

Abstract
Constitutive JAK-STAT pathway activation occurs in most myeloproliferative neoplasms as well as in a significant proportion of other hematologic malignancies, and is frequently a marker of poor prognosis. The underlying molecular alterations are heterogeneous as they include activating mutations in distinct components (cytokine receptor, JAK, STAT), overexpression (cytokine receptor, JAK) or rare JAK2 fusion proteins. In some cases, concomitant loss of negative regulators contributes to pathogenesis by further boosting the activation of the cascade. Exploiting the signaling bottleneck provided by the limited number of JAK kinases is an attractive therapeutic strategy for hematologic neoplasms driven by constitutive JAK-STAT pathway activation. However, given the conserved nature of the kinase domain among family members and the interrelated roles of JAK kinases in many physiological processes, including hematopoiesis and immunity, broad usage of JAK inhibitors in hematology is challenged by their narrow therapeutic window. Novel therapies are, therefore, needed. The development of more selective inhibitors is a questionable strategy as such inhibitors might abrogate the beneficial contribution of alleviating the cancer-related pro-inflammatory microenvironment and raise selective pressure to a threshold that allows the emergence of malignant subclones harboring drug-resistant mutations. In contrast, synergistic combinations of JAK inhibitors with drugs targeting cascades that work in concert with JAK-STAT pathway appear to be promising therapeutic alternatives to JAK inhibitors as monotherapies.
Introduction
Mature blood cells have a limited lifespan and are continuously renewed through a multi-step process called hematopoiesis, initiated in the bone marrow by the proliferation and differentiation of a small population of pluripotent hematopoietic stem cells (Figure 1). Undergoing asymmetric divisions, hematopoietic stem cells have the ability to replenish their pool by self-renewal and to differentiate into lineage-committed progenitors with increasingly restricted potential which will ultimately give rise to all specialized blood cells.1 A network of hematopoietic cytokines dictates the fate (proliferation, differentiation or apoptosis) of the various progenitors, thereby maintaining steady state levels of blood cells in the periphery or inducing amplification of specific cell types in response to particular stimuli to meet physiological needs. Abnormalities in the hematopoietic program disrupt homeostasis and drive the accumulation of intermediate progenitors and/or mature cells in the bone marrow, blood and/or peripheral lymphoid organs resulting in a variety of malignancies.2
Figure 1.
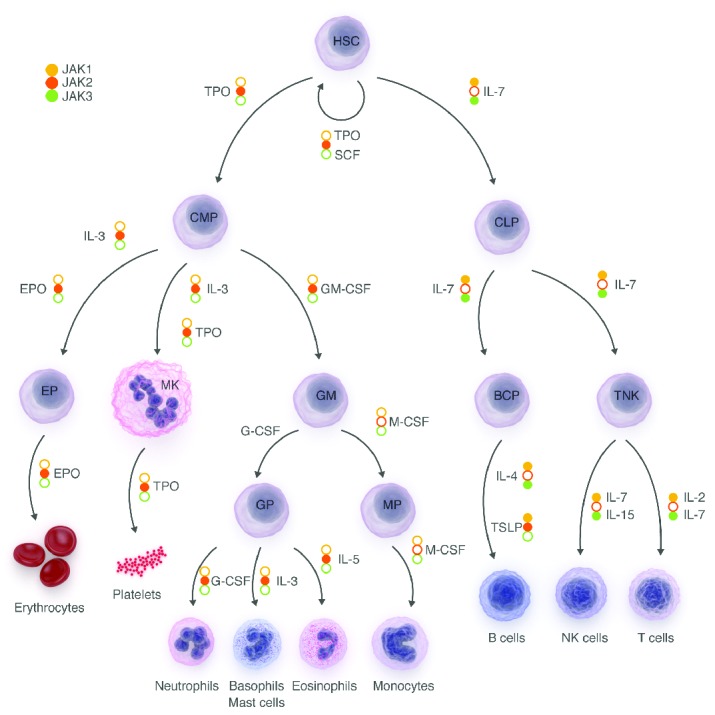
Hematopoiesis. Hematopoiesis originates from a hematopoietic stem cell, which can undergo either self-renewal or hierarchical differentiation into lineage-committed progenitors with decreasing potential that ultimately will give rise to all mature blood cells. Cytokines and their receptor-associated JAK necessary for the progenitors to pass through the different maturation steps are indicated. HSC: hematopoietic stem cell; CMP: common myeloid progenitor; CLP: common lymphoid progenitor; GM: granulocyte macrophage progenitor; BCP: B cell progenitor; TNK: T and natural killer cell progenitor; EP: erythroid progenitor; Mk: megakaryocyte; GP: granulocyte progenitor; MP: macrophage progenitor; TPO: thrombopoietin; SCF: stem cell factor; IL: interleukin; GM-CSF: granulocyte/monocyte colony-stimulating factor; G-CSF: granulocyte colony-stimulating factor; M-CSF: monocyte colony-stimulating factor; TSLP: thymic stromal-derived lymphopoietin.
Overview of the JAK-STAT pathway
Hematopoietic cytokines bind to their cognate receptors at the surface of target cells; the receptors are composed of at least two single membrane-spanning chains. Except for several tyrosine kinase receptors, such as c-kit, Fms-like tyrosine kinase 3 (FLT3) or the receptor for macrophage colony-stimulating factor (M-CSF), the intracellular part of hematopoietic receptor chains lacks intrinsic enzymatic activity. However, these receptor chains constitutively and specifically associate with a member of the Janus kinase family (JAK1, JAK2, JAK3 or TYK2) in order to form functional complexes capable of transducing ligand-induced signals. Following cytokine engagement, receptor chains re-orientate or oligomerize leading to juxtaposition, and hence transactivation of the two associated JAK. Once activated, JAK phosphorylate tyrosine residues in the cytoplasmic part of the receptor creating docking sites for downstream Src homology-2 (SH2) domain-containing adaptor and effector proteins. Depending on the amino acids surrounding the phosphotyrosine, any one or more of the seven signal transducer and activator of transcription factors (STAT-1, -2, -3, -4, -5a, -5b and -6) can be recruited and phosphorylated at the receptor, homo- or heterodimerize and translocate into the nucleus to regulate transcription of target genes.3
The JAK-STAT pathway constitutes a signal transduction system through which a large spectrum of extracellular cytokines and nearly as many cognate transmembrane receptors converge towards an intracellular code employing four JAK kinases and seven STAT factors.4 Signal specificity downstream of cytokine receptors is achieved by the nature of the STAT dimers formed at the receptor, the kinetics and intensity of STAT activation as well as the triggering of additional signaling pathways such as mitogen-activated protein kinases (MAPK) and phosphatidylinositol-3′-kinase (PI3K). Transient JAK-STAT pathway activation is guaranteed by several mechanisms of negative regulation which operate at each step of signal transduction, such as ubiquitin-mediated receptor internalization, dephosphorylation of tyrosines in the JAK activation loop by constitutive phosphatases and induction of suppressor of cytokine signaling (SOCS) proteins.5
In 2005, several groups reported a unique, acquired, somatic activating mutation of JAK2 (V617F) in 95% of patients with polycythemia vera (PV) and in about half of those with essential thombocythemia (ET) or primary myelofibrosis (MF).6–9 The discovery of JAK2V617F led to screening for JAK mutations in other hematologic neoplasms. Thanks to improvements of sequencing techniques and the conduction of massive sequencing projects, the catalogue of genetic alterations in hematologic malignancies is constantly being updated as ever more new mutational targets are discovered. This review provides an overview of the different molecular mechanisms underlying constitutive activation of the JAK-STAT pathway and their respective frequencies among hematologic neoplasms. The use of JAK inhibitors in the clinic and in ongoing trials, resistance phenomena as well as future challenges are discussed. The review begins by documenting insights into the structural organization of JAK kinases, their mode of activation as well as the role of the different family members in hematopoiesis.
