

Background: The nature of the FGFR1 signaling complex on the surface of living cells remains unclear.
Results: A TR-FRET-based method revealed a ligand-independent dimer formation of FGFR1 that is independent of the cell-surface density.
Conclusion: FGFR1 constitutively forms ligand-independent dimers that are either stabilized or undergo conformational changes in the presence of agonists.
Significance: These findings help explain the mechanistic basis of FGFR1 activation by ligands and pathogenic mutations.
Keywords: dimerization, fibroblast growth factor (FGF), fibroblast growth factor receptor (FGFR), fluorescence resonance energy transfer (FRET), receptor structure-function, receptor-tyrosine kinase, fgf21, tr-fret
Abstract
Fibroblast growth factors receptors (FGFRs) are thought to initiate intracellular signaling cascades upon ligand-induced dimerization of the extracellular domain. Although the existence of unliganded FGFR1 dimers on the surface of living cells has been proposed, this notion remains rather controversial. Here, we employed time-resolved Förster resonance energy transfer combined with SNAP- and ACP-tag labeling in COS7 cells to monitor dimerization of full-length FGFR1 at the cell-surface with or without the coreceptor βKlotho. Using this approach we observed homodimerization of unliganded FGFR1 that is independent of its surface density. The homo-interaction signal observed for FGFR1 was indeed as robust as that obtained for epidermal growth factor receptor (EGFR) and was further increased by the addition of activating ligands or pathogenic mutations. Mutational analysis indicated that the kinase and the transmembrane domains, rather than the extracellular domain, mediate the ligand-independent FGFR1 dimerization. In addition, we observed a formation of a higher order ligand-independent complex by the c-spliced isoform of FGFR1 and βKlotho. Collectively, our approach provides novel insights into the assembly and dynamics of the full-length FGFRs on the cell surface.
Introduction
Fibroblast growth factor receptors (FGFRs)3 are a family of structurally related bitopic membrane proteins belonging to the receptor-tyrosine kinase (RTK) superfamily (1). Humans possess 18 genes encoding structurally related FGF ligands and 4 genes encoding FGFRs (FGFR1–4) (2–4). FGFRs and FGFs along with heparan sulfate proteoglycans play pivotal roles in regulating tissue metabolism, cellular proliferation and differentiation, and organismal development in metazoans by transducing cell-to-cell communication as well as long range tissue-to-tissue communications (5, 6). The classical examples of FGF/FGFR interaction are between FGFR1 and high affinity ligands such as FGF1 (acidic FGF), FGF2 (basic FGF), or FGF8 that work in paracrine or autocrine manner (5, 6). In addition, some FGFR splice isoforms (1c, 2c, 3c, and 4) interact with membrane-bound coreceptor proteins, Klotho or βKlotho (KLB), to form receptor complexes for the endocrine FGFs, FGF19, -21, and -23, to regulate bile acid, energy, and phosphate metabolism, respectively (3, 7–11). Notably, gain- and loss-of-function mutations in FGFRs are implicated in a variety of pathological conditions in humans (12–15). Thus, FGFRs are attractive targets for therapeutic interventions for various pathological conditions (3, 12, 16, 17).
The composition of FGFR proteins was first revealed over 25 years ago with the cloning of a full-length cDNA encoding the chicken FGFR1 (18). Like other RTKs, each FGFR molecule contains an N-terminal extracellular domain (ECD), a single-pass transmembrane domain (TMD), and a C-terminal intracellular tyrosine kinase domain (TKD) (1). The FGFR ECD contains three immunoglobulin (Ig)-like domains, called D1, D2, and D3 (3, 4). It is generally postulated that ligand-induced dimerization of ECD would lead to changes in intracellular kinase conformation, allowing trans-phosphorylation to occur (3, 4). Of the three Ig-like domains in the ECD, the N-terminal D1 is not required for ligand binding or receptor activation but is proposed to have a role in receptor autoinhibition by preventing ligand-dependent and -independent dimerization (19, 20). D2 and D3 are responsible for ligand binding and interaction with sulfated oligosaccharides and mediate receptor dimerization. X-ray crystal structures revealed the formation of 2:2 FGF·FGFR-ECD complexes in which each FGF ligand simultaneously binds to two FGFR ECD at the interface between D2 and D3, resulting in the stabilization of the 2:2 FGF·FGFR-ECD complex (21, 22). The 2:2 FGF·FGFR-ECD complex is further stabilized by binding of sulfated oligosaccharide to the basic canyon-like structure made by the two D2 molecules (23). Interestingly, interaction between the two FGFR1-ECD molecules is limited to a small patch (∼300 Å2) in D2, which involves only four amino acid residues (22). The reported x-ray crystal structures of FGFRs thus explain the observed lack of stable ligand-independent dimerization of purified FGFR-ECD proteins in solution (21–23) and support the role for ligand-induced dimerization in receptor activation (4).
Whereas dimerization of isolated ECD clearly requires ligand binding, the behavior of full-length receptors expressed on the cell surface may be more complex. The movement of membrane proteins is confined to two-dimensional space, allowing molecules to form a complex even with weak interactions. The presence of heparin sulfate proteoglycan on the cell surface could also stabilize the ECD dimer. In addition, not only the ECD but also other parts of FGFR1 may have propensity to dimerize. Dimerization of isolated TMD in liposomes has been observed (24, 25), and TKD could also contribute to receptor dimerization via interaction with intracellular signaling proteins (26, 27). Indeed, very little is known about the cell surface dynamics of full-length FGFRs. For similar reasons, cell surface interaction between FGFR and membrane-bound coreceptor Klotho or KLB are not clearly understood. For example, FGFR1 and KLB can be co-immunoprecipitated, suggesting a formation of ligand-independent complex (9, 10). However, surface plasmon resonance sensorgrams showed that interaction between FGFR ECD and KLB ECD is very weak unless stabilized by FGF21 (28).
One emerging experimental method to monitor behaviors of transmembrane proteins is time-resolved Förster resonance energy transfer (TR-FRET) in combination with SNAP tag (ST)- and ACP tag-mediated fluorescent labeling (29–32). SNAP and ACP tags are self-labeling protein tags that allow for covalent labeling of fusion proteins with diverse chemicals such as non-cell permeable fluorophores (33, 34). It is then possible to detect a direct interaction between two cell-surface proteins carrying a donor and an acceptor of energy by FRET. In contrast to bioluminescence resonance energy transfer or other classical FRET assays performed with fluorescent proteins, TR-FRET relies on a rare earth lanthanide cryptate as a donor of energy. Due to its unique intrinsic properties, the TR-FRET signal is independent of the relative orientation between the donor and acceptor of energy (35, 36), and the efficiency of the energy transfer is only dependent on the distance between the fluorophores. In addition, the lanthanide cryptate presents a long-lasting fluorescence, allowing the measurement of the TR-FRET signal after a time delay (50 μs) and thus avoiding the shorter-lived nonspecific fluorescent signals. These features along with optimal spectral properties make this system more sensitive and robust compared with classical FRET (37).
Here we adapted this technology to study human (h) FGFR1 dimerization on the cell surface. Our results establish that FGFR1 forms a density-independent preformed dimer. Unexpectedly, TMD and TKD, rather than ECD, are important for the ligand-independent dimerization at the cell surface. Together with additional findings using ligand addition and mutagenesis, our study provides novel insights into the assembly and dynamics of the FGFRs on the surface of living cells.
Experimental Procedures
Recombinant Proteins and Other Cell Culture Reagent
hFGF7, hFGF10, hFGF1, hFGF2, and hFGF21 were purchased from R&D Systems (Minneapolis, MN). Anti-FGFR1 blocking Fab (R1MAb2) was produced in Escherichia coli using standard methods. Epitope characterization of R1MAb2 has been described (17). Heparin and heparinases I, II, and III were purchased from Sigma.
