

Background: Little is known about the effect of hyperosmotic hepatocyte shrinkage on Na+-taurocholate cotransporting polypeptide (Ntcp) regulation.
Results: Hyperosmolarity induces a Fyn-dependent retrieval of Ntcp, which is reversed by tauroursodeoxycholate via a β1-integrin-mediated formation of cAMP.
Conclusion: Fyn and β1-integrins are novel regulators of Ntcp localization.
Significance: The study provides new insights into the regulation of bile salt transport.
Keywords: bile acid, cyclic AMP (cAMP), integrin, protein kinase C (PKC), Src, Fyn, Ntcp, tauroursodeoxycholate, transporter retrieval
Abstract
In perfused rat liver, hepatocyte shrinkage induces a Fyn-dependent retrieval of the bile salt export pump (Bsep) and multidrug resistance-associated protein 2 (Mrp2) from the canalicular membrane (Cantore, M., Reinehr, R., Sommerfeld, A., Becker, M., and Häussinger, D. (2011) J. Biol. Chem. 286, 45014–45029) leading to cholestasis. However little is known about the effects of hyperosmolarity on short term regulation of the Na+-taurocholate cotransporting polypeptide (Ntcp), the major bile salt uptake system at the sinusoidal membrane of hepatocytes. The aim of this study was to analyze hyperosmotic Ntcp regulation and the underlying signaling events. Hyperosmolarity induced a significant retrieval of Ntcp from the basolateral membrane, which was accompanied by an activating phosphorylation of the Src kinases Fyn and Yes but not of c-Src. Hyperosmotic internalization of Ntcp was sensitive to SU6656 and PP-2, suggesting that Fyn mediates Ntcp retrieval from the basolateral membrane. Hyperosmotic internalization of Ntcp was also found in livers from wild-type mice but not in p47phox knock-out mice. Tauroursodeoxycholate (TUDC) and cAMP reversed hyperosmolarity-induced Fyn activation and triggered re-insertion of the hyperosmotically retrieved Ntcp into the membrane. This was associated with dephosphorylation of the Ntcp on serine residues. Insertion of Ntcp by TUDC was sensitive to the integrin inhibitory hexapeptide GRGDSP and inhibition of protein kinase A. TUDC also reversed the hyperosmolarity-induced retrieval of bile salt export pump from the canalicular membrane. These findings suggest a coordinated and oxidative stress- and Fyn-dependent retrieval of sinusoidal and canalicular bile salt transport systems from the corresponding membranes. Ntcp insertion was also identified as a novel target of β1-integrin-dependent TUDC action, which is frequently used in the treatment of cholestatic liver disease.
Introduction
Disturbances of transcellular transport of bile acids in hepatocytes can result in the accumulation of potentially toxic bile acids inside the hepatocyte and can lead to cell damage and liver dysfunction (2–6). It is therefore critical that hepatocellular uptake and secretion of bile acids are coordinately regulated. At the canalicular membrane, bile salt excretion is mediated by the bile salt export pump (Bsep)2 (7) and to a lesser extent by the multidrug resistance-associated protein 2 (Mrp2) (8). The expression of Bsep and Mrp2 in the canalicular membrane of the hepatocyte is regulated by oxidative stress (9–11), lipopolysaccharide (LPS) (12–14), drugs (15, 16), and ambient osmolarity (1, 10, 17–19). The cellular hydration state is a dynamic parameter that can change physiologically within minutes depending on the ambient osmolarity, nutrient supply, influence of hormones, and oxidative stress. Alterations in the cellular hydration represent an important mechanism for the metabolic control and a messenger linking cell function to hormonal and environmental alterations. Liver cell hydration also controls biliary excretion through activation of osmosensing and osmosignaling pathways (20–22). Hyperosmotic liver cell shrinkage is cholestatic and induces a Fyn-dependent (1) retrieval of Bsep (10) and Mrp2 (17) from the canalicular membrane, whereas hypoosmotic cell swelling increases the transport capacity for bile acids in a microtubule- and mitogen-activated protein kinase (MAPK)-dependent fashion (19, 23, 24). Choleretic agents, such as cAMP and tauroursodeoxycholate (TUDC), also stimulate insertion of canalicular transporters from an intracellular compartment into the membrane via activation of phosphatidylinositide 3-kinase (PI3K)/protein kinase C (PKC)- and MAPK-dependent pathways (19, 25–29).