Janus kinases
JAK1, JAK2 and TYK2 tyrosine kinases are ubiquitously expressed and non-covalently bound to a distinct, mostly non-overlapping repertory of receptor chains. By contrast, JAK3 expression is restricted to the hematopoietic lineage and it associates exclusively with the common gamma-chain (γc). Based on their primary sequences, JAK kinases were initially divided into seven highly conserved JAK homology regions (JH1-7, starting from the C-terminal end) but were subsequently organized into four functional domains.10
The N-terminal moiety of JAK kinases constitutes the receptor-binding module with a FERM (band 4.1, ezrin, radixin, moesin) domain (JH4-7) and an atypical SH2 domain (JH3).11 It has been experimentally established that both domains do not exist as individual functional entities but structurally cooperate for non-covalent anchoring to two membrane-proximal regions of defined receptors.12,13 This was corroborated by a recent crystal structure study showing that the N-terminal part of TYK2 forms a contiguous, Y-shaped receptor-binding module that interacts with the interferon alpha receptor 1 (IFNAR1) via a composite interface formed by the FERM and SH2 domains.14 The selectivity of the JAK member engaged by a particular receptor chain does not exclusively depend on intrinsic sequences within its cytoplasmic domain but also on the stoichiometry of the receptor. For example, forced homodimers of chimeric receptors encompassing the intracellular part of interleukin (IL)-2 receptor (R) β subunit fused to the extracellular part of erythropoietin (EPO)-R activated JAK2 despite the fact that the naturally activated kinase was JAK1.15 Of note, JAK-receptor association is needed to ensure proper trafficking and localization of the complex to the plasma membrane.16–18
The C-terminal moiety of JAK contains the catalytic kinase domain (JH1), whose active conformation is stabilized upon phosphorylation of tandem tyrosine residues in its activation loop. Inappropriate kinase activity of JH1 in the absence of stimulus is prevented by a directly adjacent pseudo-kinase domain (JH2).19 The precise molecular mechanism by which JH2 regulates JAK activity remains incompletely understood but insights have been provided by recent research.20 The pseudokinase domain has long been considered as catalytically inactive because it lacks critical conserved residues needed for phosphate coordination and transfer such as the nearly invariant aspartic acid in the catalytic site and the third glycine of the GxGxϕG motif in the ATP-binding loop. However, JAK2 JH2 crystal structure analysis revealed that the pseudo-kinase domain adopts a classical bilobal kinase fold and binds ATP in a non-canonical mode.21 The precise function of the nucleotide binding of JH2 is still debated. JH2 has been demonstrated to phosphorylate two negative regulatory sites of JAK2, namely Ser523 in the SH2-JH2 linker and Tyr570 in JH2 itself.22 Whether this weak kinase activity is relevant for autoinhibition is still a matter of controversy since those residues are not conserved among other family members and disruption of JH2 nucleotide binding does not lead to constitutive JAK2 activation. Rather, it has been proposed that JH2 constitutively binds ATP without hydrolysis, making ATP an essential structural stabilizer of JH2.23
Studies of recombinant JH1 and JH1-JH2 constructs from JAK2 showed that the presence of JH2 drastically decreases JH1 kinase activity by increasing the Km for ATP possibly via an interdomain interaction that changes the conformation of JH1.24 Using the recently solved crystal structure of JH1-JH2 from TYK2, this intramolecular inhibitory interface has been localized near the JH1 catalytic site and is predominantly mediated by the N-lobes of each domain.25 The JH1-JH2 crystal structure of TYK2 supports an in cis mode of inhibition with the pseudokinase domain inhibiting the directly adjacent kinase domain within the same protein. Nevertheless, it is unclear whether this intramolecular JH1-JH2 interplay is operative in the contest of a physiological receptor. Because cytokine receptor complexes crucially comprise two or more subunits, each associated with a JAK monomer, it cannot be excluded that there is an in trans mechanism of inhibition in which the pseudokinase domain of one JAK molecule locks the kinase domain of the opposing JAK. Recently, Brooks et al. used fluorescence resonance energy transfer (FRET) to monitor the movements of JAK2 JH1 and JH2 domains during stimulation of the homodimeric growth hormone receptor. Activation resulted in an increase in distance between the two JH2 while bringing the two JH1 in closer vicinity, consistent with the in trans inhibition model.26 Evidence supporting both the in cis and in trans inhibition models could be reconciled by the hypothesis that distinct mechanisms operate between different JAK family members or that the type of associated receptor governs the mode of regulation.27
Role of JAK kinases in hematopoiesis
As outlined in Figure 1, JAK2 plays a role in the function and maintenance of hematopoietic stem cells28 by participating directly in signal transduction of thrombopoietin (TPO)29 and by contributing to signaling downstream of the stem cell factor.30 JAK2 is also crucial at various stages of myelopoiesis through binding to receptors for EPO, TPO, granulocyte colony-stimulating factor (G-CSF), granulocyte/macrophage colony-stimulating factor (GM-CSF), IL-3 and IL-5. EPO-R exists as a preformed homodimer,31 while the receptors for TPO and G-CSF are believed to homodimerize upon ligand binding. Receptors for GM-CSF, IL-3 and IL-5 consist of heterodimers of a cytokine-specific α chain and the common β chain that assemble as ligand-induced tetramers. In all cases, signal transduction results from transactivation of a pair of juxtaposed JAK2 molecules.
By contrast, JAK1 cooperates with JAK3 for lymphopoiesis by binding to heterodimeric receptors of the common γc family, IL-2R, IL-4R, IL-7R and IL-15R with JAK1 anchored to the specific chain and JAK3 to the common γc. Both kinases play obligatory and non-redundant functions as cells lacking either JAK1 or JAK3 are unable to respond to γc cytokines.32,33 However, the catalytic activity of each partner is not of equal importance since a kinase-dead form of JAK3 but not JAK1 still allows signal transduction via these receptors.34 Thus, in the context of γc-containing receptors, JAK1 functions as the primary signaling effector whereas JAK3 serves as a scaffold and partner of JAK1. JAK2 in concert with JAK1 could possibly be implicated in late development of immature B-lymphocytes through signaling downstream of thymic stromal-derived lymphopoietin (TSLP).35 With respect to their implication in the maturation and amplification of the different myeloid and lymphoid lineages (Figure 2), genetic alterations leading to oncogenic activation of particular hematopoietic cytokine receptor complexes give rise to neoplasms of corresponding cell types.
Figure 2.
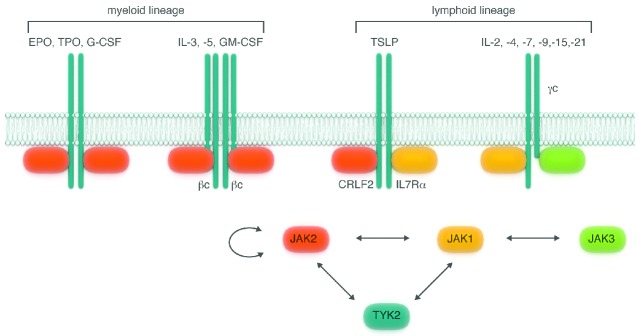
Hematopoietic cytokine-receptor complexes. Hetero- or homo-oligomerized receptor chains specifically engage a member of the JAK kinases in order to form signaling-competent hematopoietic receptor complexes. Myelopoiesis is driven by cytokines whose receptors signal through the transactivation of a pair of juxtaposed JAK2 molecules. Lymphopoiesis is mostly driven by interleukins that bind to the shared γc-receptor family, which signals through the cooperative action of JAK1 and JAK3. JAK2 and JAK1 participate in the signaling of TSLP, a cytokine involved in late development of B-lymphocytes. JAK1, JAK2 and TYK2 can work as partners whereas JAK3 works exclusively in concert with JAK1. JAK2 is the only family member that can function with itself as opposing JAK. EPO: erythropoietin; TPO: thrombopoietin; G-CSF: granulocyte colony-stimulating factor; IL: interleukin; GM-CSF: granulocyte/monocyte colony-stimulating factor; TSLP: thymic stromal-derived lymphopoietin.
Activating alterations of JAK kinases
Hematologic malignancies have been associated with activating alterations in the four members of the JAK family; the frequencies of these alterations differ, with the majority of them being point mutations. Contrasting with the dominance of the JAK2V617F mutation in myeloproliferative neoplasms (MPN), the mutations described later in other hematologic malignancies in JAK1, JAK2, JAK3 and more recently in TYK2 are much more heterogeneous both regarding the mutated residues and the frequency of mutation in a given disease. The JAK2 locus can also be involved in translocations resulting in the overexpression of wild-type JAK2 or the generation of constitutively active fusion proteins. Figure 3 gives a general overview of the different myeloid and lymphoid neoplasms for which the overall frequency of analyzed cases with genetic alterations affecting any component of the cytokine receptor-JAK-STAT axis reached at least 10%.
Figure 3.
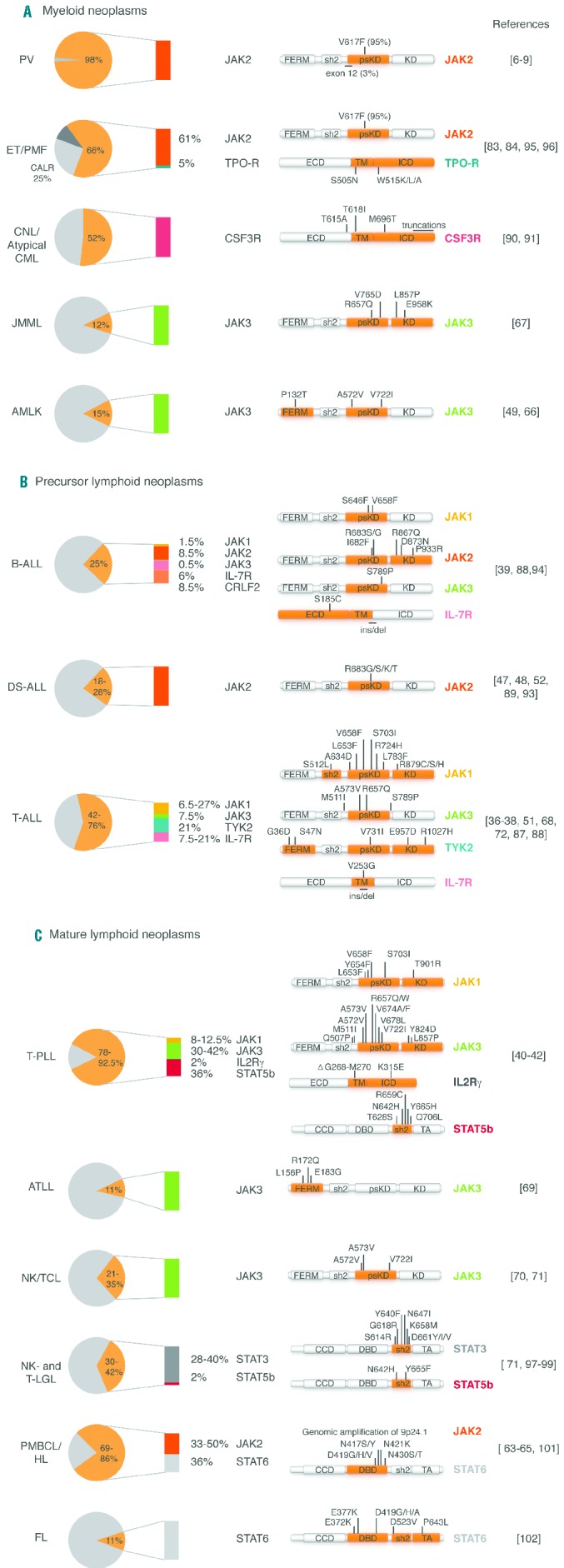
Frequencies of genetic alterations affecting the receptor-JAK-STAT axis among hematologic malignancies. Diagrams give an overview of the maximal overall frequency of cases with genetic alterations of the receptor-JAK-STAT axis in (A) myeloid lineage, (B) lymphoid progenitors and (C) lymphoid mature cells disorders (see next page). The overall frequency (i.e. the sum of the individual mutation frequencies for each component among the different references) is indicated in yellow while the absolute contributions of each are detailed using a specific color code (yellow for JAK1, orange for JAK2, green for JAK3, turquoise for TYK2, pink for cytokine receptors, black for STAT3, red for STAT5b and gray for STAT6). Schematic protein representations are used to localize the identified point mutations with the affected functional domains indicated in red. In some cases, other genetic lesions and clinical associations are described. MPN: myeloproliferative neoplasms; PV: polycythemia vera; ET: essential thrombocythemia; MF: myelofibrosis; CNL: chronic neutrophilic leukemia; CML: chronic myeloid leukemia; JMML: juvenile myelomonocytic leukemia; AMKL: acute megakaryoblastic leukemia; ALL: acute lymphoblastic leukemia; DS: Down-syndrome; T-PLL: T-prolymphocytic leukemia; ATLL: adult T-cell lymphoma; NKTCL: NK/T-cell lymphoma; NK- and T-LGL: NK-and T-large granular lymphocytic leukemia; PMBL: primary mediastinal B-cell lymphoma; HL: Hodgkin lymphoma. For abbreviations and explanations see footnote.
