosenbaum D, Haber RS, Dunaif A. Insulin resistance in polycystic ovary syndrome: decreased expression of GLUT-4 glucose transporters in adipocytes. Am J Physiol. 1993;264(2):E197–E202. [DOI] [PubMed] [Google Scholar]
- 339. Chen YH, Heneidi S, Lee JM, et al. miRNA-93 inhibits GLUT4 and is overexpressed in adipose tissue of polycystic ovary syndrome patients and women with insulin resistance. Diabetes. 2013;62(7):2278–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30(12):1000–1007. [DOI] [PubMed] [Google Scholar]
- 341. Minokoshi Y, Kahn CR, Kahn BB. Tissue-specific ablation of the GLUT4 glucose transporter or the insulin receptor challenges assumptions about insulin action and glucose homeostasis. J Biol Chem. 2003;278(36):33609–33612. [DOI] [PubMed] [Google Scholar]
- 342. Barber TM, Franks S. Adipocyte biology in polycystic ovary syndrome. Mol Cell Endocrinol. 2013;373:68–76. [DOI] [PubMed] [Google Scholar]
- 343. Lim SS, Norman RJ, Davies MJ, Moran LJ. The effect of obesity on polycystic ovary syndrome: a systematic review and meta-analysis. Obes Rev. 2013;14(2):95–109. [DOI] [PubMed] [Google Scholar]
- 344. Björntorp P. Hyperandrogenicity in women–a prediabetic condition? J Intern Med. 1993;234(6):579–583. [DOI] [PubMed] [Google Scholar]
- 345. Lovejoy JC, Bray GA, Bourgeois MO, et al. Exogenous androgens influence body composition and regional body fat distribution in obese postmenopausal women–a clinical research center study. J Clin Endocrinol Metab. 1996;81(6):2198–2203. [DOI] [PubMed] [Google Scholar]
- 346. Elbers JM, Asscheman H, Seidell JC, Megens JA, Gooren LJ. Long-term testosterone administration increases visceral fat in female to male transsexuals. J Clin Endocrinol Metab. 1997;82(7):2044–2047. [DOI] [PubMed] [Google Scholar]
- 347. Gambineri A, Patton L, Vaccina A, et al. Treatment with flutamide, metformin, and their combination added to a hypocaloric diet in overweight-obese women with polycystic ovary syndrome: a randomized, 12-month, placebo-controlled study. J Clin Endocrinol Metab. 2006;91(10):3970–3980. [DOI] [PubMed] [Google Scholar]
- 348. Maliqueo M, Sun M, Johansson J, et al. Continuous administration of a P450 aromatase inhibitor induces polycystic ovary syndrome with a metabolic and endocrine phenotype in female rats at adult age. Endocrinology. 2013;154(1):434–445. [DOI] [PubMed] [Google Scholar]
- 349. Crosignani PG, Colombo M, Vegetti W, Somigliana E, Gessati A, Ragni G. Overweight and obese anovulatory patients with polycystic ovaries: parallel improvements in anthropometric indices, ovarian physiology and fertility rate induced by diet. Hum Reprod. 2003;18(9):1928–1932. [DOI] [PubMed] [Google Scholar]
- 350. Huber-Buchholz MM, Carey DG, Norman RJ. Restoration of reproductive potential by lifestyle modification in obese polycystic ovary syndrome: role of insulin sensitivity and luteinizing hormone. J Clin Endocrinol Metab. 1999;84(4):1470–1474. [DOI] [PubMed] [Google Scholar]
- 351. Holte J, Bergh T, Berne C, Wide L, Lithell H. Restored insulin sensitivity but persistently increased early insulin secretion after weight loss in obese women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1995;80(9):2586–2593. [DOI] [PubMed] [Google Scholar]
- 352. Naderpoor N, Shorakae S, de Courten B, et al. Metformin and lifestyle modification in polycystic ovary syndrome: systematic review and meta-analysis. Hum Reprod Update. 2015;21:560–574. [DOI] [PubMed] [Google Scholar]
- 353. Dolfing JG, Stassen CM, van Haard PM, Wolffenbuttel BH, Schweitzer DH. Comparison of MRI-assessed body fat content between lean women with polycystic ovary syndrome (PCOS) and matched controls: less visceral fat with PCOS. Hum Reprod. 2011;26(6):1495–1500. [DOI] [PubMed] [Google Scholar]
