

Background: The magnesium position in the active site of GTPases varies among x-ray structures of Ran and related GTPases.
Results: The magnesium in both Ras and Ran is always coordinated to the β- and γ-phosphate.
Conclusion: Other factors like steric hindrance by the Tyr-39 side chain influence the hydrolysis rate in Ran.
Significance: The new information allows an improved view on the catalytic mechanism of GTPases.
Keywords: computer modeling, crystal structure, Fourier transform IR (FTIR), infrared spectroscopy (IR spectroscopy), nuclear transport, small GTPase, spectroscopy, X-ray crystallography, QM/MM simulations, reaction mechanism
Abstract
Small GTPases regulate key processes in cells. Malfunction of their GTPase reaction by mutations is involved in severe diseases. Here, we compare the GTPase reaction of the slower hydrolyzing GTPase Ran with Ras. By combination of time-resolved FTIR difference spectroscopy and QM/MM simulations we elucidate that the Mg2+ coordination by the phosphate groups, which varies largely among the x-ray structures, is the same for Ran and Ras. A new x-ray structure of a Ran·RanBD1 complex with improved resolution confirmed this finding and revealed a general problem with the refinement of Mg2+ in GTPases. The Mg2+ coordination is not responsible for the much slower GTPase reaction of Ran. Instead, the location of the Tyr-39 side chain of Ran between the γ-phosphate and Gln-69 prevents the optimal positioning of the attacking water molecule by the Gln-69 relative to the γ-phosphate. This is confirmed in the RanY39A·RanBD1 crystal structure. The QM/MM simulations provide IR spectra of the catalytic center, which agree very nicely with the experimental ones. The combination of both methods can correlate spectra with structure at atomic detail. For example the FTIR difference spectra of RasA18T and RanT25A mutants show that spectral differences are mainly due to the hydrogen bond of Thr-25 to the α-phosphate in Ran. By integration of x-ray structure analysis, experimental, and theoretical IR spectroscopy the catalytic center of the x-ray structural models are further refined to sub-Å resolution, allowing an improved understanding of catalysis.
Introduction
Small GTPases are molecular switches, which regulate many cellular processes (1, 2). The “on” state is characterized by a specific conformation of the switch region of the proteins, which is induced by GTP binding, whereas the “off” state is obtained with GDP in the binding pocket. The “switch off” is performed by GTP hydrolysis, which can be further accelerated by GTPase activating proteins (3). The “switch on” is facilitated by nucleotide exchange, usually effected by guanine nucleotide exchange factors. The small GTPase Ras is the central regulator in cell growth and differentiation (4). Ras is mutated in about 25% of human tumors. The related small GTPase Ran regulates nuclear transport (5, 6) and is involved in spindle formation and nuclear envelope formation of dividing cells (7). The GTP form of Ran is stabilized by Ran-binding proteins (RanBP1 or the structurally similar RanBD domains of the RanBP2 protein) (8).
The reaction rate of the intrinsic hydrolysis reaction among GTPases varies. Especially Ran has a very slow reaction rate compared with other GTPases. This is important to maintain the Ran gradient: Ran·GTP should be concentrated mainly in the nucleoplasm and Ran·GDP in the cytoplasm (9). Fast intrinsic GTP hydrolysis by Ran in the nucleoplasm (i.e. without GTPase activating protein) would decrease the gradient and impact the nuclear transport, which can lead to severe diseases like the Hutchinson-Gilford Progeria Syndrome (10). Indeed, GTP hydrolysis is roughly 26 times slower in Ran and about 13 times slower in Ran·RanBD1 than in Ras (11). Here, we address at the atomic level the mechanism underlying the difference in the Ran and Ras hydrolysis rate.
One potential candidate for the difference is the position of the magnesium ion. FTIR experiments have shown Mg2+ to be one of the key players for catalyzing GTP hydrolysis due to its ability to shift negative charge within the GTP (12). The corresponding conformational change and charge shift of the triphosphate could be a reason for the different hydrolysis rates. Indeed, the position of the Mg2+ varies among available GTPase crystal structures. Fig. 1 shows a comparison of the Mg2+ coordination in Ras and Ran with bound GTP or GTP analogues based on x-ray structural models. The x-ray structure of H-Ras·GTP (Protein Data Bank (PDB))4 (PDB code 1QRA, Fig. 1a) (13) and Ran·Gpp(NH)p·RanBD1 (PDB code 1RRP, Fig. 1b) (14) indicate a difference in the coordination of the Mg2+. In Ras (Fig. 1a) the Mg2+ is coordinated by the two hydroxyl groups of Ser-17 and Thr-35, two water molecules, an oxygen atom of the β-phosphate, and an oxygen atom of the γ-phosphate. This coordination will be labeled with respect to the GTP-Mg2+ interactions “βγ-form” in the following. In Ran·RanBD1 (Fig. 1b) five of the coordinating groups are analogues to their Ras counterparts: two hydroxyl groups (Thr-24 and Thr-42), one water molecule, one β- and one γ-phosphate oxygen atom. However, instead of the second water molecule, the Mg2+ is coordinated by one α-phosphate oxygen atom. This coordination will be labeled “αβγ-form.” GTP·Mg2+ conformation in water has also an αβγ-form (12) and shows a very slow hydrolysis rate (5 × 10−8 s−1 at 303 K) (15). The different GTP·Mg2+ conformation might explain why the hydrolysis rate of GTP·Mg2+ bound to Ran in the RanBP1 complex (3.5 × 10−5 s−1 at 303 K) is slower than for GTP·Mg2+ bound to H-Ras (6.8 × 10−4 s−1 at 303 K) (Fig. 2) (11). FTIR spectroscopy is sensitive to very small changes in the nucleotide environment, and indeed, the spectrum of the GTP-phosphates in Ran is significantly different to GTP in Ras (Fig. 3) (11).
FIGURE 1.
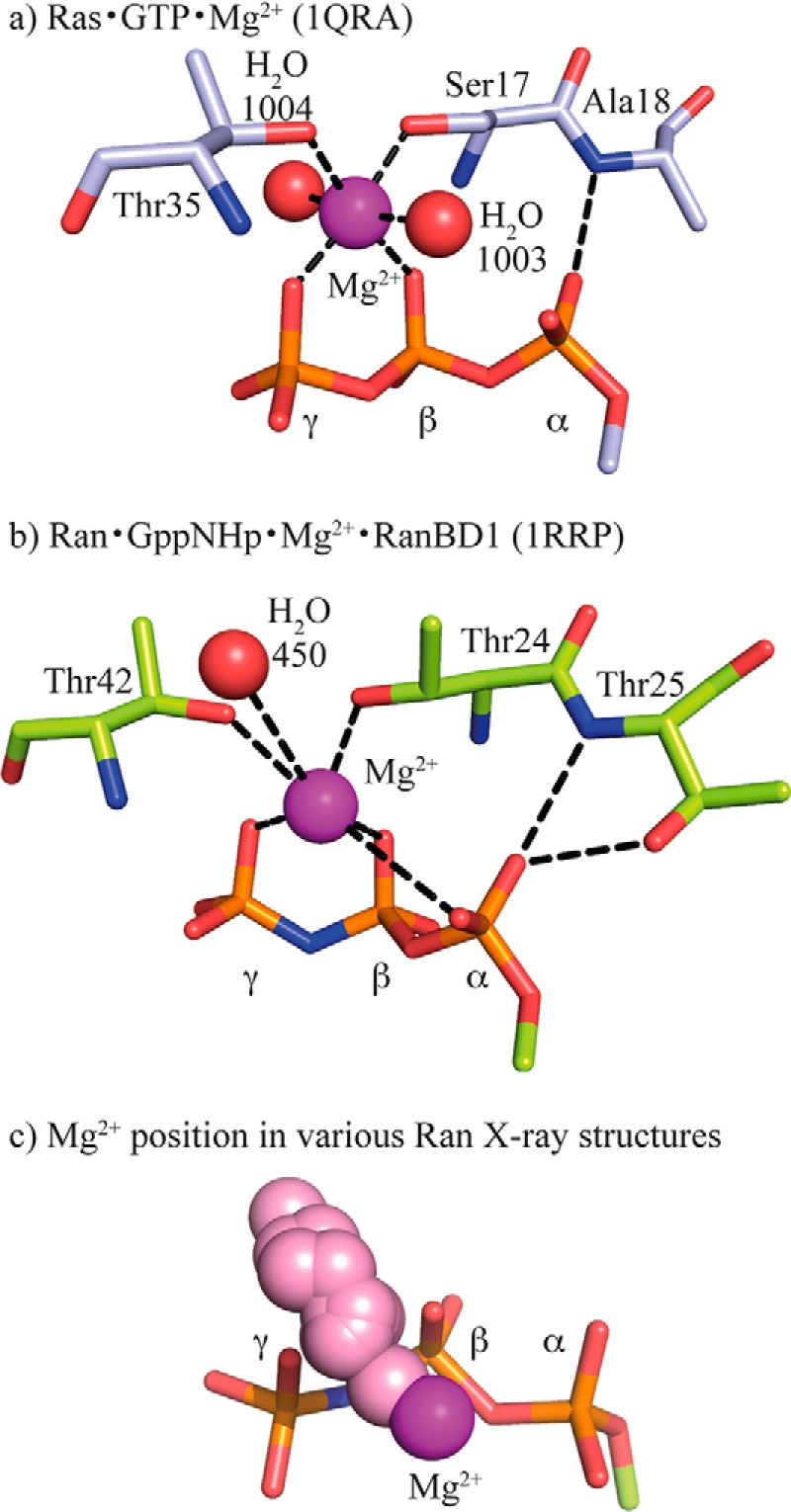
Mg2+ positions in Ras and Ran x-ray structures. Comparison of the Mg2+ coordination in the x-ray structures of (a) Ras·GTP (PDB code 1QRA, light blue carbon atoms), (b) Ran·Gpp(NH)p·RanBD1 (PDB code 1RRP, light green carbon atoms), and (c) various available Ran x-ray structures with their nucleotides aligned to Gpp(NH)p of PDB code 1RRP (Ran·Gpp(NH)p·RanBD1, Mg2+ in magenta) and the Mg2+ ions shown as pink spheres. Both, the βγ- and αβγ-form are found. A similar distribution is found for Ras complexes (Fig. 4). The used x-ray structures in c can be found in supplemental Section S2.