Plasmid Construction
The sequences encoding the SNAP tag and ACP tag were amplified by PCR from the pT8-SNAP (Cisbio, Codolet, France) and the pACP-tag(m)-2 (New England BioLabs, Ipswich, MA), respectively, to produce a CMV-based vector pRK.FLAG.SNAP or pRK.HA.ACP vectors. hFGFR1c, hFGFR1b, hEGFR and hKLB genes were PCR-amplified and cloned into these vectors to express N-terminally tagged proteins. To generate hFGFR1c-ΔTKD constructs, an octahistidine tag and stop codon were introduced immediately upstream of TKD. The resulting protein encodes the following C-terminal amino acid sequence: -IPLRRQVTHHHHHHHH. For hFGFR1c-TMD-KLB constructs, a stretch encompassing TMD (-LYLEIIIYCTGAFLISCMVGSVIVY-) containing both Tyr-372 and Cys-379 (underlined) was replaced by a stretch in hKLB of the same length (25 amino acids) encompassing TMD (-LIFLGCCFFSTLVLLLSIAIFQRQK-).
Cell Culture and Transfection
COS7 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS (Hyclone, Logan, UT). Cells were transiently transfected using Lipofectamine 2000 according to the manufacturer's instructions (Life Technologies, Inc.).
TR-FRET between SNAP Tags or between the SNAP Tag and ACP Tag
COS7 cells were co-transfected with either SNAP-tagged or ACP-tagged hFGFR1 and hKLB and seeded in a white-bottom 96-well plate (Costar, Tewksbury, MA) at 100,000 cells per well. For SNAP labeling, cells were labeled at 24 h post-transfection by incubating with 100 nm donor-conjugated benzyl-guanine SNAP-Lumi4-Tb (Cisbio) and 1 μm acceptor-conjugated benzyl-guanine SNAP-A467 (New England BioLabs) diluted in DMEM containing fetal bovine serum for 1 h at 37 °C. After 3 washes in PBS, the Lumi4-Tb emission and the TR-FRET signal were recorded at 620 and 665 nm, respectively, for 400 μs after a 60-μs delay after laser excitation at 343 nm using a Safire2 plate reader (Tecan, San Jose, CA). The emission signal of the A647 was detected at 682 nm after excitation at 640 nm using the same plate reader. For ligand-induced dimerization experiments, the TR-FRET signal was recorded at t = 0 and t = 15 min after ligand addition. The TR-FRET intensity was calculated as follows: (signal at 665 nm from cells labeled with SNAP donor and SNAP acceptor) − (signal at 665 nm from the same population of transfected cells labeled with SNAP donor and non labeled SNAP). The TR-FRET ratio represents the TR-FRET intensity divided by the donor emission at 620 nm. For SNAP-ACP labeling, cells were incubated with a 200 nm concentration of donor-conjugated benzyl-guanine SNAP-Lumi4-Tb (Cisbio) for 1 h at 37 °C, washed twice in PBS, and subsequently labeled with a 3 μm concentration of acceptor-conjugated coenzyme A CoA-A647 (New England BioLabs) in DMEM, 10 mm MgCl2, 50 mm Hepes, 1 μm Sfp phosphopantetheinyl transferase (New England BioLabs) for 1 h at 37 °C. In this case TR-FRET intensity was calculated as: (signal at 665 nm from cells labeled with SNAP donor and ACP acceptor) − (signal at 665 nm from the same population of transfected cells labeled with SNAP donor only).
Western Blot Analysis
COS7 cells were plated in 96-well plates at 10,000 cells per well. The next day cells were transfected using Lipofectamine 2000 according to the manufacturer's instructions. Plasmids encoding wild type and mutants hFGFR1c were transfected at concentrations that were determined to generated equivalent levels of cell surface receptor expression. Twenty-four hours after transfection cells were lysed in buffer containing 150 nm NaCl, 20 mm Tris (pH 7.5), 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, and 0.1% SDS supplemented with phosphatase and protease inhibitors. 5 μg of lysates were loaded on each lane of a 3–12% Bis-Tris gel, separated by electrophoresis, and transferred to nitrocellulose membranes (Life Technologies). Membranes were immunoblotted with anti-phospho ERK1/2 or anti-ERK1/2 (Cell Signaling Technology, Danvers, MA).
Enzyme-linked Immunosorbent Assay (ELISA)
ELISA on intact cells was performed as previously described (32). Briefly, cells were fixed with 4% paraformaldehyde, washed twice, and blocked in phosphate-buffered saline containing 1% FBS. Cells were then incubated with anti-HA monoclonal antibody (clone 3F10, Roche Applied Science) or anti-FLAG M2 monoclonal antibody (Sigma), both conjugated with horseradish peroxidase. After washing, cells were incubated with a SuperSignal ELISA substrate (Pierce), and chemiluminescence was detected on a Safire2 plate reader (Tecan, San Jose, CA). Specific signal was calculated by subtracting the signal recorded on mock-transfected cells.
GAL-ELK1 Luciferase Reporter Assay
COS7 cells were transfected with expression vectors encoding hFGFR1 and/or hKLB, Renilla luciferase (pRL-SV40, Promega, Madison, WI), a transcriptional activator (pFA2-ELK1, Stratagene, La Jolla, CA), and a firefly luciferase reporter gene driven by GAL4 binding sites (pFR-luc, Stratagene) (17). 24 h after transfection, culture medium was replaced by fresh serum-free media containing FGF ligand at various concentrations. After 6–8 h, the cellular activity was determined using a Dual-Glo Luciferase Assay System (Promega) and an EnVision Multilabel Reader (PerkinElmer Life Sciences) as previously described (17). Firefly luciferase activity was normalized to Renilla luciferase activity and expressed as relative luciferase unit.
Statistical Analysis
Statistical tests were conducted using either unpaired two-tailed Student's t test or one-way analysis of variance with post-hoc Dunnett's test. Error bars depict the S.E.). A p value of <0.05 was considered to be statistically significant.
Results
Unliganded FGFR1 Exist as Dimers on Cell Surface
To evaluate the dimerization state of hFGFR1 on the cell surface, we generated N-terminally SNAP-tagged hFGFR1b and hFGFR1c constructs (ST-hFGFR1b and ST-hFGFR1c). The presence of the tag did not interfere with cell surface protein expression (Fig. 1A) and hFGF2-induced signal activation (Fig. 1B), as assessed by FACS and using a GAL-ELK1 luciferase reporter assay, respectively (17). We then expressed increasing amounts of ST-hFGFR1b or ST-hFGFR1c in COS7 cells and labeled the tagged proteins with a mixture of non-cell-permeable SNAP substrates conjugated with fluorophores compatible with TR-FRET (Fig. 1C). We chose Lumi4-Tb and A647 as the donor and acceptor, respectively, because of their excellent spectral compatibilities and their Förster radius (R0) equal to 58 Å (32), which is consistent with the range of distance between two FGFR molecules in the reported crystal structures of ligand-bound FGFR dimers (21–23). The concentration of each substrate was optimized to ensure an equivalent labeling of ST-hFGFR1 with each fluorophore in order to reach the maximal TR-FRET efficiency (Fig. 1, D and E).
FIGURE 1.

Optimization of the SNAP-tag-based TR-FRET conditions. A, flow cytometry histograms (left) and the median fluorescent intensity (MFI) (right) showing binding of anti-hFGFR1 antibody on the surface of COS7 cells expressing untagged hFGFR1c, ST-hFGFR1c, or ST-hFGFR1b. B, COS7 cells expressing the indicated hFGFR1 constructs along with FA2-Elk1, SV40-Renilla luciferase, and a GAL-responsive firefly luciferase reporter gene were stimulated with increasing concentrations of hFGF2 for 6 h. The transcriptional activity was assessed in triplicate using a luciferase assay. C, schematic representation of the TR-FRET experiment design to study receptor homodimerization. D and E, COS7 cells expressing a constant amount of ST-hFGFR1c were labeled with 100 nm SNAP-Lumi4-Tb (donor) and increasing concentrations SNAP-A647 (acceptor). The fluorescence emissions of the donor and acceptor were recorded at 620 and 682 nm, respectively, and plotted according to the log concentration of SNAP-A647 (D). The TR-FRET signal was also measured in parallel (E). A, B, D, and E, data are representative of three independent experiments and are shown as the means ± S.E.
Using this optimized experimental setting, we measured a significant TR-FRET signal for both hFGFR1b and hFGFR1c isoforms, which was proportional to their cell surface expression (Fig. 2A). Notably, the TR-FRET ratio, which represents the TR-FRET intensity normalized by the receptor expression as measured by the donor emission, was constant over a wide range of receptor density (Fig. 2B). This suggests a specific and density-independent protein-protein interaction rather than a signal arising from random collisions due to receptor overexpression. The range of hFGFR1 levels expressed by transfected COS7 cells was comparable with less than that of the CAL120 cell line endogenously expressing hFGFR1 (Fig. 2, C and D), suggesting that the observed interactions do not originate from an artificial clustering due to overexpression but, rather, have a biological relevance.
FIGURE 2.

Density-independent dimerization of hFGFR1 at the cell surface. A, TR-FRET intensity measured on COS7 cells expressing increasing amounts of SNAP-tagged proteins ST-hFGFR1c (black circle), ST-hFGFR1b (blue square), or ST-hFGFR1c-ΔD1 (red triangle) plotted against the expression level of the SNAP-tagged receptor (ExpST), as measured by the donor emission at 620 nm. TR-FRET intensity and donor emission are expressed as counts per second (c.p.s.). B, comparison of TR-FRET ratio between ST-hFGFR1c, ST-hFGFR1b or ST-hFGFR1c-ΔD1 as a function of ST-hFGFR1 expression level, as measured by the donor emission at 620 nm. For A and B, data are the mean ± S.E. of three independent experiments (n = 3). C and D, flow cytometry histograms (left) and MFI (right) representative of hFGFR1 expression by COS7 cells transfected with increasing amounts of ST-hFGFR1c DNA (C) or by untransfected CAL120 cells (D). Data are representative of three independent experiments.
hFGFR1c construct lacking D1 and the flexible linker between D1 and D2 (ST-hFGFR1c-ΔD1) also exhibited density-independent TR-FRET ratios that were higher than the full-length construct (Fig. 2, A and B). This observation is consistent with the previously proposed autoinhibitory role of D1 (19, 20). Alternatively, the increased TR-FRET ratio observed with the hFGFR1c-ΔD1 could be due to a shorter distance between two N termini in the resulting homodimer. Although we cannot distinguish between these possibilities, the results above support the use of N-terminal SNAP tag to study hFGFR1 dimerization by TR-FRET.
To evaluate the specificity of the observed interactions, we sought to compare the TR-FRET signal resulting from hFGFR1c homo-interaction with that from hetero-interaction between hFGFR1c and hEGFR, which is not expected to occur to a significant degree. To that aim we designed an orthogonal labeling strategy. A constant amount of SNAP-tagged receptor was co-expressed with increasing amounts of ACP-tagged receptor, and the tags were labeled with non-permeable SNAP-Lumi-4Tb and coenzyme A-A647 (Fig. 3A). In this experimental setting, a specific protein-protein interaction would give rise to a saturation curve, as the increasing amount of acceptor-labeled ACP-tagged protein progressively associates with the constant amount of donor-labeled-ST protein until reaching a 1:1 donor:acceptor ratio, at which point the TR-FRET ratio becomes saturated (31). In contrast, a nonspecific interaction due to random collisions would yield only a linear increase of the TR-FRET ratio as the abundance of ACP-tagged protein increases. As expected from a specific protein-protein interaction, we observed similar saturation curves for ST-hFGFR1c/ACP-hFGFR1c (FRETmax = 8.00 ± 0.18, FRET50 = 1.99 ± 0.11, and R2 = 0.96) and ST-hEGFR/ACP-hEGFR combinations (FRETmax = 10.27 ± 0.68, FRET50 = 3.28 ± 0.48, and R2 = 0.97) (Fig. 3B). In contrast, no saturation of the TR-FRET ratio was obtained for the ST-hFGFR1c/ACP-hEGFR combination. Thus, the self-association of FGFR1c on the cell surface is specific and as efficient as the well documented ligand-independent EGFR dimerization (38–40).
FIGURE 3.

Characterization of hFGFR1 preformed dimer. A, schematic representation of the TR-FRET experiment design for an orthogonal labeling strategy. B, TR-FRET saturation curves of ST-hFGFR1c with ACP-hFGFR1c (black) or ST-hEGFR in combination with either ACP-hEGFR (red) or ACP-hFGFR1c (blue). COS7 cells were co-transfected with a constant amount of ST-hFGFR1c or ST-hEGFR and increasing amounts of ACP-hFGFR1c or ACP-hEGFR (ACP-RTK). After labeling, the TR-FRET signal along with the expressions of SNAP (ExpST)- and ACP- (ExpACP)-tagged constructs, given by the fluorescence emission of each fluropohore, were measured. The TR-FRET ratio is shown as a function of the ExpACP/ExpST ratio. C, COS7 cells were transfected with reporter constructs along with expression vectors encoding hFGFR1c and/or hFGF8b to activate hFGFR1c in an autocrine manner. Cells were then incubated for 6 h with 100 μg/ml concentrations of either blocking anti-FGFR1 Fab (hatched bar) or unrelated control Fab (black bar), and transcriptional activity was assessed using a luciferase assay. RLU, relative light units. D, TR-FRET ratio between ST-FGFR1c measured on COS7 cells treated with PBS, 100 milliunits of heparinase I, II, or III for 3 h at 37 °C before TR-FRET reading. E, TR-FRET ratio between ST-hFGFR1c after the addition of PBS (white bars) or 1 μg/ml heparin (red bars). TR-FRET ratio was recorded 5 min and 15 min after each treatment and normalized to the signal obtained after PBS addition. For A–E, representative data of four independent experiments are shown as the means ± S.E. ** = p < 0.01, ns = no significance.
The above results strongly suggest that FGFR1 can exist as preformed dimers in the absence of FGF ligand addition. To address whether COS7 cells secrete FGF agonists that can act in an autocrine manner to induce FGFR1 dimerization, we measured FGFR1 activity using a GAL-ELK1 luciferase assay in the presence and absence of an anti-FGFR1 blocking Fab (R1MAb2) that binds to the ligand binding site within D2 (17). When cells were transfected to produce hFGF8b, anti-FGFR1 Fab exhibited an ability to suppress hFGF8b-induced luciferase activity, demonstrating the anticipated blocking activity against the hFGFR1-hFGF8b interaction. However, anti-FGFR1 failed to decrease basal luciferase activity (Fig. 3C). Thus, COS7 cells do not release a significant amount of FGFR1 agonist.
We also studied the possible influence of sulfated oligosaccharides on the ligand-independent FGFR1 dimerization. First, we pretreated COS7 cells with heparinase I, II, or III to digest heparan sulfate accessible on the surface of cells before performing the TR-FRET assay. None of these treatments significantly altered the TR-FRET ratio between ST-hFGFR1c (Fig. 3D). Second, the addition of heparin had no effect on the observed hFGFR1c dimerization (Fig. 3E). Thus, we found no evidence for the role of sulfated oligosaccharides in ligand-independent hFGFR1c dimerization on the surface of COS7 cells.
FGFR1 Ligands and Activating Mutations Stabilize the Homodimers and/or Induce a Conformational Change within the Pre-existing Dimers
Next, we performed a TR-FRET homo-interaction assay to test the effects of different FGF ligands. The addition of hFGF1 or hFGF2 to COS7 cells expressing a constant amount of ST-FGFR1c significantly increased TR-FRET signal in a concentration-dependent manner (Fig. 4, A and B). The ligand-induced increases of the TR-FRET signal were observed within 5 min of ligand addition and did not further increase beyond this incubation time. No increase in signal was measured with hFGF7 and hFGF10, two FGF ligands known to have no affinity for FGFR1c (2) (Fig. 4, A and B).