- 354. Huang ZH, Manickam B, Ryvkin V, et al. PCOS is associated with increased CD11c expression and crown-like structures in adipose tissue and increased central abdominal fat depots independent of obesity. J Clin Endocrinol Metab. 2013;98(1):E17–E24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Toulis KA, Goulis DG, Farmakiotis D, et al. Adiponectin levels in women with polycystic ovary syndrome: a systematic review and a meta-analysis. Hum Reprod Update. 2009;15(3):297–307. [DOI] [PubMed] [Google Scholar]
- 356. Tan BK, Chen J, Lehnert H, Kennedy R, Randeva HS. Raised serum, adipocyte, and adipose tissue retinol-binding protein 4 in overweight women with polycystic ovary syndrome: effects of gonadal and adrenal steroids. J Clin Endocrinol Metab. 2007;92(7):2764–2772. [DOI] [PubMed] [Google Scholar]
- 357. Tan BK, Chen J, Digby JE, Keay SD, Kennedy CR, Randeva HS. Increased visfatin messenger ribonucleic acid and protein levels in adipose tissue and adipocytes in women with polycystic ovary syndrome: parallel increase in plasma visfatin. J Clin Endocrinol Metab. 2006;91(12):5022–5028. [DOI] [PubMed] [Google Scholar]
- 358. Diamanti-Kandarakis E, Paterakis T, Kandarakis HA. Indices of low-grade inflammation in polycystic ovary syndrome. Ann NY Acad Sci. 2006;1092:175–186. [DOI] [PubMed] [Google Scholar]
- 359. Wentworth JM, Naselli G, Brown WA, et al. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. 2010;59(7):1648–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Faulds G, Rydén M, Ek I, Wahrenberg H, Arner P. Mechanisms behind lipolytic catecholamine resistance of subcutaneous fat cells in the polycystic ovarian syndrome. J Clin Endocrinol Metab. 2003;88(5):2269–2273. [DOI] [PubMed] [Google Scholar]
- 361. Rebuffé-Scrive M, Cullberg G, Lundberg PA, Lindstedt G, Björntorp P. Anthropometric variables and metabolism in polycystic ovarian disease. Horm Metab Res. 1989;21(7):391–397. [DOI] [PubMed] [Google Scholar]
- 362. Adler GK, Bonyhay I, Failing H, Waring E, Dotson S, Freeman R. Antecedent hypoglycemia impairs autonomic cardiovascular function: implications for rigorous glycemic control. Diabetes. 2009;58(2):360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Fagius J. Sympathetic nerve activity in metabolic control–some basic concepts. Acta Physiol Scand. 2003;177(3):337–343. [DOI] [PubMed] [Google Scholar]
- 364. Landsberg L. Diet, obesity and hypertension: an hypothesis involving insulin, the sympathetic nervous system, and adaptive thermogenesis. Q J Med. 1986;61(236):1081–1090. [PubMed] [Google Scholar]
- 365. Landsberg L. Insulin-mediated sympathetic stimulation: role in the pathogenesis of obesity-related hypertension (or, how insulin affects blood pressure, and why). J Hypertens. 2001;19:523–528. [DOI] [PubMed] [Google Scholar]
- 366. Alvarez GE, Beske SD, Ballard TP, Davy KP. Sympathetic neural activation in visceral obesity. Circulation. 2002;106(20):2533–2536. [DOI] [PubMed] [Google Scholar]
- 367. Grassi G, Dell'Oro R, Facchini A, Quarti Trevano F, Bolla GB, Mancia G. Effect of central and peripheral body fat distribution on sympathetic and baroreflex function in obese normotensives. J Hypertens. 2004;22(12):2363–2369. [DOI] [PubMed] [Google Scholar]
- 368. Lansdown A, Rees DA. The sympathetic nervous system in polycystic ovary syndrome: a novel therapeutic target? Clin Endocrinol (Oxf). 2012;77(6):791–801. [DOI] [PubMed] [Google Scholar]