FIGURE 2.
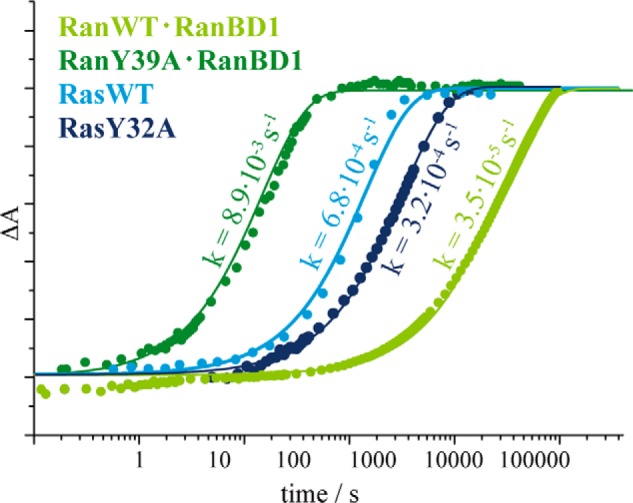
Comparison of the kinetics of Ran·RanBD1, RanY39A·RanBD1, Ras, and RasY32A. The normalized absorbance differences of adjacent bands of the GTP and GDP state are shown as a measure for the progress of the hydrolysis. The fits to single exponential functions are shown as continuous lines.
FIGURE 3.
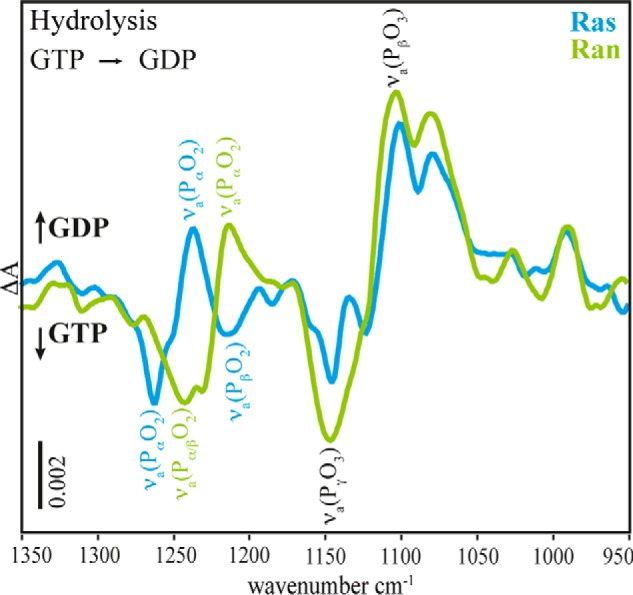
Comparison of the FTIR difference spectra of the hydrolysis reaction ahyd of Ras (blue) and Ran·RanBD1 (green, scaled by factor 1.7). The main differences occur in the νa(PαO2)-vibrational modes, the remaining part of the phosphate region is very similar.
However, available x-ray structures reveal a more complicated picture than described above. In particular, an analysis of the Mg2+ position in various GTPase x-ray structures reveals an uncertainty in the Mg2+ location. There are also Ran structures available indicating a βγ-form (Fig. 1c). In contrast, all structures of Ras alone show a βγ-form (Fig. 4d). However, if Ras complexes are also considered, the position of the Mg2+ is again shifting quite systematically (Fig. 4a). The same behavior is also observed for Rab GTPases (Fig. 4, c and e). To investigate this unexpected variability of Mg2+ location and obtain additional information about its position in solution, we combine in this study new crystal structures of Ran complexes with improved resolution with theoretical and experimental vibrational spectroscopy.
FIGURE 4.
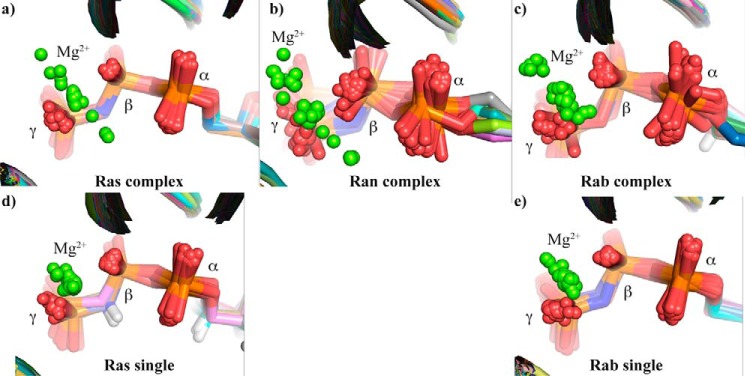
Mg2+ positions in small GTPases. Comparison of the Mg2+ coordination in the x-ray structures with bound GTP or GTP analogue of (a) H-Ras in complex with other proteins (averaged resolution over 14 structures: 2.4 ± 0.4 Å), (b) Ran in complex with other proteins (averaged resolution: 2.7 ± 0.5 Å), (c) Rab in complex with other proteins (averaged resolution over 21 structures: 2.3 ± 0.5 Å), (d) uncomplexed H-Ras (averaged resolution over 67 structures: 1.8 ± 0.4 Å), and (e) uncomplexed Rab (averaged resolution over 39 structures: 1.9 ± 0.4 Å). The used x-ray structures in this figure can be found in supplemental Section S3.
It has been shown that spectral changes can be decoded into structural changes by the combination of FTIR spectroscopy with biomolecular simulations (16). QM/MM calculations allow the accurate calculation of the vibrational spectrum (17–21). If experimental and theoretical spectra agree, the theoretical structural model is validated and the detailed simulated structural models can be used for interpreting reaction mechanisms and catalysis. Bond length can be calculated with a resolution of 0.01 Å, well below the resolution of x-ray analysis. However, these changes are significant for catalysis. The change of a C-C single bond by 0.1 Å corresponds to 6 kJ/mol, which is about one-quarter of the change in free energy of activation for GTP hydrolysis in water compared with GTP in Ras. A combined QM/MM and FTIR analysis was performed for H-Ras and it was found that a combination of charge shifts, an increase in Pβ-Pγ distance, and a strained, staggered conformation of the GTP induced by the Ras and GAP seem to be responsible for catalysis (16).
Here, we report on an atomic structural model of the triphosphate environment of Ran obtained by the combination of x-ray structure analysis, biomolecular simulations, and FTIR spectroscopy. Our model allows for the elucidation of the correct Mg2+ position in solution and permits an investigation of the dramatic difference in Ran and Ras hydrolysis rates.
Materials and Methods
Biomolecular Simulations
Molecular mechanics (MM) simulations were performed with Gromacs 4.0.7 (22, 23), and the hybrid quantum mechanics/molecular mechanics (QM/MM) simulations with Gromacs/Gaussian03 (23–25). The used simulation systems are described in detail in supplemental Table S1. Hydrogen atoms were added to the x-ray structures by the MAXIMOBY algorithm (26). Afterward the protonated structures were checked and amended by the same algorithm. Parameters for the MM and QM/MM simulations were as described previously (12). To calculate theoretical IR features and structural details we used the same workflow as described in an earlier paper (16), but with an enlarged quantum region.
For the QM/MM equilibration run the triphosphate, ribose, Mg2+, Ala-18 (Ras)/Thr-25 (Ran), the backbone carboxyl and oxygen atom of Ser-17/Thr-24, a dummy atom between the carboxyl atom, and the α carboxyl atom of Ser-17/Thr-24, the backbone nitrogen atom and its bound hydrogen atom of Leu-19/Phe-26, and the dummy atom between the nitrogen atom and the α carboxyl atom of Leu-19/Phe-26 were treated quantum mechanically. For the first QM/MM energy optimization run the triphosphate, the ribose, the Mg2+, Ala-18/Thr-25, the backbone of Ser-17/Thr-24, the backbone of Lys-16/Lys-23, the backbone carboxyl and oxygen atom of Gly-15/Gly-22, a dummy atom between the carboxyl atom and the α carboxyl atom of Gly-15/Gly-22, the backbone nitrogen atom and its bound hydrogen atom of Leu-19/Phe-26, and the dummy atom between the nitrogen atom and the α carboxyl atom of Leu-19/Phe-26 were treated quantum mechanically. For the second QM/MM energy optimization run and the following normal mode analysis the triphosphate, ribose, Mg2+, Ala-18 (Ras)/Thr-25 (Ran), backbone carboxyl and oxygen atom of Ser-17/Thr-24, a dummy atom between the carboxyl atom and the α carboxyl atom of Ser-17/Thr-24, the backbone nitrogen atom and its bound hydrogen atom of Leu-19/Phe-26, and the dummy atom between the nitrogen atom and the α carboxyl atom of Leu-19/Phe-26 were treated quantum mechanically.
Averaged structures were generated by the tool developed by Fischer and Kandt (27). Thereby an iterative scheme of calculating an average conformation and re-aligning the trajectory to that average structure to compute a new average structure was performed. This procedure was repeated until the average structure stopped changing and the root mean square deviation was below 0.001 Å. Then the structure with the closest root mean square deviation to the generated average structure was extracted from the trajectory as the run-average structure. The calculation of the B-factors from the MD simulations is described in supplemental Table S2.
Caged Compounds
The P3–1-(2-nitrophenyl)-ethylester of GTP (NPE-GTP) was synthesized in two steps from 2-nitrophenylacetylhydrazone and GTP with subsequent purification via anion exchange chromatography according to a procedure by Walker and Trentham (28). The P3-para-hydroxyphenacyl ester of GTP (pHP-GTP) was synthesized by coupling GDP and pHP-caged Pi. The latter was obtained in five steps from para-hydroxyacetophenone and dibenzylphosphate and according to a procedure from Park and Givens (29).