Frequencies of alterations in JAK kinase genes among hematologic malignancies
In accordance with its role in lymphopoiesis, activating mutations of JAK1 have been described in lymphoid neoplasms with their frequency being highest in T-cell acute lymphoblastic leukemia (ALL) (6.5–27%)36–38 and lower in B-cell ALL (1.5%)39 and T-cell prolymphocytic leukemia (8–12.5%).40–42 JAK1 mutants have also been reported in very rare cases of acute myeloid leukemia.43
As already mentioned, the JAK2V617F mutation is highly prevalent in classical MPN so that screening for this mutation has become a standard diagnostic procedure. The same JAK2V617F mutation was described later in a small proportion of patients with atypical MPN,44 and in some with myelodysplastic syndromes, particularly in patients suffering from refractory anemia with ringed sideroblasts and thrombocythemia.45 In addition, mutations in the exon 12 of JAK2 were described in JAK2V617F-negative PV patients who typically had isolated erythrocytosis.46
JAK2 mutations were also found in lymphoid malignancies such as high-risk B-ALL (8.5%)39 and B-ALL associated with Down syndrome (18–28%).47–53 Notably, the vast majority of lymphoid lineage JAK2 mutations affect the R683 residue in JH2. Interestingly, JAK2 R683 variants are frequently associated with the overexpression of CRLF2, suggesting that this cytokine receptor chain is involved in the functional signaling platform of these mutants.54 CRLF2 is a JAK2-binding chain that together with IL-7Rα and JAK1 forms the functional receptor complex for TSLP. The rationale for the specific association of JAK2V617F with MPN and JAK2R683 with ALL is speculative. The JAK2V617F mutant might be more efficient in transactivating another JAK2 in homodimeric cytokine receptor complexes such as EPO-R or TPO-R, whereas conversely the JAK2R683 variants might preferentially activate a JAK1 molecule in the heterodimeric TSLP receptor complex. Alternatively, the two mutants might differ in their affinity for a particular receptor chain, substrate specificity or intensity of signal transduction.
Rare chromosomal rearrangements juxtaposing the kinase domain-coding portion of the JAK2 gene to genomic regions coding for the oligomerization domain of either TEL, BCR, PCM1, Pax5 or ETV6 were described essentially in ALL and several cases of atypical chronic myeloid leukemia.51,55–59 The resultant fusion proteins oligomerize, facilitating transphosphorylation of the JAK2 activation loops, and constitutively trigger several downstream signal transduction pathways, such as STAT5,55 PI3K60 or the MAP kinase,61 independently of the presence of anchoring receptors. Interestingly, although artificial chimeric TEL-JAK1, TEL-JAK3 and TEL-TYK2 proteins are able to sustain cytokine-independent growth of Ba/F3 cells,62 there is no patient-derived chromosomal translocation that fuses the kinase domain of JAK1, JAK3 or TYK2 to a dimerizer described so far. This is probably related to an intrinsic genetic instability of the JAK2 locus, which can otherwise also be subject to amplifications in 30–50% of Hodgkin lymphomas and primary peripheral B-cell lymphomas.63–65
Gain-of-function mutations in JAK3 are scattered throughout the different functional domains and are found in diverse malignancies of both myeloid and lymphoid origins. JAK3 mutations were initially described in acute megakaryoblastic leukemia samples (15%)66 and subsequently identified in juvenile myelomonocytic leukemias (12%).67 With regards to malignancies of lymphoid lineage, JAK3 mutations were reported in a significant proportion of T-ALL,68 especially in an aggressive subtype called early T-cell precursor-ALL,38 as well as in diverse mature T-cell neoplasms.40,42,69–71
More recently, TYK2 mutants were discovered in T-ALL (21%) and seem to promote cell survival via activation of STAT1 downstream of IL-10R and upregulation of the anti-apoptotic BCL2 protein.72
Characterization of JAK mutants
Functional characterization of the different JAK variants revealed that they display common features as well as mutant-specific particularities. As demonstrated for JAK1, JAK2 and JAK3, mutants require a functional FERM domain and the presence of anchoring homo- or heterodimeric cytokine receptors to promote signaling and cell transformation.73–75 One notable exception is the JAK1L910P mutant that exhibits residual activity upon inactivating mutation in its FERM-domain.75 JAK1 mutants can trigger STAT activation via IL2-Rβ or IL-9Rα homodimers without need for JAK3 and γc.74 By contrast, JAK3 mutants (with the exception of JAK3L857Q) are strictly dependent on JAK1 kinase activity.76 Intrinsic specificity among distinct mutants of the same JAK is illustrated by qualitative and quantitative differences in STAT activation, FERM-domain requirement, susceptibility to JAK inhibitors and, concerning the JAK1 mutants, sensitivity to type-I interferons.75–78
The overwhelming majority of activating JAK mutations occur in the pseudokinase domain (JH2). The mechanism through which such mutations increase the activity of the kinase domain was almost exclusively studied for JAK2V617F. Superimposing the crystal structures of the pseudokinase domains of JAK2V617F and the analogous JAK1V658F mutants with their wild-type counterparts highlighted a stiffening and extension of the αC helix mediated by the action of a phenylalanine triad.21,79 The aromatic ring of the phenylalanine at position 617 in JAK2 and 658 in JAK1 fills in the site normally occupied by that of a highly conserved phenylalanine residue in the SH2-JH2 linker (F537 in JAK2 and F575 in JAK1) thereby inducing the rotation of a third phenylalanine located in the aC helix (F595 in JAK2 and F636 in JAK1). Replacement of either partner of this F-F-F triad by non-aromatic residues diminishes constitutive kinase activity.79,80 Interestingly, JAK2V617F is transferable to JAK1 and TYK2 but not JAK3 evoking a different autoinhibition mechanism of JAK3 compared to the three others.81
Besides JAK2V617F, most of the cancer-associated mutations in the pseudokinase domain of JAK1, JAK2 and JAK3 are clustered in the N-lobe and the SH2-JH2 linker fragment. Mapping of the analogous affected residues on the crystal structure of TYK2 revealed that they localize in or near the JH1-JH2 interface and probably relieve the inhibitory influence.25 On the other side of the interface, all known kinase N-lobe mutations participate directly in the JH1-JH2 interaction.
Activating alterations in cytokine receptors
The first mechanism of constitutive receptor activation involves point mutations or small insertions/deletions of residues located in and around the transmembrane domain which result in reorientation or dimerization of unliganded chains. Several examples are listed hereunder. TPO-R has an intracytoplasmic juxtamembrane amphipathic motif necessary for preventing its activation in the absence of ligand.82 Mutations in this motif (W515K/L/A) were described in 5% of patients with JAK2V617F-negative MF or ET83,84 and were shown to change the spatial conformation of the receptor enabling TPO-independent JAK2 transphosphorylation.85 Additionally, mutations in the transmembrane domain of TPO-R (S505N and S505A) cause familial ET.86 Small in-frame insertions/deletions in the transmembrane domain of IL-7Rα were found in about 10% of T- and B-ALL.87,88 These sequence modifications often introduce an unpaired cysteine in the transmembrane domain and promote formation of intermolecular disulfide bonds between IL-7Rα mutant subunits, thereby driving constitutive signaling via JAK1 and independently of IL-7, γc or JAK3.87 Point mutations inserting a cysteine into the extracellular domain of IL-7Rα (S185C) and CRLF2 (F232C) were identified in a few B-ALL cases and could act via the same activation mechanism.89 Membrane-proximal mutations of CSF3R (T615A and T618I), the receptor for G-CSF, are highly prevalent in atypical chronic myeloid leukemia and in chronic neutrophilic leukemia, and induce ligand-independent activation of the receptor by a mechanism that remains to be elucidated.90,91 More recently, a whole-exome sequencing study provided the first report of two concomitant sequence modifications in γc (a small deletion in the trans-membrane domain and the K315E mutation in the intracellular part) detected in a sample of T-cell prolymphocytic leukemia.41 The significance of this finding is not yet clear as only one patient in a series of 50 analyzed harbored these γc mutations.
