

Background: Helicases are implicated in fork remodeling.
Results: Mycobacterium tuberculosis RecG helicase/translocase remodels stalled replication forks.
Conclusion: M. tuberculosis RecG but not RuvAB or RecA is efficient in fork reversal activity.
Significance: This study identifies a fork remodeling enzyme and provides insights into fork restart mechanisms in M. tuberculosis.
Keywords: DNA damage, DNA helicase, DNA replication, genomic instability, Mycobacterium tuberculosis
Abstract
Aberrant DNA replication, defects in the protection, and restart of stalled replication forks are major causes of genome instability in all organisms. Replication fork reversal is emerging as an evolutionarily conserved physiological response for restart of stalled forks. Escherichia coli RecG, RuvAB, and RecA proteins have been shown to reverse the model replication fork structures in vitro. However, the pathways and the mechanisms by which Mycobacterium tuberculosis, a slow growing human pathogen, responds to different types of replication stress and DNA damage are unclear. Here, we show that M. tuberculosis RecG rescues E. coli ΔrecG cells from replicative stress. The purified M. tuberculosis RecG (MtRecG) and RuvAB (MtRuvAB) proteins catalyze fork reversal of model replication fork structures with and without a leading strand single-stranded DNA gap. Interestingly, single-stranded DNA-binding protein suppresses the MtRecG- and MtRuvAB-mediated fork reversal with substrates that contain lagging strand gap. Notably, our comparative studies with fork structures containing template damage and template switching mechanism of lesion bypass reveal that MtRecG but not MtRuvAB or MtRecA is proficient in driving the fork reversal. Finally, unlike MtRuvAB, we find that MtRecG drives efficient reversal of forks when fork structures are tightly bound by protein. These results provide direct evidence and valuable insights into the underlying mechanism of MtRecG-catalyzed replication fork remodeling and restart pathways in vivo.
Introduction
Accurate transmission of genetic information requires error-free duplication of chromosomal DNA during every round of cell division. Defects associated with replication are considered as a major source of genome instability in all organisms (1–3). Normal DNA replication is hampered when the fork encounters road blocks that have the potential to stall or collapse a replication fork. The types of lesions that potentially block replication fork include lesions on the template DNA, various secondary structures, R-loops, or DNA bound proteins (2, 4–7). Experimental evidence from various model organisms, including bacteria, suggests that stalled fork undergoes remodeling through replication fork reversal (RFR)5 to facilitate restart of stalled replication. RFR leads to the annealing of nascent strands to generate a four-stranded structure called “chicken foot” that resembles a Holliday junction (HJ) (4, 5, 8, 9).
Studies carried out over several decades propose various mechanisms of replication restart upon fork reversal. In Escherichia coli, recombination-dependent and -independent pathways of replication restart have been identified (5, 10–14). Cleavage of reversed forks results in the generation of DNA double strand breaks (DSBs), and processing of DSBs by nucleases and subsequently by recombination enzymes leads to the formation of D-loops. E. coli PriA binds to D-loops and facilitates replisome reassembly to restart the replication in an origin (oriC)-independent fashion (15). In addition, recombination-mediated replication restart can occur before the cleavage of the reversed fork. Degradation of nascent duplexes by exonucleases can generate ssDNA substrate for initiation of recombination followed by D-loop formation. Reassembly of the replisome at the D-loop can generate an active replication fork with two upstream HJs (5, 10, 15). However, replication can resume only after resolution/dissolution of HJs by endonucleases or helicases/topoisomerases. RFR can also provide a template switching mechanism for replication restart in a recombination-independent manner. When there is a template lesion and if it is not bypassed by repriming, replication forks continue to stall and may be refractory to NER/BER because of the lack of complementary strand. In this scenario, upon fork reversal, nascent lagging strand can serve as a template for DNA synthesis from the 3′ end of nascent leading strand. This template switching reaction may allow bypass of the lesion after restoration of the reversed fork to resume the replication. The lesion on the template strand can be repaired later by NER/BER machinery (5, 10, 15). In an alternative mechanism, RFR at the site of template lesion allows reannealing of the parent strands such that NER/BER factors can eliminate the lesion. Later, reversal of the regressed fork can facilitate reassembly of the replisome to restart the replication. When there are road blocks created due to DNA-bound proteins, RFR can provide a mechanism for fork restoration. In addition, action of nucleases on the nascent duplex DNA at the reversed fork restores the fork structure to which the replisome can be reassembled to resume the replication (5, 10, 15).
Investigations into the search for enzymes that catalyze RFR have led to the identification of various helicases/translocases having the potential to remodel stalled replication forks (4, 5, 10). A number of biochemical studies show that E. coli RecG helicase/translocase (10, 16–20) and RuvAB proteins that catalyze HJ branch migration promote RFR (21–23). The recG or ruv mutations in E. coli lead to UV light-induced sensitivity (24, 25), implying that RecG and RuvAB participate in the processing of replication forks arrested by UV light-induced template damage. Interestingly, the combined mutations of recG with ruv lead to exacerbated sensitivity of E. coli cells to UV light and severe growth defects compared with either recG or ruv single mutants (24–27), indicating the likely overlapping functions of RecG and RuvAB in processing the stalled replication forks. The bacterial RecA and its orthologs in eukaryotes play a central role in DNA repair and recombination (28–38). In addition to RecG and RuvAB, E. coli RecA has been implicated in mediating RFR after UV irradiation (39) and denaturation of thermosensitive DnaB replicative helicase (40). A biochemical study by Robu et al. (41) demonstrated that RecA can catalyze reversal of model replication forks. Although these proteins are known to act on stalled forks, the participation of a specific enzyme varies depending on the type of damage that is used to arrest the replication fork (12, 42). Moreover, model substrates that were used to analyze the fork reversal activities of RecG, RuvAB, or RecA did not have the lesion on the leading strand template DNA. Recently, RFR activities of E. coli RecG and RuvAB have been demonstrated with model substrates containing template damage (22). Using a similar approach, a parallel study showed that SSB prevents RecA-catalyzed RFR (43). However, further studies are required to gain insights into the mechanism of RecG-, RuvAB-, or RecA-mediated fork reversal with the model substrates that mimic the type of template damage expected to occur in in vivo.
Mycobacterium tuberculosis, the causative agent of tuberculosis, is responsible for the highest number of deaths worldwide by a single infectious agent (44). M. tuberculosis cells are extremely slow growing with a generation time of ∼24 h. In the host macrophage, this pathogen is exposed to reactive oxygen species (ROS) and reactive nitrogen intermediates (RNI), which are known to cause various types of DNA damage (45, 46). Moreover, M. tuberculosis genome is GC-rich (65%) and hence is susceptible to excessive oxidative as well as alkylating damage (47, 48). In addition, because of the GC-rich genome, this pathogen is known to contain >10,000 G-quadruplex-forming motifs (49, 50). Such secondary structures as well as oxidative and alkylating lesions induced by ROS and RNI are known to cause replicative stress. Under these conditions, it is unclear how this pathogen deals with circumventing replicative stress. Moreover, the enzymes that participate in RFR and restart of stalled/collapsed replication forks in this pathogen are obscure. Although orthologs of E. coli RecG, RuvAB, and RecA are conserved in this pathogen (51, 52), their role in remodeling the stalled replication forks and subsequent restart of stalled forks is elusive. Our previous study showed that M. tuberculosis RecG catalyzes unwinding of DNA repair/recombination substrates and is able to rescue E. coli ΔrecG cells from DNA damage-induced toxicity (53). Another report demonstrated that M. tuberculosis RuvAB catalyzes fork reversal of model replication forks (54). However, the mechanism underlying RFR of DNA substrates having lesions on the template DNA has not been addressed. The template lesions are expected to occur in the latently infected mycobacteria or during its active replication in the host macrophage cells. In this study, we show that M. tuberculosis RecG (MtRecG) rescues E. coli ΔrecG cells from HU-induced replicative stress. Notably, our comparative studies with fork structures containing leading strand template lesion and with an in vitro template switching mechanism of lesion bypass reveal that MtRecG but not MtRuvAB or MtRecA is proficient in driving the fork reversal. Finally, we demonstrate that MtRecG drives efficient reversal of forks when fork structures are bound by protein. Our data suggest that MtRecG might participate in the replication restart pathways via reversal of stalled forks and provide insights into the possible mechanisms of replication restart in mycobacteria.
Experimental Procedures
Reagents
Fine chemicals were purchased from Sigma and GE Healthcare. T4 DNA ligase, polymerase I Klenow fragment, and restriction enzymes were purchased from New England Biolabs. T4 polynucleotide kinase, Gene Ruler 50-bp DNA ladder (SM0371), and Gene Ruler Ultra low range DNA ladder (SM01211) were purchased from Thermo Scientific. Wheat germ topoisomerase I was from Promega. [γ-32P]ATP was purchased from Bhabha Atomic Research Center, Mumbai, India.
Proteins, Plasmids, and DNA
MtRecG and K321A MtRecG proteins were purified as described previously (53). MtRuvA (55), MtRuvB (54), MtRecA (56), EcSSB (57), and MtSSB (58) proteins were purified as described. KpnI H149A mutant enzyme was purified as described previously (59). All oligonucleotides used in this study were purchased from Sigma, and the oligonucleotide containing iso-cytosine (iso-C) modification as used in an earlier report (60) was purchased from IDT, Inc. The sequences of oligonucleotides are shown in Table 1, and the structures of substrates and respective oligonucleotides used to assemble these substrates are shown in Table 2. Oligonucleotides were further purified on denaturing PAGE, estimated, and stored in −20 °C.
TABLE 1.