- 369. Hagbarth KE, Vallbo AB. Pulse and respiratory grouping of sympathetic impulses in human muscle-nerves. Acta Physiol Scand. 1968;74(1):96–108. [DOI] [PubMed] [Google Scholar]
- 370. Sverrisdóttir YB, Mogren T, Kataoka J, Janson PO, Stener-Victorin E. Is polycystic ovary syndrome associated with high sympathetic nerve activity and size at birth? Am J Physiol Endocrinol Metab. 2008;294(3):E576–E581. [DOI] [PubMed] [Google Scholar]
- 371. Stener-Victorin E, Jedel E, Janson PO, Sverrisdottir YB. Low-frequency electroacupuncture and physical exercise decrease high muscle sympathetic nerve activity in polycystic ovary syndrome. Am J Physiol Regul Integr Comp Physiol. 2009;297(2):R387–R395. [DOI] [PubMed] [Google Scholar]
- 372. Pastore LM, Williams CD, Jenkins J, Patrie JT. True and sham acupuncture produced similar frequency of ovulation and improved LH to FSH ratios in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2011;96(10):3143–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Johansson J, Redman L, Veldhuis PP, et al. Acupuncture for ovulation induction in polycystic ovary syndrome: a randomized controlled trial. Am J Physiol Endocrinol Metab. 2013;304(9):E934–E943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Giallauria F, Palomba S, Manguso F, et al. Abnormal heart rate recovery after maximal cardiopulmonary exercise stress testing in young overweight women with polycystic ovary syndrome. Clin Endocrinol (Oxf). 2008;68(1):88–93. [DOI] [PubMed] [Google Scholar]
- 375. Tasali E, Chapotot F, Leproult R, Whitmore H, Ehrmann DA. Treatment of obstructive sleep apnea improves cardiometabolic function in young obese women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2011;96(2):365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Yildirir A, Aybar F, Kabakci G, Yarali H, Oto A. Heart rate variability in young women with polycystic ovary syndrome. Ann Noninvasive Electrocardiol. 2006;11(4):306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Tekin G, Tekin A, Kiliçarslan EB, et al. Altered autonomic neural control of the cardiovascular system in patients with polycystic ovary syndrome. Int J Cardiol. 2008;130(1):49–55. [DOI] [PubMed] [Google Scholar]
- 378. Di Domenico K, Wiltgen D, Nickel FJ, Magalhães JA, Moraes RS, Spritzer PM. Cardiac autonomic modulation in polycystic ovary syndrome: does the phenotype matter? Fertil Steril. 2013;99(1):286–292. [DOI] [PubMed] [Google Scholar]
- 379. Semenova I. Adrenergic innervation of the ovaries in Stein-Leventhal syndrome [in Russian]. Vestn Akad Med Nauk SSSR. 1969;24:58–62. [PubMed] [Google Scholar]
- 380. Heider U, Pedal I, Spanel-Borowski K. Increase in nerve fibers and loss of mast cells in polycystic and postmenopausal ovaries. Fertil Steril. 2001;75(6):1141–1147. [DOI] [PubMed] [Google Scholar]
- 381. Garcia-Rudaz C, Armando I, Levin G, Escobar ME, Barontini M. Peripheral catecholamine alterations in adolescents with polycystic ovary syndrome. Clin Endocrinol (Oxf). 1998;49(2):221–228. [DOI] [PubMed] [Google Scholar]
- 382. Barria A, Leyton V, Ojeda SR, Lara HE. Ovarian steroidal response to gonadotropins and β-adrenergic stimulation is enhanced in polycystic ovary syndrome: role of sympathetic innervation. Endocrinology. 1993;133(6):2696–2703. [DOI] [PubMed] [Google Scholar]
- 383. Dissen GA, Garcia-Rudaz C, Paredes A, Mayer C, Mayerhofer A, Ojeda SR. Excessive ovarian production of nerve growth factor facilitates development of cystic ovarian morphology in mice and is a feature of polycystic ovarian syndrome in humans. Endocrinology. 2009;150(6):2906–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Stener-Victorin E, Jedel E, Mannerås L. Acupuncture in polycystic ovary syndrome: current experimental and clinical evidence. J Neuroendocrinol. 2008;20(3):290–298. [DOI] [PubMed] [Google Scholar]
- 385. Lara HE, Dissen GA, Leyton V, et al. An increased intraovarian synthesis of nerve growth factor and its low affinity receptor is a principal component of steroid-induced polycystic ovary in the rat. Endocrinology. 2000;141(3):1059–1072. [DOI] [PubMed] [Google Scholar]
- 386. Manni L, Holmäng A, Cajander S, Lundeberg T, Aloe L, Stener-Victorin E. Effect of anti-NGF on ovarian expression of α1- and β2-adrenoceptors, TrkA, p75NTR, and tyrosine hydroxylase in rats with steroid-induced polycystic ovaries. Am J Physiol Regul Integr Comp Physiol. 2006;290(3):R826–R835. [DOI] [PubMed] [Google Scholar]
- 387. Govind A, Obhrai MS, Clayton RN. Polycystic ovaries are inherited as an autosomal dominant trait: analysis of 29 polycystic ovary syndrome and 10 control families. J Clin Endocrinol Metab. 1999;84(1):38–43. [DOI] [PubMed] [Google Scholar]
- 388. Pall M, Stephens K, Azziz R. Family size in women with polycystic ovary syndrome. Fertil Steril. 2006;85(6):1837–1839. [DOI] [PubMed] [Google Scholar]
- 389. Carey AH, Chan KL, Short F, White D, Williamson R, Franks S. Evidence for a single gene effect causing polycystic ovaries and male pattern baldness. Clin Endocrinol (Oxf). 1993;38(6):653–658. [DOI] [PubMed] [Google Scholar]
- 390. Sam S, Coviello AD, Sung YA, Legro RS, Dunaif A. Metabolic phenotype in the brothers of women with polycystic ovary syndrome. Diabetes Care. 2008;31(6):1237–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. West S, Vähäsarja M, Bloigu A, et al. The impact of self-reported oligo-amenorrhea and hirsutism on fertility and lifetime reproductive success: results from the Northern Finland Birth Cohort 1966. Hum Reprod. 2014;29(3):628–633. [DOI] [PubMed] [Google Scholar]
- 392. Goodarzi MO. Looking for polycystic ovary syndrome genes: rational and best strategy. Semin Reprod Med. 2008;26(1):5–13. [DOI] [PubMed] [Google Scholar]
- 393. Parker R, Ming PM, Rajan R, Goodner DM, Remé G. Clinical and cytogenetic studies of patients with polycystic ovary disease. Am J Obstet Gynecol. 1980;137(6):656–660. [DOI] [PubMed] [Google Scholar]
- 394. Sengoku K, Tamate K, Takuma N, Yoshida T, Goishi K, Ishikawa M. The chromosomal normality of unfertilized oocytes from patients with polycystic ovarian syndrome. Hum Reprod. 1997;12(3):474–477. [DOI] [PubMed] [Google Scholar]
- 395. Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 1994;265(5181):2037–2048. [DOI] [PubMed] [Google Scholar]
- 396. Gallo V, Egger M, McCormack V, et al. STrengthening the Reporting of OBservational studies in Epidemiology–Molecular Epidemiology (STROBE-ME): an extension of the STROBE Statement. PLoS Med. 2011;8(10):e1001117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Kosova G, Urbanek M. Genetics of the polycystic ovary syndrome. Mol Cell Endocrinol. 2013;373:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Jahanfar S, Eden JA, Warren P, Seppälä M, Nguyen TV. A twin study of polycystic ovary syndrome. Fertil Steril. 1995;63(3):478–486. [PubMed] [Google Scholar]
- 399. Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab. 2006;91(6):2100–2104. [DOI] [PubMed] [Google Scholar]
- 400. Kuijper EA, Vink JM, Lambalk CB, Boomsma DI. Prevalence of polycystic ovary syndrome in women from opposite-sex twin pairs. J Clin Endocrinol Metab. 2009;94(6):1987–1990. [DOI] [PubMed] [Google Scholar]