A second mechanism of receptor activation has been described in patients with chronic neutrophilic leukemia or atypical chronic myeloid leukemia and involves truncations of the cytoplasmic tail of CSF3R, leading to escape of negative regulation.90 The truncated proteins are the result of frameshift or nonsense mutations and increase the pro-liferative signal by impairing receptor internalization and altering the interaction with negative regulators such as SOCS3. Interestingly, proliferation of cells transformed with these mutants was relatively insensitive to JAK2 inhibitors, but could be efficiently blocked by SRC inhibitors, such as dasatinib, indicating that SRC signaling could be preferentially activated by these truncations. CSF3R truncating mutations were very recently identified in 2% of pediatric acute myeloid leukemias.92
The last reported cytokine receptor alteration is overexpression of CRLF2, which has already been mentioned in this review. CRLF2 is aberrantly expressed in 50% of patients with Down syndrome ALL and in 10% of non-Down syndrome ALL,54,89,93,94 essentially due to chromosomal rearrangements juxtaposing the CRLF2 locus to the immunoglobulin heavy chain enhancer or P2RY8 promoter. In B-ALL, CRLF2 overexpression in frequently associated with JAK2R683 variants or IL-7Rα mutations, suggesting that concomitant genetic events affecting components of the TSLP-receptor complex are required for its oncogenic potency.
Taken together, gain-of-function cytokine receptor alterations are diverse and encompass mutations in the transmembrane region which induce ligand-independent receptor dimerization/activation, cytoplasmic tail truncations that abolish negative regulation and chromosomal translocations that enhance protein expression. At present, such genetic modifications are well established for TPO-R, CRLF2, IL-7Rα and CSF3R but it is not excluded that whole-exome sequencing studies will identify alterations in other cytokine receptor chains as suggested by the recent finding of very rare mutations in γc.
Calreticulin mutations in JAK2 and thrombopoietin receptor mutation-negative essential thrombocythemia and myelofibrosis
In December 2013, the molecular diagnostic gap that remained after the discovery of TPO-R mutations in JAK2-negative ET and MF patients was partially filled. Two independent groups reported that 25% of ET and MF patients had small insertions/deletions in an unexpected gene coding for calreticulin (CALR), a lectin chaperone involved in the folding quality control machinery of the endoplasmic reticulum and in calcium homeostasis.95,96 All identified sequence modifications invariably provoked a +1 base shift to the same alternative open reading frame resulting in the generation of a novel C-terminus with impaired calcium-binding and loss of the endoplasmic reticulum-retention motif, although there was no difference in the pattern of subcellular localization of wild-type CALR and mutant forms.95 Expression of the most frequent CALR mutant caused transformation to cytokine-independent cells with phosphorylation of STAT5 by means of a still unknown mechanism but the homogeneity within the frameshifted variants evokes a strong selective pressure towards newly acquired functionalities rather than loss-of-function alterations.96 JAK2, TPO-R and CALR mutations were mutually exclusive genetic events in MPN with the 10% of triple-negative cases having the worse prognosis.
Activating mutations in STAT factors
At present, STAT3 and STAT5b mutations have only been demonstrated in T-cell and natural killer (NK)-cell leukemias, while mutations of STAT6 have been associated with B-cell lymphomas. However, given the crucial function of STAT factors in stages of hematopoiesis, it can be expected that further sequencing studies will reveal a role for mutations of STAT family genes in the pathogenesis of other hematologic malignancies of different origins.
STAT3 mutations
The occurrence of STAT3 mutations stressed the common molecular pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia, as similar percentages (27–30%) of patients with these two disease entities carry these somatic mutations.97,98 Altogether, eight different substitutions clustered in the exon 21 coding for the SH2 domain have been identified and all affected residues map at the interface of STAT dimers. In vitro studies showed that Y640F and D661V mutants are constitutively phosphorylated and localize in the nucleus. Patients with STAT3 mutations presented more often with disease-related autoimmune phenomena, such as rheumatoid arthritis and autoimmune hemolytic anemia, than did patients without these mutations.97,98 An overproduction of immune cytokines or a better resistance to apoptosis in the STAT3-mutated large granular lymphocyte leukemia cell population, including auto-reactive T cells, might explain this clinical association.
STAT5b mutations
Two different proven gain-of-function STAT5b mutations (Y665F and N642H) were found in 2% of STAT3 mutation-negative patients with T-large granular lymphocyte leukemia with the N642H substitution being associated with an aggressive, fatal disease course.99 These same mutations and other new ones were later found in 36% of cases of T-cell prolymphocytic leukemia, with this being a higher frequency than that of all other mutated JAK-STAT components (JAK1, JAK3, IL-7Rα and γc) in the analyzed cohort.41 STAT5b mutations are uncommon in T-ALL but their occurrence underlines the significance of the IL7R-JAK-STAT5 pathway in the pathogenesis of T-ALL.100 Interestingly, while STAT5b mutant blasts were resistant to JAK inhibitors, the cells were highly sensitive to inhibitors of STAT5b targets such as anti-apoptotic BCL-2 proteins.100
STAT6 mutations
Identification of heterozygous point mutations in the DNA binding domain of STAT6 in 36% of PMBCL represented the first observation of recurrent STAT mutations in human cancers.101 Very recently, additional activating STAT6 substitutions were described in 11% of follicular lymphomas and were demonstrated to prolong residency of STAT6 in the nucleus, resulting in an exaggerated transcriptional response to the IL-4 highly present in the tumor microenvironment.102
Loss of negative regulators
The activation of the JAK-STAT pathway is limited in time and amplitude by several regulatory mechanisms. For instance, constitutive phosphatases serve as quenchers of phosphotyrosines involved in activation and recruitment. Additionally, inducible regulators of the SOCS family mediate ubiquitin-tagging for proteasomal degradation of the receptor complex, suppression of JAK catalytic activity or competition with STAT factors for docking sites on receptors. Loss-of-function alterations or epigenetic silencing affecting genes involved in the repression process are expected to result in more intense and sustained cytokine receptor signaling. Some illustrative examples are documented in this section.
PTPN2 is a protein tyrosine phosphatase (PTP) able to dephosphorylate tyrosine residues located in the activation loop of JAK1 and JAK3.103 Mono- or bi-allelic inactivation of PTPN2 through deletion of the entire gene locus was found in 6% of T-ALL cases,104 sometimes together with JAK1-activating mutations.104 Down-regulation of PTPN2 sensitizes to JAK1- but not JAK2-mediated transformation and reduces the sensitivity of transformed cells to JAK inhibitors.105 Similarly, loss-of-function mutations of CD45, another JAK-regulating phosphatase encoded by the PTPRC gene, were detected in rare cases of T-ALL.106 Remarkably, all CD45 mutations occurred in combination with activating mutations in IL-7Rα, JAK1, or LCK. Deletions or methylation of the PTPRK locus coding for a phosphatase that directly binds to and dephosphorylates STAT3, were recently reported in 55% of nasal NK/T-cell lymphomas.107
LNK is an adaptor protein that inhibits JAK2 activation downstream of TPO-R and EPO-R.108 Mutations or deletions in the pleckstrin-homology (PH) domain of LNK are reported in rare cases of MPN.109,110 They occur either as an isolated event in chronic phase diseases109 or concomitantly with JAK2V617F mutation in patients who progress to develop acute myeloid leukemia.110
SOCS1 mutations have been described in Hodgkin lymphomas and primary mediastinal B-cell lymphomas (PMBCL).111 Reduced expression of SOCS1 and SOCS3, related to hypermethylation of their promoter, has been described in different lymphoid hematologic malignancies,112 including MPN, in association113 or not114 with the JAK2V617F mutation.
Taken together, these observations underline the role played by deficiency of JAK-STAT negative regulation processes in the pathogenesis of diverse hematologic neoplasms. Of note, such alterations can occur in concomitance with activating receptor or JAK mutations meaning that they are not mutually exclusive genetic events.104,106 Hence, it is postulated that loss of suppression does not drive tumorigenesis directly, but rather contributes to hyperactivation of the JAK-STAT pathway. Furthermore, as tyrosine phosphatases are not JAK-restricted but have multiple targets, their lack could exert broader oncogenic effects on intracellular signaling, beyond the JAK-STAT pathway. Finally, some of these alterations, such as PTPN2 deletions, reduce the sensitivity of transformed cells to JAK inhibitors, so screening patients for such deletions could be relevant for the prediction of treatment response.
JAK inhibition in hematologic malignancies
The oncogene addiction concept postulates that, despite the large number of genetic lesions conveyed by a single cancer cell, some tumors rely on a single dominant oncogene for growth and survival, so that inhibition of this specific Achilles’ heel is sufficient to halt the neoplastic phenotype.115 At the clinical level, translation of the oncogene addiction model into the rationale and effective design of targeted therapeutics against individual oncoproteins is best exemplified by the success story of imatinib, an ATP-competitive tyrosine kinase inhibitor of BCR-ABL, which dramatically prolonged the life expectancy of patients with chronic myeloid leukemia.116 The finding of aberrant JAK-STAT activation in a great majority of MPN and a significant portion of other hematologic malignancies raised the hope for an imatinib-like tyrosine kinase inhibitor therapy. The heterogeneity in the nature of the underlying genetic mechanisms could be overcome by exploiting the signaling bottleneck provided by the limited number of JAK kinases (Figure 4). However, it should be noted that activating mutations in STAT factors are relatively insensitive to JAK inhibitors.
Figure 4.
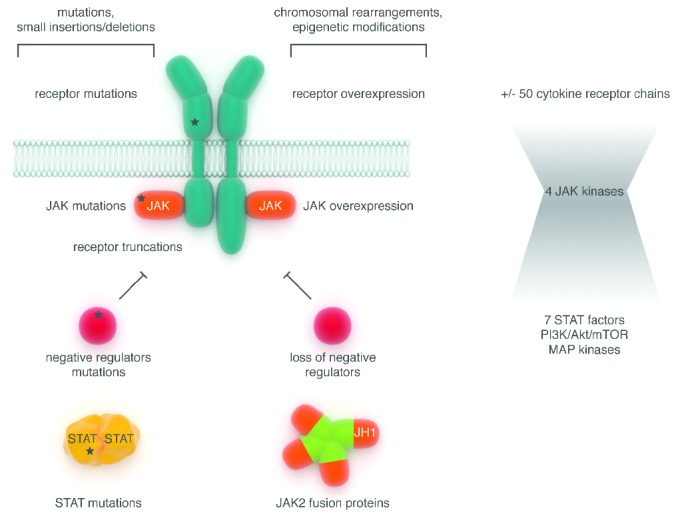
Mechanisms of constitutive JAK-STAT pathway activation in hematologic malignancies. The molecular mechanisms underlying constitutive activation of the JAK-STAT pathway are depicted and can affect all the components of the cascade (receptors, JAK, STAT, negative regulators). They can result from small nucleotide sequence modifications (mutations, insertions, deletions) or larger changes at the chromosomal level (translocations and epigenetic changes).