Sequence of oligonucleotides used in this study
| Oligonucleotide | Length | Sequences 5′–3′ | Ref. or source |
|---|---|---|---|
| a | 50 | GTCGGATCCTCTAGACAGCTCCATGATCACTGGCACTGGTAGAATTCGGC | 17 |
| b | 50 | CAACGTCATAGACGATTACATTGCTACATGGAGCTGTCTAGAGGATCCGA | 17 |
| c | 26 | TGCCGAATTCTACCAGTGCCAGTGAT | 17 |
| d | 25 | TAGCAATGTAATCGTCTATGACGTT | 17 |
| A | 94 | TCCTTTTGATAAGAGGTCATTTTTGCGGATGGCTTAGAGCTTAATTGCTAAATCTGGTGCTGTAGGTCAACATGTTGTAAATATGCAGCTAAAG | 54 |
| B | 94 | CTTTAGCTGCATATTTACAACATGTTGACCTACAGCACCAGATTCAGCAATTAAGCTCTAAGCCATCCGCAAAAATGACCTCTTATCAAAAGGA | 54 |
| C | 64 | CTTTAGCTGCATATTTACAACATGTTGACCTACAGCACCAGATTTAGCAATTAAGCTCTAAGCC | This study |
| D | 64 | GGCTTAGAGCTTAATTGCTAAATCTGGTGCTGTAGGTCAACATGTTGTAAATATGCAGCTAAAG | This study |
| E | 34 | CTTTAGCTGCATATTTACAACATGTTGACCTACA | This study |
| F | 34 | TGTAGGTCAACATGTTGTAAATATGCAGCTAAAG | This study |
| GC | 81 | CTTTAGCTGCATATTTACAACATGTTGACCTTCAGTA/isodC/AATCTGCTCTGATGCCGCATAGTGTCATGCCAGAGCTTTGTAC | 60 |
| G | 81 | CTTTAGCTGCATATTTACAACATGTTGACCTTCAGTACAATCTGCTCTGATGCCGCATAGTGTCATGCCAGAGCTTTGTAC | This study |
| H | 81 | GTACAAAGCTCTGGCATGACACTATGCGGCATCAGAGCAGATTGTACTAAAGGTCAACATGTTGTAAATATGCAGCTAAAG | This study |
| I | 43 | GTACAAAGCTCTGGCATGACACTATGCGGCATCAGAGCAGATT | This study |
| J | 48 | AGTACAATCTGCTCTGATGCCGCATAGTGTCATGCCAGAGCTTTGTAC | This study |
| K | 81 | CGGGTGTCGGGGCGCATGACACTATGCGGCATCAGAGCAGATTGTACTGAAGGTCAACATGTTGTAAATATGCAGCTAAAG | 60 |
| L | 30 | GTACAAAGCTCTGGCATGATACTATGCGGC | 60 |
| M | 50 | TCAGTACAATCTGCTCTGATGCCGCATAGTATCATGCGCCCCGACACCCG | 60 |
| N | 75 | AGCTACCATGCCTGCCTCAAGGTACCGTAATATGCCTACACTGGAGTACCGGAGCATCGTCGTGACTGGGAAAAC | This study |
| O | 75 | GTTTTCCCAGTCACGACGATGCTCCGGTACTCCAGTGTAGGCATATTACGAATTCTTGAGGCAGGCATGGTAGCT | This study |
| P | 40 | TGTAGGCATATTACGGTACCTTGAGGCAGGCATGGTAGCT | This study |
| Q | 40 | AGCTACCATGCCTGCCTCAAGGTACCGTAATATGCCTACA | This study |
| R | 40 | AATATGCCTACACTGGAGTACCGGAGCATCGTCGTGACTGGGAAAAC | This study |
| S | 40 | GTTTTCCCAGTCACGACGATGCTCCGGTACTCCAGTGTAGGCATATT | This study |
TABLE 2.
DNA substrates used in this study
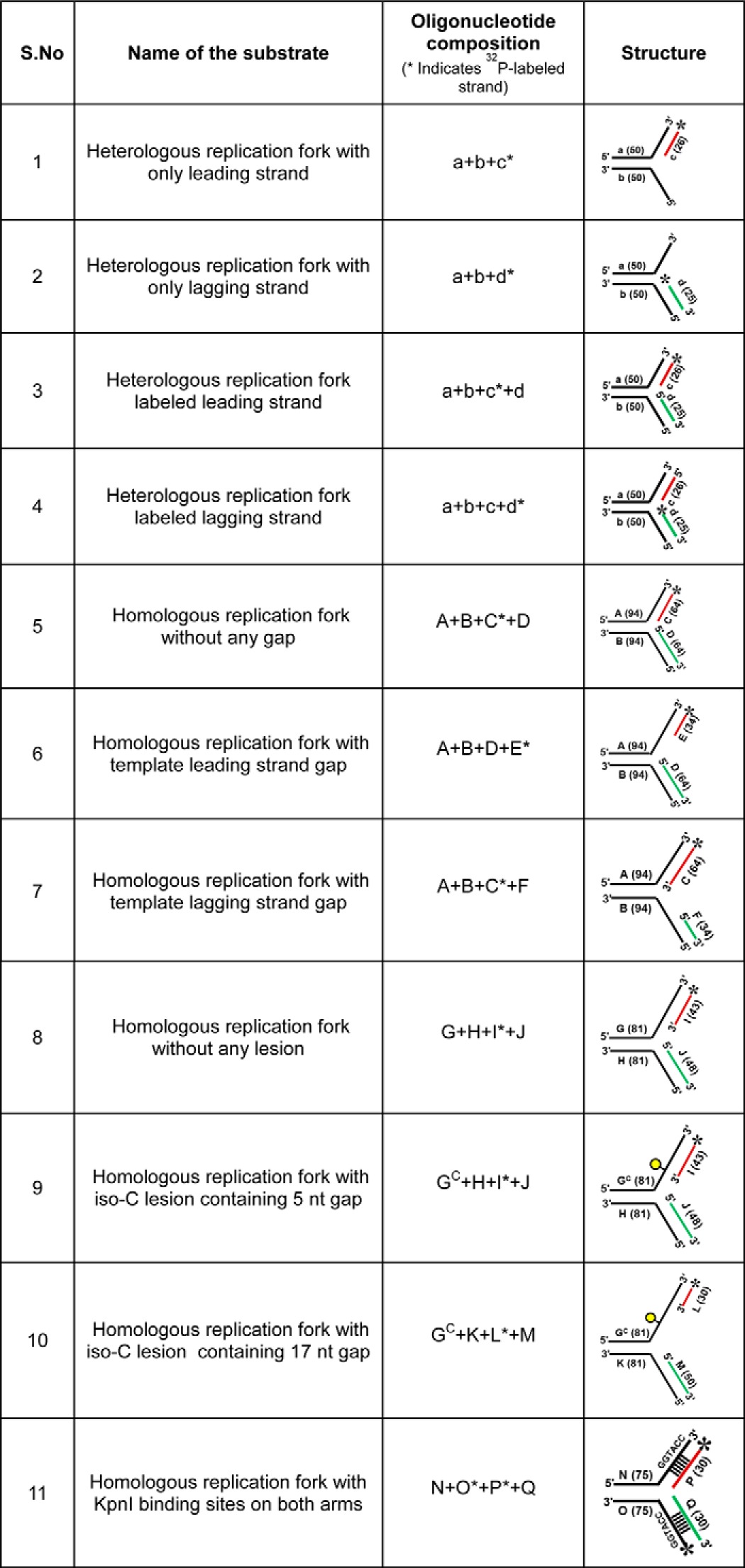
Construction of DNA Substrates
Heterologous replication fork substrates were prepared as described previously (53). Briefly, labeled oligonucleotides were annealed with unlabeled oligonucleotides in a molar ratio of 1:1.5 in an annealing buffer containing 1× SSC (150 mm sodium chloride and 15 mm sodium citrate). The reaction mixture was incubated 95 °C for 5 min and then cooled slowly in the thermal cycler. The assembled substrates were purified by resolving on 8% native polyacrylamide gel in 1× TBE buffer. Identities of assembled substrates were tested by mobility shift and restriction digestion. Homologous substrates were assembled as described previously (54) with few modifications. In two-step annealing reactions, the first labeled leading strand was annealed with unlabeled template strand (Table 2) in an annealing buffer containing 1× SSC, 5 mm MgCl2, and 50 mm KCl. In an identical annealing reaction, lagging strands and the respective complementary strands were taken to anneal. Reactions were denatured 95 °C for 5 min and then cooled slowly in the thermal cycler. Annealed products were mixed together and incubated at 37 °C for 1 h and later maintained at room temperature for 1 h. Finally, annealed substrates were loaded onto 6% native polyacrylamide gel, separated in a buffer containing 1× Tris borate buffer containing 50 mm KCl and 2 mm MgCl2, and visualized by autoradiography. Identities of assembled substrates were tested by mobility shift and restriction digestion. Plasmid-based replication fork substrates were prepared as described previously (61). Briefly, gapped pG46-B plasmid was radiolabeled with 32P and annealed with gapped pG68-A in buffer containing 50 mm Tris-HCl (pH 7.5) and 10 mm MgCl2. Reaction was incubated at 80 °C for 15 min and then gradually cooled to room temperature. Reactions were further treated with wheat germ topoisomerase I in a reaction buffer containing 50 mm Tris-HCl (pH 7.5), 50 mm NaCl, 0.1 mm EDTA, 1 mm DTT, and 10% glycerol at 37 °C for 30 min. Finally, assembled substrates were resolved on 0.8% agarose gel using 0.5× TBE and purified. For preparation of joint molecule substrate, radiolabeled gapped pG46-B plasmid was annealed with XhoI-digested gapped pG68-A plasmid in an annealing buffer containing 50 mm Tris-HCl (pH 7.5) and 10 mm MgCl2 at 55 °C for 15 min.
DNA Damage Sensitivity Assays
Sensitivity to HU was determined by growing wild-type E. coli AB1157 and N3793 (ΔrecG) strains carrying either vector alone or pGS772 E. coli recG plasmid or pGS772MtrecG plasmid (subcloned from pET28aMtrecG) at 37 °C to an A600 of 0.6 in LB broth containing 100 μg/ml ampicillin. Appropriate dilutions were made in M9 minimal media, and aliquots were either plated (∼ 300 cells for quantitative measurement) or spotted onto LB agar plates (for qualitative assay) supplemented with 100 μg/ml ampicillin and indicated concentrations of HU. Following overnight incubation in the dark at 37 °C, colonies were counted, and images were acquired using FUJIFILM LAS3000 gel documentation system.
Fork Reversal Assays
Plasmid-based fork reversal assays were performed as described previously (61). Briefly, 2 nm replication fork or joint molecule substrates were incubated in a buffer containing 50 mm Tris-HCl (pH 7.5), 1 mm DTT, 10 mm KCl, 5 mm MgCl2, 1 mm ATP, 100 μg/ml BSA. Reactions were initiated by addition of the indicated concentration of MtRecG or K321A MtRecG and incubated at 37 °C for 30 min. Reactions were terminated by addition of excess ATPγS and subsequently digested with indicated restriction enzymes for 30 min at 37 °C. Reactions were deproteinized by incubation with SDS (0.4%) and proteinase K (0.5 μg/μl) at 37 °C for 30 min. Following addition of 2 μl of gel loading dye (0.1% (w/v) of bromphenol blue and xylene cyanol in 20% glycerol), samples were electrophoresed on 10% polyacrylamide gel in 1× TBE (90 mm Tris borate buffer (pH 8.3) containing 1.0 mm EDTA) at 150 V for 2 h at room temperature. Gels were dried and exposed to a phosphorimaging screen and visualized using Fuji FLA-5000 phosphorimager. Reaction products were quantified using FUJIFILM MultiGauge software (version 3.0), and the data were subjected to nonlinear regression analysis and plotted using GraphPad Prism (version 5.0).
Homologous replication fork reversal assays were performed in a reaction mixture (10 μl) containing 50 mm Tris-HCl (pH 7.5), 1 mm DTT, 10 mm KCl, 5 mm MgCl2, 1 mm ATP, 100 μg/ml BSA, and 10 nm of indicated 32P-labeled homologous replication fork substrate. Reactions were initiated with the addition of MtRecG, MtRuvAB, or MtRecA and incubated at 37 °C for 30 min followed by addition of termination buffer containing 25 mm EDTA, 0.4% SDS, 0.5 mg/ml proteinase K. After incubation for 15 min at 37 °C, 1 μl of gel loading dye (0.1% (w/v) of bromphenol blue and xylene cyanol in 20% glycerol) was added, and samples were resolved on 8% polyacrylamide gel in 1× TBE (90 mm Tris borate buffer (pH 8.3) containing 1.0 mm EDTA) at 135 V for 1.5 h at room temperature. Gels were dried and exposed to phosphorimaging screen and visualized using a Fuji FLA-5000 phosphorimager. Reaction products were quantified using FUJIFILM MultiGauge software (version 3.0), and data were subjected to nonlinear regression analysis and plotted using GraphPad Prism (version 5.0). For reactions in the presence of Mt/EcSSB,10 nm homologous replication fork substrate containing a 50-nt gap was incubated with increasing concentrations of Mt/EcSSB in a buffer containing 50 mm Tris-HCl (pH 7.5), 1 mm DTT, 10 mm KCl, 5 mm MgCl2, 1 mm ATP, and 100 μg/ml BSA for 15 min at 37 °C. Later, fork reversal reaction was initiated by addition of MtRecG or MtRuvAB, and incubation was continued for 15 min at 37 °C. Reaction products were resolved and analyzed as described above. Fork unwinding with heterologous substrates was carried out as described previously (53).