FIGURE 4.
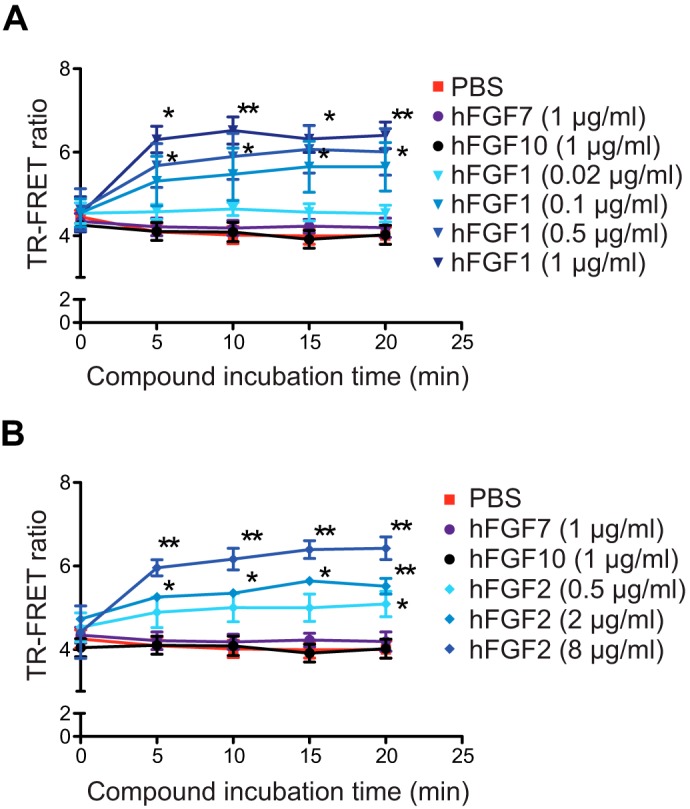
TR-FRET monitoring of ligand-induced hFGFR1c dimerization. A and B, TR-FRET variation kinetics measured on COS7 cells expressing a constant amount of ST-hFGFR1c. The indicated ligands were added to the cells at t = 0 after labeling of ST-hFGFR1c. Representative data of four independent experiments are shown as the means ± S.E. ** = p < 0.01; * = p < 0.05.
In addition to FGF ligand addition, we also evaluated the effect of activating mutations on the dimerization signal. Germ line-activating mutations located in ECD (N330I) and TMD (C379R) cause an autosomal dominant disorder called osteoglophonic dysplasia (13) (Fig. 5A). Analogous mutations were also found in FGFR2 and -3 in various human genetic syndromes and in human tumor samples (14, 41, 42). To compare the signaling properties of wild type (WT) and mutant FGFR1c constructs, we expressed each of them as a SNAP-tagged protein. The amount of transfected DNA for each construct was adjusted to achieve equivalent expression levels on the cell surface, as measured by cell-based ELISA (Fig. 5B). Under this condition, each mutant construct exhibited a high degree of constitutive activity, as shown using the GAL-ELK1 luciferase assay (Fig. 5C) or by analysis of ERK1/2 phosphorylation using Western blot (Fig. 5D). The constitutive activity observed for the mutants C379R and N330I was correlated with an increased TR-FRET signal as compared with the WT when expressed at similar levels (Fig. 5, E and F).
FIGURE 5.

Impact of activating mutations on hFGFR1c preformed dimer. A, topological organization of FGFR1c. The alternatively spliced region in D3 is indicated in brown. The red box represents the acidic box. The locations of the activating mutations examined are indicated by arrows. B, cell surface expression of hFGFR1 assessed by anti-FLAG ELISA. A FLAG tag is present on the N terminus of the SNAP tag. Data are expressed as arbitrary units (a.u.). C, COS7 cells expressing the indicated ST-hFGFR1 constructs along with the GAL-ELK1:luciferase reporter. Luciferase activity is shown as relative luciferase units (RLU). D, Western blot analysis of ERK phosphorylation in COS7 cells transfected with WT or mutated hFGFR1c constructs. Each construct was tested in duplicate. E, cell surface expression of the indicated tagged hFGFR1 constructs, as measured by SNAP-donor emission. F, TR-FRET ratio measured on COS7 cells expressing the indicated ST-hFGFR1 constructs. G, COS7 cells expressing the indicated ST-hFGFR1 constructs along with the GAL-ELK1:luciferase reporter. Luciferase activity is shown as relative luciferase units. H, cell surface expression of hFGFR1 assessed by anti-FLAG ELISA. Data are expressed as arbitrary units. I, cell surface expression of the indicated tagged hFGFR1 constructs, as measured by SNAP-donor emission. J, TR-FRET ratio measured on COS7 cells expressing the indicated ST-hFGFR1 constructs. In B–E and G–I), data are representative of three independent experiments, each performed in triplicate and shown as the means ± S.E. For F and J, data are the means ± S.E. of six independent experiments (n = 6). *** = p < 0.001; ns = no significance.
In addition to the constitutive activating mutants in ECD and TMD, we also generated two ST-FGFR1c constructs with constitutively active mutations in the TKD, N546K, or K656E (Fig. 5A). These mutations are infrequently found in malignant cells (43, 44) and are thought to affect the conformational dynamics of the kinase domain and increase intrinsic kinase activity (45, 46). As expected, N546K and K656E mutants showed strong constitutive activity for a similar cell-surface expression level (Fig. 5, G and H). However, for an equivalent expression level (Fig. 5I), the TR-FRET signals were similar for the mutants N546K, K656E, and the wild type, suggesting that these activating mutations in the TKD do not affect the basal level of FGFR1c homodimerization (Fig. 5J).
TMD and TKD Play a Major Role in FGFR1c Ligand-independent Dimerization
Previous studies suggested that each of the ECD, TMD, and TKD plays a role in ligand-independent dimerization via direct protein-protein interactions or indirect interactions with heparin or intracellular signaling proteins (25, 26, 47, 48). We decided to test this notion using the TR-FRET system. First, we generated a ST-FGFR1c construct in which the TMD was replaced with that of the coreceptor KLB (ST-FGFR1c-TMD-KLB), whereas maintaining its length (Fig. 6A). The TR-FRET efficiency for WT and TMD-KLB constructs was similar, suggesting that a specific interaction between the two FGFR1 TMD is not essential for ligand-independent FGFR1 dimerization (Fig. 6B). The ST-FGFR1c-ΔTKD construct lacking the entire TKD (Fig. 6A) also displayed a density-independent TR-FRET ratio (Fig. 6C), suggesting that TKD is also dispensable for the ligand-independent FGFR1 dimerization. In contrast to the two aforementioned constructs, the TR-FRET ratio for the ST-FGFR1c-TMD-KLB-ΔTKD construct lacking the TKD and harboring KLB TMD in place of WT TMD (Fig. 6A) increased proportionally to its expression and was weak at low density of receptors (Fig. 6C). This density-dependent TR-FRET ratio indicates nonspecific interactions likely due to random collisions. Thus, unlike a previous proposal (48), hFGFR1 ECD alone is not sufficient to support specific dimerization when tethered to a plasma membrane via a heterologous TMD. Instead, hFGFR1 TMD and TKD play a redundant role in FGFR1c ligand-independent dimerization.
FIGURE 6.

Structure-function analysis of basal hFGFR1c homo-dimerization. A, schematic representation of WT and mutant hFGFR1c receptor constructs. TMD from hFGFR1c and hKLB are represented in black and red, respectively. B and C, TR-FRET ratio between each construct as a function of ST-hFGFR1 expression level. Data are representative of four independent experiments, each performed in quadruplicate and shown as the means ± S.E.