The Na+-taurocholate cotransporting polypeptide (Ntcp) (30) is the major uptake system for conjugated bile salts from the blood into liver parenchymal cells. Loss of Ntcp results in increased plasma levels of conjugated bile acids, as also confirmed by a recent report on human NTCP deficiency (31), which is clinically characterized by mild hypotonia, growth retardation, and delayed motor milestones. Functional studies show that the underlying mutation results in a markedly reduced uptake activity of taurocholate (TC), which is due to an absence of the mutant protein in the plasma membrane of the hepatocyte (31). Recent studies suggest that the cholestatic bile salt taurolithocholate inhibits hepatic TC uptake and decreases plasma membrane levels of Ntcp (32). Also, taurochenodeoxycholate (TCDC) inhibits TC uptake at the sinusoidal membrane in a chelerythrine- and cypermethrin-dependent way (33). Ntcp is a serine/threonine phosphorylated protein (34) and underlies complex regulation (35). cAMP can induce an increase in TC uptake by insertion of Ntcp into the plasma membrane (36) via increases in intracellular Ca2+ and subsequent activation of protein phosphatase 2B (PP2B) via Ca2+-calmodulin kinase (35). This is associated with a dephosphorylation of Ntcp at Ser-226 (37). Hepatocyte swelling stimulates rapid Ntcp insertion into the basolateral membrane and increases TC uptake in hepatocytes (38), but nothing is known about a regulation of Ntcp by hyperosmotic cell shrinkage and the hydrophilic nontoxic bile acid TUDC.
The hydrophilic bile acid ursodeoxycholic acid, which in vivo is rapidly converted to its taurine conjugate TUDC (39), is a mainstay in the treatment of cholestatic liver disease, such as primary biliary cirrhosis or intrahepatic cholestasis of pregnancy (40–44). Intrahepatic cholestasis results in an accumulation of potentially toxic bile acids inside the hepatocyte that can cause hepatocyte death due to activation of CD95-dependent apoptosis (45, 46). TUDC has potent anticholestatic and antiapoptotic properties. Experimental and clinical evidence suggests that at least three mechanisms are responsible for the hepatoprotective properties of TUDC in cholestatic disorders (47) as follows: 1) stimulation of hepatobiliary secretion; 2) cytoprotection of hepatocytes against bile acid-induced apoptosis and cytokine-induced injury; and 3) protection of cholangiocytes against cytotoxicity of hydrophobic bile acids. We could recently demonstrate that both the choleretic and antiapoptotic effects of TUDC are mediated by an activation of α5β1-integrins (48, 49). TUDC-induced integrin activation leads to the formation of cAMP, which triggers inactivation of the CD95 and induction of a MAPK phosphatase (49). The stimulation of biliary excretion is also triggered via TUDC-induced integrin activation and subsequent activation of the MAPKs p38MAPK and ERKs (19, 50). This dual MAPK activation results in a TUDC-induced stimulation of biliary excretion by rapid targeting and insertion of intracellularly stored transport ATPases, such as Bsep or Mrp2 into the canalicular membrane (25, 29). However, nothing is known about the effects of TUDC on the localization of the basolateral Ntcp, the major uptake system for conjugated bile acids into the hepatocyte. This study shows that TUDC triggers the insertion of the bile salt transporter Ntcp into the plasma membrane and that integrins are the sensor for this mechanism. Our study also shows that hyperosmotic hepatocyte shrinkage triggers an oxidative stress-,PKCζ-, and Fyn-dependent internalization of Ntcp, which is reversed by TUDC in a β1-integrin- and cAMP-dependent manner. The findings are not only of interest for the understanding of hepatocellular bile acid handling and TUDC action in liver, but may also be relevant for hepatitis B virus entry into the hepatocyte, which was recently shown to be mediated by NTCP (51). Indeed, recent data suggest that interleukin-6 inhibits hepatitis B virus entry into the hepatocyte through down-regulation of NTCP (52).
Experimental Procedures
Materials
The materials used were purchased as follows: apocynin, SU6656, PP-2, H-Gly-Arg-Gly-Asp-Ser-Pro-OH (GRGDSP) and H-Gly-Arg-Ala-Asp-Ser-Pro-OH (GRADSP) were from Merck-Millipore (Darmstadt, Germany); N-acetylcysteine (NAC), dibutyryl-cAMP (Bt2cAMP) sodium salt, collagenase, insulin, chelerythrine, and TUDC were from Sigma (Munich, Germany); H89 dihydrochloride, PKCζ pseudosubstrate, penicillin/streptomycin, and Fluoromount-G were from Tocris/Biozol (Eching, Germany); fetal bovine serum (FBS), 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), Lipofectamine 2000, G418 geneticin, and Dulbecco's modified Eagle's medium Nutrimix F-12 were from Life Technologies, GmbH (Darmstadt, Germany); Gö 6850 (bisindolylmaleimide I) and Gö 6976 were from Enzo Life Sciences GmbH (Lörrach, Germany); cOmplete-protease inhibitor mixture tablets and PhosSTOP-phosphatase inhibitor mixture tablets were from Roche Diagnostics (Mannheim, