

The FATC domain of Tel1 is studied via introduction of substitution and truncation mutations. It is found to be required for localization to sites of DNA damage and is essential for phosphorylation of exogenous substrates but dispensable for the intrinsic kinase activity.
Abstract
Two large phosphatidylinositol 3-kinase–related protein kinases (PIKKs), ATM and ATR, play a central role in the DNA damage response pathway. PIKKs contain a highly conserved extreme C-terminus called the FRAP-ATM-TRRAP-C-terminal (FATC) domain. In budding yeast, ATM and ATR correspond to Tel1 and Mec1, respectively. In this study, we characterized functions of the FATC domain of Tel1 by introducing substitution or truncation mutations. One substitution mutation, termed tel1-21, and a truncation mutation, called tel1-ΔC, did not significantly affect the expression level. The tel1-21 mutation impaired the cellular response to DNA damage and conferred moderate telomere maintenance defect. In contrast, the tel1-ΔC mutation behaved like a null mutation, conferring defects in both DNA damage response and telomere maintenance. Tel1-21 protein localized to DNA ends as effectively as wild-type Tel1 protein, whereas Tel1-ΔC protein failed. Introduction of a hyperactive TEL1-hy mutation suppressed the tel1-21 mutation but not the tel1-ΔC mutation. In vitro analyses revealed that both Tel1-21 and Tel1-ΔC proteins undergo efficient autophosphorylation but exhibit decreased kinase activities toward the exogenous substrate protein, Rad53. Our results show that the FATC domain of Tel1 mediates localization to DNA ends and contributes to phosphorylation of target proteins.
INTRODUCTION
The maintenance of genomic stability relies on a network of cellular processes, including DNA replication, DNA repair, and cell-cycle progression (Elledge, 1996; Harper and Elledge, 2007). Two phosphatidylinositol 3-kinase–related protein kinases (PIKKs), ATM and ATR, coordinate the cellular processes and govern the DNA damage response pathway. The DNA damage response pathway, also called the DNA damage checkpoint pathway, is highly conserved in eukaryotes (Harper and Elledge, 2007; Cimprich and Cortez, 2008). In budding yeast, ATM and ATR correspond to Tel1 and Mec1, respectively (Elledge, 1996). ATR/Mec1 recognizes many different types of DNA damage. By contrast, ATM/Tel1 acts specifically in response to DNA double-strand breaks (DSBs; Harper and Elledge, 2007; Cimprich and Cortez, 2008). Telomeres—the ends of linear eukaryotic chromosomes—have a specialized chromatin structure that discriminates chromosome ends from DSBs (Smogorzewska and de Lange, 2004; Wellinger and Zakian, 2012). ATM/Tel1 also maintains telomere length homeostasis (Greenwell et al., 1995; Metcalfe et al., 1996).
Evidence indicates that the Mre11-Rad50-Nbs1 (MRN) complex, which corresponds to the Mre11-Rad50-Xrs2 (MRX) complex in budding yeast, is a sensor that activates ATM/Tel1 (Lee and Paull, 2007). MRN/MRX localizes to DNA ends and plays multiple roles in DNA end processing (Symington and Gautier, 2011). In addition, MRN/MRX recruits ATM/Tel1 to DNA ends via the C-terminus of Nbs1/Xrs2 (Nakada et al., 2003a; Falck et al., 2005). The recruitment of ATM/Tel1 to DSBs is required for ATM/Tel1-mediated DNA damage response in vivo (Nakada et al., 2003a; Falck et al., 2005). Furthermore, purified MRN/MRX complex increases catalytic activity of ATM/Tel1 in the presence of DNA fragments in vitro (Lee and Paull, 2005; Fukunaga et al., 2011). Thus MRN/MRX interacts with and activates ATM/Tel1 at DNA ends. ATM/Tel1 phosphorylates the checkpoint mediators, including MDC1 in mammals and Rad9 in budding yeast, and in turn stimulates the downstream targets, such as protein kinase Chk2 in mammals and protein kinase Rad53 in budding yeast (Vialard et al., 1998; Schwartz et al., 2002; Pellicioli and Foiani, 2005; Harper and Elledge, 2007). In budding yeast, MRX also recognizes short telomeres and recruits Tel1 to the DNA ends (Bianchi and Shore, 2007; Chang et al., 2007; Hector et al., 2007; Sabourin et al., 2007; Viscardi et al., 2007), promoting telomere addition at short telomeres. Recent evidence suggests that Tel1 phosphorylates Cdc13 and stimulates telomerase recruitment (Tseng et al., 2006; Shen et al., 2014). However, this proposed mechanism has been under debate (Gao et al., 2010; Wu and Zakian, 2011; Wellinger and Zakian, 2012).
ATM/Tel1 and other PIKKs, including ATR, DNA-PKcs, mammalian target of rapamycin (mTOR; FRAP), and TRRAP, share a similar domain organization (Lempiainen and Halazonetis, 2009; Figure 1A). The kinase domains of PIKKs are located near their carboxyl termini and are flanked by the conserved FRAP-ATM-TRRAP (FAT) and FAT C-terminal (FATC) domains (Bosotti et al., 2000). Large amino-terminal and internal regions of PIKKs are composed of numerous α-helical Huntington-elongation factor 3-A subunit of protein phosphatase 2A-TOR1 (HEAT) repeats (Perry and Kleckner, 2003). PIKKs share an ∼30-residue FATC domain at the far C-terminus. However, the FATC domain is not completely unique to PIKKs; structure analyses indicate that its N-terminal half forms a helix that is also found in phosphatidylinositol 3-kinases (PI3Ks), whereas the C-terminal half forms another helix, which is unique to PIKKs (Walker et al., 1999; Miller et al., 2010; Yang et al., 2013).
FIGURE 1:

Substitution and truncation mutations of Tel1 at the conserved extreme C-terminal region. (A) Schematic of tel1 mutations at the FATC domain. Tel1 possesses a kinase domain (KD) within the C-terminal portion. The aligned sequences are derived from the FATC domains of ATM-family proteins (Tel1, human ATM, and fission yeast Tel1), ATR family proteins (Mec1, human ATR, and fission yeast Rad3), TOR proteins (budding yeast Tor1 and human mTOR), and human DNA-PK. Identical amino acid residues are boxed in black. Related amino acid residues are highlighted in gray. The α-helix structures kα11 and kα12 (12a, 12b, 12c) are adopted from Yang et al. (2013). The tel1-21, tel1-22, and tel1-23 mutations change the amino acid residues into alanine at positions 2779–2780, 2782–2783, and 2786–2787 (indicated by asterisks), respectively. The tel1-ΔC mutation introduces the termination codon for the amino acid residue at 2778 (indicated by X), truncating the amino acid residues from 2778 through 2787. The HEAT repeats and the FAT domains are shown as well. The residue numbers of the FAT and KD domains for Tel1 are indicated along with the overall Tel1 structure. The mutation site of TEL1-hy385 is shown on the overall structure (Baldo et al., 2008). (B) Expression level of Tel1 in tel1-21, tel1-22, tel1-23, and tel1-ΔC mutants. Top, cells expressing Tel1-HA (KSC1709), Tel1-21-HA (KSC3087), Tel1-22-HA (KSC3088), Tel1-23-HA (KSC3089), or Tel1-ΔC-HA (KSC3549) were subjected to immunoblotting analysis with anti-HA or anti-tubulin antibodies. Bottom, protein levels of Tel1 were normalized to tubulin and expressed relative to the wild type.
In this study, we characterized the PIKK-specific FATC function of Tel1 by introducing substitution or truncation mutations. A substitution mutation (tel1-21) and a truncation mutation (tel1-ΔC) affected phosphorylation of exogenous substrates but not autophosphorylation. Moreover, the tel1-ΔC mutation caused a defect in localization to DSBs. These results show that the FATC domain of Tel1 plays a key role in recognition of target proteins, as well as in accumulation at DNA ends.
RESULTS
Substitution and truncation mutations in the FATC domain of Tel1
PIKKs, including Tel1, share the FATC domain at the C-terminus (Bosotti et al., 2000). Whereas the N-terminal half of the FATC domain forms a helix that is also found in PI3Ks, the C-terminal half that forms another helix (kα12) is unique to PIKKs (Figure 1A; Walker et al., 1999; Miller et al., 2010; Yang et al., 2013). To understand the significance of the FATC domain, we focused on the last 15 amino acids, which consist of the kα12 helix. We constructed three substitution mutations in which the amino acid residues 2779–2780, 2782–2783, or 2786–2787 were all replaced with alanine. These mutations were termed tel1-21, tel1-22, and tel1-23, respectively (Figure 1A). The human R3047X ATM mutant lacking the last 10 amino acids activates checkpoint signaling in response to DSB induction but fails to respond to oxidative stress (Guo et al., 2010). We also generated an equivalent truncation mutation named tel1-ΔC (Figure 1A). Because the FATC domain is required for proper expression of ATM and ATR family proteins (Gilad et al., 1998; Nakada et al., 2005), we first examined the expression level of their gene products (Figure 1B). The tel1-22 and tel1-23 mutations decreased the expression level of Tel1 protein (∼35 and ∼50% of that of wild-type protein, respectively). Unlike the tel1-22 and tel1-23 mutations, the tel1-21 and tel1-ΔC mutations did not significantly affect the expression level.
Effect of FATC mutations on the cellular responses to DSB induction
Tel1, like other ATM family proteins, plays a role in the cellular response to DNA damage (Morrow et al., 1995; Sanchez et al., 1996). Although tel1Δ mutation by itself does not confer apparent sensitivity to DNA damage, it does exacerbate the sensitivity of mec1 mutants to DNA damage (Morrow et al., 1995; Sanchez et al., 1996). We therefore asked whether these tel1 mutations confer a similar effect (Figure 2, A and B). Because the mec1Δ tel1Δ double mutation results in a loss of cell proliferation (Chan et al., 2001), we used a weak allele of MEC1, mec1-81, to assess the effect of tel1 mutations (Nakada et al., 2003b). As found previously, mec1-81 single mutants were sensitive to DSB-inducing phleomycin and camptothecin, but introduction of a tel1Δ mutation enhanced the sensitivities to these damaging agents in mec1-81 mutants (Nakada et al., 2003a). Each of the tel1-22, tel1-23, and tel1-ΔC mutations increased the sensitivities similar to the tel1Δ mutation. However, the tel1-21 mutation caused a milder effect than the tel1-22, tel1-23 and tel1-ΔC mutations.
FIGURE 2:

DNA damage response of the FATC mutants. (A, B) DNA damage sensitivity of tel1-21, tel1-22, tel1-23, and tel1-ΔC mutants. Tenfold serial dilutions of cultures were spotted on yeast extract/peptone/dextrose (YEPD) medium with or without phleomycin (PHL; A) or camptothecin (CPT; B). Plates were incubated at 30°C for 2 or 3 d. Strains used were the wild type (KSC1560), mec1-81 (KSC1662), mec1-81 tel1Δ (KSC1564), mec1-81 tel1-21 (KSC3102), mec1-81 tel1-22 (KSC3100), mec1-81 tel1-23 (KSC3101), and mec1-81 tel1-ΔC (KSC3552). All the strains contain an sml1Δ mutation, which suppresses the lethality of mec1 mutants. (C) Effect of tel1 mutations on DNA damage checkpoint signaling. Cells expressing Rad53-HA were arrested with nocodazole (15 μg/ml) at G2/M and exposed to phleomycin (20 μg/ml) for 1 h. Cells were harvested and subjected to immunoblotting analysis with anti-HA antibodies. Strains used were the wild type (KSC1560), mec1-81 (KSC1662), mec1-81 sae2Δ (KSC3140), mec1-81 sae2Δ tel1Δ (KSC3141), mec1-81 sae2Δ tel1-21 (KSC3142), mec1-81 sae2Δ tel1-22 (KSC3143), mec1-81 sae2Δ tel1-23 (KSC3144), and mec1-81 sae2Δ tel1-ΔC (KSC3555). All the strains contain an sml1Δ mutation.
Tel1 controls phosphorylation and activation of Rad53 after DNA damage, and its phosphorylation status is well correlated with activation of DNA damage checkpoint (Elledge, 1996; Longhese et al., 1998). We next determined the effect of these mutations on Rad53 phosphorylation after DNA damage (Figure 2C). To detect Tel1-specific signaling, we monitored Rad53 phosphorylation in a mec1-81 sae2Δ background. Mutations in SAE2 enhance Tel1-mediated checkpoint signaling (Usui et al., 2001; Fukunaga et al., 2011). Cells expressing hemagglutinin (HA)-tagged Rad53 were grown to log phase and then arrested at G2/M with nocodazole. After arrest, cells were exposed to phleomycin and subjected to immunoblotting analysis with anti-HA antibodies. Rad53 phosphorylation occurred in wild-type cells, whereas phosphorylation decreased in mec1-81 mutants. However, Rad53 phosphorylation became readily detectable in mec1-81 sae2Δ mutants. Consistent with the current view that the sae2Δ mutation enhances Tel1-mediated checkpoint signaling (Usui et al., 2001; Fukunaga et al., 2011), introduction of the tel1Δ mutation decreased Rad53 phosphorylation in mec1-81 sae2Δ mutants. The tel1-21, tel1-22, tel1-23 or tel1-ΔC mutation also decreased Rad53 phosphorylation in mec1-81 sae2Δ mutants.
Telomere maintenance in FATC mutants
In budding yeast, Tel1 plays an essential role in telomere length maintenance (Wellinger and Zakian, 2012). We further determined the effect of tel1-21, tel1-22, tel1-23, or tel1-ΔC mutation on telomere length (Figure 3A). Telomeres become shorter in tel1Δ mutants than in wild-type cells (Greenwell et al., 1995; Wellinger and Zakian, 2012). Telomeres in tel1-22, tel1-23, or tel1-ΔC mutants were as short as those in tel1Δ mutants. In contrast, telomeres became slightly shorter in tel1-21 mutants than in wild-type cells. Although MEC1 is not essential for telomere homeostasis in TEL1 cells, Mec1 plays a key role in telomere length regulation in the absence of Tel1 (Wellinger and Zakian, 2012). It is therefore possible that Mec1 contributes to telomere addition in tel1-21 mutants. We then examined the effect of mec1Δ mutation on telomere length in tel1-21 mutants (Figure 3B). The telomere length was very similar in tel1-21 single and mec1Δ tel1-21 double mutants. Thus the tel1-21 mutation largely eliminates Tel1 function in DNA damage response but retains some telomere maintenance function. In contrast, the tel1-22, tel1-23, or tel1-ΔC mutation behaves like a tel1-null mutant.