FGFR1c and KLB Form a Heterotetrameric Complex in the Absence of FGF21
Next, we decided to test the physical interactions between FGFR1c and the coreceptor KLB on the cell surface in the absence or the presence of the natural ligand FGF21. We first ensured that the cell-surface expression and activity of SNAP-tagged hKLB (ST-hKLB) were similar to that of WT hKLB (Fig. 7, A and B). Then we applied the orthogonal labeling strategy to test hFGFR1c·hKLB interaction. In this experiment, a fixed amount of ST-hKLB was co-expressed with increasing amounts of ACP-tagged hFGFR1c or hEGFR in COS7 cells. Transfected cells were then incubated with SNAP-Lumi-4Tb and coenzyme A-A647 for labeling of the SNAP and ACP tags, respectively, before TR-FRET reading. We obtained a saturation curve for hKLB and hFGFR1c (FRETmax = 4.92 ± 0.27, FRET50 = 0.49 ± 0.065, and R2 = 0.93), indicative of a specific and saturable interaction (Fig. 7C). In contrast, co-expression of ST-hKLB with ACP-hEGFR gave rise to a non-saturating curve expected for a nonspecific interaction due to random collisions (Fig. 7C). The blocking anti-FGFR1 Fab reduced luciferase activity in the GAL-ELK1 luciferase assay when cells were transfected to produce hFGF21 (Fig. 7D). However, it failed to decrease basal activity (Fig. 7D); thus, the observed hFGFR1c·hKLB association in COS7 cells is likely ligand-independent.
FIGURE 7.

Analysis of hFGFR1c-hKLB interaction on the cell surface. A, cell surface expression of hKLB assessed in the presence of the indicated FGFR1 constructs by anti-FLAG ELISA. A FLAG tag is present on the N terminus of the KLB constructs. Data are expressed as arbitrary units (a.u.). B, COS7 cells expressing the indicated hFGFR and hKLB constructs along with the GAL-ELK1:luciferase reporter were stimulated with increasing concentrations of hFGF21 for 6 h. The transcriptional activity was assessed using a luciferase assay. RLU, relative light units. C, TR-FRET saturation curves of ST-hKLB with ACP-hFGFR1c or ACP-hEGFR. COS7 cells were co-transfected with a constant amount of ST-hKLB (5 ng: light blue or gray; 10 ng, medium blue or gray; 20 ng, dark blue or gray) and increasing amounts of ACP-hFGFR1c or ACP-hEGFR. After labeling, the TR-FRET signal along with the expression of ST-hKLB (ExpST) and ACP-hRTK (ExpACP), given by the fluorescence emission of each fluropohore, were measured. TR-FRET ratio is shown as a function of the ExpACP/ExpST ratio. D, COS7 cells were co-transfected with expression vectors encoding hFGFR1c and hKLB along with GAL-ELK1:luciferase reporter. Some cells were also transfected with an expression vector encoding hFGF21 as indicated to activate FGFR1c·KLB complex in an autocrine manner. Cells were then incubated for 6 h with 100 μg/ml concentrations of either blocking anti-FGFR1 Fab (hatched bar) or unrelated control Fab (black bar), and transcriptional activity was assessed using a luciferase assay. E, competition experiment between ST-hFGFR1c expressed at a constant level and labeled with TR-FRET donor and acceptor and increasing amounts of unlabeled ACP-hKLB. The x axis represents the expression level of ACP-hKLB measured by anti-HA ELISA. F, cell surface expression of ST-hFGFR1 and ACP-hKLB in E, assessed by anti-FLAG (for ST-FGFR1) or anti-HA (for ACP-hKLB) ELISA. Data are expressed as arbitrary units. G and H, TR-FRET ratio between indicated proteins after incubation with PBS (white bars) or hFGF21 (green bars) for 15 min. TR-FRET ratio is normalized to the TR-FRET signal obtained in presence of PBS. Data in C–E and G–H are the means ± S.E. of three independent experiments (n = 3). Data in A–B and F are representative of three independent experiments and shown as the means ± S.E. *** = p < 0.001; ns = no significance.
To better understand how the hFGFR1c·hKLB complex is organized on the cell surface, we monitored the TR-FRET signal between labeled ST-hFGFR1c protomers in the presence of increasing amounts of non-labeled hKLB. The TR-FRET ratio remained constant independently of hKLB expression, indicating that hKLB does not promote or interfere with the basal level of hFGFR1 homodimerization (Fig. 7, E and F). Altogether, these results are consistent with a heteromeric organization composed of a central FGFR1c dimer flanked by one or two KLB proteins.
We also assessed the ability of hFGF21 to modulate hFGFR1c·hKLB hetero-interaction or hFGFR1 homo-interaction. We first analyzed the effect of hFGF21 on the TR-FRET signal between ST-hFGFR1c and ACP-hKLB (Fig. 7G). FGF21 did not alter the TR-FRET signal between these two proteins, consistent with the idea that hFGFR1c and hKLB heterodimer is not impacted or does not undergo any significant conformational change in the presence of hFGF21. However, hFGF21 enhanced the level of the TR-FRET signal observed for the interaction between ST-hFGFR1c protomers in the presence of hKLB (Fig. 7H). No such modulation was observed for ST-hFGFR1b, consistent with hFGF21 being unable to signal via hFGFR1b. These data together support a model in which FGFR1c·KLB would pre-exist as a trimetric or tetrameric complex on the cell surface even in the absence of FGF21 ligand, and FGF21 would activate this complex likely by further stabilizing or promoting FGFR1c/FGFR1c interaction within the heteromeric complex (Fig. 8).
FIGURE 8.
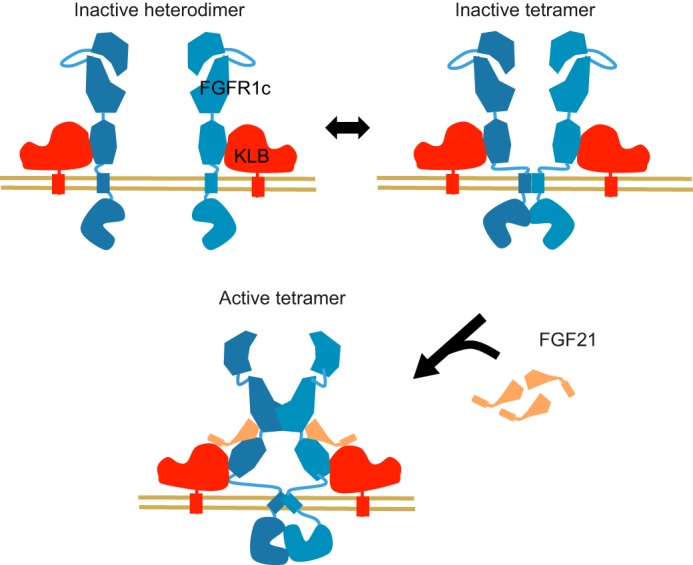
Proposed model for the organization and activation of FGFR1c·KLB receptor complex at the cell surface. In the absence of agonist ligands, FGFR1c has the ability to form homodimers but most likely oscillates between inactive monomeric and dimeric forms. Ligand-independent FGFR1c homodimerization is mediated by the TMD and through (direct or indirect) associations via TKD. In addition, FGFR1c forms a stable, ligand-independent heterodimer with KLB. Ligand-induced homodimerization of FGFR1c ECD in the presence or in absence of KLB induces a conformational rearrangement in TMD, allowing for the formation of an asymmetric active TKD dimer and subsequent transphosphorylation.