Proteins (Cloning, Overexpression, and Preparation)
Point mutations in Ras and Ran were obtained by site-directed mutagenesis using the overlap extension PCR method. Wild-type and A18T H-Ras(1–166) were prepared with Escherichia coli (CK600K) using the ptac-expression system as described (30). For Ran wild-type, RanT25A, and RanY39A(1–216) the pET3d-expression system with E. coli (BL21(DE3)) was used (8). Human RanBP1(1–201) and NF1–333 were both overexpressed as a GST (glutathione S-transferase) fusion protein using the plasmid pGEX-4T-1mod and E. coli (BL21(DE3)). The pGEX-4T-1mod plasmid has an additional tobacco etch virus protease recognition site. Purification was done by GSH affinity chromatography, on column digestion with tobacco etch virus protease, and subsequent gel filtration. Overexpression and purification of Rna1p from Schizosaccharomyces pombe (1–386) was done as previously described (31). RanBD1(1155–1321) (first Ran-binding domain of human RanBP2) was prepared as described by Vetter et al. (14).
Nucleotide Exchange and Manganese Loading with Different GTPases
For FTIR measurements Ras, Ran, and their mutants were loaded with caged GTP according to John et al. (32). A minimum 1 mg of GTPase was incubated with a 1.5 m excess of pHP-GTP and 1 unit of alkaline phosphatase in reaction buffer (50 mm Tris-HCl, 10 μm ZnSO4, 200 mm (NH4)2SO4, 1 mm DTT, pH 7.5). The reactions were observed via HPLC. For Ran, a 1:1 complex of Ran·RanBP1 was used.
The exchange to Ran·NPE-GTP·Mn2+ was a two-step process. First, nucleotide-free Ran:RanBP1 was produced according to John et al. (32) and second, following a 10-min EDTA treatment (1 mm), the GTPase was loaded with 20 mm Mn2+ and 1.5 m excess NPE-GTP. The use of NPE-GTP instead of pHP-GTP was necessary because pHP-GTP is not stable in the presence of Mn2+ ions. After nucleotide exchange all samples were subsequently applied to a NAP-5 gel filtration column to change to low salt buffer for FTIR sample preparation (Ras: 500 μm Hepes buffer, 50 μm MgCl2, 50 μm DTT, pH 7.4; Ran: 1 mm Hepes buffer, 100 μm MgCl2/MnCl2, 100 μm DTT, pH 7.6) and removal of excess nucleotide.
FTIR Spectroscopy and Sample Preparation
Samples were prepared between CaF2 windows as described (33) and measured on a Bruker Vertex 80V spectrometer. Ran measurements were done with 5 mm concentration of the GTPase in complex with RanBP1 and 5 mol % Rna1p (0.25 mm), Ras measurements with 10 mm and 1 mol % NF1–333 (0.1 mm) using a buffer composed of 200 mm Hepes buffer, pH 7.5, 20 mm MgCl2, and 20 mm DTT. Intrinsic reactions with Ran, Ras, and mutants were carried out at 283 K, and manganese measurements with Ran at 278 K. Photolysis of the caged compounds and data acquisition was performed as described (34). The data were analyzed between 1800 and 950 cm−1 with a global fit method (35). In this analysis, the absorbance changes ΔA during the hydrolysis reaction were analyzed by fitting the data to a one exponential function with an apparent rate constant khyd and the amplitude ahyd(ν) (Equation 1).
The photolysis spectrum aph(ν) is a difference spectrum comparing the state before triggering with the state after flashing, extrapolated to t = 0. In the figures, the disappearing bands are shown face downwards and the appearing bands face upwards. The absolute absorbance differences depend on the exact sample thickness, the laser intensity, and the amount of caged GTP after nucleotide exchange. Thus, for the comparison of two spectra, one can be scaled to match the absolute absorbance difference of the other.
Protein Preparation for X-ray Crystallography
Exchange of bound GDP to Gpp(NH)p, a nonhydrolysable GTP analogue, on RanY39A was performed by incubating 1 μmol of each RanY39A and RanBD1 with a 2.5 excess of Gpp(NH)p and 20 units of alkaline phosphatase in a volume of 2.7 ml for 20 h at room temperature in 50 mm Tris buffer, pH 7.6, 200 mm (NH4)2SO4, 5 mm dithiothreitol. The formed complex was purified on a prepacked Superdex 75 pg 16/60 column (GE Healthcare) used at an ÄKTAprime System (GE Healthcare) in 20 mm Tris buffer, pH 7.4, 2 mm MgCl2, and 2 mm DTT. Peak fractions were collected; purity was checked by SDS-PAGE, pooled, and concentrated (Amicon Ultra-15 centrifugal filter unit with molecular mass cutoff 30,000 Da, Millipore) up to 24.4 g/liter, shock frozen, and stored at −80 °C.
Protein Crystallization and Structure Solution
RanY39A(1–216)·Gpp(NH)p·RanBD1 was prepared at 24.4 mg/ml in 20 mm Tris, pH 7.4, 2 mm MgCl2, and 2 mm DTT. Hanging drops were set up at 20 °C by mixing 1.5 μl of protein with 1.5 μl of reservoir consisting of 1.8 m ammonium sulfate, 2.5% PEG 1500, and 100 mm Hepes, pH 7.5. Crystals were flash frozen in 1.8 m ammonium sulfate, 2% PEG 1000, 100 mm Hepes, pH 7.5, and 30% glycerol. Data were collected at a wavelength of 1.0039 using a Pilatus 6M detector. The crystals in space group C2221 diffracted to 3.2 Å, contained 2 complexes per asymmetric unit and were solved using PHASER with the RanΔ191·BeF structure as a template. Refinement in PHENIX with local NCS and secondary structure constraints as well as RanΔ191·BeF as a reference structure (i.e. dihedral angles are constrained to the reference structure to get better geometries at this low resolution) converged to a final R and Rfree of 22.2 and 25.5%, respectively, with 0.76% Ramachandran outliers. Comparison of the model refined with and without using constraints from the reference structure showed no major differences except for better geometries in the former case.
RanΔ191GDP·BeF3·RanBD1 was prepared in buffer “A” (20 mm KHPO4, pH 7.4, 2 mm MgCl2, 1 mm DTT, 5% (w/v) sucrose) by adding 67.8 μl of RanΔ191 (8 mg/ml) + 36.3 μl of RanBD1 (10.8 mg/ml) + 5 μl of BeFx stock (1 m BeF2 + 3 m NaF). The hanging drops were set up at 18 °C by mixing 3 μl of the protein complex with 3 μl of reservoir consisting of 18% PEG 4000, 250 mm ammonium sulfate, and 100 mm MES, pH 6.25. Crystals were flash frozen in the reservoir solution plus 10% glycerol. The crystals in space group P21 had 2 complexes in the asymmetric unit and diffracted to 2.4 Å. Data were collected at a wavelength of 0.9797 using a MarCCD detector.
The structure was solved using PHASER from the CCP4 suite with the Ran·RanBD1 complex 1RRP from the Protein Data Bank as a template, built in COOT, and refined with REFMAC5 using TLS with four segments per domain and tight NCS constraints to a final R factor of 24.64% and Rfree of 27.80%. Relaxing the NCS constraints increased Rfree. The geometry is excellent with only 0.63% outliers in the Ramachandran plot. All following crystals were set up as hanging drop at 18 °C with the protein dissolved in the buffer “A” supplemented by 50 mm BeF2 and 150 mm NaF as described above and collected using a MarCCD detector (all at 0.984 Å except RanY39A in P1 at 1.000 Å).
For RanWT(1–216)·GDP·BeF3, 1.2 μl of protein 18 mg/ml was mixed with 3 μl of reservoir (30% PEG 1000, 50 mm Tris, pH 8, 20 mm MgCl2) and 1.8 μl of water. Crystals were flash frozen without cryoprotectant and diffracted to 1.7 Å in space group P41. A similar setup but with 50 mm Hepes, pH 7.2, instead of the Tris buffer, with 32% PEG 1000 and using 3 μl of protein plus 3 μl of reservoir yielded crystals of space group P21 that diffracted to 1.8 Å and were flash frozen in reservoir containing 10% glycerol. Both space groups had 2 molecules per asymmetric unit.
3 μl of RanY39A(1–216)·GDP·BeF3 at 11.4 mg/ml in buffer A mixed with 3 μl of 35% PEG 1000, 50 mm Tris, pH 8.5, 20 mm MgCl2, 1 mm GDP, 1 mm BeF2 gave P1 crystals with 8 molecules per asymmetric unit in space group P1 that diffracted to 1.75 Å and could be frozen without cryoprotectant. A similar setup with 50 mm Hepes, pH 7.2, instead of the Tris buffer and 34% PEG 1000 yielded P21 crystals that diffracted to 1.75 Å with 2 molecules per asymmetric unit and were flash frozen in reservoir containing 10% glycerol. The P1 data could also be processed in P21, but molecular replacement showed clearly that this is a case of pseudosymmetry, and the structure could be solved correctly only in P21. All datasets were collected at 100 K at the SLS synchrotron in Villigen, Switzerland, at beamline X10SA (PXII). The PDB codes of the x-ray structures solved within this manuscript are 5CIQ and 5CIT (RanWT(1–216)-GDP-BeF3, space groups P41 and P21, only GDP visible), 5CIW and 5CJ2 (RanY39A(1–216)-GDP-BeF3, space groups P21 and P1, only GDP visible), 5CLL (Ran(1–191)-GDP-BeF3-RanBD1(1155–1321) and 5CLQ (RanY39A(1–216)-GPPNHP-RanBD1(1155–1321). Details are found in supplemental Table S3.
Results and Discussion
Influence of the Magnesium Positions on the Phosphate Vibrations
As introduced above, one reason for the well known difference of the phosphate absorptions in the FTIR spectra of GTP bound to Ras compared with Ran·RanBD1 (see Ref. 11 and Fig. 3) could be a different Mg2+ coordination of the triphosphate according to the corresponding x-ray structures (PDB codes 1QRA (Ras) and 1RRP (Ran·RanBD1)) called βγ- or αβγ-form in the following. To investigate whether this might be the cause of the band shifts, we performed biomolecular simulations of H-Ras·GTP as well as of Ran·GTP·RanBD1 to be able to calculate the theoretical infrared spectra for comparison with the experimentally determined phosphate vibrations. The high resolution x-ray structure 1QRA (βγ-form) was employed as a starting structure for H-Ras, for Ran·GTP·RanBD1 the complex of Ran·Gpp(NH)p with RanBD1 (x-ray structure 1RRP) was used (αβγ-form). Additionally, we simulated both H-Ras in a Ran-like αβγ-form, and Ran with a Ras-like βγ-form. In PDB code 1QRA a crystal contact leads to an artificially “flipped out” conformation of Tyr-32. As discussed previously (12), this tyrosine moves in our simulations into the canonical position close to the γ-phosphate that is found in the majority of Ras crystal structures. This structure corresponding to the state 2 observed in NMR spectroscopy (36) with the canonical position of Tyr-32 was used as starting structure for all simulations. All four systems were stable during 50-ns simulations. The four structures, averaged over the equilibrated last 25 ns, are shown in Fig. 5.
FIGURE 5.
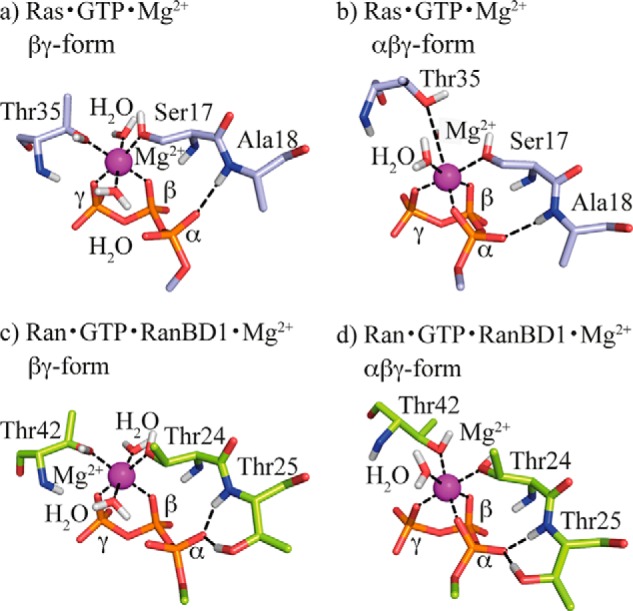
Structures from molecular dynamics simulations. Comparison of the averaged structures of the equilibrated last 25 ns of 50-ns molecular dynamics simulations of solvated Ras·GTP·Mg2+ (light blue carbon atoms) in the βγ-form (a) and αβγ-form (b) as well as Ran·GTP·Mg2+·RanBD1 (light green carbon atoms) in the βγ-form (c) and αβγ-form (d). In c the second water molecule HOHX does not exist in the x-ray structure of RanWT (PDB code 1RRP) and has been added referring to the Ras structure of the βγ-form. In all four 50-ns simulation trajectories of Ran and Ras the Mg2+ remained stably tridentately or bidentately coordinated by the triphosphate depending on the starting structure. Oxygen atoms are red, nitrogen atoms are blue, phosphate atoms are orange, hydrogen atoms are light gray, and the magnesium ion is purple.
Next, we did QM/MM simulations of six snapshots of each system and calculated the theoretical infrared spectra. To obtain reliable spectra, we treated a large part of the active site by quantum mechanics, i.e. besides the triphosphate also the ribose, Mg2+, and the amino acid that forms hydrogen bonds to the α-phosphate, Ala-18 for Ras or Thr-25 for Ran. Thus, all binding partners of the α-phosphate are treated quantum mechanically and the calculated energy of the α-phosphate vibration should be very accurate. The results of these calculations are summarized in Fig. 6.
FIGURE 6.
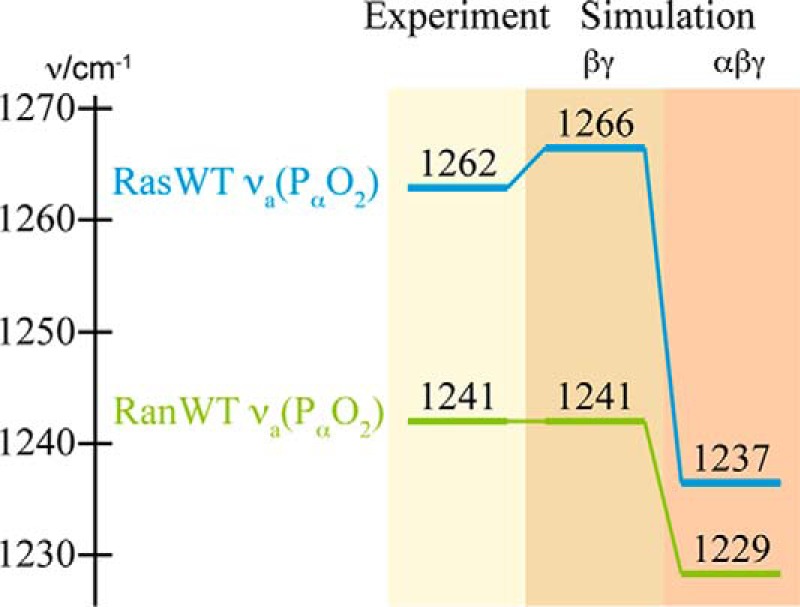
Comparison of the asymmetrical α-phosphate stretching modes of GTP bound to Ras or Ran, respectively. The experimental values of the hydrolysis difference spectra of Ras and Ran are compared with the calculated ones with both βγ- and αβγ-coordination of the Mg2+. The corresponding IR difference spectra are shown in Fig. 7.
The calculated frequencies for νa(PαO2) of Ras are 1266 cm−1 for the βγ-form and 1237 cm−1 for the αβγ-form, respectively. As expected, the coordination of the α-phosphate by the Mg2+ leads to a strong red shift of the vibration. Although the value of the βγ-form is very close to the experimental value of 1266 cm−1, the αβγ-form deviates by 25 cm−1. For Ran·RanBD1 the corresponding vibration is already red shifted compared with Ras for the βγ-form and calculated to be at 1241 cm−1. For the αβγ-form a further red shift to 1229 cm−1 is calculated. Thus, the value for the βγ-form coincides with the experimental value, whereas the value of the αβγ-form deviates by 12 cm−1. Accordingly, the calculations suggest the βγ-form for both proteins, Ras and Ran·RanBD1.
To obtain further experimental evidence for a similar Mg2+ coordination of Ras and Ran·RanBD1 in solution we performed an exchange of Mg2+ to Mn2+. Mn2+ is known to coordinate GTP in Ras in the same manner as Mg2+ and selective shifts of the vibrations of coordinated phosphate groups can be expected (37). The spectra obtained via photolysis of caged GTP are shown in Fig. 7 and the shifts are summarized in Table 1. The observed shifts are comparable for Ras and Ran·RanBD1, again suggesting a similar coordination of the metal ion. In both cases no significant shift of the νa(PαO2) vibration is observed, indicating no direct interaction of the Mn2+ with the α-phosphate. According to our experience, the α-phosphate should clearly show a shift upon changes in its environment, whereas the effect on the γ-phosphate is usually less clear. Furthermore, the γ-phosphate environment in the MD simulations was not treated quantum mechanically so that the predictions would be not as accurate as for the α-phosphate. In summary, theory and experiment consistently suggest Mg2+ coordination to the β- and γ-phosphates (βγ-form) for both proteins, Ran and Ras.
FIGURE 7.
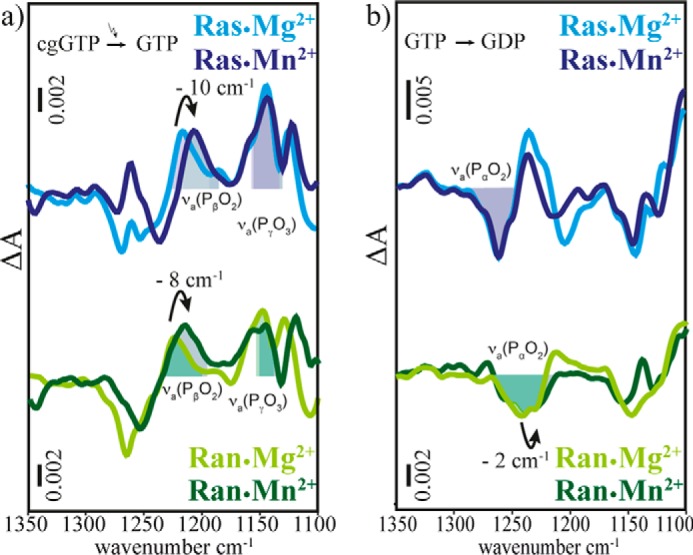
Scaled FTIR difference spectra of (a) photolysis aph and (b) hydrolysis ahyd of RasWT·GTP·Mg2+ (light blue), RasWT·GTP·Mn2+ (dark blue), RanWT·GTP·Mg2+·RanBD1 (light green), and RanWT·GTP·Mn2+·RanBD1 (dark green). The observed band shifts are summarized in Table 1.
TABLE 1.