FIGURE 3:
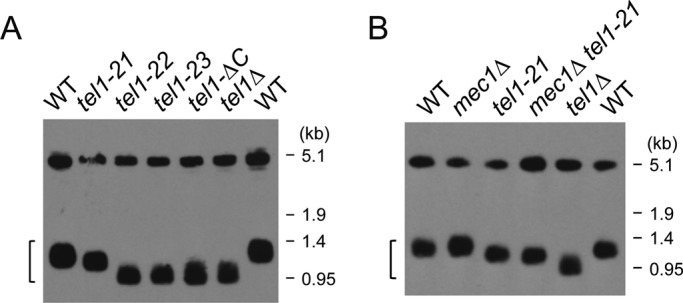
Telomere length of the FATC mutants. (A) Genomic DNA prepared from cells was digested with XhoI and analyzed by Southern blots to monitor the telomere length. The hybridization probe was a DNA fragment containing ∼0.9-kb Y′ element and ∼120–base pair TG repeat sequence. The bracket shows DNA fragments containing the telomere. Strains used were the wild type (KSC006), tel1Δ (KSC1057), tel1-21 (KSC3378), tel1-22 (KSC3379), tel1-23 (KSC3377), and tel1-ΔC (KSC3556). (B) Southern blotting analysis performed as in A. Strains used were the wild type (KSC1560), tel1Δ (KSC1661), mec1Δ (KSC1561), tel1-21 (KSC3629), and mec1Δ tel1-21 (KSC3630). All the strains contain an sml1Δ mutation.
Defective localization of Tel1-ΔC protein to DNA ends
Tel1 localizes to DNA ends to activate the signaling pathway and elongate short telomeres (Nakada et al., 2003a; Bianchi and Shore, 2007; Chang et al., 2007; Hector et al., 2007; Sabourin et al., 2007). We thus examined the effect of tel1-21 or tel1-ΔC mutation on localization of Tel1 to HO-induced DSBs by chromatin immunoprecipitation (ChIP) assay (Figure 4A). In budding yeast, HO endonuclease introduces a sequence-specific DSB. We used an experimental system in which cells contain a single HO cleavage site at the ADH4 locus, and HO is expressed from the GAL-HO plasmid after incubation with galactose (Nakada et al., 2003a). Cells expressing Tel1-HA, Tel1-21-HA, or Tel1-ΔC-HA were transformed with the GAL-HO plasmid. Transformed cells were grown initially in sucrose to repress HO expression and then transferred to medium containing nocodazole to arrest at G2/M. After arrest, galactose was added to induce HO expression. Cells were then subjected to the ChIP assay. Tel1-21 mutant protein associated with HO-induced DSBs as efficiently as Tel1 wild-type protein did. In contrast, association of Tel1-ΔC protein with DSBs was significantly decreased compared with that of wild-type Tel1 protein.
FIGURE 4:

Effect of tel1-21 or tel1-ΔC mutation on association with DNA ends. (A) Effect of tel1-21 or tel1-ΔC mutation on Tel1 localization to an HO-induced DSB. Wild-type (KSC1709), tel1-21 (KSC3087) or tel1-ΔC (KSC3549) cells expressing Tel1-HA were transformed with the YCpA-GAL-HO plasmid. Transformed cells were grown in sucrose and treated with nocodazole. After arrest at G2/M, the culture was incubated with galactose for 3 h to induce HO expression. Top, the strains contain an HO cleavage site, marked with HIS2, at the ADH4 locus on chromosome (Chr.) VII. The HO1 primer pair amplifies a region 1 kb away from the HO cleavage site. An arrow represents the telomere. Bottom, cells were subjected to chromatin immunoprecipitation with anti-HA antibodies. Association of Tel1 with an HO-induced DSB was analyzed by real-time PCR. Relative enrichment was determined from three independent experiments. (B) Effect of tel1-ΔC mutation on the intracellular distribution. TEL1-HA (KSC1709) or tel1-ΔC-HA (KSC3549) cells were grown to mid log phase, harvested, and spheroplasted. Spheroplasts were homogenized to prepare whole-cell extracts (W) and then separated into the cytoplasmic (C) and nuclear (N) fractions. Samples from each fraction were separated by SDS–PAGE and immunoblotted with anti-HA, anti-Zwf1 (glucose-6-phosphate dehydrogenase; G6PDH) or anti-nuclear pore complex (NPC) antibodies. (C) Effect of tel1-ΔC mutation on Tel1–Tel2 interaction. Whole-cell extracts (WCE) prepared from cells expressing Tel1-HA (KSC1709), Tel2-FLAG Tel1-HA (KSC3633), or Tel2-FLAG tel1-ΔC-HA (KSC3634) were subjected to immunoprecipitation (IP) with anti-FLAG antibodies. Samples were immunoblotted with anti-HA or anti-FLAG antibodies. (D) Effect of tel1-ΔC mutation on Tel1–Xrs2 interaction. FLAG-Tel1 or FLAG-Tel1-ΔC was incubated with GST-Xrs2C–bound glutathione beads. Proteins bound to glutathione beads were subjected to immunoblotting analysis with anti-FLAG antibodies. The binding assay used 0.5 pmol of FLAG-Tel1 proteins and 70 pmol of GST-Xrs2C.
Because Tel1-ΔC protein does not efficiently accumulate at DNA ends, we determined the effect of tel1-ΔC mutation on cellular localization (Figure 4B). Cellular fractionation analysis indicated that Tel1 exists in both nuclear and cytoplasmic fractions. However, the tel1-ΔC mutation did not significantly affect the intracellular distribution. Tel1 interacts with the Tel2-Tti1-Tti2 (TTT) complex, which promotes protein stability of PIKKs (Hayashi et al., 2007; Takai et al., 2007, 2010; Anderson et al., 2008; Hurov et al., 2010; Stirling et al., 2011). The TTT complex also controls localization of Tel1 to DNA ends (Anderson et al., 2008). We next examined the effect of tel1-ΔC on Tel1-TTT interaction by coimmunoprecipitation assay (Figure 4C). However, Tel1 and Tel1-ΔC were found to interact similarly with Tel2.