In Vitro DNA Lesion Bypass by Template Switching Assay
Stalled replication forks containing iso-C lesion on leading strand template DNA was generated as described above (60). 10 nm homologous replication fork substrate was incubated in a reaction buffer containing 50 mm Tris-HCl (pH 7.5), 1 mm DTT, 10 mm KCl, 5 mm MgCl2, 1 mm ATP, 100 μg/ml BSA, and 25 μm dNTP mix. Reactions were initiated by addition of DNA polymerase I Klenow fragment (0.05 units), and incubation was continued for 10 min at 37 °C. Later, the indicated proteins were added, and incubation was continued for an additional 20 min. Reactions were terminated by addition of formamide containing 0.1% bromphenol blue and 0.1% xylene cyanol, incubated for 5 min at 95 °C, and analyzed by 15% denaturing PAGE.
Electrophoretic Mobility Shift Assays
Reactions were performed as described previously (53). Briefly, 0.5 nm heterologous or 10 nm homologous replication fork substrates were incubated in a binding buffer containing 25 mm Tris-HCl (pH 7.5), 5 mm MgCl2, 10 mm KCl, 1 mm DTT, 100 μg/ml BSA, and 3% glycerol. Reactions were started with an addition of increasing concentrations of protein and terminated with addition of 1× DNA loading dye (0.1% (w/v) bromphenol blue and xylene cyanol in 20% glycerol). Samples were resolved on 6% polyacrylamide gel in 0.25 × TBE (22.5 mm Tris borate buffer (pH 8.3) and 0.25 mm EDTA) at 150 V for 4 h at 4 °C. Gels were dried, and the DNA-protein complexes were visualized using a Fuji FLA-5000 phosphorimager. Reaction products were quantified using FUJIFILM MultiGauge software (version 3.0), data were subjected to nonlinear regression analysis and plotted using GraphPad Prism (version 5.0).
Homologous Replication Fork Reversal Assay in the Presence of H149A KpnI
Homologous replication fork substrate (10 nm) containing the KpnI-binding site was incubated with 100 nm H149A KpnI in a buffer containing 50 mm Tris-HCl (pH 7.5), 1 mm DTT, 10 mm KCl, 5 mm MgCl2, 1 mm ATP, and 100 μg/ml BSA for 15 min at 37 °C. Later, fork reversal reaction was initiated by addition of MtRecG or MtRuvAB, and incubation was continued for 15 min at 37 °C. Reaction products were resolved and analyzed as described above.
Results
M. tuberculosis recG Rescues E. coli ΔrecG Cells from Replicative Stress
Genotoxic agents such as MMS and UV light induce template damage and lead to fork stalling when the replisome encounters lesions on the template DNA. E. coli ΔrecG cells exhibit sensitivity to MMS and UV light (24, 25), suggesting that RecG participates in replication fork repair and restart of stalled forks. Previously, we have shown that M. tuberculosis recG rescues E. coli ΔrecG cells from replicative stress induced by UV light and MMS (53). Here, we extended our studies to test whether E. coli ΔrecG cells exhibit hypersensitivity to HU, which depletes dNTPs, thereby causing replication arrest. E. coli ΔrecG cells (N3793) transformed with empty vector showed a modest growth defect in the absence of HU compared with the wild-type E. coli (AB1157) (Fig. 1A). When examined with HU, the surviving fraction of E. coli ΔrecG cells declined with increasing doses of HU, and this defect could be rescued by expression of EcRecG (Fig. 1, B and C). Strikingly, ectopic expression of MtRecG was able to significantly rescue the hypersensitivity of E. coli ΔrecG cells from HU-induced replication stress (Fig. 1, B and C). These data are in agreement with our previous study (53) and imply that M. tuberculosis RecG might have a similar function as that of E. coli RecG in replication fork repair and restart of stalled forks.
FIGURE 1.
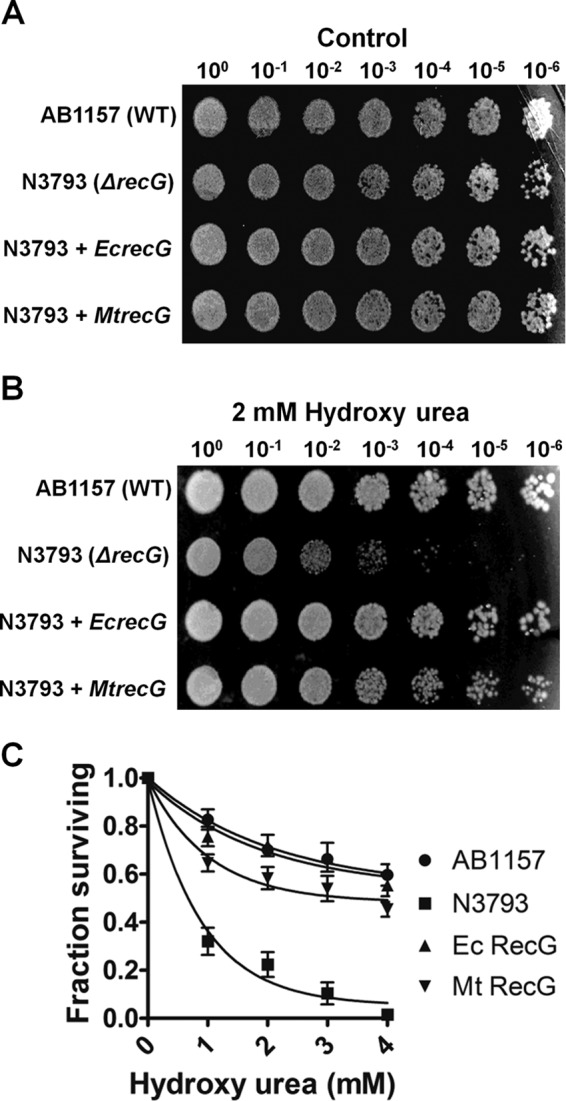
M. tuberculosis recG functionally complements E. coli ΔrecG cells from replicative stress induced by HU. Survival of wild-type E. coli (AB1157) and E. coli ΔrecG (N3793) mutant strains were transformed with either empty vector or plasmids encoding E. coli recG and M. tuberculosis recG without (A) and with HU treatment (B). For qualitative analysis, the indicated dilutions of cells were plated and incubated overnight. The images of the plates were captured by the gel documentation system. For quantitative measurement, serially diluted cells were plated and counted after overnight incubation (C). The error bars represent standard error of the mean (S.E.) from three independent experiments.
M. tuberculosis RecG Promotes Regression of Model Replication Fork
In response to replication stress, replisome stalls and fork reversal catalyzed by helicases/translocases are a prominent mechanism by which replication restarts (4, 5, 10, 62). To test whether purified M. tuberculosis RecG helicase/translocase can drive RFR, we used a previously characterized replication fork substrate (61). The substrates were prepared as described previously using two plasmids (pG68-A and pG46-B) that contain 68- and 46-nt gaps (61). Annealing of these two plasmids followed by incubation with topoisomerase I results in a replication fork structure with a plectonemic joint (Fig. 2A). Because of the differences in the gap of these two plasmids, it provides a 14-nt gap on the leading strand template that can arise due to uncoupling of leading and lagging strand synthesis. Fork reversal by RecG is expected to transfer daughter strand chromosome-containing restriction endonuclease sites into the regressed arm of the fork (Fig. 2B). Thus, fork regression can be monitored by the generation of linear fragments after incubation with restriction endonucleases. To test the ability of M. tuberculosis RecG (MtRecG)-mediated reversal of the model replication fork, we incubated the substrate with 10 nm MtRecG, followed by incubation with AvrII, BamHI, EcoRI, PvuII, and AlwnI restriction endonucleases. The products were resolved on 10% polyacrylamide gel and analyzed as described under “Experimental Procedures.” As can be seen in Fig. 2C, release of linear fragments (36–266 bp) corresponding to the 5′ end of the reversed arm was evident in the reactions that contained MtRecG, suggesting that MtRecG can catalyze reversal of the model replication fork. The presence of 266-bp PvuII product suggests that MtRecG can promote fork regression efficiently for distances of >250 bp. The Walker motif MtRecG K321A mutant was devoid of fork reversal activity (Fig. 2C), indicating that fork reversal catalyzed by MtRecG requires ATP hydrolysis.
FIGURE 2.

M. tuberculosis RecG catalyzes efficient fork reversal with plasmid-based replication fork structure. A, schematic representation of assembly of plasmid-based replication fork structure. Annealing of the 5′-phosphate-labeled pG46-B with pG68-A followed by topoisomerase I treatment results in catenation of the two plasmids through the plectonemic joint. B, schematic diagram of RecG-mediated fork reversal of the substrate shown in A. Fork reversal activity leads to annealing of the two strands (regressed arm) that contain restriction endonuclease sites (A, AvrII; B, BamHI; E, EcoRI; P, PvuII and N, AlwnI). Expected products of indicated restriction endonucleases are shown below. C, analysis of fork reversal products with wild-type MtRecG and ATPase-deficient MtRecG. Reaction mixture contained 2 nm replication fork substrate in a reaction buffer containing 1 mm ATP and 5 mm MgCl2 in the absence (lanes 1–5) or presence of either 10 nm K321A MtRecG (lanes 6–10) or presence of 10 nm MtRecG (lanes 11–15). Reactions were stopped with addition of ATPγS and subsequently digested with indicated restriction enzymes. Products were resolved on 10% native polyacrylamide gel and visualized by autoradiography. Lane 16, ultra-low range (10–300 bp) DNA ladder.
M. tuberculosis RecG and RuvAB Promotes Reversal of Homologous Replication Fork Structures
E. coli RecG and RuvAB proteins are known to catalyze regression of model replication forks (16, 17, 21–23). To gain insights into the RFR activities of M. tuberculosis RecG in comparison with M. tuberculosis RuvAB (MtRuvAB), we prepared oligonucleotide-derived homologous replication fork substrates as described under “Experimental Procedures.” RFR activity results in the formation of HJ (chicken foot) structure, and subsequent branch migration of these structures results in the generation of two linear dsDNA products (Fig. 3A). Radiolabeled substrates were incubated with increasing concentrations of M. tuberculosis RecG and RuvAB, and the products were resolved on 8% polyacrylamide gel and visualized as described under “Experimental Procedures.” Incubation with increasing concentrations of MtRecG (0.005–1 nm) resulted in an increase in the reversal of forks to generate linear dsDNA products (Fig. 3B). Quantitative data reveal that MtRecG was able to reverse up to ∼80% substrates (Fig. 3C). MtRecG activity remained unchanged even at higher concentrations of protein (up to 5 nm) (data not shown). In a parallel reaction, we examined the ability of MtRuvAB to reverse the replication fork structures by incubating with increasing concentrations of MtRuvAB (50 nm RuvA and 25–300 nm RuvB). At concentrations between 100 and 300 nm MtRuvB, the MtRuvAB complex was able to exhibit optimal (∼65%) reversal of model replication fork structures (Fig. 3, B and C), and this activity did not alter with a further increase in MtRuvAB (data not shown). To determine the rate of RFR by MtRecG and MtRuvAB, we measured kinetics of fork reversal promoted by MtRecG and MtRuvAB. As shown in Fig. 3D, MtRecG exhibited a robust activity to reverse up to 80% of substrates in about 5 min. In contrast, MtRuvAB was able to reverse only ∼40% of substrates (Fig. 3D), suggesting that MtRuvAB is catalytically less efficient in promoting fork reversal.
FIGURE 3.

RFR activity of M. tuberculosis RecG and RuvAB proteins. A, schematic representation of oligonucleotide-based replication fork reversal and outcome of the fork reversal reaction. The asterisk indicates the 32P label. Oligonucleotides are indicated by letters, and the length of each oligo is shown in parentheses. B, analysis of RFR in the presence of increasing concentrations of MtRecG and MtRuvAB. Reaction mixture contained 10 nm 32P-labeled substrates, 1 mm ATP, and 5 mm MgCl2. Reactions were initiated by addition of 0.005, 0.01, 0.05, 0.1, 0.5, and 1.0 nm MtRecG (lanes 3–8, respectively) and 50 nm MtRuvA with 25, 50, 100, 200, and 300 nm MtRuvB (lanes 9–13, respectively). Lane 1, reaction mixture in the absence of any protein; lane 2, 10 nm K321A MtRecG; lane 14 shows heat-denatured substrate, and lanes 15 (M1) and lane 16 (M2) represents markers. Reaction products were resolved on 8% native polyacrylamide gel and analyzed by autoradiography. C, quantitative data for the ability of MtRecG- and MtRuvAB-mediated RFR. D, kinetics of fork reversal catalyzed by MtRecG and MtRuvAB proteins. The reactions were initiated by addition of 0.1 nm MtRecG and 100/300 nm MtRuvA/RuvB, respectively, in a reaction buffer containing 10 nm substrates with 1 mm ATP and 5 mm MgCl2. Reactions were terminated at indicated time intervals, and products were resolved on 8% native polyacrylamide gel and visualized by autoradiography. The error bars represent standard error of the mean (S.E.) from three independent experiments.
Fork Reversal Activities of M. tuberculosis RecG, RuvAB, and RecA Proteins on Model Substrates That Contain Leading and Lagging Strand Gaps
Lesions on the leading strand template DNA cause replication stalling and result in the generation of leading strand gaps. Because lagging strand synthesis occurs in a discontinuous fashion, the replisome can bypass the lesions that are present on the lagging strand template DNA. In this scenario, repriming ahead of the lesion leads to the generation of a gap in the lagging strand. The presence of a lesion on the leading strand template DNA causes replication arrest, and it has been implicated that such stalled forks are acted upon by specialized helicases/translocases to resume the replication via reversal of stalled forks (4, 7, 10, 12, 42). In addition to E. coli RecG and RuvAB, RecA has also been reported to promote RFR (41). To investigate whether MtRecG, MtRuvAB, and MtRecA can mediate RFR on the substrates that contain the leading strand gap, we assembled radiolabeled model replication fork substrates with a leading strand gap (Fig. 4A) and examined the activities of MtRecG, MtRuvAB, and MtRecA. Incubation of increasing concentrations of MtRecG resulted in an increase in reversal of gapped fork structures (Fig. 4B). Under similar conditions, MtRuvAB and MtRecA also exhibited increasing activity at different concentrations of tested MtRuvAB and MtRecA (Fig. 4B). Quantitative data show that the extent of MtRuvAB-catalyzed fork reversal was near similar to MtRecG (Fig. 4C). In contrast, MtRecA displayed lower activity compared with MtRecG- and MtRuvAB-promoted fork reversal (Fig. 4C). To measure the rate of fork reversal with leading strand gap structures, we performed kinetic analyses. As shown in Fig. 4D, MtRecG was proficient and exhibited a higher rate of fork reversal compared with MtRuvAB and MtRecA.
FIGURE 4.

Comparative fork reversal activities of M. tuberculosis RecG, RuvAB, and RecA with homologous replication fork containing a leading strand gap. A, schematic representation of homologous replication fork containing leading strand gap and outcome of fork reversal reaction. The asterisk indicates 32P label. B, analysis of RFR promoted by MtRecG, MtRuvAB, and MtRecA. Reaction mixture contained 10 nm 32P-labeled substrate in the presence of 1 mm ATP and 5 mm MgCl2. Reactions were initiated by addition of the following: 0.005, 0.01, 0.05, 0.1, 0.5, and 1.0 nm MtRecG (lanes 3–8, respectively);50 nm MtRuvA, 25, 50, 100, 200, and 300 nm MtRuvB (lanes 9–13, respectively); and 31.25, 62.5, 125, 250, 500, and 1000 nm MtRecA (lanes 18–23, respectively). Lanes 14 and 24 represent heat-denatured substrate. Lanes 15 and 25 (M1) denote a marker of leading and lagging strand annealed products, respectively. Lanes 16 and 26 (M2) represent a marker for products that are generated by helicase unwinding of the parental strands. Lanes 1 and 17 denote reactions in the absence of any protein. Lane 2 represents reaction with 10 nm K321A MtRecG. C, quantitative data for the efficiency of MtRecG-, MtRuvAB-, and MtRecA-catalyzed fork reversal. D, kinetics of fork reversal activities of MtRecG, MtRuvAB, and MtRecA proteins. The reactions were initiated by addition of 0.1 nm MtRecG, 100/300 nm MtRuvA/MtRuvB, and 250 nm MtRecA in a reaction buffer containing 10 nm substrates with 1 mm ATP and 5 mm MgCl2. Reactions were terminated at indicated time intervals, and products were resolved on 8% native polyacrylamide gel and analyzed by autoradiography. The error bars represent standard error of the mean (S.E.) from three independent experiments.
To determine whether reversal of leading strand gap structures by MtRecG requires the presence of lagging strand at the fork junction, we assembled heterologous replication structures with either only leading or lagging strands. Interestingly, MtRecG failed to unwind the substrates that lacked the lagging strand (Fig. 5, A and C), and this was not due to its inability to bind to these structures (Fig. 5, E and G). To validate our observation, we prepared heterologous replication fork structures by labeling either leading or lagging strands, and we examined the unwinding activity of these structures by MtRecG. Similar to our previous data, MtRecG was able to efficiently unwind lagging strand-labeled substrates (Fig. 5, B and D). As expected, binding of MtRecG to these substrates was similar (Fig. 5, F and H). These data suggest that MtRecG targets the lagging strand containing stalled forks for its fork reversal activity. In an earlier study, it was shown that the decrease in the length of lagging strand results in a decrease in the unwinding of replication fork structures by E. coli RecG (17). A similar observation has been made with MtRecG (data not shown).
FIGURE 5.

M. tuberculosis RecG prefers replication fork substrates containing lagging strand at the fork junction. A, reactions contained 1 nm 32P-labeled substrate (shown on top of each panel) in the absence (lanes 1 and 8) or presence of 0.5, 5, 50, 100, and 200 nm MtRecG (lanes 2–6 and lanes 9–13, respectively). Reactions were terminated and resolved on 8% native polyacrylamide gel and visualized by autoradiography. Lanes 7 and 14 show heat-denatured substrates. B, rates of unwinding of replication fork with indicated time points (shown on top) with 100 nm MtRecG. C and D, quantitative data for the experiments shown in A and B, respectively. Electrophoretic mobility shift assay showing binding affinity of MtRecG to partial (E) or complete (F) replication fork structures. Reactions contained 0.5 nm 32P-labeled substrate (shown on top of each panel) in the absence (lanes 1and 8) or presence of 10, 20, 50, 100, 200, and 400 nm MtRecG (lanes 2–7 and lanes 9–14, respectively). G and H, graphical representation of the amount of DNA substrate bound by MtRecG for experiments shown in E and F, respectively.
Furthermore, we analyzed the ability of MtRecG-, MtRuvAB-, and MtRecA-mediated RFR with substrate containing the lagging strand gap. Fig. 6A shows the schematic representation of lagging strand gap fork structure. Unlike substrates with a leading strand gap, MtRecG-mediated RFR was evident only at higher concentrations of protein with lagging strand gap (Fig. 6, B and C). In a similar reaction, MtRuvAB exhibited lower activity (Fig. 6, B and C), and MtRecA failed to reverse the lagging strand gap fork structures (Fig. 6, B and C).
FIGURE 6.

Comparative RFR activities of M. tuberculosis RecG, RuvAB, and RecA on homologous replication fork containing lagging strand gap. A, schematic representation and outcome of RFR of homologous replication fork containing lagging strand gap. The asterisk indicates 32P label. B, analysis of RFR catalyzed by MtRecG, MtRuvAB, and MtRecA. Reaction mixture contained 10 nm 32P-labeled substrate in the presence of 1 mm ATP and 5 mm MgCl2. Reactions were initiated by addition of 0.005, 0.01, 0.05, 0.1, 0.5, and 1.0 nm MtRecG (lanes 3–8, respectively), 50 nm MtRuvA, and 25, 50, 100, 200, and 300 nm MtRuvB (lanes 9–13, respectively) and 31.25, 62.5, 125, 250, 500, and 1000 nm MtRecA (lanes 18–23, respectively). Lanes 14 and 24 represent heat-denatured substrate. Lanes 15 and 25 (M1) show a marker of leading and lagging strand annealed products, respectively. Lanes 16 and 26 (M2) represent a marker for products that are generated by helicase unwinding of the parental strands. Lanes 1 and 17 denote reactions in the absence of any protein. Lane 2 represents reaction with 10 nm K321A MtRecG. C, quantitative data for the MtRecG-, MtRuvAB-, and MtRecA-catalyzed RFR of substrates that contain lagging strand gap. The error bars represent standard error of the mean (S.E.) from three independent experiments.
Role of M. tuberculosis SSB in Modulating the RFR Activities of MtRecG and MtRuvAB
SSB or RPA proteins are abundant ssDNA-binding proteins that load onto the gaps generated during replication stalling (63–65). Binding of SSB/RPA proteins is known to stabilize the forks, and it has been shown that RPA modulates the activity of eukaryotic helicases/translocases that participate in RFR (4). To examine whether MtSSB has any role in modulating the activity of MtRecG- and MtRuvAB-mediated RFR with a leading strand gap, we carried out fork reversal activity in the presence of MtSSB. In our leading strand gap substrate, we maintained a 50-nt gap, and SSB was used in a stoichiometric ratio corresponding to the ssDNA gap. MtSSB showed efficient binding to this substrate but not to the fork structure that lacked ssDNA gap (data not shown). MtSSB did not affect the activity of MtRecG even at the tested highest concentration (Fig. 7, A and B). In contrast, increasing concentrations of MtSSB caused moderate reduction in the fork reversal activity of MtRuvAB (Fig. 7, A and B). In a parallel study, we also examined the role of E. coli SSB in modulating the MtRecG or MtRuvAB-mediated fork reversal activity. However, in our assays, E. coli SSB behaved similar as that of MtSSB (Fig. 7, A and B).