Band shifts in Ras and Ran
FTIR spectroscopic measurements of Mg2+ to Mn2+ exchange in Ras and Ran show that the shifts between Ran·GTP·RanBD1·Mg2+ and Ran·GTP·RanBD1·Mn2+ are comparable to the shifts between Ras·GTP·Mg2+ and Ras·GTP·Mn2+. The corresponding IR difference spectra are shown in Fig. 7.
| νa(PαO2) | νa(PβO2) | νa(PγO3) | |
|---|---|---|---|
| Ran·GTP·RanBD1·Mg2+ | 1241 | 1224 | 1148 |
| Ran·GTP·RanBD1·Mn2+ | 1239 | 1216 | 1145 |
| Shift | −2 | −8 | −3 |
| Ras·GTP·Mg2+ | 1262 | 1216 | 1145 |
| Ras·GTP·Mn2+ | 1262 | 1206 | 1145 |
| Shift | 0 | −10 | 0 |
A New X-Ray Structure of Ran·RanBD1
To further investigate the discrepancy between the Mg2+ position in the structure of the (low resolution) Ran·Gpp(NH)p·RanBD1 complex (PDB code 1RRP) and the findings described above, we searched for crystal forms with better diffraction and managed to crystallize a complex of RanΔ191·GDP·BeF3·RanBD1 that diffracted to 2.4 Å. The GDP·BeF3 complexes of small GTPases are usually good mimics of the GTP state (38). Although the construct lacks the C terminus of Ran starting from residue 192, the overall arrangement of the Ranand RanBD1 domains in the structure of Ran(1–191)·GDP·BeF3·RanBD1(1155–1321) (called Ran·RanBD1 in the following) are very similar to PDB code 1RRP. The deletion in Ran has removed the helix that wraps around the RanBD1 domain together with the negatively charged 211DEDDDL216 motif that confers a large part of the binding affinity between Ran and the Ran binding domains (39). However, the remaining affinity is sufficiently large to ensure formation of the canonical complex. Concurrently, in the second complex of the asymmetric unit in the original Ran·RanBD1 structure (PDB code 1RRP), the C-terminal helix and 211DEDDDL216 motif (residue 187 to the end) are disordered, indicating a flexible position of this region. Lack of this C-terminal moiety might thus have helped to improve resolution of the Ran·RanBD1 complex crystals (2.4 Å in our structure compared with 2.96 Å in code 1RRP). Further effects of the missing Ran C terminus are discussed below.
When comparing our new Ran·GDP·BeF3·RanBD1 structure to code 1RRP, it was apparent that the magnesium ion was shifted by 1.9 Å (Fig. 8). In our higher resolution structure, the magnesium is almost exactly in the same position as in the Ras structures that usually have a very good resolution and quality (e.g. PDB codes 5P21 and 2RGE). In 1RRP, the magnesium ion occupies a position where other structures have a water molecule that coordinates the magnesium ion. Indeed, analysis of all available structures of Ran (Fig. 4b) and Ras (Fig. 4, a and d) reveals that the magnesium ions are found spread out along a line that connects two of the axial water molecules and the Mg2+. It appears that, especially in lower quality structures, the Mg ion often accidentally wanders off into the water positions during refinement. This is nicely seen in Fig. 4 where high resolution x-ray structure of uncomplexed GTPases (Fig. 4, second row) reveal a clear βγ-form, whereas in the usually lower resolved x-ray structures of complexed GTPases (Fig. 4, upper row) the Mg2+ coordination varies. In summary, this would suggest that the βγ-form is the correct magnesium position for Ras and Ran, and the αβγ-form would be a crystallographic artifact due to low resolution data.
FIGURE 8.
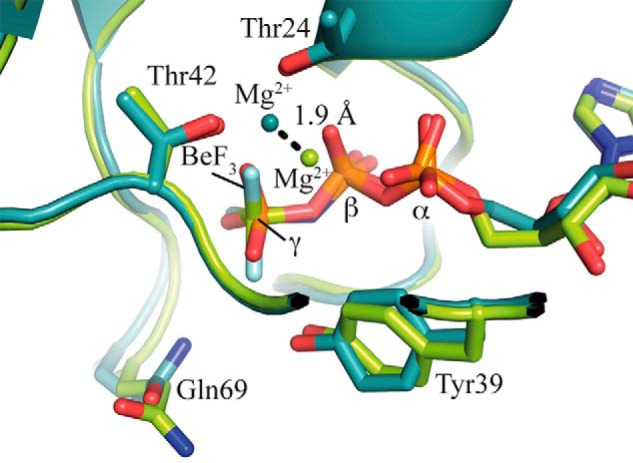
Superimposition of Ran·Gpp(NH)p·RanBD1 (PDB code 1RRP, light green) and RanΔ191·GDP·BeF3·RanBD1 (this work, PDB code 5CLL, cyan). The side chains of Thr-24, Tyr-39, Thr-42, and Gln-69 and the nucleotides and BeF3 are shown as sticks, the magnesium atoms as spheres. The distance between the two magnesium positions is approximately 1.9 Å, and the light green magnesium ion of PDB code 1RRP occupies the position of a water molecule in RanΔ191·GDP·BeF3·RanBD1 (not shown for clarity).
In our new Ran·RanBD1 structure, the beryllium fluoride superimposes nicely with the γ-phosphate oxygens of the Gpp(NH)p in 1RRP, and is only slightly rotated around an axis from the beryllium to the closest β-oxygen of the GDP (Fig. 8). The switch regions are, as expected, very similar to 1RRP, just the tip of switch II with Gln-69 is slightly more retracted from the γ-phosphate position, perhaps due to the slightly bulkier beryllium fluoride. As usual, switch II shows slightly elevated temperature factors. However, Gln-69 has a well defined electron density with distances of 5.4 and 5.73 Å between the GlnCδ atom and γ-phosphate.
Efforts to obtain a Ran·GDP·BeF3 structure without RanBD1 were not successful: two structures of Ran wild-type in different space groups were solved (supplemental Table S3) that grew in a GDP·BeF3 containing buffer, but contained Ran only in the canonical GDP form, no trace of density at the γ-phosphate position that could correspond to BeF3− was visible, corroborating the hypothesis that RanBD1 stabilizes the GTP conformation of Ran (14) and enables binding of BeF3−.
Influence of Thr-25 of Ran on the α-Phosphate Vibrations
If the Mg2+ coordination is not the cause for the spectral difference between Ras and Ran·RanBD1, what is the reason for the red shift of the α-phosphate vibration in Ran·RanBD1 relative to Ras, found in both experiment and theory? One explanation could be the additional hydrogen bond of the Thr-25 side chain to an α-phosphate oxygen in Ran. This hydrogen bond is not present in Ras because an alanine is in the corresponding position.
To prove this idea, we performed the FTIR spectroscopic measurements of the corresponding mutants H-RasA18T with an additional hydrogen bond to the α-phosphate and RanT25A that lacks this bond, respectively. The mutation A18T in N-Ras has been found in tumors with very good prognosis (40). Demunter and co-workers (40) measured the KD of the nucleotide for this mutant and found it unchanged compared with wild-type (supplemental Fig. S2 shows infrared difference spectra of Ras wild-type compared with RasA18T obtained via photolysis of caged GTP (cgGTP) bound as the nucleotide). In the photolysis spectrum bands facing downward (disappearing bands) are due to Ras·cgGTP and bands facing upward (appearing bands) are due to Ras·GTP. In the hydrolysis spectrum bands facing downward are due to Ras·GTP and the bands facing upward are due to Ras·GDP + Pi. All bands that do not change during the reaction are cancelled out in these difference spectra. It can be seen that the spectra of wild-type and mutant are very similar.
The corresponding experiments for Ran wild-type and RanT25A are shown in supplemental Fig. S3. In all experiments, the almost unchanged spectra in the amide region between 1300 and 1700 cm−1 indicate that the observed processes are not influenced by the mutations. Only in the region of the phosphate absorptions are some changes observed. This region is shown enlarged in Fig. 9. The asymmetric stretching vibrations of the β-phosphate (νa(PβO2)) at 1216 cm−1 and γ-phosphate (νa(PγO3)) at 1143 cm−1, observed as positive bands in the photolysis spectrum, are unchanged. At the same time the asymmetric stretching vibration of the α-phosphate (νa(PαO2)), observed as a negative band in the hydrolysis spectrum, is downshifted by 21 cm−1 upon the mutation A18T in Ras. Thus, this band is now almost in the position of the νa(PαO2) band of Ran. Conversely, the νa(PαO2) band of Ran shifts upwards by 19 cm−1 upon T25A mutation, close to its position in Ras. Similar to Ras, no significant changes are found for β- and γ-phosphate vibrations upon mutation of T25A in Ran. In summary, the phosphate bands of RasA18T resemble very precisely the corresponding spectra of Ran wild-type. Vice versa, the RanT25A spectra are very similar to Ras wild-type. Thus, the FTIR spectroscopic experiment reveals that the different α-phosphate band positions in Ran compared with Ras are not due to a different coordination of Mg2+ but due to the additional hydrogen bond of Thr-25 of Ran that is substituted by an alanine in Ras wild-type.
FIGURE 9.
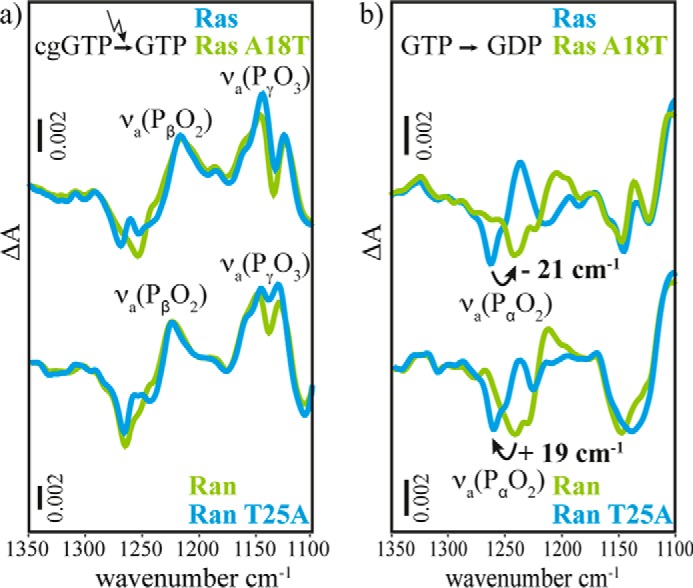
FTIR difference spectra of photolysis aph (a) and hydrolysis ahyd (b) of RasWT·GTP·Mg2+, RasA18T·GTP·Mg2+, RanWT·GTP·Mg2+·RanBD1, and RanT25A·GTP·Mg2+·RanBD1. The intensities are scaled to the intensity of WT proteins by multiplication with factors between 1.3 and 3. The νa(PβO2) vibrational mode does not shift, as observed best in the photolysis spectrum aph. The νa(PαO2) vibrational mode is downshifted 21 cm−1 by the A18T mutation in Ras and upshifted 19 cm−1 by the T25A mutation in Ran, observed best in the hydrolysis spectrum ahyd. The spectrum of RasA18T is very similar to the one of RanWT·RanBD1 and the spectrum of RanT25A·RanBD1 is very similar to the one of RasWT. By these mutations the Ras spectra can be transduced into Ran-like spectra and vice versa. The main spectral difference between Ras and Ran·RanBD1 are due to the additional hydrogen bond of the hydroxyl group of Thr-25 in Ran. The spectra of the entire mid-infrared region are shown as supplemental Fig. S2 (Ras) and supplemental Fig. S3 (Ran).