Tel1 localizes to DNA ends through an Xrs2-dependent mechanism (Nakada et al., 2003a). We previously showed that Tel1 interacts directly with the C-terminus of Xrs2 in vitro (Hirano et al., 2009). We further examined the effect of tel1-ΔC mutation on Tel1-Xrs2 interaction by the pull-down assay (Figure 4D). A glutathione S-transferase (GST) fusion of the Xrs2 C-terminus (GST-Xrs2C) was captured on glutathione–Sepharose beads and incubated with purified FLAG-Tel1 or Tel1-ΔC proteins. Bound proteins were then analyzed by immunoblotting with anti-FLAG antibodies. Although Xrs2 C-terminus was able to bind to the added Tel1 protein, efficient Tel1–Xrs2 interaction did not occur with Tel1-ΔC mutant protein. These results indicate that the FATC domain of Tel1 mediates Tel1–Xrs2 interaction.
Intragenic suppression of tel1-21 by a hyperactive TEL1 mutation
Previous studies identified TEL1-hy mutations that increase basal catalytic activities (Baldo et al., 2008). Tel1-21 mutant protein localized efficiently to DNA ends, whereas Tel1-ΔC did not. We addressed whether any TEL1-hy mutation suppresses the tel1-21 or tel1-ΔC mutation (Figure 5). To this end, we introduced TEL1-hy mutations into the mec1-81 tel1-21 and mec1-81 tel1-ΔC strains. One substitution mutation (N2692D) within the kinase domain, termed TEL1-hy385 (Figure 1A), increased resistance to camptothecin and methylmethane sulfonate (MMS) in tel1-21 mutant cells but not in tel1-ΔC mutants (Figure 5, A and B). We further determined whether the TEL1-hy385 mutation suppresses telomere defects of tel1-21 and tel1-ΔC mutants. Similar to the mec1Δ mutation (Figure 3B), the mec1-81 mutation had little effect on telomere length regardless of the presence or absence of Tel1 function (Figure 5C). The TEL1-hy385 mutation elongated telomeres in mec1-81 single- and mec1-81 tel1-21 double-mutant cells (Figure 5D). However, the TEL1-hy385 mutation did not affect telomere length in mec1-81 tel1-ΔC cells (Figure 5D). These results are consistent with the finding that Tel1-ΔC protein does not efficiently associate with DNA ends and raise the possibility that Tel1-21 mutant protein has decreased catalytic activity.
FIGURE 5:
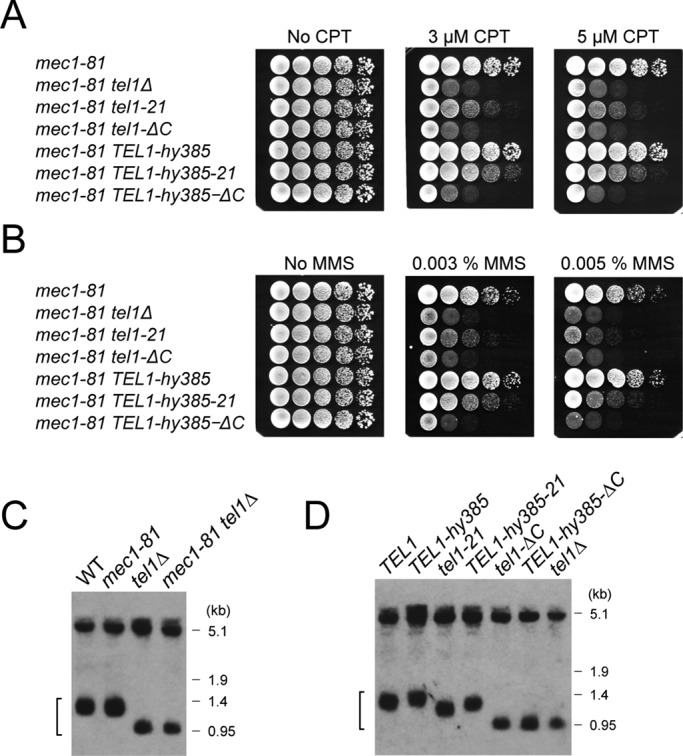
Intragenic suppression of tel1-21 by a hyperactive TEL1 mutation. (A, B) Effect of TEL1-hy385 mutation on DNA damage sensitivities of tel1-21 and tel1-ΔC mutants. Tenfold serial dilutions of cultures were spotted on YEPD medium with or without camptothecin (CPT) (A) or MMS (B). Plates were incubated at 30°C for 2 or 3 d. Strains used were mec1-81 (KSC1662), mec1-81 tel1Δ (KSC1564), mec1-81 tel1-21 (KSC3102), mec1-81 tel1-ΔC (KSC3552), mec1-81 TEL1-hy385 (KSC3635), mec1-81 TEL1-hy385-21 (KSC3636), and mec1-81 TEL1-hy385-ΔC (KSC3637). All the strains contain an sml1Δ mutation. (C) Telomere length in mec1-81 mutants. Telomere length was analyzed by Southern blots as in Figure 3. Strains used were wild type (KSC1560), mec1-81 (KSC1662), tel1∆ (KSC1661), and mec1-81 tel1∆ (KSC1564). All the strains contain an sml1Δ mutation. (D) Effect of TEL1-hy385 mutation on telomere length of tel1-21 and tel1-ΔC mutants. The same strains used in A were subjected to Southern blotting analysis to monitor telomere length as in C.
Catalytic activity of Tel1-21 and Tel1-ΔC proteins
Mec1 and Tel1 phosphorylate Rad9 to transiently recruit Rad53 nearby at sites of DNA damage (Vialard et al., 1998; Schwartz et al., 2002). In turn, Mec1 and Tel1 phosphorylate Rad53, thereby increasing the kinase activity of Rad53 (Pellicioli and Foiani, 2005). Several lines of evidence suggest that Mec1 and Tel1 phosphorylate the Rad53 C-terminus after DNA damage (Schwartz et al., 2002; Lee et al., 2003; Smolka et al., 2005; Sweeney et al., 2005). As discussed earlier, the tel1-21 and the tel1-ΔC mutations did not significantly affect the expression level of Tel1. We next tested whether Tel1-21 or Tel1-ΔC protein phosphorylates Rad53 in vitro by using GST-Rad53 C-terminus fusion as a substrate (Fukunaga et al., 2011; Figure 6A). Human ATM undergoes autophosphorylation at serine 1981 in response to DNA damage (Bakkenist and Kastan, 2003). S1981 phosphorylation is detected in cells carrying the ATM R3047X gene (Guo et al., 2010), a mutation that is equivalent to the tel1-ΔC mutation. We also examined whether Tel1-21 or Tel1-ΔC protein undergoes autophosphorylation (Figure 6A). We used the strain expressing HA-tagged kinase-negative Tel1-KN protein as a negative control (Nakada et al., 2003b). We prepared extracts from cells expressing Tel1-HA, Tel1-21-HA, Tel1-ΔC-HA, or Tel1-KN-HA and immunoprecipitated them with anti-HA antibodies. Immunoprecipitates were then subjected to an in vitro kinase assay, in which radioactive phosphorus (32P) was incorporated into GST-Rad53 by immunoprecipitates containing Tel1-HA. The same reactions were also analyzed for 32P incorporation into Tel1 protein. Both Tel1-21 and Tel1-ΔC mutant proteins were significantly defective in phosphorylating Rad53 compared with wild-type Tel1. However, autophosphorylation of Tel1-21 and Tel1-ΔC mutant proteins occurred similarly to that of wild-type Tel1 protein. Autophosphorylation was not saturated in the reaction condition (20-min incubation), as more-pronounced phosphoincorporation was detected after a prolonged incubation (40-min incubation; Figure 6B). Thus the FATC domain is critical for phosphorylation of Rad53 protein but dispensable for autophosphorylation. Taken together, our results support a model in which the FATC domain of Tel1 mediates recognition of target proteins, as well as localization to DNA ends.