Ruxolitinib in myeloid malignancies
The discovery of the MPN JAK2V617F mutation in 2005 has been rapidly exploited in the clinic. No more than 6 years later, ruxolitinib, a dual JAK1/2-specific ATP-competitive inhibitor, was approved by the US Food and Drug Administration for the treatment of intermediate- and high-risk MF, a disease for which allogeneic stem cell transplantation was the only curative option. In clinical trials, ruxolitinib treatment was more effective at controlling splenomegaly and alleviating MF-associated symptoms than either placebo (COMFORT-I)117 or best available therapy (COMFORT-II).118 Surprisingly, MF patients benefit from ruxolitinib therapy regardless of their JAK2 mutational status. Consistent with this unexpected observation, gene expression profiling data showed that MPN samples are characterized by a shared transcriptional signature of activated JAK2-STAT5 signaling, irrespective of disease phenotype or somatic genotype.119 Subsequent identification of mutations in TPO-R and calreticulin, which result in activation of the JAK2-STAT5 pathway as discussed above, provided an explanation for this observation. The central role played by aberrant JAK-STAT pathway activation in MPN gave a rationale for the unrestricted efficacy of ruxolitinib and raised the hope that patients with mutation-positive and -negative PV, ET or MF patients could efficiently respond to JAK inhibitors.
However, ruxolitinib exerted only limited salutary effects in terms of reversion of bone marrow fibrosis and reduction of allele burden, suggesting that the concentration of the drug required to kill neoplastic cells is impossible to reach in patients without inducing serious adverse events due to total inhibition of physiological cytokine signaling. Indeed, because of its lack of specificity towards JAK2-mutated forms, ruxolitinib impedes physiological hematopoiesis leading to unwanted adverse events, the most frequent being anemia and thrombocytopenia (incidence >20%).120
Alternatively, the MPN cells might not be JAK2 oncogene-addicted and the benefit observed regarding constitutional symptoms might be related to alleviation of the pro-inflammatory tumor microenvironment rather than selective suppression of the disease clone.117 The fact that ruxolitinib also inhibits JAK1, an important player in innate and specific immune responses, might contribute to this anti-inflammatory activity. However, the drawback of this JAK1 inhibition is reduced control of silent infections and an increased incidence of opportunistic diseases, occasionally observed upon long-term administration.121–124
Notwithstanding those side effects, the substantial improvement in quality of life of MF patients encouraged the conduction of additional clinical trials, with the purpose of broadening the indications for ruxolitinib or testing other JAK2 inhibitors. Recently, a phase III clinical study aiming at evaluating the efficacy of ruxolitinib as second-line treatment in PV patients intolerant to hydroxy urea led to the conclusion that JAK2 inhibition was superior to standard therapy in controlling hematocrit, reducing spleen size and improving PV-associated symptoms.125 Ongoing phase III clinical trials in MF patients are assessing the efficacy and safety of less myelosuppressive novel JAK2 inhibitors such as momelotinib (CYT387) versus ruxolitinib (NCT01969838) and pacritinib (SB1518) versus best available therapy (NCT01773187). Besides classical MPN, a phase II trial is currently estimating the potential of ruxolitinib in chronic neutrophilic leukemia and atypical chronic myeloid leukemia, in which genetic activating alterations of CSF3R are encountered in a significant proportion of cases, as described above, and are associated with a poor outcome (NCT02092324). Ruxolitinib has already proven effective in a CSF3RT618I bone marrow transplant mouse model126 and one patient with CSF3RT618I-positive atypical chronic myeloid leukemia has been reported to have experienced a significant clinical response to ruxolitinib.127 Further encouragement can be derived from reports of two patients with PCM1-JAK2-positive chronic eosinophilic leukemia who achieved complete hematologic and cytogenic responses on ruxolitinib,128,129 although, in two other described cases, rapid remission was only of limited duration and relapse occurred within 24 months.130
Ruxolitinib in lymphoid malignancies
In pre-clinical experiments, ruxolitinib treatment yielded a significant decrease of circulating blast counts in murine xenograft models of Philadelphia chromosome (Ph)-like B-ALL131 and early T-cell precursor (ETP)-ALL,132 two recently described high-risk subtypes of ALL with aberrant activation of the JAK-STAT pathway for which novel therapies are needed. In the ETP-ALL model, the efficacy of treatment was independent of the presence of JAK-STAT mutations, raising the possibility that the therapeutic potential of ruxolitinib extends beyond the cases with JAK-STAT mutations.132 As suggested for MPN, a JAK activation footprint may be more significant than the presence of a mutation for predicting response to therapy. This notion greatly increases the numbers of patients that could potentially benefit from JAK inhibitors and will probably motivate the setting up of future clinical trials in lymphoid malignancies. A recent clinical report described a unique case of a patient with refractory PMBCL with an activating JAK3 mutation (A573V) who experienced partial disease remission upon treatment with tofacitinib, a JAK1/3-specific inhibitor, in combination with immuno-chemotherapy.133
JAK inhibitor resistance in hematologic malignancies
Although ruxolitinib has proven effective in patients with MF, some of them experienced treatment failure.134 Besides discontinuation due to drug-related toxicity (such as early-onset cytopenias), ruxolitinib treatment failure in MF manifests as primary refractoriness or secondary resistance. Primary refractoriness can be defined as no or only minimal clinical response (<35% reduction of spleen volume compared to baseline), whereas secondary resistance is indicated by the loss of a previously confirmed clinical response (such as splenic relapse) or progression to leukemia.120 In a cohort of 39 ruxolitinib-treated MF patients, resistance was observed in 41% of cases, mostly as a late event (after a median exposure of 1 year) and rarely as primary refractoriness (<10%). The same study identified a significant correlation between resistance and the absence of mutations in JAK2 (V617F), MPL, TET2, and SRSF2 at diagnosis.135
Based on clinical experiences with other tyrosine kinase inhibitor therapies, it could be expected that resistant patients would have acquired mutations at the drug-binding site of the target kinase, as illustrated by the emergence of BCR-ABL mutations in patients with chronic myeloid leukemia.116 A dozen different point mutations conferring resistance to ruxolitinib were discovered using in vitro random mutagenesis screens of JAK2 and in vitro selection of spontaneous resistant cell lines.78,136–138 However, no mutations in JAK2 could be detected in patients treated with ruxolitinib, indicating that this mechanism is rarely involved in clinical resistance.135 Tolerable doses of ruxolitinib provide only partial inhibition of the kinase, so the therapeutic pressure may be insufficient for the selection of resistant mutants. Rather, chronic exposure to suboptimal inhibitor concentrations might favor less drastic escape mechanisms. A reversible stage of so-called “inhibitor persistence” has been observed in ruxolitinib-treated patients and in JAK2V617F-transformed cell lines cultured with gradually increasing doses of ruxolitinib in vitro.139 Cells were allowed to persist despite JAK2 inhibition thanks to restoration of JAK–STAT signaling via overexpression of JAK2, which facilitates its heterodimeric transactivation with JAK1 or TYK2. Interestingly, inhibitors of HSP90, a chaperone known to stabilize JAK2, synergize with JAK2 inhibitors and overcome TKI persistence as well as genetic resistance.137,139,140 A phase II study is currently evaluating the potency of a HSP90 inhibitor in MF and refractory PV/ET patients (NCT01668173).
Perspectives for the use of JAK inhibitors in hematologic malignancies
At present, clinical data revealed that the targeted therapy concept prompted by the success story of imatinib would be more complex to apply with JAK inhibitors. The incapacity of ruxolitinib to reduce allele burden is potentially due to the absence of strong JAK2 oncogene addiction of MPN cells as discussed before. Nevertheless, broader investigations are encouraged by the substantial benefit of JAK2 inhibition irrespective of mutational status in MPN patients, preclinical evidence for ruxolitinib indications in other malignancies (chronic neutrophilic leukemia, ETP-ALL, Ph-like ALL) and significant clinical responses to JAK inhibitors in rare case reports (CSF3RT618I-positive or PCM1-JAK2-positive MPN and a JAK3A573V-positive PMBCL).
A second hurdle is the lack of selectivity of the currently used inhibitors, which implies that the tolerable doses lie within a narrow therapeutic window because of the collateral inhibition of other JAK members. Specific inhibitors for one of the four closely related JAK kinases could partially tackle this drawback but is difficult to achieve because of the marked similarity in the active site within the family. Ruxolitinib (and other JAK inhibitors under clinical evaluation) falls within the so-called type I family, which means that it precisely targets the well-conserved ATP-binding pocket of JAK1 and JAK2 in both active and inactive conformations.141,142 By contrast, inhibitors that have a type II binding-mode (such as imatinib) specifically engage and stabilize inactive kinases by exploiting an additional, less conserved allosteric site directly adjacent to the ATP binding pocket, providing another handle for tuning selectivity.141 Type II inhibition of JAK2 with NVP-CHZ868 demonstrated potent disease-modifying activity with significant reduction of allele burden in different in vivo MPN models.143 Alternatively, a covalent inhibitor exploiting a non-conserved cystein residue in the active site of JAK3 has been shown to have strong target specificity.144 Very recently, a new class of inhibitors was found to bind to and lock the pseudokinase domain of TYK2 in a conformation that stabilizes the autoinhibitory interaction with the adjacent kinase domain, preventing its receptor-mediated activation.145 These JH2 stabilizers occupy a site analogous to the ATP-binding site in catalytic kinases by forming hydrogen bonds with non-conserved residues, providing a novel approach for the design of highly selective inhibitors.145
Paradoxically, increasing specificity by development of type II, covalent or JH2 inhibitors could result in a decrease in the overall clinical benefit because of loss of collateral inhibition of cancer-related inflammatory processes. In addition, one can speculate that more potent inhibitors would raise the selective pressure to a threshold that allows the emergence of tumor subclones harboring drug-resistant mutations, as observed with imatinib therapy.