Discussion
Although numerous biochemical and structural studies revealed dimerization-mediated activation mechanisms of various RTKs including FGFRs, the details of the mechanism are still elusive. Two prevalent conceptual models, a “diffusion-based model” and a “preformed dimer model,” have been proposed for ligand-mediated activation of FGFRs (27, 49). In the classical diffusion-based model, a receptor mostly exists as a monomer, freely diffusing in the plasma membrane lipid bilayer, and ligand binding induces receptor dimerization and activation by simply placing two cytoplasmic kinase domains into sufficient proximity, allowing transphosphorylation to occur. This model matches with the previously observed ligand-independent FGFR activation by artificial dimerizers (50, 51). In the preformed dimer model, binding of a ligand to a preformed dimer would induce a conformational change in the cytoplasmic kinase domain complex, leading to the formation of active kinase domain conformation (38, 39). EGFR is a classical example of an RTK that exists as both a monomer and ligand-independent dimer, and both of the aforementioned mechanisms are likely being employed for this receptor (38–40, 52). By combining SNAP and ACP tags with TR-FRET technology, we demonstrate here that, like EGFR, full-length FGFR1 can efficiently form ligand-independent oligomers, most likely dimers, on the cell surface. This confirms the previously proposed but rather controversial idea that FGFR1 forms preformed dimers in the absence of ligand (48, 53). Structure-function analysis supports the role for TMD and TKD, rather than ECD, in ligand-independent FGFR1 dimerization.
There have been previous attempts to use FRET to study FGFR dimerization in the context of plasma membrane (25, 54, 55). Because our approach utilizes non-cell-permeable fluorescent probes, it enabled a FRET-based approach to study FGFR interactions specifically on the cell surface in the context of physiological membrane environment and in the presence of intracellular scaffolding proteins that might affect FGFR dimerization. Importantly, our approach utilizes full-length FGFR composed of ECD, TMD, and TKD, unlike previous FRET-based studies using shorter fragments lacking TKD (25, 54, 55). The importance of this was revealed by our structure-function analysis that supports the role for both TMD and TKD in ligand-independent FGFR1 dimerization. In addition, our approach enabled correlation of receptor dimerization and signaling activity, which was not possible with methods utilizing fragments lacking TKD. A limitation of our approach (and other FRET-based approaches) is that a TR-FRET signal increase caused by an activating ligand or an activating mutation is the result of a change in distance between fluorophores that could originate from either 1) stabilization of pre-existing interactions, 2) conformational change within pre-existing dimers, or 3) an increase of the dimeric versus monomeric population. Because we record the signal on a cell population over a defined period of time, we cannot discriminate between these three possibilities. Addressing this question would require a single molecule visualization approach, such as the quantum-dot-based optical tracking method that has been employed for EGFR (40). Also, it is important to note that we cannot determine the stoichiometry of the complexes formed, and thus we cannot discriminate between dimerization and oligomerization, although the previous studies support the formation of FGFR dimers.
A particularity of the FGFR signaling system is the involvement of heparan sulfate proteoglycans. FGFRs are likely associated with heparin sulfate chain of proteoglycan or free heparin on the cell surface (49). Heparan sulfate proteoglycan is indeed essential in ligand-induced activation of FGFRs, at least in part by increasing affinity of FGF ligands for FGFRs (56). In addition, heparin appears to have a ligand-independent role as well, as it can, for example, activate FGFR4, but not FGFR1 (57). A role for heparan sulfate/heparin in ligand-independent FGFR1 dimerization has also been proposed (49). However, under the condition of our experiments, FGFR1 ECD alone is not sufficient to support specific dimerization when tethered to a plasma membrane via a heterologous TMD. This is consistent with the conclusion by Chen et al. (25) that FGFR3 ECD inhibits rather than promotes ligand-independent dimerization when expressed on the surface of liposomes. Indeed, our structure-function analysis suggests that TMD and TKD play dominant and redundant roles in the formation of pre-existing dimers on the surface of COS7 cells.
Another complexity of FGFR signaling is the requirement of the co-receptor Klotho or KLB for activation by the endocrine FGFs (FGF19, -21, and -23) (7, 8). Previously, co-immunoprecipitation was used to demonstrate the ligand-independent formation of stable FGFR1c·KLB complex (9, 10, 58). However, a model has been recently proposed in which FGF21 would facilitate FGFR1c·KLB interaction by suppressing intramolecular interactions between FGFR1c D1 and the rest of the ECD (28). Stabilization of the interaction between FGFR1c and KLB by FGF21 was also demonstrated utilizing recombinant protein fragments (28). Using the cell-surface TR-FRET approach, we demonstrate here unequivocally that FGFR1c and KLB form a stable complex in the absence of FGF21. The addition of FGF21 does not affect the interaction between FGFR1c and KLB but increases the homo-interaction of FGFR1c on the cell surface in the presence of KLB. Thus, the ability of FGF21 to stabilizing the FGFR1-ECD/KLB-ECD interaction previously observed in solution may not have a major impact in the activation of FGFR/KLB signaling pathway.
There has been a previous attempt to study behaviors of C-terminally tagged full-length FGFR1c and KLB in the context of plasma membrane using a fluorescent imaging-based method (“the number and brightness analysis”) in living HeLa cells (53). This study by Ming et al. (53) has observed that full-length FGFR1c molecules exist as dimers in the absence of ligands, consistent with our conclusion. FGF21 further enhanced dimerization (i.e. >1.5-fold increase in relative brightness) of FGFR1 but only when KLB is present. The study also found that KLB existed as monomers in the absence of ligand addition and, oddly, that FGF21 addition induced dimerization of KLB only when FGFR1 was absent (but presumably in the presence of endogenously expressed FGFRs). Taking together, Ming et al. (53) suggested that FGF21 affects KLB in two independent ways; on one hand, FGF21 induces KLB dimerization without FGFR1, and on the other, FGF21 induces formation of an active 2:1 FGFR1·KLB signaling complex. Taking all the existing data together, we favor a model in which KLB and FGFR1c form a 2:1 FGFR1:KLB heterotrimeric complex or 2:2 FGFR1:KLB heterotetrameric complex in the absence of FGF21, depending on the abundance of KLB. KLB-dependent FGF21 binding to the receptor complex further increases FGFR1 dimerization, resulting in signal transduction (Fig. 8).
In conclusion, using TR-FRET combined with SNAP- and ACP-tag technologies, we provided compelling evidence for the existence of ligand-independent dimers of FGFR1 that is by and large independent of surface density. TMD and TKD, but not ECD, are responsible for this interaction. Activating ligands and some, but not all, activating mutations affect dimerization state and/or dimer conformation. In addition, our data demonstrate the presence of stable FGFR1c·KLB hetero-complex at the cell surface in the absence of FGF21. These results provide novel insights into the assembly and dynamics of the FGFRs in the context of a physiological membrane environment. Furthermore, we could envision extending this study to monitor FGFR dimerization in native tissues (e.g. tumor samples) and fully characterize the RTK activation mechanism beyond the FGFR protein family.
Author Contributions
L. C.-A., D. E., and J. S. designed the study and wrote the paper. L. C.-A. performed and analyzed TR-FRET assays and other cell based assays. D. R. D. performed the Western blots. J. S. produced plasmid constructs. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Seth Harris, Damien Maurel, and Katharina Stengel for helpful discussions. We also thank Michelle Chan and Mark Chen for help on plasmid vector construction. We acknowledge colleagues in Antibody Engineering, ES Cell Culture, and Protein Chemistry Departments for anti-FGFR1 antibody production.
This work was supported by Genentech, Inc. All the authors were employees of Genentech, Inc. The authors declare that they have no conflicts of interest with the contents of this article.
- FGFR
- fibroblast growth factor receptor
- hFGFR
- human FGFR
- RTK
- receptor-tyrosine kinase
- ECD
- extracellular domain
- TMD
- transmembrane domain
- TKD
- tyrosine kinase domain
- TR-FRET
- time-resolved FRET
- ST
- SNAP tag
- ST-hKLB
- SNAP-tagged hKLB
- KLB
- βKlotho
- hKLB
- human KLB
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl) propane-1,3-diol.