Complementary, we performed MD simulations on the corresponding mutated proteins, i.e. RanT25A·GTP·RanBD1 and H-RasA18T (supplemental Fig. S1, b and d), and compared the results with the respective wild-types. We calculated the IR spectra and predicted the band shifts between wild-type and mutation (Table 2). Such relative changes are much more precise than the absolute values because they are less dependent on the method used and extent of the quantum mechanically treated region as shown already for isotopic shifts in Ras (41). We calculated the βγ-form for both mutants but the αβγ-form only for the RanT25A mutant because in Ras the Mg2+ coordination is undisputed.
TABLE 2.
Comparison of the shifts of the asymmetrical phosphate stretching modes of the α- (νa(PαO2)) and β-phosphate (νa(PβO2)) group of GTP bound to Ras upon A18T mutation and Ran upon T25A mutation, respectively
The experimental value is compared to the calculated ones with both βγ- and αβγ-form. The corresponding IR difference spectra are shown in Fig. 5.
| Mg2+ coordination | RasA18T shift |
RanT25A shift |
|||||
|---|---|---|---|---|---|---|---|
| νa(PαO2) | νa(PβO2) | νa(PγO3) | νa(PαO2) | νa(PβO2) | νa(PγO3) | ||
| Experiment | −21 | 0 | 2 | 19 | 0 | −3 | |
| Simulation | βγ | −16 | −3 | 7 | 17 | 4 | 2 |
| Simulation | aβγ | 12 | 10 | −1 | |||
The results from the MD simulations underline the experimental FTIR findings. The theoretically predicted shifts induced by the mutation (Table 2) fit well to the measurement if the βγ-form is assumed in the MD simulations: only the α-phosphate band shifts significantly, by 17 cm−1 in theory and 19 cm−1 in the experiment. If the αβγ-form is assumed in RanT25A the calculated shifts deviate much stronger from the experiment: the νa(PαO2) vibrational mode shifts by only 12 cm−1 upon RanT25A mutation in the αβγ-form compared with 19 cm−1 in the experiment and more importantly a similar shift of 10 cm−1 was found for the νa(PβO2) vibrational mode, in contrast to the experiment, where it remains unchanged upon mutation.
Merging the results from the experimental and theoretical mutagenesis studies it clearly shows that the Mg2+ in Ran·GTP·RanBD1 is in the βγ-form like it is in Ras. All spectral changes of the phosphate vibrations between Ras and Ran can be explained by the additional hydrogen bond of the hydroxyl group of Thr-25.
Structural Details of the Triphosphate: Distance of β- and γ-Phosphate
If the differences in the hydrolysis rate of H-Ras and Ran·RanBD1 cannot be explained correctly by a different Mg2+ coordination, what else can be the reason? Now that we have dynamic computational models of Ras·GTP·Mg2+ and Ran·GTP·Mg2+·RanBD1 with atomic resolution, which are validated by the agreement of experimental and calculated infrared spectra, we can examine them with regard to structural parameters far beyond the resolution of x-ray crystallography and with GTP instead of being limited to analogues like Gpp(NH)p or GDP·BeF3. As shown above this high resolution is essential to elucidate catalytic effects. Fig. 10 shows the small structural differences between GTP in water, bound to Ran and bound to Ras. Interestingly, the distance of the γ-phosphate to the β-phosphate increases from 2.99 Å in water to 3.00 Å in Ran but to 3.03 Å in Ras. The faster the reaction the larger is this distance. Similarly the PβOPγ angle increases. This could be one cause for the slower GTPase reaction of Ran compared with Ras.
FIGURE 10.
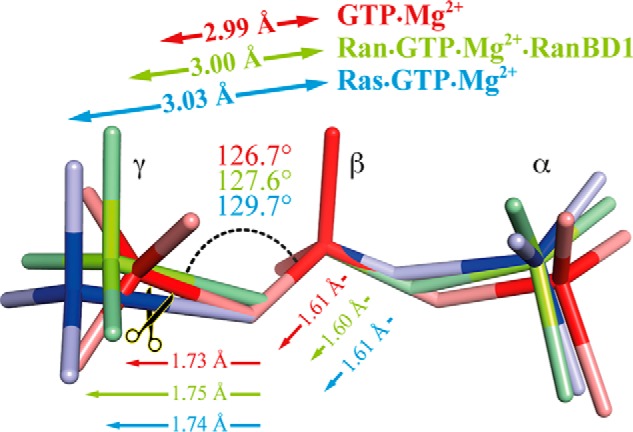
Structural changes of the triphosphate in water, bound to Ran, and Ras. The educt states are destabilized by the elongation of the distance between the γ- and β-phosphorous atom. The educt state in Ran·RanBD1 is less destabilized than in Ras so the energy barrier for hydrolysis in Ran is higher than in Ras. The values are the average over six 2.5-ps QM/MM simulations. Changes are exaggerated for clarity. The triphosphate structures in water and Ras are obtained by Rudack et al. (12).
Structure of RanY39A·RanBD1
Another observation hints to the influence of the side chain of Tyr-39. The Y39A mutant of Ran was found to catalyze GTP hydrolysis about 2 orders of magnitude faster compared with Ran wild-type, whereas in Ras the corresponding Y32A mutation has almost no influence on GTP hydrolysis compared with wild-type (Fig. 2) (11). We therefore tried to solve the structure of RanY39A to elucidate the reason of the faster hydrolysis. As for RanWT, we first tried to obtain a structure of RanY39A alone (without binding partner) in complex with GDP·BeF3 and solved two 1.75-Å crystal structures in different space groups (supplemental Table S3), but only GDP was observed in the active site, similar to wild-type Ran where a Ran binding domain is needed to force Ran·GDP into the GTP conformation and thus enables BeF3− binding. The region around the mutated Tyr-39 did not show any significant changes relative to wild-type, and the same is true for the remainder of the protein.
In contrast, we could crystallize the RanY39A·Gpp(NH)p·RanBD1 complex (RanY39A·RanBD1 in the following) and solve the structure with 3.2-Å resolution. The magnesium ions are located in a position very close to the Ran·RanBD1 and Ras positions, although at a resolution of 3.2 Å this is not very reliable as mentioned above.
Close to the active site, switch II shows a relatively large deviation of residues 70–75 relative to 1RRP and Ran·RanBD1. This is most likely due to crystal packing because a neighboring molecule is so close that it “flattens” the switch II helix and pushes Phe-72 and Gln-69 slightly forward in direction of the axis of the switch II helix. In contrast, the switch I region is almost completely unaltered compared with Ran wild-type except for the missing side chain of Tyr-39. As in the original Ran-RanBD1 structure (PDB code 1RRP) and as well as in our Ran-RanBD1 structure, the temperature factors for the switch II region are elevated, in the case of RanY39A·RanBD1 more than in the other two structures. Gln-69 has well defined electron density in both RanY39A chains with distances of 6.07 and 6.22 Å from the Cδ atom of Gln-69 to the γ-phosphate. Further details are given in the supplemental Section S1.
In summary, there are no significant changes in the active site that could directly explain the different reaction rates of Ran wild-type and Y39A. This prompted us to investigate the possibility that the wild-type of Ran has a slower hydrolysis rate than the mutant because the side chain of Tyr-39 blocks access of the catalytic glutamine to the γ-phosphate as described in the following.
The role of Tyr-39 and Gln-69
The side chain of Tyr-39 is found in a position close to the γ-phosphate in all known Ran·GTP complex structures, whereas structures of related GTPases show a greater variability of the tyrosine orientation, ranging from positions relatively similar to Ran (called “closed” in the following) to positions where the tyrosine side chain is far away from the γ-phosphate (“away” position). Interestingly, there seems to be a tendency that in small GTP-binding proteins with a fast GTP hydrolysis this tyrosine is preferentially in the away position according to x-ray and NMR data, and the mutation to, e.g. alanine, does not have a large effect on the hydrolysis rate (e.g. Cdc42 (42, 43)), whereas in GTPases with a slow hydrolysis the tyrosine is close to the γ-phosphate (e.g. Rab6a, which is approximately 4 times slower than Ran·GTP and 8 times slower than Ran·GTP·RanBD1) (44). Fig. 11a shows the side chains of Gln-69 and Tyr-39 in Ran·RanBD1 (PDB code 1RRP) compared with the structure of RanY39A·RanBD1 solved in this work. In the available x-ray structures of Ran wild-type, including the Ran·GDP·BeF3·RanBD1 structure solved here, Tyr-39 is contacting the γ-phosphate (Fig. 8). FTIR spectroscopic measurements on RanWT·GTP·RanBD1 and RanY39A·GTP·RanBD1 confirm a direct hydrogen bond between Tyr-39 and the γ-phosphate, which can be observed by the blue shift of the γ-phosphate vibration upon Y39A mutation by 12 cm−1 (Fig. 11b). This shift is of a similar magnitude as the one found for removing one hydrogen bond from the α-phosphate upon T25A mutation as discussed above. How does Tyr-39 hamper GTP hydrolysis? A possible explanation is the steric hindrance depicted in Fig. 11c. It is known that Gln-61(Ras)/Gln-69(Ran) is important for the hydrolysis reaction because it holds the nucleophilic water molecule in the attacking position (45, 46) and mutations of this residues are often found in tumors (47). The tyrosine side chain intrudes between the Gln and γ-phosphate. The distance of the side chain oxygen of Tyr-39 from a line between the GlnCδ and γ-phosphor atom can be taken as a measure for the steric hindrance. This distance is 2.6 Å in Ran, but 3.8 Å in Ras, which could also explain why in Ras this effect is not seen and, accordingly, the mutation Y32A has no significant effect on the hydrolysis rate (Fig. 2). As expected, this steric hindrance is removed in the Ran·RanGAP complex (46) where the corresponding distance is 3.3 Å, corroborating the idea that the tyrosine side chain interferes with the hydrolysis.