MPN disorders stem from the constitutive activation of JAK2 at the apex of downstream signal propagation mediated by interconnected cascades of effectors (STAT3/5, PI3K/Akt/mTOR, MAPK) that synergistically act on cell proliferation.146 Activation of the PI3K/Akt/mTOR pathway is required for JAK2V617F-induced transformation of cells as well as JAK2V617F-induced tumorigenesis in mice147 and CD34+ cells derived from MPN patients showed increased levels of phospho-mTOR.148 Thus, simultaneous intervention at both arms of the JAK-STAT and PI3K signaling cascades became a rational option for intervention. Two independent groups demonstrated that combining ruxolitinib with a PI3K inhibitor synergistically inhibited proliferation of in vitro MPN cell models and reduced spleen weight in preclinical MPN models.149,150 A similar synergistic action was also observed between mTOR and JAK2 inhibitors.151 A translational phase I clinical study is currently evaluating the safety and maximal tolerated doses of the combination of ruxolitinib and BKM120, a PI3K inhibitor, in MF patients (NCT01730248). In addition to further clinical benefit, simultaneous blockade of oncogenic signaling at multiple levels by combined therapy approaches (such as ruxolinitib with PI3K inhibitors or HSP90 inhibitors) might avoid the emergence of resistance by distributing the selective pressure between distinct targets.
Conclusion
A promising future is awaiting JAK inhibitors in the therapeutic arsenal of hematologic malignancies provided that appropriate indications are identified. Contrasting with the direct connection between imatinib and BCR-ABL-positive leukemias, the path to determining relevant applications for JAK inhibitors is more sinuous. First, the genetic alterations converging towards constitutive JAK activation are diverse and sometimes unexpected, such as the recently discovered calreticulin mutations. Furthermore, certain subtypes of ALL display an activated JAK transcriptional signature in the absence of known genetic abnormalities and respond to JAK inhibitor treatment in preclinical models, which makes the identification of suitable clinical indications even more complex. Second, once targetable candidate diseases have been selected, their JAK oncogene-addiction will remain to be validated clinically. Finally, the optimal therapeutic window is narrowed by the collateral inhibition of non-mutated JAK, with a fine balance between, on the one hand, adverse side effects due to impairments of physiological processes such as hematopoiesis or immunity and, on the other hand, beneficial contributions of alleviating the cancer-related pro-inflammatory microenvironment. It is, therefore, very likely that JAK inhibitors combined with other targeted therapies will be more efficient in synergistically modifying the natural history of the disease, overcoming inhibitor persistence and preventing the emergence of resistant subclones.
Acknowledgments
This work was supported by Foundation Salus Sanguinis. LK is a MD Postdoctoral Fellow of the Fonds National de la Recherche Scientifique (FNRS), Belgium.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.
References
- 1.Gottgens B. Regulatory network control of blood stem cells. Blood. 2015;125(17):2614–2620. [DOI] [PubMed] [Google Scholar]
- 2.Warr MR, Pietras EM, Passegue E. Mechanisms controlling hematopoietic stem cell functions during normal hematopoiesis and hematological malignancies. Wiley Interdiscip Rev Syst Biol Med. 2011;3(6):681–701. [DOI] [PubMed] [Google Scholar]
- 3.Ward AC, Touw I, Yoshimura A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood. 2000;95(1):19–29. [PubMed] [Google Scholar]
- 4.Liongue C, Ward AC. Evolution of the JAK-STAT pathway. JAKSTAT. 2013;2(1):e22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valentino L, Pierre J. JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol. 2006;71(6):713–721. [DOI] [PubMed] [Google Scholar]
- 6.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. [DOI] [PubMed] [Google Scholar]
- 7.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. [DOI] [PubMed] [Google Scholar]
- 8.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. [DOI] [PubMed] [Google Scholar]
- 9.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. [DOI] [PubMed] [Google Scholar]
- 10.Yamaoka K, Saharinen P, Pesu M, Holt VE, 3rd, Silvennoinen O, O’Shea JJ. The Janus kinases (Jaks). Genome Biol. 2004;5(12):253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haan C, Is’harc H, Hermanns HM, et al. Mapping of a region within the N terminus of Jak1 involved in cytokine receptor interaction. J Biol Chem. 2001;276(40):37451–37458. [DOI] [PubMed] [Google Scholar]
- 12.Chen M, Cheng A, Chen YQ, et al. The amino terminus of JAK3 is necessary and sufficient for binding to the common gamma chain and confers the ability to transmit interleukin 2-mediated signals. Proc Natl Acad Sci USA. 1997;94(13):6910–6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radtke S, Haan S, Jorissen A, et al. The Jak1 SH2 domain does not fulfill a classical SH2 function in Jak/STAT signaling but plays a structural role for receptor interaction and upregulation of receptor surface expression. J Biol Chem. 2005;280(27):25760–25768. [DOI] [PubMed] [Google Scholar]
- 14.Wallweber HJ, Tam C, Franke Y, Starovasnik MA, Lupardus PJ. Structural basis of recognition of interferon-alpha receptor by tyrosine kinase 2. Nat Struct Mol Biol. 2014;21(5):443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrag F, Pezet A, Chiarenza A, et al. Homodimerization of IL-2 receptor beta chain is necessary and sufficient to activate Jak2 and downstream signaling pathways. FEBS Lett. 1998;421(1):32–36. [DOI] [PubMed] [Google Scholar]
- 16.Huang LJ, Constantinescu SN, Lodish HF. The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol Cell. 2001;8(6):1327–1338. [DOI] [PubMed] [Google Scholar]
- 17.Radtke S, Hermanns HM, Haan C, et al. Novel role of Janus kinase 1 in the regulation of oncostatin M receptor surface expression. J Biol Chem. 2002;277(13):11297–11305. [DOI] [PubMed] [Google Scholar]
- 18.Royer Y, Staerk J, Costuleanu M, Courtoy PJ, Constantinescu SN. Janus kinases affect thrombopoietin receptor cell surface localization and stability. J Biol Chem. 2005;280(29):27251–27261. [DOI] [PubMed] [Google Scholar]
- 19.Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002;277(49):47954–47963. [DOI] [PubMed] [Google Scholar]
- 20.Babon JJ, Lucet IS, Murphy JM, Nicola NA, Varghese LN. The molecular regulation of Janus kinase (JAK) activation. Biochem J. 2014;462(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bandaranayake RM, Ungureanu D, Shan Y, Shaw DE, Silvennoinen O, Hubbard SR. Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat Struct Mol Biol. 2012;19(8):754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ungureanu D, Wu J, Pekkala T, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol. 2011;18(9):971–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammaren HM, Ungureanu D, Grisouard J, Skoda RC, Hubbard SR, Silvennoinen O. ATP binding to the pseudokinase domain of JAK2 is critical for pathogenic activation. Proc Natl Acad Sci USA. 2015;112(15):4642–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanz A, Ungureanu D, Pekkala T, et al. Analysis of Jak2 catalytic function by peptide microarrays: the role of the JH2 domain and V617F mutation. PLoS One. 2011;6(4):e18522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lupardus PJ, Ultsch M, Wallweber H, Bir Kohli P, Johnson AR, Eigenbrot C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc Natl Acad Sci USA. 2014;111(22):8025–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks AJ, Dai W, O’Mara ML, et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science. 2014;344(6185):1249783. [DOI] [PubMed] [Google Scholar]
- 27.Waters MJ, Brooks AJ. JAK2 activation by growth hormone and other cytokines. Biochem J. 2015;466(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akada H, Akada S, Hutchison RE, Sakamoto K, Wagner KU, Mohi G. Critical role of Jak2 in the maintenance and function of adult hematopoietic stem cells. Stem Cells. 2014;32(7):1878–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Solar GP, Kerr WG, Zeigler FC, et al. Role of c-mpl in early hematopoiesis. Blood. 1998;92(1):4–10. [PubMed] [Google Scholar]
- 30.Radosevic N, Winterstein D, Keller JR, Neubauer H, Pfeffer K, Linnekin D. JAK2 contributes to the intrinsic capacity of primary hematopoietic cells to respond to stem cell factor. Exp Hematol. 2004;32(2):149–156. [DOI] [PubMed] [Google Scholar]
- 31.Livnah O, Stura EA, Middleton SA, Johnson DL, Jolliffe LK, Wilson IA. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science. 1999;283(5404):987–990. [DOI] [PubMed] [Google Scholar]
- 32.Rodig SJ, Meraz MA, White JM, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93(3):373–383. [DOI] [PubMed] [Google Scholar]
- 33.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995;270(5237):794–797. [DOI] [PubMed] [Google Scholar]
- 34.Haan C, Rolvering C, Raulf F, et al. Jak1 has a dominant role over Jak3 in signal transduction through gammac-containing cytokine receptors. Chem Biol. 2011;18(3):314–323. [DOI] [PubMed] [Google Scholar]
- 35.Astrakhan A, Omori M, Nguyen T, et al. Local increase in thymic stromal lymphopoietin induces systemic alterations in B cell development. Nat Immunol. 2007;8(5): 522–531. [DOI] [PubMed] [Google Scholar]
- 36.Flex E, Petrangeli V, Stella L, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 2008;205(4):751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeong EG, Kim MS, Nam HK, et al. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res. 2008;14(12):3716–3721. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mullighan CG, Zhang J, Harvey RC, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2009;106(23):9414–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bellanger D, Jacquemin V, Chopin M, et al. Recurrent JAK1 and JAK3 somatic mutations in T-cell prolymphocytic leukemia. Leukemia. 2013;28(2):417–419. [DOI] [PubMed] [Google Scholar]