References
- 1. Lemmon M. A., Schlessinger J. (2010) Cell signaling by receptor-tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mohammadi M., Olsen S. K., Ibrahimi O. A. (2005) Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 16, 107–137 [DOI] [PubMed] [Google Scholar]
- 3. Beenken A., Mohammadi M. (2009) The FGF family: biology, pathophysiology, and therapy. Nat. Rev. Drug Discov. 8, 235–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goetz R., Mohammadi M. (2013) Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 14, 166–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Itoh N., Ornitz D. M. (2004) Evolution of the Fgf and Fgfr gene families. Trends Genet. 20, 563–569 [DOI] [PubMed] [Google Scholar]
- 6. Itoh N., Ornitz D. M. (2011) Fibroblast growth factors: from molecular evolution to roles in development, metabolism, and disease. J. Biochem 149, 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beenken A., Mohammadi M. (2012) The structural biology of the FGF19 subfamily. Adv. Exp. Med. Biol. 728, 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuro-o M. (2012) Klotho and βKlotho. Adv. Exp. Med. Biol. 728, 25–40 [DOI] [PubMed] [Google Scholar]
- 9. Kurosu H., Choi M., Ogawa Y., Dickson A. S., Goetz R., Eliseenkova A. V., Mohammadi M., Rosenblatt K. P., Kliewer S. A., Kuro-o M. (2007) Tissue-specific expression of βKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 282, 26687–26695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kharitonenkov A., Dunbar J. D., Bina H. A., Bright S., Moyers J. S., Zhang C., Ding L., Micanovic R., Mehrbod S. F., Knierman M. D., Hale J. E., Coskun T., Shanafelt A. B. (2008) FGF-21/FGF-21 receptor interaction and activation is determined by βKlotho. J. Cell Physiol. 215, 1–7 [DOI] [PubMed] [Google Scholar]
- 11. Kolumam G., Chen M. Z., Tong R., Zavala-Solorio J., Kates L., van Bruggen N., Ross J., Wyatt S. K., Gandham V. D., Carano R. A., Dunshee D. R., Wu A. L., Haley B., Anderson K., Warming S., Rairdan X. Y., Lewin-Koh N., Zhang Y., Gutierrez J., Baruch A., Gelzleichter T. R., Stevens D., Rajan S., Bainbridge T. W., Vernes J. M., Meng Y. G., Ziai J., Soriano R. H., Brauer M. J., Chen Y., Stawicki S., Kim H. S., Comps-Agrar L., Luis E., Spiess C., Wu Y., Ernst J. A., McGuinness O. P., Peterson A. S., Sonoda J. (2015) Sustained brown fat stimulation and insulin sensitization by a humanized bispecific antibody agonist for fibroblast growth factor receptor 1/βKlotho complex. EBioMedicine 2, 730–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Turner N., Grose R. (2010) Fibroblast growth factor signalling: from development to cancer. Nat. Rev. Cancer 10, 116–129 [DOI] [PubMed] [Google Scholar]
- 13. White K. E., Cabral J. M., Davis S. I., Fishburn T., Evans W. E., Ichikawa S., Fields J., Yu X., Shaw N. J., McLellan N. J., McKeown C., Fitzpatrick D., Yu K., Ornitz D. M., Econs M. J. (2005) Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am. J. Hum. Genet. 76, 361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kelleher F. C., O'Sullivan H., Smyth E., McDermott R., Viterbo A. (2013) Fibroblast growth factor receptors, developmental corruption and malignant disease. Carcinogenesis 34, 2198–2205 [DOI] [PubMed] [Google Scholar]
- 15. Du X., Xie Y., Xian C. J., Chen L. (2012) Role of FGFs/FGFRs in skeletal development and bone regeneration. J. Cell Physiol. 227, 3731–3743 [DOI] [PubMed] [Google Scholar]
- 16. Zhang J., Li Y. (2014) Fibroblast growth factor 21, the endocrine FGF pathway and novel treatments for metabolic syndrome. Drug Discov. Today 19, 579–589 [DOI] [PubMed] [Google Scholar]
- 17. Wu A. L., Kolumam G., Stawicki S., Chen Y., Li J., Zavala-Solorio J., Phamluong K., Feng B., Li L., Marsters S., Kates L., van Bruggen N., Leabman M., Wong A., West D., Stern H., Luis E., Kim H. S., Yansura D., Peterson A. S., Filvaroff E., Wu Y., Sonoda J. (2011) Amelioration of type 2 diabetes by antibody-mediated activation of fibroblast growth factor receptor 1. Sci. Transl. Med. 3, 113ra126. [DOI] [PubMed] [Google Scholar]
- 18. Lee P. L., Johnson D. E., Cousens L. S., Fried V. A., Williams L. T. (1989) Purification and complementary DNA cloning of a receptor for basic fibroblast growth factor. Science 245, 57–60 [DOI] [PubMed] [Google Scholar]
- 19. Kiselyov V. V., Bock E., Berezin V., Poulsen F. M. (2006) NMR structure of the first Ig module of mouse FGFR1. Protein Sci. 15, 1512–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Olsen S. K., Ibrahimi O. A., Raucci A., Zhang F., Eliseenkova A. V., Yayon A., Basilico C., Linhardt R. J., Schlessinger J., Mohammadi M. (2004) Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc. Natl. Acad. Sci. U.S.A. 101, 935–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Plotnikov A. N., Hubbard S. R., Schlessinger J., Mohammadi M. (2000) Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 101, 413–424 [DOI] [PubMed] [Google Scholar]
- 22. Plotnikov A. N., Schlessinger J., Hubbard S. R., Mohammadi M. (1999) Structural basis for FGF receptor dimerization and activation. Cell 98, 641–650 [DOI] [PubMed] [Google Scholar]
- 23. Schlessinger J., Plotnikov A. N., Ibrahimi O. A., Eliseenkova A. V., Yeh B. K., Yayon A., Linhardt R. J., Mohammadi M. (2000) Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 6, 743–750 [DOI] [PubMed] [Google Scholar]
- 24. Bocharov E. V., Lesovoy D. M., Goncharuk S. A., Goncharuk M. V., Hristova K., Arseniev A. S. (2013) Structure of FGFR3 transmembrane domain dimer: implications for signaling and human pathologies. Structure 21, 2087–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen L., Placone J., Novicky L., Hristova K. (2010) The extracellular domain of fibroblast growth factor receptor 3 inhibits ligand-independent dimerization. Sci. Signal 3, ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin C. C., Melo F. A., Ghosh R., Suen K. M., Stagg L. J., Kirkpatrick J., Arold S. T., Ahmed Z., Ladbury J. E. (2012) Inhibition of basal FGF receptor signaling by dimeric Grb2. Cell 149, 1514–1524 [DOI] [PubMed] [Google Scholar]
- 27. Belov A. A., Mohammadi M. (2012) Grb2, a double-edged sword of receptor-tyrosine kinase signaling. Sci. Signal. 5, pe49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yie J., Wang W., Deng L., Tam L. T., Stevens J., Chen M. M., Li Y., Xu J., Lindberg R., Hecht R., Véniant M., Chen C., Wang M. (2012) Understanding the physical interactions in the FGF21/FGFR/β-Klotho complex: structural requirements and implications in FGF21 signaling. Chem. Biol. Drug Des. 79, 398–410 [DOI] [PubMed] [Google Scholar]
- 29. Maurel D., Comps-Agrar L., Brock C., Rives M. L., Bourrier E., Ayoub M. A., Bazin H., Tinel N., Durroux T., Prézeau L., Trinquet E., Pin J. P. (2008) Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat. Methods 5, 561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Monnier C., Tu H., Bourrier E., Vol C., Lamarque L., Trinquet E., Pin J. P., Rondard P. (2011) Trans-activation between 7TM domains: implication in heterodimeric GABAB receptor activation. EMBO J. 30, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Doumazane E., Scholler P., Zwier J. M., Trinquet E., Rondard P., Pin J. P. (2011) A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J. 25, 66–77 [DOI] [PubMed] [Google Scholar]