FIGURE 6:
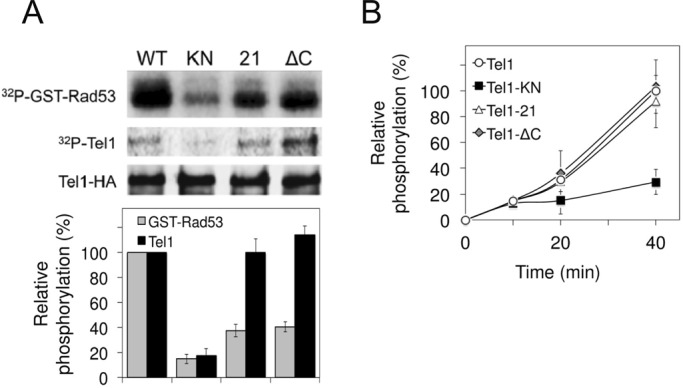
Effect of tel1-21 or tel1-ΔC mutation on protein kinase activity in vitro. (A) Effect of tel1-21 or tel1-∆C mutation on Rad53 phosphorylation and Tel1 autophosphorylation. Extracts were prepared from cells expressing Tel1-HA (KSC1709), Tel1-KN-HA (KSC1751), Tel1-21-HA (KSC3087), or Tel1-ΔC-HA (KSC3549) and subjected to immunoprecipitation with anti-HA antibodies. Immunoprecipitates were assayed for kinase reactions using GST-Rad53 C-terminus as a substrate. Top, Rad53 phosphorylation and Tel1 autophosphorylation were detected by autoradiography, and the amount of Tel1 protein was analyzed by immunoblotting with anti-HA antibodies. Bottom, phosphoincorporation was normalized relative to that from wild-type Tel1 protein (100%). The average intensity calculated from three independent experiments is shown, and the error bars represent the SD derived from these experiments. (B) Tel1 autophosphorylation after incubation for various lengths of time. Tel1 proteins were immunoprecipitated and subjected to the in vitro kinase assay as in A, but the reaction mixtures were incubated for various lengths of time (0, 10, 20, and 40 min). Phosphoincorporation into Tel1 was normalized relative to that for wild-type Tel1 protein at 0 (0%) and 40 min (100%).
DISCUSSION
The C-terminal half of the FATC domain is specifically conserved among PIKKs, including ATM, ATR, DNA-PK, and mTOR (Bosotti et al., 2000; Yang et al., 2013). We examined roles of the FATC domain of Tel1 by introducing substitution and truncation mutations in the domain. One substitution, termed tel1-21, and a truncation mutation, termed tel1-ΔC, did not significantly alter the expression level of Tel1. The tel1-ΔC mutation behaved like a null mutation and caused defects in both DNA damage response and telomere homeostasis. The tel1-21 mutation caused less-pronounced defects than the tel1-ΔC mutation. Whereas Tel1-21 protein localized to DNA ends as efficiently as wild-type Tel1 protein, Tel1-ΔC did not. Both Tel1-21 and Tel1-ΔC proteins exhibited decreased kinase activities toward Rad53 C-terminus, although they underwent autophosphorylation efficiently. These results suggest that the FATC domain of Tel1 plays a key role in DNA damage recognition, as well as in phosphorylation of target proteins.
Tel1-21 and Tel1-ΔC proteins are defective in phosphorylating Rad53, although they retain activities to undergo overall autophosphorylation. Thus the C-terminal half of the FATC domain is critical for phosphorylating exogenous substrates but dispensable for autophosphorylation. Our finding supports a model in which the FATC domain contributes to recognition of exogenous target proteins. Indeed, crystal structures of mTOR reveal that the FATC domain is integrated into the kinase domain structure, and the substrate-holding groove consists of portions of the FATC domain (Yang et al., 2013). Tel1-dependent phosphorylation stimulates telomerase recruitment to short telomeres, although the targets of Tel1 at telomeres have not been precisely determined (Gao et al., 2010; Wu and Zakian, 2011; Wellinger and Zakian, 2012; Shen et al., 2014). The tel1-21 mutation behaves almost like a null mutation with regard to DNA damage response, whereas it causes a modest defect in telomere length homeostasis. One explanation could be that Tel1-dependent phosphorylation is an essential but not a rate-limiting step during telomere extension. Because telomeres lose only three to five nucleotides per cell division (Wellinger and Zakian, 2012), cells could largely maintain telomere length even if Tel1-21–dependent phosphorylation occurred very slowly. Alternatively, Tel1-21 might largely retain catalytic activities to phosphorylate targets at telomeres. Previous studies showed that ATM R3047X, lacking the C-terminal half of the FATC domain, undergoes autophosphorylation after DNA damage in vivo (Guo et al., 2010) but exhibits decreased kinase activity toward exogenous substrates in vitro (Banin et al., 1998). These observations also support the idea that the FATC domain contributes to target protein recognition, although we cannot exclude the possibility that the tel1-21 mutation stimulates kinase-independent functions in telomere maintenance (Ma and Greider, 2009). The C-terminal half of the FATC domain is unique to PIKKs, although the N-terminal half is found in PI3K structures (Walker et al., 1999; Miller et al., 2010; Yang et al., 2013). We found that the C-terminal half of the Tel1 FATC domain is not essential for autophosphorylation in vitro. PI3Ks phosphorylate the 3′-position of the inositol ring in phosphatidylinositol lipids (Fruman et al., 1998). In addition to lipid kinase activities, PI3Ks possess an intrinsic protein kinase activity directed toward the adaptors and the catalytic subunits themselves (Stack and Emr, 1994; Vanhaesebroeck et al., 1999; Foukas et al., 2004). Thus PIKKs might have evolved from PIK3s, acquiring the additional helix domain to phosphorylate exogenous protein substrates.
Tel1 localizes to DNA ends by interacting with the C-terminus of Xrs2 (Nakada et al., 2003a). The current model suggests that the C-terminus of Xrs2/Nbs1 and the HEAT repeats of Tel1/ATM interact with each other (You et al., 2005). Supporting this model, we isolated the C-terminus of Xrs2 from a two-hybrid screen using the N-terminus of Tel1 as bait (Nakada et al., 2003a). In this report, we showed that the FATC domain of Tel1 also mediates Tel1–Xrs2 interaction and contributes to the localization to DNA ends. Similar to Tel1-21 protein, Tel1-ΔC protein retains weak kinase activities. However, unlike Tel1-21 protein, Tel1-ΔC protein did not efficiently accumulate at DNA ends. Consistently, we found that Tel1-ΔC protein interacts with the C-terminus of Xrs2 less efficiently than does wild-type Tel1. Defective localization of Tel1-ΔC to DNA ends should not result from decreased kinase activities, because Tel1 localization to DNA ends does not depend on kinase activity (Fukunaga et al., 2011). The hyperactive TEL1-hy385 mutation suppressed the tel1-21 mutation but not the tel1-ΔC mutation, supporting the view that Tel1 localization to DNA ends is critical for DSB response and telomere maintenance (Nakada et al., 2003a; Hector et al., 2007; Sabourin et al., 2007). Of note, the FATC domain of Mec1 also has been implicated in localization to sites of DNA damage (Nakada et al., 2005). Mec1 forms a complex with Ddc2 that interacts directly with RPA-coated single-stranded DNA (Zou and Elledge, 2003). Whereas the N-terminal domain of Mec1 interacts with Ddc2 (Wakayama et al., 2001), the FATC domain of Mec1 appears to mediate RPA-Ddc2 interaction (Nakada et al., 2005). How does the FATC domain collaborate with the N-terminal HEAT repeat to localize Tel1 and Mec1 to sites of DNA damage? The crystal structure analysis of mTOR revealed that the FATC domain is integrated into the kinase domain (Yang et al., 2013), although this analysis did not include most of the N-terminal HEAT repeats. For DNA-PKcs, the 6.6-Å x-ray diffraction data suggest that the kinase domain is adjacent to the N-terminal HEAT repeat ring (Sibanda et al., 2010). Thus the FATC domain of PIKKs could affect the overall structure of the HEAT repeats, which may position near the kinase domain.
The other two mutations, tel1-22 and tel1-23, decreased the expression level of Tel1 more significantly than did tel1-21 or tel1-ΔC. We therefore did not extensively characterize molecular details of the Tel1-22 and Tel1-23 proteins. Tel2 interacts with other conserved proteins, Tti1 and Tti2, and controls stability, perhaps by mediating maturation of PIKKs (Hayashi et al., 2007; Takai et al., 2007, 2010; Anderson et al., 2008; Hurov et al., 2010; Stirling et al., 2011). One possibility is that these mutations affect the interaction of Tel1 with the Tel2-Tti1-Tti2 (TTT) complex. However, the tel1-ΔC mutation did not affect Tel1–Tel2 interaction, suggesting that the FATC domain is dispensable for Tel1–Tel2 interaction. Previous studies indicated that Tel2 interacts with PIKKs through the HEAT domain (Takai et al., 2007). It is thus less likely that the tel1-22 or tel1-23 mutation could interfere with the Tel1–TTT interaction. The TTT complex could control protein stability of PIKKs through the FATC domain; indeed, tti2 mutations suppress an FATC mutation of TRA1 (Genereaux et al., 2012). However, the same tti2 mutation failed to suppress an equivalent mutation of MEC1 (Genereaux et al., 2012). More-comprehensive studies would be required to determine whether the FATC domain mediates TTT-dependent function. Although the tel1-22 or tel1-23 mutation did not completely abolish the expression, these mutations conferred defects in both DNA damage response and telomere maintenance. Similar to Tel1-ΔC protein, Tel1-22 and Tel1-23 proteins might not efficiently localize to DNA ends.
In summary, our results support a model in which the FATC domain of Tel1 plays a key role in localization to DNA ends and exogenous substrate phosphorylation. Of note, the FATC domain of Mec1 mediates similar functions. Further structure and function studies would help to elucidate the precise molecular mechanism of how PIKKs assemble at specific locations and phosphorylate target proteins.
MATERIALS AND METHODS
Strains and plasmids
The C-terminal region of the tel1-21, tel1-22, tel1-23, tel1-ΔC, and TEL1-hy385 mutations was amplified and fused to the URA3 marker by PCR using the oligonucleotides KS948 and KS1845, KS1846, KS1140, KS2807, or KS3262, respectively, and integrated into the TEL1 locus (Reid et al., 2002). The mec1-81, mec1Δ, sae2Δ, tel1-KN, and tel1Δ mutations have been described (Nakada et al., 2003b; Hirano et al., 2009; Fukunaga et al., 2011). The TEL1-HA construct was introduced into the TEL1 locus as described (Fukunaga et al., 2011). The TEL2-FLAG strain was generated by a PCR-based method using the pKL258 plasmid (a gift from M. Kanemaki and K. Labib, University of Manchester, UK) using the primer pair KS2789 and KS2790. To create pRS316-GAL-FLAG-TEL1, the GAL-FLAG-TEL1 construct from pGAL-FLAG-TEL1 (Hirano et al., 2009) was introduced into SacII-SalI–treated pRS316 (Sikorski and Hieter, 1989). The pRS316-GAL1-FLAG-TEL1-ΔC plasmid was generated from pRS316-GAL1-FLAG-TEL1 by replacing the SphI-SalI fragment with that containing the tel1-ΔC mutation. All of the strains and oligonucleotides used in this study are listed in Supplemental Tables S1 and S2, respectively. The GST-Rad53 (containing the C-terminal Rad53) and GST-Xrs2C plasmid have been described (Nakada et al., 2003b; Hirano et al., 2009; Fukunaga et al., 2011). YCpT-RAD53-HA has been described elsewhere (Fukunaga et al., 2011).
Tel1 kinase assay
HA-tagged Tel1 protein was immunoprecipitated using protein A–Sepharose beads as described (Fukunaga et al., 2011; Bandhu et al., 2014). Kinase reactions were conducted in 40 μl of the reaction buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid–KOH, pH 7.5, 4 mM MgCl2, 4 mM MnCl2, 50 μM ATP) containing 3 μCi of [γ-32P]ATP (3000 Ci/mmol, PerkinElmer, Melville, NY) for 20 min at 30°C. GST-tagged Rad53 C-terminus (GST-Rad53) was used as a substrate (1 μg for each reaction) for the in vitro kinase assay (Wakayama et al., 2001; Nakada et al., 2003b). Details of the kinase reaction have been described previously (Fukunaga et al., 2011; Bandhu et al., 2014). The reaction mixtures were electrophoresed on 6% SDS–polyacrylamide gels (30:1) and transferred to polyvinylidene fluoride membranes for immunoblotting analysis to determine the amount of Tel1 proteins. The same membranes were then subjected to autoradiography to detect Tel1 autophosphorylation. To monitor 32P incorporation into GST-Rad53, aliquots of the reaction mixtures were run on 10% gels. The autophosphorylation time-course experiment was carried out without addition of GST-Rad53. Because the expression level of Tel1-ΔC was lower than that of wild-type cells, the amount of Tel1-ΔC in the immune complex was less than that of wild-type Tel1 protein if immunoprecipitated from the same amount of cells. Amounts of extracts used for immunoprecipitation of Tel1-HA, Tel1-KN-HA, and Tel1-21-HA were adjusted by including the extract from the isogenic nontagged strain. Phosphorylation was quantified with a phosphorimager system (Typhoon 8600; GE Healthcare, Buckinghamshire, UK).
Telomere Southern blots
Telomere length was monitored by Southern blotting analysis (Hirano et al., 2009; Fukunaga et al., 2012). Genomic DNA was digested with XhoI, electrophoresed, and transferred to nylon membranes. Membranes were hybridized with a DNA fragment containing a Y′ element and telomeric TG repeat. The DNA probe was labeled by the DIG labeling system (Roche, Basel, Switzerland).
GST-Xrs2 pull-down assay
The pull-down assay was done as described previously (Hirano et al., 2009), with some modifications. GST-Xrs2C was bound to glutathione–Sepharose beads and then incubated with purified FLAG-Tel1 protein in the binding buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.01% Triton X-100) at 4°C for 90 min. FLAG-tagged Tel1 or Tel1-ΔC protein was purified from cells carrying pRS316-GAL-FLAG-TEL1 or pRS316-GAL-FLAG-TEL1-ΔC plasmid, respectively, as previously described (Hirano et al., 2009; Fukunaga et al., 2012).
Other methods
Chromatin immunoprecipitation assay was carried out as described (Nakada et al., 2003a; Fukunaga et al., 2011). The DNA damage sensitivity assay was as described (Nakada et al., 2003a). Cellular fractionation, immunoprecipitation, and immunoblotting were performed as described previously (Wakayama et al., 2001; Fukunaga et al., 2012). Immunoblots were quantified using an Odyssey imager (LI-COR, Lincoln, NE). Anti-Zwf1 (glucose-6-phosphate dehydrogenase) and anti–nuclear pore complex antibodies were purchased from Sigma-Aldrich (St. Louis, MO) and Covance (Princeton, NJ), respectively.
Supplementary Material
Acknowledgments
We thank Hengyao Niu, Masato Kanemaki, Karim Labib, Maria Pia Longhese, and Patrick Sung for providing materials and John Kang and Carol Newlon for critical reading of the manuscript. This work was supported by National Institutes of Health Grants GM073876 and CA148939.
Abbreviations used:
- FAT
FRAP-ATM-TRRAP
- FATC
FRAP-ATM-TRRAP-C-terminal
- HEAT
Huntington-elongation factor 3-A subunit of protein phosphatase 2A-TOR1
- PI3K
phosphatidylinositol 3-kinase
- PIKK
phosphatidylinositol 3-kinase–related protein kinase
- TTT
Tel2-Tti1-Tti2.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-05-0259) on August 5, 2015.
REFERENCES
- Anderson CM, Korkin D, Smith DL, Makovets S, Seidel JJ, Sali A, Blackburn EH. Tel2 mediates activation and localization of ATM/Tel1 kinase to a double-strand break. Genes Dev. 2008;22:854–859. doi: 10.1101/gad.1646208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Baldo V, Testoni V, Lucchini G, Longhese MP. Dominant TEL1-hy mutations compensate for Mec1 lack of functions in the DNA damage response. Mol Cell Biol. 2008;28:358–375. doi: 10.1128/MCB.01214-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandhu A, Kang J, Fukunaga K, Goto G, Sugimoto K. Ddc2 mediates Mec1 activation through a Ddc1- or Dpb11-independent mechanism. PLoS Genet. 2014;10:e1004136. doi: 10.1371/journal.pgen.1004136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Bianchi A, Shore D. Increased association of telomerase with short telomeres in yeast. Genes Dev. 2007;21:1726–1730. doi: 10.1101/gad.438907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosotti R, Isacchi A, Sonnhammer EL. FAT: a novel domain in PIK-related kinases. Trends Biochem Sci. 2000;25:225–227. doi: 10.1016/s0968-0004(00)01563-2. [DOI] [PubMed] [Google Scholar]
- Chan SW, Chang J, Prescott J, Blackburn EH. Altering telomere structure allows telomerase to act in yeast lacking ATM kinases. Curr Biol. 2001;11:1240–1250. doi: 10.1016/s0960-9822(01)00391-8. [DOI] [PubMed] [Google Scholar]
- Chang M, Arneric M, Lingner J. Telomerase repeat addition processivity is increased at critically short telomeres in a Tel1-dependent manner in Saccharomyces cerevisiae. Genes Dev. 2007;21:2485–2494. doi: 10.1101/gad.1588807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Foukas LC, Beeton CA, Jensen J, Phillips WA, Shepherd PR. Regulation of phosphoinositide 3-kinase by its intrinsic serine kinase activity in vivo. Mol Cell Biol. 2004;24:966–975. doi: 10.1128/MCB.24.3.966-975.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Hirano Y, Sugimoto K. Subtelomere-binding protein Tbf1 and telomere-binding protein Rap1 collaborate to inhibit localization of the Mre11 complex to DNA ends in budding yeast. Mol Biol Cell. 2012;23:347–359. doi: 10.1091/mbc.E11-06-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Kwon Y, Sung P, Sugimoto K. Activation of protein kinase Tel1 through recognition of protein-bound DNA ends. Mol Cell Biol. 2011;31:1959–1971. doi: 10.1128/MCB.05157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Toro TB, Paschini M, Braunstein-Ballew B, Cervantes RB, Lundblad V. Telomerase recruitment in Saccharomyces cerevisiae is not dependent on Tel1-mediated phosphorylation of Cdc13. Genetics. 2010;186:1147–1159. doi: 10.1534/genetics.110.122044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genereaux J, Kvas S, Dobransky D, Karagiannis J, Gloor GB, Brandl CJ. Genetic evidence links the ASTRA protein chaperone component Tti2 to the SAGA transcription factor Tra1. Genetics. 2012;191:765–780. doi: 10.1534/genetics.112.140459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad S, Chessa L, Khosravi R, Russell P, Galanty Y, Piane M, Gatti RA, Jorgensen TJ, Shiloh Y, Bar-Shira A. Genotype-phenotype relationships in ataxia-telangiectasia and variants. Am J Hum Genet. 1998;62:551–561. doi: 10.1086/301755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD. TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell. 1995;82:823–829. doi: 10.1016/0092-8674(95)90479-4. [DOI] [PubMed] [Google Scholar]
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Hatanaka M, Nagao K, Nakaseko Y, Kanoh J, Kokubu A, Ebe M, Yanagida M. Rapamycin sensitivity of the Schizosaccharomyces pombe tor2 mutant and organization of two highly phosphorylated TOR complexes by specific and common subunits. Genes Cells. 2007;12:1357–1370. doi: 10.1111/j.1365-2443.2007.01141.x. [DOI] [PubMed] [Google Scholar]
- Hector RE, Shtofman RL, Ray A, Chen BR, Nyun T, Berkner KL, Runge KW. Tel1p preferentially associates with short telomeres to stimulate their elongation. Mol Cell. 2007;27:851–858. doi: 10.1016/j.molcel.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Fukunaga K, Sugimoto K. Rif1 and Rif2 inhibit localization of Tel1 to DNA ends. Mol Cell. 2009;33:312–322. doi: 10.1016/j.molcel.2008.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurov KE, Cotta-Ramusino C, Elledge SJ. A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev. 2010;24:1939–1950. doi: 10.1101/gad.1934210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26:7741–7748. doi: 10.1038/sj.onc.1210872. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Schwartz MF, Duong JK, Stern DF. Rad53 phosphorylation site clusters are important for Rad53 regulation and signaling. Mol Cell Biol. 2003;23:6300–6314. doi: 10.1128/MCB.23.17.6300-6314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lempiainen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009;28:3067–3073. doi: 10.1038/emboj.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese MP, Foiani M, Muzi-Falconi M, Lucchini G, Plevani P. DNA damage checkpoint in budding yeast. EMBO J. 1998;17:5525–5528. doi: 10.1093/emboj/17.19.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Greider CW. Kinase-independent functions of TEL1 in telomere maintenance. Mol Cell Biol. 2009;29:5193–5202. doi: 10.1128/MCB.01896-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe JA, Parkhill J, Campbell L, Stacey M, Biggs P, Byrd PJ, Taylor AM. Accelerated telomere shortening in ataxia telangiectasia. Nat Genet. 1996;13:350–353. doi: 10.1038/ng0796-350. [DOI] [PubMed] [Google Scholar]
- Miller S, Tavshanjian B, Oleksy A, Perisic O, Houseman BT, Shokat KM, Williams RL. Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science. 2010;327:1638–1642. doi: 10.1126/science.1184429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DM, Tagle DA, Shiloh Y, Collins FS, Hieter P. TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell. 1995;82:831–840. doi: 10.1016/0092-8674(95)90480-8. [DOI] [PubMed] [Google Scholar]
- Nakada D, Hirano Y, Tanaka Y, Sugimoto K. Role of the C terminus of mec1 checkpoint kinase in its localization to sites of DNA damage. Mol Biol Cell. 2005;16:5227–5235. doi: 10.1091/mbc.E05-05-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Matsumoto K, Sugimoto K. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 2003a;17:1957–1962. doi: 10.1101/gad.1099003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Shimomura T, Matsumoto K, Sugimoto K. The ATM-related Tel1 protein of Saccharomyces cerevisiae controls a checkpoint response following phleomycin treatment. Nucleic Acids Res. 2003b;31:1715–1724. doi: 10.1093/nar/gkg252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Foiani M. Signal transduction: how rad53 kinase is activated. Curr Biol. 2005;15:R769–R771. doi: 10.1016/j.cub.2005.08.057. [DOI] [PubMed] [Google Scholar]
- Perry J, Kleckner N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell. 2003;112:151–155. doi: 10.1016/s0092-8674(03)00033-3. [DOI] [PubMed] [Google Scholar]
- Reid RJ, Lisby M, Rothstein R. Cloning-free genome alterations in Saccharomyces cerevisiae using adaptamer-mediated PCR. Methods Enzymol. 2002;350:258–277. doi: 10.1016/s0076-6879(02)50968-x. [DOI] [PubMed] [Google Scholar]
- Sabourin M, Tuzon CT, Zakian VA. Telomerase and Tel1p preferentially associate with short telomeres in S. cerevisiae. Mol Cell. 2007;27:550–561. doi: 10.1016/j.molcel.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. Regulation of RAD53 by the ATM-like kinase MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF. Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell. 2002;9:1055–1065. doi: 10.1016/s1097-2765(02)00532-4. [DOI] [PubMed] [Google Scholar]
- Shen ZJ, Hsu PH, Su YT, Yang CW, Kao L, Tseng SF, Tsai MD, Teng SC. PP2A and Aurora differentially modify Cdc13 to promote telomerase release from telomeres at G2/M phase. Nat Commun. 2014;5:5312. doi: 10.1038/ncomms6312. [DOI] [PubMed] [Google Scholar]
- Sibanda BL, Chirgadze DY, Blundell TL. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature. 2010;463:118–121. doi: 10.1038/nature08648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, de Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 2004;73:177–208. doi: 10.1146/annurev.biochem.73.071403.160049. [DOI] [PubMed] [Google Scholar]
- Smolka MB, Albuquerque CP, Chen SH, Schmidt KH, Wei XX, Kolodner RD, Zhou H. Dynamic changes in protein-protein interaction and protein phosphorylation probed with amine-reactive isotope tag. Mol Cell Proteomics. 2005;4:1358–1369. doi: 10.1074/mcp.M500115-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack JH, Emr SD. Vps34p required for yeast vacuolar protein sorting is a multiple specificity kinase that exhibits both protein kinase and phosphatidylinositol-specific PI 3-kinase activities. J Biol Chem. 1994;269:31552–31562. [PubMed] [Google Scholar]
- Stirling PC, Bloom MS, Solanki-Patil T, Smith S, Sipahimalani P, Li Z, Kofoed M, Ben-Aroya S, Myung K, Hieter P. The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLoS Genet. 2011;7:e1002057. doi: 10.1371/journal.pgen.1002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol. 2005;15:1364–1375. doi: 10.1016/j.cub.2005.06.063. [DOI] [PubMed] [Google Scholar]
- Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- Takai H, Wang RC, Takai KK, Yang H, de Lange T. Tel2 regulates the stability of PI3K-related protein kinases. Cell. 2007;131:1248–1259. doi: 10.1016/j.cell.2007.10.052. [DOI] [PubMed] [Google Scholar]
- Takai H, Xie Y, de Lange T, Pavletich NP. Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev. 2010;24:2019–2030. doi: 10.1101/gad.1956410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng SF, Lin JJ, Teng SC. The telomerase-recruitment domain of the telomere binding protein Cdc13 is regulated by Mec1p/Tel1p-dependent phosphorylation. Nucleic Acids Res. 2006;34:6327–6336. doi: 10.1093/nar/gkl786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T, Ogawa H, Petrini JH. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell. 2001;7:1255–1266. doi: 10.1016/s1097-2765(01)00270-2. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Higashi K, Raven C, Welham M, Anderson S, Brennan P, Ward SG, Waterfield MD. Autophosphorylation of p110delta phosphoinositide 3-kinase: a new paradigm for the regulation of lipid kinases in vitro and in vivo. EMBO J. 1999;18:1292–1302. doi: 10.1093/emboj/18.5.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialard JE, Gilbert CS, Green CM, Lowndes NF. The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J. 1998;17:5679–5688. doi: 10.1093/emboj/17.19.5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscardi V, Bonetti D, Cartagena-Lirola H, Lucchini G, Longhese MP. MRX-dependent DNA damage response to short telomeres. Mol Biol Cell. 2007;18:3047–3058. doi: 10.1091/mbc.E07-03-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakayama T, Kondo T, Ando S, Matsumoto K, Sugimoto K. Pie1, a protein interacting with Mec1, controls cell growth and checkpoint responses in Saccharomyces cerevisiae. Mol Cell Biol. 2001;21:755–764. doi: 10.1128/MCB.21.3.755-764.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–320. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- Wellinger RJ, Zakian VA. Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: beginning to end. Genetics. 2012;191:1073–1105. doi: 10.1534/genetics.111.137851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zakian VA. The telomeric Cdc13 protein interacts directly with the telomerase subunit Est1 to bring it to telomeric DNA ends in vitro. Proc Natl Acad Sci USA. 2011;108:20362–20369. doi: 10.1073/pnas.1100281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature. 2013;497:217–223. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Chahwan C, Bailis J, Hunter T, Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol. 2005;25:5363–5379. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.