- 41.Kiel MJ, Velusamy T, Rolland D, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014;124(9):1460–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergmann AK, Schneppenheim S, Seifert M, et al. Recurrent mutation of JAK3 in T-cell prolymphocytic leukemia. Genes Chromosomes Cancer. 2014;53(4):309–316. [DOI] [PubMed] [Google Scholar]
- 43.Xiang Z, Zhao Y, Mitaksov V, et al. Identification of somatic JAK1 mutations in patients with acute myeloid leukemia. Blood. 2008;111(9):4809–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zecca M, Bergamaschi G, Kratz C, et al. JAK2 V617F mutation is a rare event in juvenile myelomonocytic leukemia. Leukemia. 2007;21(2):367–369. [DOI] [PubMed] [Google Scholar]
- 45.Malcovati L, Della Porta MG, Pietra D, et al. Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood. 2009;114(17):3538–3545. [DOI] [PubMed] [Google Scholar]
- 46.Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bercovich D, Ganmore I, Scott LM, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372(9648):1484–1492. [DOI] [PubMed] [Google Scholar]
- 48.Kearney L, Gonzalez De Castro D, Yeung J, et al. Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood. 2009;113(3):646–648. [DOI] [PubMed] [Google Scholar]
- 49.Malinge S, Ragu C, Della-Valle V, et al. Activating mutations in human acute megakaryoblastic leukemia. Blood. 2008;112(10):4220–4226. [DOI] [PubMed] [Google Scholar]
- 50.Loh ML, Zhang J, Harvey RC, et al. Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children’s Oncology Group TARGET Project. Blood. 2013;121(3):485–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaikwad A, Rye CL, Devidas M, et al. Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol. 2009;144(6):930–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kratz CP, Boll S, Kontny U, Schrappe M, Niemeyer CM, Stanulla M. Mutational screen reveals a novel JAK2 mutation, L611S, in a child with acute lymphoblastic leukemia. Leukemia. 2006;20(2):381–383. [DOI] [PubMed] [Google Scholar]
- 54.Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114(13): 2688–2698. [DOI] [PubMed] [Google Scholar]
- 55.Lacronique V, Boureux A, Valle VD, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278(5341):1309–1312. [DOI] [PubMed] [Google Scholar]
- 56.Reiter A, Walz C, Watmore A, et al. The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res. 2005;65(7):2662–2667. [DOI] [PubMed] [Google Scholar]
- 57.Murati A, Gelsi-Boyer V, Adelaide J, et al. PCM1-JAK2 fusion in myeloproliferative disorders and acute erythroid leukemia with t(8;9) translocation. Leukemia. 2005;19(9): 1692–1696. [DOI] [PubMed] [Google Scholar]
- 58.Griesinger F, Hennig H, Hillmer F, et al. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosomes Cancer. 2005;44(3):329–333. [DOI] [PubMed] [Google Scholar]
- 59.Peeters P, Raynaud SD, Cools J, et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90(7): 2535–2540. [PubMed] [Google Scholar]
- 60.Nguyen MH, Ho JM, Beattie BK, Barber DL. TEL-JAK2 mediates constitutive activation of the phosphatidylinositol 3′-kinase/protein kinase B signaling pathway. J Biol Chem. 2001;276(35):32704–32713. [DOI] [PubMed] [Google Scholar]
- 61.Ho JM, Nguyen MH, Dierov JK, et al. TEL-JAK2 constitutively activates the extracellular signal-regulated kinase (ERK), stress-activated protein/Jun kinase (SAPK/JNK), and p38 signaling pathways. Blood. 2002;100(4):1438–1448. [PubMed] [Google Scholar]
- 62.Lacronique V, Boureux A, Monni R, et al. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood. 2000;95(6):2076–2083. [PubMed] [Google Scholar]
- 63.Joos S, Kupper M, Ohl S, et al. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+ Hodgkin cells. Cancer Res. 2000;60(3):549–552. [PubMed] [Google Scholar]
- 64.Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198(6):851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lenz G, Wright GW, Emre NC, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA. 2008;105(36):13520–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walters DK, Mercher T, Gu TL, et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. 2006;10(1):65–75. [DOI] [PubMed] [Google Scholar]
- 67.Sakaguchi H, Okuno Y, Muramatsu H, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 2013;45(8):937–941. [DOI] [PubMed] [Google Scholar]
- 68.Bains T, Heinrich MC, Loriaux MM, et al. Newly described activating JAK3 mutations in T-cell acute lymphoblastic leukemia. Leukemia. 2012;26(9):2144–2146. [DOI] [PubMed] [Google Scholar]
- 69.Elliott NE, Cleveland SM, Grann V, Janik J, Waldmann TA, Dave UP. FERM domain mutations induce gain of function in JAK3 in adult T-cell leukemia/lymphoma. Blood. 2011;118(14):3911–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koo GC, Tan SY, Tang T, et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012;2(7):591–597. [DOI] [PubMed] [Google Scholar]
- 71.Bouchekioua A, Scourzic L, de Wever O, et al. JAK3 deregulation by activating mutations confers invasive growth advantage in extranodal nasal-type natural killer cell lymphoma. Leukemia. 2013;28(2):338–348. [DOI] [PubMed] [Google Scholar]
- 72.Sanda T, Tyner JW, Gutierrez A, et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer Discov. 2013;3(5):564–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu X, Levine R, Tong W, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci USA. 2005;102(52):18962–18967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hornakova T, Staerk J, Royer Y, et al. Acute lymphoblastic leukemia-associated JAK1 mutants activate the Janus kinase/STAT pathway via interleukin-9 receptor alpha homodimers. J Biol Chem. 2009;284(11): 6773–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gordon GM, Lambert QT, Daniel KG, Reuther GW. Transforming JAK1 mutations exhibit differential signalling, FERM domain requirements and growth responses to interferon-gamma. Biochem J. 2010;432(2):255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Degryse S, de Bock CE, Cox L, et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood. 2014;124(20):3092–3100. [DOI] [PubMed] [Google Scholar]
- 77.Hornakova T, Chiaretti S, Lemaire MM, et al. ALL-associated JAK1 mutations confer hypersensitivity to the antiproliferative effect of type I interferon. Blood. 2010;115(16):3287–3295. [DOI] [PubMed] [Google Scholar]
- 78.Hornakova T, Springuel L, Devreux J, et al. Oncogenic JAK1 and JAK2-activating mutations resistant to ATP-competitive inhibitors. Haematologica. 2011;96(6):845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Toms AV, Deshpande A, McNally R, et al. Structure of a pseudokinase-domain switch that controls oncogenic activation of Jak kinases. Nat Struct Mol Biol. 2013;20(10): 1221–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dusa A, Mouton C, Pecquet C, Herman M, Constantinescu SN. JAK2 V617F constitutive activation requires JH2 residue F595: a pseudokinase domain target for specific inhibitors. PLoS One. 2010;5(6):e11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Staerk J, Kallin A, Demoulin JB, Vainchenker W, Constantinescu SN. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: cross-talk with IGF1 receptor. J Biol Chem. 2005;280(51): 41893–41899. [DOI] [PubMed] [Google Scholar]
- 82.Staerk J, Lacout C, Sato T, Smith SO, Vainchenker W, Constantinescu SN. An amphipathic motif at the transmembrane-cytoplasmic junction prevents autonomous activation of the thrombopoietin receptor. Blood. 2006;107(5):1864–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472–3476. [DOI] [PubMed] [Google Scholar]
- 85.Defour JP, Itaya M, Gryshkova V, et al. Tryptophan at the transmembrane-cytosolic junction modulates thrombopoietin receptor dimerization and activation. Proc Natl Acad Sci USA. 2013;110(7):2540–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ding J, Komatsu H, Iida S, et al. The Asn505 mutation of the c-MPL gene, which causes familial essential thrombocythemia, induces autonomous homodimerization of the c-Mpl protein due to strong amino acid polarity. Blood. 2009;114(15):3325–3328. [DOI] [PubMed] [Google Scholar]
- 87.Zenatti PP, Ribeiro D, Li W, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;43(10):932–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shochat C, Tal N, Bandapalli OR, et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lym-phoblastic leukemias. J Exp Med. 2011;208(5):901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hertzberg L, Vendramini E, Ganmore I, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood. 2010;115(5):1006–1017. [DOI] [PubMed] [Google Scholar]
- 90.Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pardanani A, Lasho TL, Laborde RR, et al. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia. 2013;27(9):1870–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sano H, Ohki K, Park MJ, et al. CSF3R and CALR mutations in paediatric myeloid disorders and the association of CSF3R mutations with translocations, including t(8; 21). Br J Haematol. 2015;170(3):391–397. [DOI] [PubMed] [Google Scholar]
- 93.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41(11):1243–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yoda A, Yoda Y, Chiaretti S, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2010;107(1):252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. [DOI] [PubMed] [Google Scholar]
- 97.Jerez A, Clemente MJ, Makishima H, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012;120(15):3048–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koskela HL, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rajala HL, Eldfors S, Kuusanmaki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121(22):4541–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kontro M, Kuusanmaki H, Eldfors S, et al. Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia. 2014;28(8):1738–1742. [DOI] [PubMed] [Google Scholar]