- 32. Comps-Agrar L., Kniazeff J., Brock C., Trinquet E., Pin J. P. (2012) Stability of GABAB receptor oligomers revealed by dual TR-FRET and drug-induced cell surface targeting. FASEB J. 26, 3430–3439 [DOI] [PubMed] [Google Scholar]
- 33. Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H., Johnsson K. (2003) A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21, 86–89 [DOI] [PubMed] [Google Scholar]
- 34. George N., Pick H., Vogel H., Johnsson N., Johnsson K. (2004) Specific labeling of cell surface proteins with chemically diverse compounds. J. Am. Chem. Soc. 126, 8896–8897 [DOI] [PubMed] [Google Scholar]
- 35. Selvin P. R. (2002) Principles and biophysical applications of lanthanide-based probes. Annu. Rev. Biophys. Biomol. Struct. 31, 275–302 [DOI] [PubMed] [Google Scholar]
- 36. Zwier J. M., Bazin H., Lamarque L., Mathis G. (2014) Luminescent lanthanide cryptates: from the bench to the bedside. Inorg. Chem. 53, 1854–1866 [DOI] [PubMed] [Google Scholar]
- 37. Mathis G. (1995) Probing molecular interactions with homogeneous techniques based on rare earth cryptates and fluorescence energy transfer. Clin. Chem. 41, 1391–1397 [PubMed] [Google Scholar]
- 38. Arkhipov A., Shan Y., Das R., Endres N. F., Eastwood M. P., Wemmer D. E., Kuriyan J., Shaw D. E. (2013) Architecture and membrane interactions of the EGF receptor. Cell 152, 557–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Endres N. F., Das R., Smith A. W., Arkhipov A., Kovacs E., Huang Y., Pelton J. G., Shan Y., Shaw D. E., Wemmer D. E., Groves J. T., Kuriyan J. (2013) Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 152, 543–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chung I., Akita R., Vandlen R., Toomre D., Schlessinger J., Mellman I. (2010) Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 464, 783–787 [DOI] [PubMed] [Google Scholar]
- 41. Haugsten E. M., Wiedlocha A., Olsnes S., Wesche J. (2010) Roles of fibroblast growth factor receptors in carcinogenesis. Mol. Cancer Res. 8, 1439–1452 [DOI] [PubMed] [Google Scholar]
- 42. Li E., Hristova K. (2006) Role of receptor-tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry 45, 6241–6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rand V., Huang J., Stockwell T., Ferriera S., Buzko O., Levy S., Busam D., Li K., Edwards J. B., Eberhart C., Murphy K. M., Tsiamouri A., Beeson K., Simpson A. J., Venter J. C., Riggins G. J., Strausberg R. L. (2005) Sequence survey of receptor-tyrosine kinases reveals mutations in glioblastomas. Proc. Natl. Acad. Sci. U.S.A. 102, 14344–14349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jones D. T., Hutter B., Jäger N., Korshunov A., Kool M., Warnatz H. J., Zichner T., Lambert S. R., Ryzhova M., Quang D. A., Fontebasso A. M., Stütz A. M., Hutter S., Zuckermann M., Sturm D., Gronych J., Lasitschka B., Schmidt S., Seker-Cin H., Witt H., Sultan M., Ralser M., Northcott P. A., Hovestadt V., Bender S., Pfaff E., Stark S., Faury D., Schwartzentruber J., Majewski J., Weber U. D., Zapatka M., Raeder B., Schlesner M., Worth C. L., Bartholomae C. C., von Kalle C., Imbusch C. D., Radomski S., Lawerenz C., van Sluis P., Koster J., Volckmann R., Versteeg R., Lehrach H., Monoranu C., Winkler B., Unterberg A., Herold-Mende C., Milde T., Kulozik A. E., Ebinger M., Schuhmann M. U., Cho Y. J., Pomeroy S. L., von Deimling A., Witt O., Taylor M. D., Wolf S., Karajannis M. A., Eberhart C. G., Scheurlen W., Hasselblatt M., Ligon K. L., Kieran M. W., Korbel J. O., Yaspo M. L., Brors B., Felsberg J., Reifenberger G., Collins V. P., Jabado N., Eils R., Lichter P., Pfister S. M. (2013) Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 45, 927–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lew E. D., Furdui C. M., Anderson K. S., Schlessinger J. (2009) The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci. Signal 2, ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Webster M. K., D'Avis P. Y., Robertson S. C., Donoghue D. J. (1996) Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Mol. Cell. Biol. 16, 4081–4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kan M., Wang F., To B., Gabriel J. L., McKeehan W. L. (1996) Divalent cations and heparin/heparan sulfate cooperate to control assembly and activity of the fibroblast growth factor receptor complex. J. Biol. Chem. 271, 26143–26148 [DOI] [PubMed] [Google Scholar]
- 48. Wang F., Kan M., McKeehan K., Jang J. H., Feng S., McKeehan W. L. (1997) A homeo-interaction sequence in the ectodomain of the fibroblast growth factor receptor. J. Biol. Chem. 272, 23887–23895 [DOI] [PubMed] [Google Scholar]
- 49. McKeehan W. L., Wang F., Kan M. (1998) The heparan sulfate-fibroblast growth factor family: diversity of structure and function. Prog. Nucleic Acid Res. Mol. Biol. 59, 135–176 [DOI] [PubMed] [Google Scholar]
- 50. Ballinger M. D., Shyamala V., Forrest L. D., Deuter-Reinhard M., Doyle L. V., Wang J. X., Panganiban-Lustan L., Stratton J. R., Apell G., Winter J. A., Doyle M. V., Rosenberg S., Kavanaugh W. M. (1999) Semirational design of a potent, artificial agonist of fibroblast growth factor receptors. Nat. Biotechnol. 17, 1199–1204 [DOI] [PubMed] [Google Scholar]
- 51. Welm B. E., Freeman K. W., Chen M., Contreras A., Spencer D. M., Rosen J. M. (2002) Inducible dimerization of FGFR1: development of a mouse model to analyze progressive transformation of the mammary gland. J. Cell Biol. 157, 703–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lemmon M. A., Schlessinger J., Ferguson K. M. (2014) The EGFR family: not so prototypical receptor-tyrosine kinases. Cold Spring Harb. Perspect. Biol. 6, a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ming A. Y., Yoo E., Vorontsov E. N., Altamentova S. M., Kilkenny D. M., Rocheleau J. V. (2012) Dynamics and Distribution of Klothoβ (KLB) and fibroblast growth factor receptor-1 (FGFR1) in living cells reveal the fibroblast growth factor-21 (FGF21)-induced receptor complex. J. Biol. Chem. 287, 19997–20006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sarabipour S., Hristova K. (2015) FGFR3 unliganded dimer stabilization by the juxtamembrane domain. J. Mol. Biol. 427, 1705–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Placone J., Hristova K. (2012) Direct assessment of the effect of the Gly380Arg achondroplasia mutation on FGFR3 dimerization using quantitative imaging FRET. PLoS ONE 7, e46678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mohammadi M., Olsen S. K., Goetz R. (2005) A protein canyon in the FGF-FGF receptor dimer selects from an a la carte menu of heparan sulfate motifs. Curr. Opin. Struct. Biol. 15, 506–516 [DOI] [PubMed] [Google Scholar]
- 57. Gao G., Goldfarb M. (1995) Heparin can activate a receptor-tyrosine kinase. EMBO J. 14, 2183–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ogawa Y., Kurosu H., Yamamoto M., Nandi A., Rosenblatt K. P., Goetz R., Eliseenkova A. V., Mohammadi M., Kuro-o M. (2007) βKlotho is required for metabolic activity of fibroblast growth factor 21. Proc. Natl. Acad. Sci. U.S.A. 104, 7432–7437 [DOI] [PMC free article] [PubMed] [Google Scholar]