FIGURE 7.

SSB does not abolish M. tuberculosis RecG or RuvAB mediated fork reversal of leading strand gap structures. A, role of MtSSB and EcSSB proteins in the MtRecG- and MtRuvAB-mediated fork reversal of leading strand gap substrates. Reaction mixture contained 10 nm 32P-labeled substrate in the presence of 1 mm ATP and 5 mm MgCl2. Reactions were carried out either in the presence or absence of indicated proteins, and the products were resolved on 8% polyacrylamide gel and analyzed by autoradiography. Lanes 15 and 16 represent markers. B, quantitative data for the reactions shown in A. The error bars represent standard error of the mean (S.E.) from three independent experiments.
Next, we examined whether MtSSB has any influence on MtRecG- and MtRuvAB-mediated fork reversal activity with lagging strand gap structures. MtSSB was proficient in binding to lagging strand gap structures (data not shown) and by itself was devoid of fork reversal activity with this substrate (Fig. 8, A and B). Strikingly, incubation of 25 nm MtSSB, which is in stoichiometric amounts in relation to the 50-nt gap in the substrate followed by addition of 0.5 nm, MtRecG caused significant reduction (p < 0.05) in MtRecG-catalyzed fork reversal activity (Fig. 8, A and B). This inhibition further increased with higher concentrations of MtSSB (Fig. 8, A and B). Interestingly, this inhibition was also found with E. coli SSB (Fig. 8, A and B). Strikingly, a stoichiometric amount (25 nm) of MtSSB was sufficient to maximally inhibit MtRuvAB-mediated fork reversal on the lagging strand gap fork substrates (Fig. 8, A and B). A similar inhibition was also noted with E. coli SSB (Fig. 8, A and B).
FIGURE 8.

SSB suppresses M. tuberculosis RecG- and RuvAB-mediated fork reversal of substrates that contain lagging strand gap. A, effect of MtSSB and EcSSB proteins in the MtRecG- and MtRuvAB-mediated fork reversal of lagging strand gap substrates. Reaction mixture contained 10 nm 32P-labeled substrate in the presence of 1 mm ATP and 5 mm MgCl2. Reactions were carried out either in the presence or absence of indicated proteins, and the products were resolved on 8% polyacrylamide gel and analyzed by autoradiography. Lanes 15 and 16 represents markers. B, quantitative data for the reactions shown in A. The error bars represent standard error of the mean (S.E.) from three independent experiments.
M. tuberculosis RecG but Not RuvAB or RecA Is Proficient in Remodeling the Fork with a Template Lesion
Helicases/translocases are implicated in reversing the stalled forks in vivo when there is a lesion on the leading strand template DNA, and this activity has been proposed for the restart of stalled forks from the sites of DNA lesions (4, 10). To mimic a DNA lesion that stalls replication fork, we utilized a previously reported (60) oligonucleotide with a single iso-C that represents leading strand template DNA. Using the other complementary strand, we assembled a replication fork substrate with lesion on the leading strand template with a single base pair heterology at the junction. In addition, our assembled substrate contains a 5-nt leading strand gap (Fig. 9A). As a control, we assembled a similar substrate without a lesion on the template DNA. Using these substrates, we examined the ability of MtRecG-, MtRuvAB-, or MtRecA-mediated reversal of forks. Data presented in Fig. 9, B–D, shows that MtRecG was proficient in reversing the forks with and without a lesion on the leading strand template. Compared with homologous fork structure with leading strand gap (Fig. 4B), it required higher concentrations of MtRecG (1–10 nm) for its activity with these substrates. This could be due to of the presence of single base pair heterology at the fork junction. In contrast to MtRecG, MtRuvAB was less efficient in reversing the forks without lesion, and this activity was further reduced with fork substrate containing leading strand template lesion (Fig. 9, B, C, and E). In a similar reaction, MtRecA was devoid of reversing the fork with and without a lesion (Fig. 9, B and C). However, in our earlier reactions, MtRecA was able to promote fork reversal with a substrate that contained a 50-nt leading strand gap. It is possible that the 50-nt gap provides a better loading site for RecA nucleation and filament formation, which can drive fork reversal. Together, these data suggest that unlike MtRuvAB or MtRecA, MtRecG is proficient in reversing the fork with a lesion on the template strand.
FIGURE 9.

M. tuberculosis RecG but not RuvAB promotes efficient reversal of fork structures that contain a lesion on the leading strand template DNA. A, schematic representation of a homologous replication fork containing iso-C lesion on the leading strand template and the outcome of RFR. The asterisk indicates 32P label. Reaction mixture contained 10 nm either normal homologous replication fork substrate (B) or replication fork with an iso-C lesion on the leading strand template DNA (C) in a reaction buffer containing 1 mm ATP and 5 mm MgCl2 in the absence (lanes 1, 6, and 11) or presence of 1, 2.5, 5, and 10 nm MtRecG (lanes 2–5, respectively), 250 nm MtRuvA with 250, 500, 750, and 1000 nm MtRuvB (lanes 7–10, respectively), and 125, 250, 500, and 1000 nm MtRecA (lanes 12–15, respectively). Lane 16 represents marker. Quantitative data for the MtRecG- (D) and MtRuvAB (E)-mediated fork reversal of substrates with and without template lesion. The error bars represent standard error of the mean (S.E.) from three independent experiments.
M. tuberculosis RecG but Not RuvAB or RecA Promotes Lesion Bypass via Template Switch Mechanism in Vitro
Template switching after fork reversal is one of the proposed mechanisms for lesion bypass and restart of replication (5, 7, 10). To examine the role of MtRecG, MtRuvAB, and MtRecA in the in vitro model of lesion bypass and template switch mechanism of replication restart, we used previously characterized substrates (60) that contained lesion (iso-C) on the leading strand template (Fig. 10A). Data presented in the Fig. 10B show that DNA polymerase I (Klenow) can extend the radiolabeled leading strand to synthesize 13 nt up to the iso-C lesion on the template strand (43-nt product). However, Klenow failed to bypass the lesion to synthesize further. Interestingly, Klenow could extend its synthesis beyond the lesion when the reactions were incubated with increasing concentrations of MtRecG (Fig. 10B). These data suggest that MtRecG reverses the forks such that the leading strand anneals with the lagging strand and thereby Klenow can extend leading strand synthesis using lagging strand as a template (50-nt product). Such an extension by Klenow was not observed when the reaction was incubated with ATPase mutant of MtRecG (Fig. 10B). Notably, Klenow extended leading strand synthesis was insignificant when MtRuvAB was incubated along with Klenow (Fig. 10, B and C). However, at higher concentrations of RuvB (750 and 1000 nm), mild activity was observed (data not shown). These results suggest that MtRuvAB is less efficient in promoting fork reversal compared with MtRecG. In a similar reaction when MtRecA was present, Klenow-synthesized 50-nt product was not evident compared with control reactions, suggesting that MtRecA was devoid of fork reversal activity (Fig. 10B). Quantitative data clearly show that MtRecG is proficient in reversing the stalled fork, whereas MtRuvAB was inefficient in promoting fork reversal (Fig. 10C). Interestingly, in our assays with MtRecG, a small amount of full-length product (81 nt) (Fig. 10B) was also evident, and this product marginally increased with increasing concentrations of MtRecG. This product is expected to arise when RecG catalyzes restoration of the reversed fork followed by unwinding of the parental strands.
FIGURE 10.

M. tuberculosis RecG but not RuvAB promotes lesion bypass via template switching mechanism. A, schematic representation of lesion bypass via template switching mechanism that involves fork reversal and DNA synthesis using lagging strand as a template and fork restoration. The asterisk indicates 32P label and circle indicates lesion. Dashed lines represent polymerase extension. B, polymerase reactions with10 nm stalled replication fork substrate with DNA polymerase I Klenow fragment (0.05 units) in a reaction buffer containing 1 mm ATP and 5 mm MgCl2 (lane 2 and 10). In a similar reaction mixture, after addition of Klenow polymerase, indicated concentrations of MtRecG, K321A MtRecG, MtRuvA, MtRuvB, and MtRecA were added, and reactions were monitored separately. Lanes 1 and 9 represent stalled replication fork in the absence of any proteins. Polymerase extension products were resolved on 15% denaturing polyacrylamide gel and visualized by autoradiography. C, quantitative analysis of polymerase extension products generated in the presence of MtRecG and MtRuvAB. The error bars represent standard error of the mean (S.E.) from three independent experiments.
M. tuberculosis RecG but Not RuvAB Efficiently Reverses Model Replication Fork Bound by a Protein
Stalled forks contain many ssDNA- or dsDNA-bound proteins such as SSBs, DNA polymerases, and other replisome proteins. In this scenario, these DNA-bound proteins may impede the fork reversal activities of helicases/translocases. To examine the efficiency of MtRecG- or MtRuvAB-mediated fork reversal of substrates that are bound by protein, we designed homologous replication fork substrates having KpnI restriction enzyme-binding sites. A scheme of the assay and the resulting products after protein displacement-coupled fork reversal is shown in Fig. 11A. In our assays, we used a KpnI H149A mutant that binds to its specific dsDNA sequences with a higher affinity but lacks endonuclease activity (59). We investigated the DNA binding ability of variant KpnI to the fork structures that contain KpnI recognition sites in comparison with control substrates. Variant KpnI exhibited sequence-specific binding to the fork structures where the KpnI binding sequences were placed either on the arms or on the parental duplex (data not shown). Using these substrates, we monitored the ability of MtRecG or MtRuvAB to reverse the KpnI-bound fork structures in comparison with the KpnI-free substrates. In the absence of KpnI, both MtRecG and MtRuvAB were able to reverse the forks at their optimal concentrations (Fig. 11, B and C). Interestingly, MtRecG promoted efficient reversal of the forks that were bound by variant KpnI (Fig. 11, B and C). In contrast, MtRuvAB exhibited decreased activity with KpnI-bound fork structures (Fig. 11, B and C). The shorter duplex DNA generated after fork reversal and annealing of nascent strands was quantified, and data in Fig. 11C clearly show that 1 nm MtRecG was able to reverse the KpnI-bound fork structures with an efficiency of up to ∼100% when compared with control substrates. In contrast, ∼30–50% of reduction was evident with KpnI-bound versus -unbound substrates at all tested concentrations of MtRuvAB (Fig. 11C). These data clearly suggest that MtRecG but not MtRuvAB is proficient in reversing the stalled forks that are bound by the KpnI enzyme. Recent study shows that RPA stimulates protein displacement activities of FANCJ or RecQ1 helicase (66). However, such stimulation was not observed with MtSSB when MtRecG/MtRuvAB-catalyzed fork reversal was tested with KpnI-bound fork structures (data not shown).
FIGURE 11.

M. tuberculosis RecG but not RuvAB drives efficient reversal of forks bound by protein. A, schematic representation of a fork bound by protein and the expected products after fork reversal reaction catalyzed by MtRecG and MtRuvAB. Asterisk indicates 32P label; oval represents KpnI H149A mutant; and dashed lines represent binding site for H149A KpnI enzyme. B, fork reversal activities of MtRecG and MtRuvAB with homologous replication fork structures either in the absence (lanes 1–8) or presence (lanes 9–16) of 100 nm H149A KpnI. Reactions were initiated by addition of indicated concentrations MtRecG (lanes 3–5 and 11–13) and MtRuvA + MtRuvB (lanes 6–8 and 14–16). Lanes 2 and 10 represent reactions with K321A MtRecG in the absence or presence of H149A KpnI. Reaction products were separated on 8% native polyacrylamide gel and analyzed by autoradiography. C, quantitative analysis of MtRecG- and MtRuvAB-driven fork reversal of homologous fork structures either bound by H149A KpnI or naked substrates. The error bars represent standard error of the mean (S.E.) from three independent experiments.
Discussion
Multiple studies show that replisome efficiently bypasses the lesion on the lagging strand template, whereas damage on the leading strand template presents a serious problem for the error-free duplication of the genome (7, 10). Although replication can resume by repriming downstream to the lesion on leading strand template, fork reversal is emerging as an evolutionarily conserved physiological response from prokaryotes to eukaryotes for restart of stalled forks (7, 10). Evidence from various studies indicates that fork reversal plays a central role in the multiple mechanisms of replication restart in bacteria (5, 10, 12, 42, 67). Defects in the processing of stalled forks can lead to genetic instability and cell death. Thus, enzymes that catalyze fork reversal play a crucial role in the stabilization and restart of stalled replication forks. Consistent with our previous data (53), this study demonstrates that MtRecG rescues the hypersensitivity of E. coli ΔrecG cells from agents that cause replicative stress via depletion of the dNTP pool. In agreement with these observations, our biochemical studies show that MtRecG catalyzes remodeling of fork structures that resemble stalled forks with leading strand gap and template damage. MtSSB suppresses MtRecG- and MtRuvAB-mediated fork reversal activity with substrates that contain lagging strand gap. Strikingly, our comparative studies show that MtRecG is more efficient in promoting fork reversal than MtRuvAB or MtRecA. Moreover, MtRecG but not MtRuvAB was proficient in exhibiting a template switch mechanism of fork restart in vitro. Finally, unlike MtRuvAB, our data show that MtRecG drives efficient reversal of forks when fork structures are bound by protein. These data suggest that MtRecG might participate in replication fork stabilization and restart pathways in vivo.
E. coli ΔrecG cells exhibit sensitivity to UV light, MMS, and HU, which induce genome-wide replicative stress (24, 25). The fact that MtRecG rescues the hypersensitivity of E. coli ΔrecG cells from replicative stress implies that MtRecG participates in the rescue pathways of stalled replication forks in vivo. In corroboration with these data, purified MtRecG was robust in reversing the model replication forks, and this activity was dependent on ATP hydrolysis at catalytic concentrations. The semi-discontinuous mode of DNA synthesis suggests that lagging strand synthesis continues to occur despite the presence of lesions on the lagging strand template (68). In contrast, when leading strand polymerase encounters lesions on template DNA, it generates ssDNA gaps in the leading strand as replication continues to progress beyond the lesion via lagging strand synthesis. However, the replication fork is expected to stall due to uncoupling of leading and lagging strand synthesis. Such forks need to be restarted, and fork reversal by helicases/translocases plays an important role in rescuing the stalled forks (10, 67). MtRecG was proficient in driving the reversal of model fork structures that contain leading strand gap. Notably, MtRecG was efficient in reversing the fork structures that contained leading strand template lesions and forks that are bound by protein. Indeed, a recent study showed that E. coli RecG promotes reversal of model forks that contain cyclobutane pyrimidine dimer lesion on the leading strand template DNA (22). In addition, a biochemical study with oligonucleotide DNA substrates (17) and a recent single molecule study (69) utilizing a combination of optical and magnetic tweezers show that E. coli RecG preferentially binds and unwinds forks with lagging strand. Similar to E. coli RecG, the presence of lagging strand at the fork junction was required for efficient fork reversal by MtRecG, indicating that MtRecG might target forks that are stalled by leading strand template lesion. Structural studies from Thermatoga maritima RecG bound to a replication fork structure reveal that the “wedge” domain is critical for binding to the branched DNA molecules, and the two helicase domains interact with parental duplex of the fork (70). Structural data support the notion that RecG translocates on dsDNA-catalyzing disruption of hydrogen bonding of the two daughter duplexes, resulting in reannealing of the parental duplexes and consequent annealing of two nascent strands (20, 71). The fact that MtRecG also possesses a conserved wedge domain and other helicase motifs as that of T. maritima and E. coli RecG (53), it is likely that the mechanism of the MtRecG-mediated fork reversal may be similar to that of E. coli RecG.
Previous studies implicate the participation of RuvAB proteins in replication fork repair when there is a defect in replisome components or accessory replicative helicase (72–74). Consistently, biochemical studies showed that E. coli RuvAB promotes RFR with oligonucleotide substrates (21, 23). A more recent study demonstrated that E. coli RuvAB catalyzes RFR with structures that contain cyclobutane pyrimidine dimer lesion on the leading strand template (22). Interestingly, our comparative studies reveal that only MtRecG was proficient in driving the reversal of replication forks either with leading strand template damage or when the forks are bound by protein. In contrast, although MtRuvAB was able to promote fork reversal with leading strand gap, this activity was suppressed with template damage and when the forks were bound by protein. It is likely that MtRecG preferentially acts on stalled forks to promote fork reversal and then MtRuvAB can catalyze extended branch migration of HJs formed by MtRecG. Indeed, Gupta et al. (22) showed that E. coli RecG stimulates RuvAB-catalyzed fork reversal. Moreover, it has been shown that RecG binds to fork structures with higher affinity than RuvAB, and SSB inhibits RuvAB-mediated fork reversal activity (23). Our comparative studies clearly show that MtRecG is robust in reversing the forks with lesion on the template strand and when forks are bound by protein, implying that RecG could be an evolutionarily conserved translocase that preferentially acts on stalled forks.
E. coli RecG efficiently drives fork reversal at as low as a 0.01 nm concentration (21), which correlate with its in vivo levels (<10 molecules) (75). In our assays, MtRecG was also able to drive robust fork reversal between 0.01 and 0.1 nm concentrations. Our previous study shows that RecG is present at ∼3 molecules in M. tuberculosis cells, and its level marginally increases with DNA damage (53). In contrast to RecG, the basal levels of E. coli RuvA and RuvB proteins are known to exist at ∼700 and 200 molecules, respectively (75, 76). E. coli RuvA assembles into a tetramer, whereas RuvB exists as hexamer and such oligomeric forms of RuvA/RuvB are required for its optimal activity to catalyze unwinding of HJ as well as fork structures (21, 77–79). Conceivably, because of its intrinsic property of oligomerization, E. coli RuvA/RuvB is required at higher amounts to exhibit fork reversal activity. It is likely that M. tuberculosis RuvAB might function in a similar fashion as that of E. coli RuvAB. Indeed, for optimal fork reversal activity, MtRuvAB was required at much higher concentrations (100/300 nm of RuvA/RuvB) than MtRecG. Although the basal levels of MtRuvAB need to be determined, microarray data reveal an up-regulation in MtRuvAB transcripts upon macrophage infection (80, 81).
In addition to RecG and RuvAB, E. coli RecA has been implicated in fork repair and restoration activities (39, 67). In a recent study, Gupta et al. (43) examined the role of RecA in RFR activity in the presence of SSB. In contrast to the earlier report (41), RecA was unable to catalyze reversal of stalled forks (43). In the previous report (41), substrates were incubated first with RecA, and SSB, which is an abundant protein that exhibits higher affinity to ssDNA, was added later. Our data demonstrate that MtRecA was modest in its fork reversal activity with leading strand gap substrates, and this activity was inhibited when SSB was added first in the reactions (data not shown). Moreover, with a template lesion, the fork reversal activity of RecA was completely abolished. E. coli RecFOR proteins are known to facilitate RecA filament formation onto the ssDNA (30), and it will be interesting to test whether RecFOR stimulates RecA-mediated fork reversal activity. Moreover, in our assays, the substrates with a template lesion contained a leading strand gap of 5 nt, which may be a limitation for efficient loading of RecA molecules. However, further studies are required to rule out this possibility.
Although fork reversal provides a mechanism for recoupling helicase and polymerase to resume the replication, fork reversal activity should be regulated to prevent interference with actively elongating forks and maturation of Okazaki fragments. A recent study shows that human SMARCAL1 fork reversal activity is regulated by RPA such that binding of RPA to the lagging strand gap template prevents SMARCAL1 activity, although it stimulates SMARCAL1-mediated fork reversal when there is a leading strand gap (82). Interestingly, in our assays, MtSSB suppressed MtRecG- and MtRuvAB-catalyzed fork reversal when the fork structures contain lagging strand gap. It is likely that MtSSB modulates the fork reversal activity of MtRecG by its direct interaction with MtRecG. Indeed, E. coli SSB interacts with E. coli RecG via its C-terminal domain, and this interaction facilitates RecG binding to the stalled forks (83, 84). However, a similar physical interaction between SSB and RuvAB was not found in their study (83). SSB/RPA has been shown to modulate the activity of various helicases including PriA, RecQ, and FANCJ (66, 85, 86). Moreover, heterologous SSB proteins have been shown to functionally replace cognate SSBs in RecA-catalyzed strand exchange reactions (37). Interestingly, similar to MtSSB, E. coli SSB was able to suppress the MtRecG- and MtRuvAB-mediated fork reversal of lagging strand gap structures. However, the mechanism(s) underlying SSB modulation of MtRecG- and MtRuvAB-mediated fork reversal activities require further studies.
A recent study has provided direct evidence that T4 UvsW helicase fork reversal activity is coupled to the template switching mechanism of replication restart (62). Strikingly, our data with in vitro template switching mechanism of fork start further supported the efficient fork reversal activity of MtRecG. DNA synthesis bypassing the lesion involves fork reversal by helicases/tranlocases. MtRecG was proficient in catalyzing the stalled forks such that DNA polymerase could resume its activity. In contrast, MtRuvAB was less efficient to exhibit fork reversal activity; thereby, an extended synthesis past the lesion was not observed in reactions incubated with MtRuvAB. Similarly, MtRecA was also unable to catalyze fork reversal with lesion on the template DNA as the final product of DNA synthesis was not evident when RecA was examined in our assays. Interestingly, an extended full-length product was visible in the reactions carried out with MtRecG. This product is expected to arise only when reversed forks are restored possibly by reverse branch migration catalyzed by RecG. Indeed, such an activity is documented for E. coli RecG (16, 87). The increasing appearance of the full-length final product suggests that RecG can restore the reversed forks to facilitate replication restart. However, a more specific assay, similar to what has been used for analyzing RecQ1-mediated fork restoration (88), is required for validating the fork restoration activity of MtRecG.