- 401. Kahsar-Miller MD, Nixon C, Boots LR, Go RC, Azziz R. Prevalence of polycystic ovary syndrome (PCOS) in first-degree relatives of patients with PCOS. Fertil Steril. 2001;75(1):53–58. [DOI] [PubMed] [Google Scholar]
- 402. Yildiz BO, Goodarzi MO, Guo X, Rotter JI, Azziz R. Heritability of dehydroepiandrosterone sulfate in women with polycystic ovary syndrome and their sisters. Fertil Steril. 2006;86(6):1688–1693. [DOI] [PubMed] [Google Scholar]
- 403. Joharatnam J, Barber TM, Webber L, Conway GS, McCarthy MI, Franks S. Determinants of dyslipidaemia in probands with polycystic ovary syndrome and their sisters. Clin Endocrinol (Oxf). 2011;74(6):714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Sam S, Legro RS, Bentley-Lewis R, Dunaif A. Dyslipidemia and metabolic syndrome in the sisters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90(8):4797–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Sam S, Legro RS, Essah PA, Apridonidze T, Dunaif A. Evidence for metabolic and reproductive phenotypes in mothers of women with polycystic ovary syndrome. Proc Natl Acad Sci USA. 2006;103(18):7030–7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Baillargeon JP, Carpentier AC. Brothers of women with polycystic ovary syndrome are characterised by impaired glucose tolerance, reduced insulin sensitivity and related metabolic defects. Diabetologia. 2007;50(12):2424–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Coviello AD, Sam S, Legro RS, Dunaif A. High prevalence of metabolic syndrome in first-degree male relatives of women with polycystic ovary syndrome is related to high rates of obesity. J Clin Endocrinol Metab. 2009;94(11):4361–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Legro RS, Bentley-Lewis R, Driscoll D, Wang SC, Dunaif A. Insulin resistance in the sisters of women with polycystic ovary syndrome: association with hyperandrogenemia rather than menstrual irregularity. J Clin Endocrinol Metab. 2002;87(5):2128–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Sir-Petermann T, Angel B, Maliqueo M, Carvajal F, Santos JL, Pérez-Bravo F. Prevalence of type II diabetes mellitus and insulin resistance in parents of women with polycystic ovary syndrome. Diabetologia. 2002;45(7):959–964. [DOI] [PubMed] [Google Scholar]
- 410. Yildiz BO, Yarali H, Oguz H, Bayraktar M. Glucose intolerance, insulin resistance, and hyperandrogenemia in first degree relatives of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88(5):2031–2036. [DOI] [PubMed] [Google Scholar]
- 411. Unluturk U, Harmanci A, Kocaefe C, Yildiz BO. The genetic basis of the polycystic ovary syndrome: a literature review including discussion of PPAR-γ. PPAR Res. 2007;2007:49109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Urbanek M, Woodroffe A, Ewens KG, et al. Candidate gene region for polycystic ovary syndrome on chromosome 19p13.2. J Clin Endocrinol Metab. 2005;90(12):6623–6629. [DOI] [PubMed] [Google Scholar]
- 413. Ewens KG, Stewart DR, Ankener W, et al. Family-based analysis of candidate genes for polycystic ovary syndrome. J Clin Endocrinol Metab. 2010;95(5):2306–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Tucci S, Futterweit W, Concepcion ES, et al. Evidence for association of polycystic ovary syndrome in Caucasian women with a marker at the insulin receptor gene locus. J Clin Endocrinol Metab. 2001;86(1):446–449. [DOI] [PubMed] [Google Scholar]
- 415. Villuendas G, Escobar-Morreale HF, Tosi F, Sancho J, Moghetti P, San Millán JL. Association between the D19S884 marker at the insulin receptor gene locus and polycystic ovary syndrome. Fertil Steril. 2003;79(1):219–220. [DOI] [PubMed] [Google Scholar]