- 101.Ritz O, Guiter C, Castellano F, et al. Recurrent mutations of the STAT6 DNA binding domain in primary mediastinal B-cell lymphoma. Blood. 2009;114(6):1236–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yildiz M, Li H, Bernard D, et al. Activating STAT6 mutations in follicular lymphoma. Blood. 2015;125(4):668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ. The T cell protein tyrosine phosphatase is a negative regulator of Janus family kinases 1 and 3. Curr Biol. 2002;12(6):446–453. [DOI] [PubMed] [Google Scholar]
- 104.Kleppe M, Lahortiga I, El Chaar T, et al. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42(6): 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kleppe M, Soulier J, Asnafi V, et al. PTPN2 negatively regulates oncogenic JAK1 in T-cell acute lymphoblastic leukemia. Blood. 2011;117(26):7090–7098. [DOI] [PubMed] [Google Scholar]
- 106.Porcu M, Kleppe M, Gianfelici V, et al. Mutation of the receptor tyrosine phosphatase PTPRC (CD45) in T-cell acute lymphoblastic leukemia. Blood. 2012;119(19): 4476–4479. [DOI] [PubMed] [Google Scholar]
- 107.Chen YW, Guo T, Shen L, et al. Receptor-type tyrosine-protein phosphatase kappa directly targets STAT3 activation for tumor suppression in nasal NK/T-cell lymphoma. Blood. 2015;125(10):1589–1600. [DOI] [PubMed] [Google Scholar]
- 108.Bersenev A, Wu C, Balcerek J, Tong W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J Clin Invest. 2008;118(8):2832–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oh ST, Simonds EF, Jones C, et al. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood. 2010;116(6):988–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pardanani A, Lasho T, Finke C, Oh ST, Gotlib J, Tefferi A. LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia. 2010;24(10):1713–1718. [DOI] [PubMed] [Google Scholar]
- 111.Weniger MA, Melzner I, Menz CK, et al. Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phos-pho-STAT5 accumulation. Oncogene. 2006;25(18):2679–2684. [DOI] [PubMed] [Google Scholar]
- 112.Reddy J, Shivapurkar N, Takahashi T, et al. Differential methylation of genes that regulate cytokine signaling in lymphoid and hematopoietic tumors. Oncogene. 2005;24(4):732–736. [DOI] [PubMed] [Google Scholar]
- 113.Jost E, do ON, Dahl E, et al. Epigenetic alterations complement mutation of JAK2 tyrosine kinase in patients with BCR/ABL-negative myeloproliferative disorders. Leukemia. 2007;21(3):505–510. [DOI] [PubMed] [Google Scholar]
- 114.Teofili L, Martini M, Cenci T, et al. Epigenetic alteration of SOCS family members is a possible pathogenetic mechanism in JAK2 wild type myeloproliferative diseases. Int J Cancer. 2008;123(7):1586–1592. [DOI] [PubMed] [Google Scholar]
- 115.Torti D, Trusolino L. Oncogene addiction as a foundational rationale for targeted anticancer therapy: promises and perils. EMBO Mol Med. 2011;3(11):623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Smith BD. Imatinib for chronic myeloid leukemia: the impact of its effectiveness and long-term side effects. J Natl Cancer Inst. 2011;103(7):527–529. [DOI] [PubMed] [Google Scholar]
- 117.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. [DOI] [PubMed] [Google Scholar]
- 119.Rampal R, Al-Shahrour F, Abdel-Wahab O, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014;123(22):e123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pardanani A, Tefferi A. Definition and management of ruxolitinib treatment failure in myelofibrosis. Blood Cancer J. 2014;4:e268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Goldberg RA, Reichel E, Oshry LJ. Bilateral toxoplasmosis retinitis associated with rux-olitinib. N Engl J Med. 2013;369(7):681–683. [DOI] [PubMed] [Google Scholar]
- 122.Wathes R, Moule S, Milojkovic D. Progressive multifocal leukoencephalopathy associated with ruxolitinib. N Engl J Med. 2013;369(2):197–198. [DOI] [PubMed] [Google Scholar]
- 123.Wysham NG, Sullivan DR, Allada G. An opportunistic infection associated with rux-olitinib, a novel Janus kinase 1,2 inhibitor. Chest. 2013;143(5):1478–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Caocci G, Murgia F, Podda L, Solinas A, Atzeni S, La Nasa G. Reactivation of hepatitis B virus infection following ruxolitinib treatment in a patient with myelofibrosis. Leukemia. 2013;28(1):225–227. [DOI] [PubMed] [Google Scholar]
- 125.Vannucchi AM. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(17):1670–1671. [DOI] [PubMed] [Google Scholar]
- 126.Fleischman AG, Maxson JE, Luty SB, et al. The CSF3R T618I mutation causes a lethal neutrophilic neoplasia in mice that is responsive to therapeutic JAK inhibition. Blood. 2013;122(22):3628–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dao KH, Solti MB, Maxson JE, et al. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R-T618I-positive atypical chronic myeloid leukemia. Leuk Res Rep. 2014;3(2):67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lierman E, Selleslag D, Smits S, Billiet J, Vandenberghe P. Ruxolitinib inhibits transforming JAK2 fusion proteins in vitro and induces complete cytogenetic remission in t(8;9)(p22;p24)/PCM1-JAK2-positive chronic eosinophilic leukemia. Blood. 2012;120(7): 1529–1531. [DOI] [PubMed] [Google Scholar]
- 129.Rumi E, Milosevic JD, Casetti I, et al. Efficacy of ruxolitinib in chronic eosinophilic leukemia associated with a PCM1-JAK2 fusion gene. J Clin Oncol. 2013;31(17):e269–271. [DOI] [PubMed] [Google Scholar]
- 130.Schwaab J, Knut M, Haferlach C, et al. Limited duration of complete remission on ruxolitinib in myeloid neoplasms with PCM1-JAK2 and BCR-JAK2 fusion genes. Ann Hematol. 2015;94(2):233–238. [DOI] [PubMed] [Google Scholar]
- 131.Maude SL, Tasian SK, Vincent T, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lym-phoblastic leukemia. Blood. 2012;120(17): 3510–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Maude SL, Dolai S, Delgado-Martin C, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125(11):1759–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hanna DM, Fellowes A, Vedururu R, Mechinaud F, Hansford JR. A unique case of refractory primary mediastinal B-cell lymphoma with JAK3 mutation and the role for targeted therapy. Haematologica. 2014;99(9):e156–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Quintas-Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res. 2013;19(8):1933–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Andreoli A, Verger E, Robin M, et al. Clinical resistance to ruxolitinib is more frequent in patients without MPN-associated mutations and is rarely due to mutations in the JAK2 kinase drug-binding domain. Blood. 2013; 122(21):1591–1591. [Google Scholar]
- 136.Deshpande A, Reddy MM, Schade GO, et al. Kinase domain mutations confer resistance to novel inhibitors targeting JAK2V617F in myeloproliferative neoplasms. Leukemia. 2012;26(4):708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Weigert O, Lane AA, Bird L, et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J Exp Med. 2012;209(2):259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Marit MR, Chohan M, Matthew N, et al. Random mutagenesis reveals residues of JAK2 critical in evading inhibition by a tyrosine kinase inhibitor. PLoS One. 2012;7(8):e43437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Koppikar P, Bhagwat N, Kilpivaara O, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 2012;489(7414):155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bhagwat N, Levine RL, Koppikar P. Sensitivity and resistance of JAK2 inhibitors to myeloproliferative neoplasms. Int J Hematol. 2014;97(6):695–702. [DOI] [PubMed] [Google Scholar]
- 141.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2(7):358–364. [DOI] [PubMed] [Google Scholar]
- 142.Andraos R, Qian Z, Bonenfant D, et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2012;2(6):512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Meyer SC, Keller MD, Koppikar P, et al. Type II inhibition of JAK2 with NVP-CHZ868 reverses type I JAK inhibitor persistence and demonstrates increased efficacy in MPN models. Blood. 2014;124(21):160.25013160 [Google Scholar]
- 144.Goedken ER, Argiriadi MA, Banach DL, et al. Tricyclic covalent inhibitors selectively target Jak3 through an active site thiol. J Biol Chem. 2015;290(8):4573–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tokarski JS, Zupa-Fernandez A, Tredup JA, et al. Tyrosine kinase 2-mediated signal transduction in T lymphocytes is blocked by pharmacological stabilization of its pseudokinase domain. J Biol Chem. 2015; 290(17):11061–11074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wolf A, Eulenfeld R, Gabler K, et al. JAK2-V617F-induced MAPK activity is regulated by PI3K and acts synergistically with PI3K on the proliferation of JAK2-V617F-positive cells. JAKSTAT. 2013;2(3):e24574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kamishimoto J, Tago K, Kasahara T, Funakoshi-Tago M. Akt activation through the phosphorylation of erythropoietin receptor at tyrosine 479 is required for myeloproliferative disorder-associated JAK2 V617F mutant-induced cellular transformation. Cell Signal. 2011;23(5):849–856. [DOI] [PubMed] [Google Scholar]
- 148.Vicari L, Martinetti D, Buccheri S, et al. Increased phospho-mTOR expression in megakaryocytic cells derived from CD34+ progenitors of essential thrombocythaemia and myelofibrosis patients. Br J Haematol. 2012;159(2):237–240. [DOI] [PubMed] [Google Scholar]
- 149.Bartalucci N, Tozzi L, Bogani C, et al. Co-targeting the PI3K/mTOR and JAK2 signalling pathways produces synergistic activity against myeloproliferative neoplasms. J Cell Mol Med. 2013;17(11):1385–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Choong ML, Pecquet C, Pendharkar V, et al. Combination treatment for myeloproliferative neoplasms using JAK and pan-class I PI3K inhibitors. J Cell Mol Med. 2013;17(11): 1397–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bogani C, Bartalucci N, Martinelli S, et al. mTOR inhibitors alone and in combination with JAK2 inhibitors effectively inhibit cells of myeloproliferative neoplasms. PLoS One. 2013;8(1):e54826. [DOI] [PMC free article] [PubMed] [Google Scholar]