The robust and efficient fork reversal activity of MtRecG with substrates with template lesions or when forks are bound by protein suggests that MtRecG could play an important role in the replication fork repair and restart pathways in mycobacteria. The MtRecG-catalyzed RFR may support the template switching mechanism of replication restart in mycobacteria (Fig. 12A). Indeed, in our assays MtRecG was able to facilitate DNA synthesis beyond the lesion, which supports the template switching model of replication restart. In this scenario, upon fork reversal, lesion containing template strand is reannealed to the parental complementary strand, and replication will be resumed bypassing the lesion upon DNA synthesis from the 3′ end of leading strand utilizing lagging strand as a template followed by fork restoration (Fig. 12A). The lesion can be repaired by the BER/NER pathway subsequently. Indeed, a recent study with T4 UvsW helicase supports the template switching mechanism of replication restart via fork reversal (62). Alternatively, fork reversal may provide the opportunity for NER/BER machinery to excise the lesion, and DNA synthesis with template switching followed by fork restoration can resume the replication (Fig. 12B). There are other possibilities that replication can be restarted from a stalled fork. Fork reversal generates four-stranded HJs that are substrates for cleavage by RuvC junction-specific endonuclease (Fig. 12C). Indeed, a recent study shows that RecG-driven reversed forks undergo cleavage by RuvC (22). Upon cleavage, generated DSBs can be processed by nucleases for a recombination-mediated restart of replication via D-loop formation (Fig. 12C). Finally, exonucleolytic degradation (5′ → 3′) of nascent strand leaving the complementary 3′ ssDNA provides a substrate for initiation of recombination using homologous sequences in the reannealed parental duplex DNA. Resolution/dissolution of the HJs coupled with assembly of a replisome at the D-loop can restore a replication fork (Fig. 12D).
FIGURE 12.

Model depicting possible replication restart mechanisms in M. tuberculosis via fork reversal. A, template switching; B, lesion repair by NER/BER; C, fork cleavage by endonuclease and HR-mediated restart via D-loop formation; and D, nucleolytic degradation of lagging strand and recombination mediated replication restart (see text for descriptions).
M. tuberculosis genome is GC-rich (65%), and during infection, this pathogen is exposed to macrophage-induced ROS and RNI, which cause oxidative and alkylating lesions (89). In addition, G-rich sequence motifs that have the potential to form G-quadruplex (G4) DNA secondary structures are also abundant in this pathogen (49, 50). During M. tuberculosis infection and propagation in host macrophages, the ROS- and RNI-induced DNA damage, different types of secondary structures, including G4 and ongoing transcription, exert enormous replicative stress. Under these conditions, it is intriguing and also the least understood regarding how this pathogen circumvents a high dose of replicative stress to survive, multiply, and cause pathogenesis. It is likely that M. tuberculosis could have evolved with an efficient mechanism(s) to evade the replicative stress. Thus, it is crucial to understand the mechanisms and the pathways by which M. tuberculosis deals with replication problems. Fork reversal by RecG provides one such mechanism to protect and restart stalled forks in M. tuberculosis. Our studies reveal that catalytic amounts of RecG promote efficient reversal of stalled forks when there is template damage or when forks are bound by protein. In E. coli, RecG protein levels are unchanged during SOS response (75). Notably, a microarray data with macrophage infection (80, 90) and our previous in vitro study (53) show that MtRecG levels are moderately up-regulated (∼2-fold) when DNA damage was induced. It is likely that this induction of RecG may be required for circumventing the replicative stress incurred during infection, survival, and pathogenesis. Thus, RecG in M. tuberculosis could be a potential drug target. However, further studies are required to understand the pathways and the mechanisms by which this pathogen circumvents replication problems for its survival, persistence, and pathogenesis.
Author Contributions
R. S. T. and G. N. conceived and coordinated the study. G. N. wrote the paper. R. S. T. designed and performed experiments. R. S. T. and G. N. analyzed the data. S. B. provided technical assistance. J. S. K. and K. M. provided reagents for our studies. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Drs. V. Nagaraja for providing KpnI mutant enzyme and D. N. Rao for sharing reagents. We thank Drs. Robert Lloyd and Leonard Wu for the generous gift of ΔrecG E. coli cells and of pG68 and pG46 plasmids, respectively. We thank Drs. D. N. Rao, Amit Singh, Matteo Berti, and Kumar Somyajit for their useful discussions. Sneha Saxena and Anup Mishra are acknowledged for their critical reading of the manuscript.
This work was supported by in part Department of Atomic Energy Grants 2010/37B/40/BRNS/1437 and 37(1)/14/52/2014-BRNS/1620 and DBT-IISc Partnership Program Grant DBT/BF/PR/INS/IISc/2011-12 and by the Center of Excellence and Innovation program of the Department of Biotechnology, New Delhi (to the K. M. laboratory). The authors declare that they have no conflicts of interest with the contents of this article.
- RFR
- replication fork reversal
- SSB
- single-stranded DNA-binding protein
- RPA
- replication protein A
- NER
- nucleotide excision repair
- BER
- base excision repair
- MMS
- methyl methane sulfonate
- HU
- hydroxy urea
- nt
- nucleotide
- ssDNA
- single-stranded DNA
- HJ
- Holliday junction
- iso-C
- iso-cytosine
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- ROS
- reactive oxygen species
- RNI
- reactive nitrogen intermediate
- RPA
- replication protein A.
References
- 1. Branzei D., Foiani M. (2010) Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 11, 208–219 [DOI] [PubMed] [Google Scholar]
- 2. Cox M. M., Goodman M. F., Kreuzer K. N., Sherratt D. J., Sandler S. J., Marians K. J. (2000) The importance of repairing stalled replication forks. Nature 404, 37–41 [DOI] [PubMed] [Google Scholar]
- 3. Nagaraju G., Scully R. (2007) Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair 6, 1018–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neelsen K. J., Lopes M. (2015) Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 16, 207–220 [DOI] [PubMed] [Google Scholar]
- 5. Syeda A. H., Hawkins M., McGlynn P. (2014) Recombination and replication. Cold Spring Harb. Perspect. Biol. 6, a016550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zeman M. K., Cimprich K. A. (2014) Causes and consequences of replication stress. Nat. Cell Biol. 16, 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yeeles J. T., Poli J., Marians K. J., Pasero P. (2013) Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 5, a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McGlynn P. (2004) Links between DNA replication and recombination in prokaryotes. Curr. Opin. Genet. Dev. 14, 107–112 [DOI] [PubMed] [Google Scholar]
- 9. McGlynn P., Lloyd R. G. (2002) Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 3, 859–870 [DOI] [PubMed] [Google Scholar]
- 10. Atkinson J., McGlynn P. (2009) Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 37, 3475–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Michel B., Flores M. J., Viguera E., Grompone G., Seigneur M., Bidnenko V. (2001) Rescue of arrested replication forks by homologous recombination. Proc. Natl. Acad. Sci. U.S.A. 98, 8181–8188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Michel B., Grompone G., Florès M. J., Bidnenko V. (2004) Multiple pathways process stalled replication forks. Proc. Natl. Acad. Sci. U.S.A. 101, 12783–12788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kreuzer K. N. (2005) Interplay between DNA replication and recombination in prokaryotes. Annu. Rev. Microbiol. 59, 43–67 [DOI] [PubMed] [Google Scholar]
- 14. Kuzminov A. (2001) DNA replication meets genetic exchange: chromosomal damage and its repair by homologous recombination. Proc. Natl. Acad. Sci. U.S.A. 98, 8461–8468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heller R. C., Marians K. J. (2006) Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell Biol. 7, 932–943 [DOI] [PubMed] [Google Scholar]
- 16. McGlynn P., Lloyd R. G. (2000) Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell 101, 35–45 [DOI] [PubMed] [Google Scholar]
- 17. McGlynn P., Lloyd R. G. (2001) Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc. Natl. Acad. Sci. U.S.A. 98, 8227–8234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McGlynn P., Lloyd R. G., Marians K. J. (2001) Formation of Holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: RecG stimulates regression even when the DNA is negatively supercoiled. Proc. Natl. Acad. Sci. U.S.A. 98, 8235–8240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Slocum S. L., Buss J. A., Kimura Y., Bianco P. R. (2007) Characterization of the ATPase activity of the Escherichia coli RecG protein reveals that the preferred cofactor is negatively supercoiled DNA. J. Mol. Biol. 367, 647–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bianco P. R. (2015) I came to a fork in the DNA and there was RecG. Prog. Biophys. Mol. Biol. 117, 166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McGlynn P., Lloyd R. G. (2001) Action of RuvAB at replication fork structures. J. Biol. Chem. 276, 41938–41944 [DOI] [PubMed] [Google Scholar]
- 22. Gupta S., Yeeles J. T., Marians K. J. (2014) Regression of replication forks stalled by leading-strand template damage: I. Both RecG and RuvAB catalyze regression, but RuvC cleaves the Holliday junctions formed by RecG preferentially. J. Biol. Chem. 289, 28376–28387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abd Wahab S., Choi M., Bianco P. R. (2013) Characterization of the ATPase activity of RecG and RuvAB proteins on model fork structures reveals insight into stalled DNA replication fork repair. J. Biol. Chem. 288, 26397–26409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lloyd R. G., Buckman C. (1991) Genetic analysis of the recG locus of Escherichia coli K-12 and of its role in recombination and DNA repair. J. Bacteriol. 173, 1004–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lloyd R. G. (1991) Conjugational recombination in resolvase-deficient ruvC mutants of Escherichia coli K-12 depends on recG. J. Bacteriol. 173, 5414–5418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Donaldson J. R., Courcelle C. T., Courcelle J. (2004) RuvAB and RecG are not essential for the recovery of DNA synthesis following UV-induced DNA damage in Escherichia coli. Genetics 166, 1631–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rudolph C. J., Upton A. L., Briggs G. S., Lloyd R. G. (2010) Is RecG a general guardian of the bacterial genome? DNA Repair 9, 210–223 [DOI] [PubMed] [Google Scholar]
- 28. Kowalczykowski S. C., Dixon D. A., Eggleston A. K., Lauder S. D., Rehrauer W. M. (1994) Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev. 58, 401–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cox M. M. (1999) Recombinational DNA repair in bacteria and the RecA protein. Prog. Nucleic Acid Res. Mol. Biol. 63, 311–366 [DOI] [PubMed] [Google Scholar]
- 30. Cox M. M. (2007) Motoring along with the bacterial RecA protein. Nat. Rev. Mol. Cell Biol. 8, 127–138 [DOI] [PubMed] [Google Scholar]
- 31. West S. C. (2003) Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 4, 435–445 [DOI] [PubMed] [Google Scholar]
- 32. Sung P., Krejci L., Van Komen S., Sehorn M. G. (2003) Rad51 recombinase and recombination mediators. J. Biol. Chem. 278, 42729–42732 [DOI] [PubMed] [Google Scholar]
- 33. Sung P., Klein H. (2006) Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 7, 739–750 [DOI] [PubMed] [Google Scholar]