FIGURE 11.

Summary of the main results from, x-ray crystallography, FTIR spectroscopy, and MD simulations. In a the comparison of the x-ray structures of RanWT·Gpp(NH)p·RanBD1 (PDB code 1RRP) (green) and RanY39A·Gpp(NH)p (this work, PDB code 5CIW, dark green) is depicted. In RanWT Tyr-39 is fixed between the γ-phosphate and Gln-69. b, FTIR difference spectra of photolysis aph of RanWT·GTP·Mg2+·RanBD1 (light green), and RanY39A·GTP·Mg2+·RanBD1 (dark green). The intensity of the WT measurement is scaled by the factor 1.3. The shift of 12 cm−1 of the γ-phosphate band reveals the hydrogen bond between Tyr-39 and γ-phosphate, which is absent in RanY39A. c, comparison of the key residues in Ras and Ran involved in the steric hindrance as obtained from x-ray structures and the MD simulations. In Ran the oxygen of the Tyr side chain is much closer to a line from the Gln Cδ atom to the phosphor atom of the γ-phosphate, leading to the hypothesis that the Tyr-39-OH hinders the Gln-69 side chain from assuming an optimum catalytic position close to the γ-phosphate. This could be one of the explanations why the hydrolysis rate of Ran is slower that the one of Ras. All distances and the list of averaged PDB structures used to calculate the shown atom positions are given in supplemental Fig. S4. In d the averaged structure of the catalytic glutamine of the last 25 of 50-ns MD simulations of RanWT·GTP·RanBD1, RanY39A·GTP·RanBD1, RasWT·GTP, and RasY32A·GTP are represented. The colors display the simulated B-factor averaged over the side chain and backbone atoms (supplemental Table S2). Note that the simulated B-factors are obtained from the fluctuation during the MD simulation and are not directly comparable with the crystallographic B-factors.
MD Simulations of WT and Tyr Mutants
Complementary to the analysis of the crystal structures, we analyzed our MD simulations regarding the role of the discussed Tyr and Gln residues. The distance of the side chain oxygen of Tyr-39 from a line between Gln Cδ and the γ-phosphor atom averaged over the simulation trajectory is 2.2 Å for Ran and 3.6 Å for Ras, in agreement with the x-ray data (Fig. 11c), confirming the steric hindrance. Furthermore, the simulation of RanWT·RanBD1 exhibits a stable positioning of Tyr-39 at the γ-phosphate during a 50-ns simulation (Fig. 12). Gln-69 is trapped in an unreactive conformation by a direct hydrogen bond to Tyr-39. In contrast, the analogous simulation of Ras reveals a fluctuating position of the Gln-61 of Ras with a larger calculated B-factor. Thus Gln-61 can acquire the reactive conformation from time to time. The different flexibility of the Gln side chain is also seen in Fig. 11d, which shows the Gln color coded by the calculated B-factors. The Gln side chain of the slow hydrolyzing Ran wild-type has a very low B-factor, whereas the faster hydrolyzing RanY39A has a much higher B-factor. In the case of Ras, the Gln side chain is already flexible in Ras wild-type and unchanged in RasY32A (supplemental Table S2), in line with a rate more comparable with RanY39A for both Ras wild-type and Y32A mutant. A similar mechanism might also be present in various related slow Rab GTPases. Especially the slow hydrolyzing Rab6a has a tyrosine in a similar position as Tyr-39 in Ran (44).
FIGURE 12.
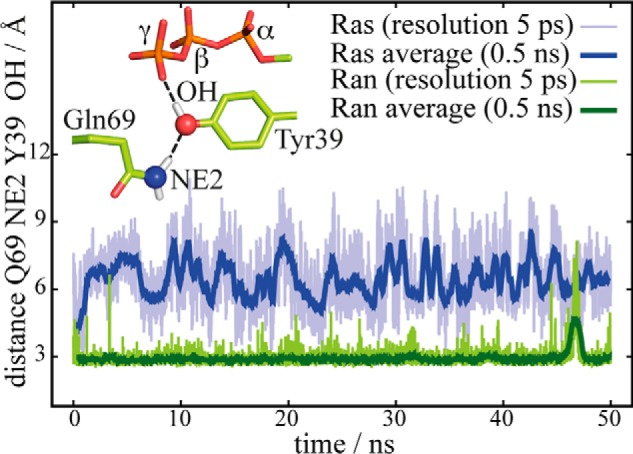
Fixation of Gln-69 in Ran. Depicted in green is the distance between the nitrogen atom of the Gln-69 and the oxygen atom of the Tyr-39 head group of Ran·RanBD1 compared with the distance between the nitrogen atom of the Gln-61 and the oxygen atom of the Tyr-32 head group of Ras. The side chain of Gln-69 of Ran is fixed in a hydrolytically unfavorable position by a stable hydrogen bond to the oxygen atom of the Tyr-39 head group, whereas the Gln-61 side chain in Ras is more flexible.
Conclusion
By a combination of theoretical IR spectroscopy and time-resolved FTIR spectroscopic measurements we have shown that Mg2+ is βγ coordinated in both proteins, Ras and Ran. An in-depth analysis of existing x-ray structures together with the new x-ray structures reported here confirm a clear βγ-coordination as well. Our structure of the Ran·GDP·BeF3·RanBD1 complex has an improved resolution compared with previous structures and thus allows a more accurate view of the nucleotide binding site. The red shift of the asymmetric α-phosphate vibration (νa(PαO2)) was shown to be due to a hydrogen bond between Thr-25 and the α-phosphate oxygens in Ran that do not exist in Ras, as proven by the agreement of theoretical and experimental mutagenesis studies. Thus, the differences in the hydrolysis rate between Ras and Ran cannot be explained by a difference in the Mg2+ coordination. We therefore suggested that Tyr-39 of Ran interfered with the catalytically favored position of Gln-69 relative to the γ-phosphate in Ran via steric hindrance, whereas in Ras the catalytic competent structure can be obtained more frequently by the more flexible and less hindered Gln-61 side chain because the Tyr-32 side chain in Ras is positioned slightly different. In agreement, the hydrolysis reaction of the Y39A mutant of Ran is accelerated by 2 orders of magnitude, whereas the analogue Y32A mutant of Ras is not accelerated (Fig. 2).
In addition, we present a proof of principle for the high sensitivity of the integration of theoretical and experimental IR spectroscopy. Here, we have predicted IR spectroscopic shifts of mutations, which afterward were verified by FTIR spectroscopic measurements. The difference of one hydrogen bond was resolved by both, theory and experiment. The integration of x-ray structure analysis, QM/MM simulations, and IR spectroscopy is a powerful tool to analyze the structure and dynamics of the catalytic center of a protein with atomic resolution. Only such atomic resolution provides the basis to understand protein catalysis.
Author Contributions
C. K., I. V., and K. G. designed the study. C. K., I. V., T. R., and S. J. wrote the paper. I. V., S. J., and S. B. expressed and purified proteins. T. R. performed the calculations, S. J. performed the FTIR measurements, I. V. and S. B. performed the x-ray crystallography.
Supplementary Material
Acknowledgments
We thank Dr. Frederick Großerüschkamp and Sara Stephan for the Ras·Mn2+ and RasY32A measurements, Claus Küpper for performing RanWT·RanBD1 MD simulations, and Stefan Tennigkeit for help in the purification of the Ran and Ras mutants. We acknowledge Dr. Jürgen Schlitter for fruitful discussions. X-ray data collection was performed at the Swiss Light Source, beamline X10SA, Paul Scherrer Institute, Villigen, Switzerland. We thank the beamline staff and Toni Meinhart, Bernhard Loll, Andrea Rocker, Wulf Blankenfeldt, Nils Schrader, and Sven Hennig for help with the data collection.
This work was supported by Deutsche Forschungsgemeinschaft Grant SFB 642. The authors declare no conflict of interest.

This article contains supplemental Tables S1–S3, Figs. S1–S4, and Sections S1–S3.
- PDB
- Protein Data Bank
- Gpp(NH)p
- guanosine 5′-(β,γ-imido)triphosphate
- QM/MM
- quantum mechanics/molecular mechanics
- cgGTP
- caged GTP
- NPE-GTP
- 1-(2-nitrophenyl)-ethylester of GTP
- pHP-GTP
- para-hydroxyphenacyl ester of GTP.