- 416. Shi Y, Zhao H, Shi Y, et al. Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat Genet. 2012;44(9):1020–1025. [DOI] [PubMed] [Google Scholar]
- 417. Ruan Y, Ma J, Xie X. Association of IRS-1 and IRS-2 genes polymorphisms with polycystic ovary syndrome: a meta-analysis. Endocr J. 2012;59(7):601–609. [DOI] [PubMed] [Google Scholar]
- 418. Biyasheva A, Legro RS, Dunaif A, Urbanek M. Evidence for association between polycystic ovary syndrome (PCOS) and TCF7L2 and glucose intolerance in women with PCOS and TCF7L2. J Clin Endocrinol Metab. 2009;94(7):2617–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Wojciechowski P, Lipowska A, Rys P, et al. Impact of FTO genotypes on BMI and weight in polycystic ovary syndrome: a systematic review and meta-analysis. Diabetologia. 2012;55(10):2636–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Louwers YV, Rayner NW, Herrera BM, et al. BMI-associated alleles do not constitute risk alleles for polycystic ovary syndrome independently of BMI: a case-control study. PLoS One. 2014;9(1):e87335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Li T, Wu K, You L, et al. Common variant rs9939609 in gene FTO confers risk to polycystic ovary syndrome. PLoS One. 2013;8(7):e66250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Claussnitzer M, Dankel SN, Kim KH, et al. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med. 2015;373(10):895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Laven JS, Mulders AG, Suryandari DA, et al. Follicle-stimulating hormone receptor polymorphisms in women with normogonadotropic anovulatory infertility. Fertil Steril. 2003;80(4):986–992. [DOI] [PubMed] [Google Scholar]
- 424. Lerchbaum E, Trummer O, Giuliani A, Gruber HJ, Pieber TR, Obermayer-Pietsch B. Susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21, and 9q33.3 in a cohort of Caucasian women. Horm Metab Res. 2011;43(11):743–747. [DOI] [PubMed] [Google Scholar]
- 425. McAllister JM, Modi B, Miller BA, et al. Overexpression of a DENND1A isoform produces a polycystic ovary syndrome theca phenotype. Proc Natl Acad Sci USA. 2014;111(15):E1519–E1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. McAllister JM, Legro RS, Modi BP, Strauss JF., 3rd Functional genomics of PCOS: from GWAS to molecular mechanisms. Trends Endocrinol Metab. 2015;26(3):118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Cui L, Zhao H, Zhang B, et al. Genotype-phenotype correlations of PCOS susceptibility SNPs identified by GWAS in a large cohort of Han Chinese women. Hum Reprod. 2013;28(2):538–544. [DOI] [PubMed] [Google Scholar]
- 428. Mutharasan P, Galdones E, Peñalver Bernabé B, et al. Evidence for chromosome 2p16.3 polycystic ovary syndrome susceptibility locus in affected women of European ancestry. J Clin Endocrinol Metab. 2013;98(1):E185–E190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Lee PJ, Patel A, Hindmarsh PC, Mowat AP, Leonard JV. The prevalence of polycystic ovaries in the hepatic glycogen storage diseases: its association with hyperinsulinism. Clin Endocrinol (Oxf). 1995;42(6):601–606. [DOI] [PubMed] [Google Scholar]
- 430. Hwang JY, Lee EJ, Jin Go M, et al. Genome-wide association study identifies GYS2 as a novel genetic factor for polycystic ovary syndrome through obesity-related condition. J Hum Genet. 2012;57(10):660–664. [DOI] [PubMed] [Google Scholar]
- 431. Hayes MG, Urbanek M, Ehrmann DA, et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun. 2015;6:7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Valkenburg O, Uitterlinden AG, Piersma D, et al. Genetic polymorphisms of GnRH and gonadotrophic hormone receptors affect the phenotype of polycystic ovary syndrome. Hum Reprod. 2009;24(8):2014–2022. [DOI] [PubMed] [Google Scholar]