- 34. San Filippo J., Chi P., Sehorn M. G., Etchin J., Krejci L., Sung P. (2006) Recombination mediator and Rad51 targeting activities of a human BRCA2 polypeptide. J. Biol. Chem. 281, 11649–11657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Somyajit K., Subramanya S., Nagaraju G. (2010) RAD51C: a novel cancer susceptibility gene is linked to Fanconi anemia and breast cancer. Carcinogenesis 31, 2031–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ganesh N., Muniyappa K. (2003) Mycobacterium smegmatis RecA protein is structurally similar to but functionally distinct from Mycobacterium tuberculosis RecA. Proteins 53, 6–17 [DOI] [PubMed] [Google Scholar]
- 37. Ganesh N., Muniyappa K. (2003) Characterization of DNA strand transfer promoted by Mycobacterium smegmatis RecA reveals functional diversity with Mycobacterium tuberculosis RecA. Biochemistry 42, 7216–7225 [DOI] [PubMed] [Google Scholar]
- 38. Kuzminov A. (1999) Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol. Mol. Biol. Rev. 63, 751–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Courcelle J., Donaldson J. R., Chow K. H., Courcelle C. T. (2003) DNA damage-induced replication fork regression and processing in Escherichia coli. Science 299, 1064–1067 [DOI] [PubMed] [Google Scholar]
- 40. Seigneur M., Ehrlich S. D., Michel B. (2000) RuvABC-dependent double-strand breaks in dnaBts mutants require recA. Mol. Microbiol. 38, 565–574 [DOI] [PubMed] [Google Scholar]
- 41. Robu M. E., Inman R. B., Cox M. M. (2001) RecA protein promotes the regression of stalled replication forks in vitro. Proc. Natl. Acad. Sci. U.S.A. 98, 8211–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Michel B., Boubakri H., Baharoglu Z., LeMasson M., Lestini R. (2007) Recombination proteins and rescue of arrested replication forks. DNA Repair 6, 967–980 [DOI] [PubMed] [Google Scholar]
- 43. Gupta S., Yeeles J. T., Marians K. J. (2014) Regression of replication forks stalled by leading-strand template damage: II. Regression by RecA is inhibited by SSB. J. Biol. Chem. 289, 28388–28398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lienhardt C., Glaziou P., Uplekar M., Lönnroth K., Getahun H., Raviglione M. (2012) Global tuberculosis control: lessons learnt and future prospects. Nat. Rev. Microbiol. 10, 407–416 [DOI] [PubMed] [Google Scholar]
- 45. Nathan C., Shiloh M. U. (2000) Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. U.S.A. 97, 8841–8848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Warner D. F., Mizrahi V. (2006) Tuberculosis chemotherapy: the influence of bacillary stress and damage response pathways on drug efficacy. Clin. Microbiol. Rev. 19, 558–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kurthkoti K., Varshney U. (2011) Base excision and nucleotide excision repair pathways in mycobacteria. Tuberculosis 91, 533–543 [DOI] [PubMed] [Google Scholar]
- 48. Kurthkoti K., Varshney U. (2012) Distinct mechanisms of DNA repair in mycobacteria and their implications in attenuation of the pathogen growth. Mech. Ageing Dev. 133, 138–146 [DOI] [PubMed] [Google Scholar]
- 49. Rawal P., Kummarasetti V. B., Ravindran J., Kumar N., Halder K., Sharma R., Mukerji M., Das S. K., Chowdhury S. (2006) Genome-wide prediction of G4 DNA as regulatory motifs: role in Escherichia coli global regulation. Genome Res. 16, 644–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thakur R. S., Desingu A., Basavaraju S., Subramanya S., Rao D. N., Nagaraju G. (2014) Mycobacterium tuberculosis DinG is a structure-specific helicase that unwinds G4 DNA: implications for targeting G4 DNA as a novel therapeutic approach. J. Biol. Chem. 289, 25112–25136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muniyappa K., Vaze M. B., Ganesh N., Sreedhar Reddy M., Guhan N., Venkatesh R. (2000) Comparative genomics of Mycobacterium tuberculosis and Escherichia coli for recombination (rec) genes. Microbiology 146, 2093–2095 [DOI] [PubMed] [Google Scholar]
- 52. Ambur O. H., Davidsen T., Frye S. A., Balasingham S. V., Lagesen K., Rognes T., Tønjum T. (2009) Genome dynamics in major bacterial pathogens. FEMS Microbiol. Rev. 33, 453–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thakur R. S., Basavaraju S., Somyajit K., Jain A., Subramanya S., Muniyappa K., Nagaraju G. (2013) Evidence for the role of Mycobacterium tuberculosis RecG helicase in DNA repair and recombination. FEBS J. 280, 1841–1860 [DOI] [PubMed] [Google Scholar]
- 54. Khanduja J. S., Muniyappa K. (2012) Functional analysis of DNA replication fork reversal catalyzed by Mycobacterium tuberculosis RuvAB proteins. J. Biol. Chem. 287, 1345–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Khanduja J. S., Tripathi P., Muniyappa K. (2009) Mycobacterium tuberculosis RuvA induces two distinct types of structural distortions between the homologous and heterologous Holliday junctions. Biochemistry 48, 27–40 [DOI] [PubMed] [Google Scholar]
- 56. Kumar R. A., Vaze M. B., Chandra N. R., Vijayan M., Muniyappa K. (1996) Functional characterization of the precursor and spliced forms of RecA protein of Mycobacterium tuberculosis. Biochemistry 35, 1793–1802 [DOI] [PubMed] [Google Scholar]
- 57. Lohman T. M., Green J. M., Beyer R. S. (1986) Large-scale overproduction and rapid purification of the Escherichia coli ssb gene product. Expression of the ssb gene under λ PL control. Biochemistry 25, 21–25 [DOI] [PubMed] [Google Scholar]
- 58. Reddy M. S., Guhan N., Muniyappa K. (2001) Characterization of single-stranded DNA-binding proteins from mycobacteria. The carboxyl-terminal of domain of SSB is essential for stable association with its cognate RecA protein. J. Biol. Chem. 276, 45959–45968 [DOI] [PubMed] [Google Scholar]
- 59. Saravanan M., Bujnicki J. M., Cymerman I. A., Rao D. N., Nagaraja V. (2004) Type II restriction endonuclease R. KpnI is a member of the HNH nuclease superfamily. Nucleic Acids Res. 32, 6129–6135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bugreev D. V., Rossi M. J., Mazin A. V. (2011) Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res. 39, 2153–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ralf C., Hickson I. D., Wu L. (2006) The Bloom's syndrome helicase can promote the regression of a model replication fork. J. Biol. Chem. 281, 22839–22846 [DOI] [PubMed] [Google Scholar]
- 62. Manosas M., Perumal S. K., Croquette V., Benkovic S. J. (2012) Direct observation of stalled fork restart via fork regression in the T4 replication system. Science 338, 1217–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wold M. S. (1997) Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 66, 61–92 [DOI] [PubMed] [Google Scholar]
- 64. Fanning E., Klimovich V., Nager A. R. (2006) A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 34, 4126–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Meyer R. R., Laine P. S. (1990) The single-stranded DNA-binding protein of Escherichia coli. Microbiol. Rev. 54, 342–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sommers J. A., Banerjee T., Hinds T., Wan B., Wold M. S., Lei M., Brosh R. M. Jr. (2014) Novel function of the Fanconi anemia group J or RECQ1 helicase to disrupt protein-DNA complexes in a replication protein A-stimulated manner. J. Biol. Chem. 289, 19928–19941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cox M. M. (2001) Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annu. Rev. Genet. 35, 53–82 [DOI] [PubMed] [Google Scholar]
- 68. Marians K. J. (1992) Prokaryotic DNA replication. Annu. Rev. Biochem. 61, 673–719 [DOI] [PubMed] [Google Scholar]
- 69. Manosas M., Perumal S. K., Bianco P. R., Bianco P., Ritort F., Benkovic S. J., Croquette V. (2013) RecG and UvsW catalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat. Commun. 4, 2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Singleton M. R., Scaife S., Wigley D. B. (2001) Structural analysis of DNA replication fork reversal by RecG. Cell 107, 79–89 [DOI] [PubMed] [Google Scholar]
- 71. Mahdi A. A., Briggs G. S., Sharples G. J., Wen Q., Lloyd R. G. (2003) A model for dsDNA translocation revealed by a structural motif common to RecG and Mfd proteins. EMBO J. 22, 724–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Baharoglu Z., Petranovic M., Flores M. J., Michel B. (2006) RuvAB is essential for replication forks reversal in certain replication mutants. EMBO J. 25, 596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Baharoglu Z., Bradley A. S., Le Masson M., Tsaneva I., Michel B. (2008) ruvA mutants that resolve Holliday junctions but do not reverse replication forks. PLoS Genet. 4, e1000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Le Masson M., Baharoglu Z., Michel B. (2008) ruvA and ruvB mutants specifically impaired for replication fork reversal. Mol. Microbiol. 70, 537–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lloyd R. G., Sharples G. J. (1991) Molecular organization and nucleotide sequence of the recG locus of Escherichia coli K-12. J. Bacteriol. 173, 6837–6843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shurvinton C. E., Lloyd R. G. (1982) Damage to DNA induces expression of the ruv gene of Escherichia coli. Mol. Gen. Genet. 185, 352–355 [DOI] [PubMed] [Google Scholar]
- 77. West S. C. (1997) Processing of recombination intermediates by the RuvABC proteins. Annu. Rev. Genet. 31, 213–244 [DOI] [PubMed] [Google Scholar]
- 78. van Gool A. J., Shah R., Mézard C., West S. C. (1998) Functional interactions between the Holliday junction resolvase and the branch migration motor of Escherichia coli. EMBO J. 17, 1838–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Eggleston A. K., Mitchell A. H., West S. C. (1997) In vitro reconstitution of the late steps of genetic recombination in E. coli. Cell 89, 607–617 [DOI] [PubMed] [Google Scholar]
- 80. Schnappinger D., Ehrt S., Voskuil M. I., Liu Y., Mangan J. A., Monahan I. M., Dolganov G., Efron B., Butcher P. D., Nathan C., Schoolnik G. K. (2003) Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198, 693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rachman H., Strong M., Ulrichs T., Grode L., Schuchhardt J., Mollenkopf H., Kosmiadi G. A., Eisenberg D., Kaufmann S. H. (2006) Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect. Immun. 74, 1233–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bétous R., Couch F. B., Mason A. C., Eichman B. F., Manosas M., Cortez D. (2013) Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 3, 1958–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Buss J. A., Kimura Y., Bianco P. R. (2008) RecG interacts directly with SSB: implications for stalled replication fork regression. Nucleic Acids Res. 36, 7029–7042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sun Z., Tan H. Y., Bianco P. R., Lyubchenko Y. L. (2015) Remodeling of RecG helicase at the DNA replication fork by SSB protein. Sci. Rep. 5, 9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Harmon F. G., Kowalczykowski S. C. (2001) Biochemical characterization of the DNA helicase activity of the Escherichia coli RecQ helicase. J. Biol. Chem. 276, 232–243 [DOI] [PubMed] [Google Scholar]
- 86. Heller R. C., Marians K. J. (2005) Unwinding of the nascent lagging strand by Rep and PriA enables the direct restart of stalled replication forks. J. Biol. Chem. 280, 34143–34151 [DOI] [PubMed] [Google Scholar]
- 87. Whitby M. C., Ryder L., Lloyd R. G. (1993) Reverse branch migration of Holliday junctions by RecG protein: a new mechanism for resolution of intermediates in recombination and DNA repair. Cell 75, 341–350 [DOI] [PubMed] [Google Scholar]
- 88. Berti M., Ray Chaudhuri A., Thangavel S., Gomathinayagam S., Kenig S., Vujanovic M., Odreman F., Glatter T., Graziano S., Mendoza-Maldonado R., Marino F., Lucic B., Biasin V., Gstaiger M., Aebersold R., et al. (2013) Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 20, 347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ehrt S., Schnappinger D. (2009) Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell. Microbiol. 11, 1170–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gorna A. E., Bowater R. P., Dziadek J. (2010) DNA repair systems and the pathogenesis of Mycobacterium tuberculosis: varying activities at different stages of infection. Clin. Sci. 119, 187–202 [DOI] [PubMed] [Google Scholar]