References
- 1. Wittinghofer A., Vetter I. R. (2011) Structure-function relationships of the G domain, a canonical switch motif. Annu. Rev. Biochem. 80, 943–971 [DOI] [PubMed] [Google Scholar]
- 2. Cherfils J., Zeghouf M. (2013) Regulation of Small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 93, 269–309 [DOI] [PubMed] [Google Scholar]
- 3. Ligeti E., Welti S., Scheffzek K. (2012) Inhibition and termination of physiological responses by GTPase activating proteins. Physiol. Rev. 92, 237–272 [DOI] [PubMed] [Google Scholar]
- 4. Cox A. D., Der C. J. (2010) Ras history: the saga continues. Small GTPases 1, 2–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stewart M. (2007) Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Biol. 8, 195–208 [DOI] [PubMed] [Google Scholar]
- 6. Jamali T., Jamali Y., Mehrbod M., Mofrad M. R. K. (2011) Nuclear pore complex. Int. Rev. Cell Mol. Biol. 287, 233–233-286 [DOI] [PubMed] [Google Scholar]
- 7. Görlich D., Kutay U. (1999) Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 15, 607–660 [DOI] [PubMed] [Google Scholar]
- 8. Kuhlmann J., Macara I., Wittinghofer A. (1997) Dynamic and equilibrium studies on the interaction of Ran with its effector, RanBP1. Biochemistry 36, 12027–12035 [DOI] [PubMed] [Google Scholar]
- 9. Görlich D., Seewald M. J., Ribbeck K. (2003) Characterization of Ran-driven cargo transport and the RanGTPase system by kinetic measurements and computer simulation. EMBO J. 22, 1088–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kelley J. B., Datta S., Snow C. J., Chatterjee M., Ni L., Spencer A., Yang C.-S., Cubeñas-Potts C., Matunis M. J., Paschal B. M. (2011) The defective nuclear lamina in Hutchinson-Gilford Progeria Syndrome disrupts the nucleocytoplasmic Ran gradient and inhibits nuclear localization of Ubc9. Mol. Cell. Biol. 31, 3378–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brucker S., Gerwert K., Kötting C. (2010) Tyr39 of Ran preserves the Ran·GTP gradient by inhibiting GTP hydrolysis. J. Mol. Biol. 401, 1–6 [DOI] [PubMed] [Google Scholar]
- 12. Rudack T., Xia F., Schlitter J., Kötting C., Gerwert K. (2012) The role of magnesium for geometry and charge in GTP hydrolysis, revealed by quantum mechanics/molecular mechanics simulations. Biophys. J. 103, 293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scheidig A. J., Burmester C., Goody R. S. (1999) The pre-hydrolysis state of p21(ras) in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure 7, 1311–1324 [DOI] [PubMed] [Google Scholar]
- 14. Vetter I. R., Nowak C., Nishimoto T., Kuhlmann J., Wittinghofer A. (1999) Structure of a Ran-binding domain complexed with Ran bound to a GTP analogue: implications for nuclear transport. Nature 398, 39–46 [DOI] [PubMed] [Google Scholar]
- 15. Kötting C., Gerwert K. (2004) Time-resolved FTIR studies provide activation free energy, activation enthalpy and activation entropy for GTPase reactions. Chem. Phys. 307, 227–232 [Google Scholar]
- 16. Rudack T., Xia F., Schlitter J., Kötting C., Gerwert K. (2012) Ras and GTPase-activating protein (GAP) drive GTP into a precatalytic state as revealed by combining FTIR and biomolecular simulations. Proc. Natl. Acad. Sci. U.S.A. 109, 15295–15300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Warshel A., Karplus M. (1972) Calculation of ground and excited state potential surfaces of conjugated molecules: I. formulation and parametrization. J. Am. Chem. Soc. 94, 5612–5625 [Google Scholar]
- 18. Mroginski M. A., Murgida D. H., Hildebrandt P. (2007) The chromophore structural changes during the photocycle of phytochrome: a combined resonance Raman and quantum chemical approach. Acc. Chem. Res. 40, 258–266 [DOI] [PubMed] [Google Scholar]
- 19. Steinbrecher T., Elstner M. (2013) QM and QM/MM simulations of proteins. in Biomolecular Simulations (Monticelli L., Salonen E., eds) pp. 91–124, Humana Press, Totowa, NJ [Google Scholar]
- 20. Yoo H.-Y., Boatz J. A., Helms V., McCammon J. A., Langhoff P. W. (2001) Chromophore protonation states and the proton shuttle mechanism in green fluorescent protein: inferences drawn from ab initio theoretical studies of chemical structures and vibrational spectra. J. Phys. Chem. B 105, 2850–2857 [Google Scholar]
- 21. Kubelka J., Keiderling T. A. (2001) Ab initio calculation of amide carbonyl stretch vibrational frequencies in solution with modified basis sets: 1. N-methyl acetamide. J. Phys. Chem. A 105, 10922–10928 [Google Scholar]
- 22. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 23. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. (2005) Gromacs: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 24. Groenhof G., Bouxin-Cademartory M., Hess B., De Visser S. P., Berendsen H. J., Olivucci M., Mark A. E., Robb M. A. (2004) Photoactivation of the photoactive yellow protein: why photon absorption triggers a trans-to-cis lsomerization of the chromophore in the protein. J. Am. Chem. Soc. 126, 4228–4233 [DOI] [PubMed] [Google Scholar]
- 25. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J. A., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A. G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al-Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., Pople J. A. (2003) Gaussian 03, Gaussian Inc., Pittsburgh PA [Google Scholar]
- 26. Höweler U. (2007) MAXIMOBY 9.07 and MOBY 3.0, CHEOPS Altenberge, Deutschland [Google Scholar]
- 27. Fischer N., Kandt C. (2011) Three ways in, one way out: water dynamics in the trans-membrane domains of the inner membrane translocase AcrB. Proteins 79, 2871–2885 [DOI] [PubMed] [Google Scholar]
- 28. Walker J. (1988) Photolabile 1-(2-nitrophenyl)ethyl phosphate esters of adenine nucleotide analogs: synthesis and mechanism of photolysis. J. Am. Chem. Soc. 110, 7170–7177 [Google Scholar]
- 29. Park C.-H., Givens R. S. (1997) New photoactivated protecting groups: 6. p-hydroxyphenacyl: a phototrigger for chemical and biochemical probes. J. Am. Chem. Soc. 119, 2453–2463 [Google Scholar]
- 30. Tucker J., Sczakiel G., Feuerstein J., John J., Goody R. S., Wittinghofer A. (1986) Expression of p21 proteins in Escherichia coli and stereochemistry of the nucleotide-binding site. EMBO J. 5, 1351–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hillig R. C., Renault L., Vetter I. R., Drell T. 4th, Wittinghofer A., Becker J. (1999) The crystal structure of rna1p: a new fold for a GTPase-activating protein. Mol. Cell 3, 781–791 [DOI] [PubMed] [Google Scholar]
- 32. John J., Sohmen R., Feuerstein J., Linke R., Wittinghofer A., Goody R. S. (1990) Kinetics of interaction of nucleotides with nucleotide-free H-ras p21. Biochemistry 29, 6058–6065 [DOI] [PubMed] [Google Scholar]
- 33. Cepus V., Scheidig A. J., Goody R. S., Gerwert K. (1998) Time-resolved FTIR studies of the GTPase reaction of H-ras p21 reveal a key role for the β-phosphate. Biochemistry 37, 10263–10271 [DOI] [PubMed] [Google Scholar]
- 34. Kötting C., Kallenbach A., Suveyzdis Y., Eichholz C., Gerwert K. (2007) Surface change of Ras enabling effector binding monitored in real time at atomic resolution. ChemBioChem. 8, 781–787 [DOI] [PubMed] [Google Scholar]
- 35. Hessling B., Souvignier G., Gerwert K. (1993) A model-independent approach to assigning bacteriorhodopsin's intramolecular reactions to photocycle intermediates. Biophys. J. 65, 1929–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Spoerner M., Hozsa C., Poetzl J. A., Reiss K., Ganser P., Geyer M., Kalbitzer H. R. (2010) Conformational states of human rat sarcoma (Ras) protein complexed with its natural ligand GTP and their role for effector interaction and GTP hydrolysis. J. Biol. Chem. 285, 39768–39778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rohrer M., Prisner T. F., Brügmann O., Käss H., Spoerner M., Wittinghofer A., Kalbitzer H. R. (2001) Structure of the metal-water complex in Ras center dot GDP studied by high-field EPR spectroscopy and P-31 NMR spectroscopy. Biochemistry 40, 1884–1889 [DOI] [PubMed] [Google Scholar]
- 38. Gavriljuk K., Gazdag E.-M., Itzen A., Kötting C., Goody R. S., Gerwert K. (2012) Catalytic mechanism of a mammalian Rab·RabGAP complex in atomic detail. Proc. Natl. Acad. Sci. U.S.A. 109, 21348–21353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Richards S. A., Lounsbury K. M., Macara I. G. (1995) The C terminus of the nuclear RAN/TC4 GTPase stabilizes the GDP-bound state and mediates interactions with RCC1, RAN-GAP, and HTF9A/RANBP1. J. Biol. Chem. 270, 14405–14411 [DOI] [PubMed] [Google Scholar]
- 40. Demunter A., Ahmadian M. R., Libbrecht L., Stas M., Baens M., Scheffzek K., Degreef H., De Wolf-Peeters C., van Den Oord J. J. (2001) A novel N-ras mutation in malignant melanoma is associated with excellent prognosis. Cancer Res. 61, 4916–4922 [PubMed] [Google Scholar]
- 41. Xia F., Rudack T., Kötting C., Schlitter J., Gerwert K. (2011) The specific vibrational modes of GTP in solution and bound to Ras: a detailed theoretical analysis by QM/MM simulations. Phys. Chem. Chem. Phys. 13, 21451–21460 [DOI] [PubMed] [Google Scholar]
- 42. Fidyk N. J., Cerione R. A. (2002) Understanding the catalytic mechanism of GTPase-activating proteins: demonstration of the importance of switch domain stabilization in the stimulation of GTP hydrolysis. Biochemistry 41, 15644–15653 [DOI] [PubMed] [Google Scholar]
- 43. Phillips M. J., Calero G., Chan B., Ramachandran S., Cerione R. A. (2008) Effector proteins exert an important influence on the signaling-active state of the small GTPase Cdc42. J. Biol. Chem. 283, 14153–14164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bergbrede T., Pylypenko O., Rak A., Alexandrov K. (2005) Structure of the extremely slow GTPase Rab6A in the GTP bound form at 1.8-Å resolution. J. Struct. Biol. 152, 235–238 [DOI] [PubMed] [Google Scholar]
- 45. Krengel U., Schlichting I., Scherer A., Schumann R., Frech M., John J., Kabsch W., Pai E. F., Wittinghofer A. (1990) Three-dimensional structures of H-ras p21 mutants: molecular basis for their inability to function as signal switch molecules. Cell 62, 539–548 [DOI] [PubMed] [Google Scholar]
- 46. Seewald M. J., Körner C., Wittinghofer A., Vetter I. R. (2002) RanGAP mediates GTP hydrolysis without an arginine finger. Nature 415, 662–666 [DOI] [PubMed] [Google Scholar]
- 47. Prior I. A., Lewis P. D., Mattos C. (2012) A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.