- 433. Jansen E, Laven JS, Dommerholt HB, et al. Abnormal gene expression profiles in human ovaries from polycystic ovary syndrome patients. Mol Endocrinol. 2004;18(12):3050–3063. [DOI] [PubMed] [Google Scholar]
- 434. Wood JR, Nelson VL, Ho C, et al. The molecular phenotype of polycystic ovary syndrome (PCOS) theca cells and new candidate PCOS genes defined by microarray analysis. J Biol Chem. 2003;278(29):26380–26390. [DOI] [PubMed] [Google Scholar]
- 435. Wood JR, Ho CK, Nelson-Degrave VL, McAllister JM, Strauss JF., 3rd The molecular signature of polycystic ovary syndrome (PCOS) theca cells defined by gene expression profiling. J Reprod Immunol. 2004;63(1):51–60. [DOI] [PubMed] [Google Scholar]
- 436. Diao FY, Xu M, Hu Y, et al. The molecular characteristics of polycystic ovary syndrome (PCOS) ovary defined by human ovary cDNA microarray. J Mol Endocrinol. 2004;33(1):59–72. [DOI] [PubMed] [Google Scholar]
- 437. Wood JR, Dumesic DA, Abbott DH, Strauss JF., 3rd Molecular abnormalities in oocytes from women with polycystic ovary syndrome revealed by microarray analysis. J Clin Endocrinol Metab. 2007;92(2):705–713. [DOI] [PubMed] [Google Scholar]
- 438. Legro RS, Barnhart HX, Schlaff WD, et al. Ovulatory response to treatment of polycystic ovary syndrome is associated with a polymorphism in the STK11 gene. J Clin Endocrinol Metab. 2008;93(3):792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439. Gambineri A, Tomassoni F, Gasparini DI, et al. Organic cation transporter 1 polymorphisms predict the metabolic response to metformin in women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 2010;95(10):E204–E208. [DOI] [PubMed] [Google Scholar]
- 440. Simoni M, Tempfer CB, Destenaves B, Fauser BC. Functional genetic polymorphisms and female reproductive disorders: part I: polycystic ovary syndrome and ovarian response. Hum Reprod Update. 2008;14(5):459–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Mifsud A, Ramirez S, Yong EL. Androgen receptor gene CAG trinucleotide repeats in anovulatory infertility and polycystic ovaries. J Clin Endocrinol Metab. 2000;85(9):3484–3488. [DOI] [PubMed] [Google Scholar]
- 442. Hickey T, Chandy A, Norman RJ. The androgen receptor CAG repeat polymorphism and X-chromosome inactivation in Australian Caucasian women with infertility related to polycystic ovary syndrome. J Clin Endocrinol Metab. 2002;87(1):161–165. [DOI] [PubMed] [Google Scholar]
- 443. Wang R, Goodarzi MO, Xiong T, Wang D, Azziz R, Zhang H. Negative association between androgen receptor gene CAG repeat polymorphism and polycystic ovary syndrome? A systematic review and meta-analysis. Mol Hum Reprod. 2012;18(10):498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Xu N, Azziz R, Goodarzi MO. Epigenetics in polycystic ovary syndrome: a pilot study of global DNA methylation. Fertil Steril. 2010;94(2):781–783.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Dunaif A, Fauser BC. Renaming PCOS–a two-state solution. J Clin Endocrinol Metab. 2013;98(11):4325–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Kuang H, Li Y, Wu X, et al. Acupuncture and clomiphene citrate for live birth in polycystic ovary syndrome: study design of a randomized controlled trial. Evid Based Complement Alternat Med. 2013;2013:527303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Legro RS, Brzyski RG, Diamond MP, et al. Letrozole versus clomiphene for infertility in the polycystic ovary syndrome. N Engl J Med. 2014;371(2):119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]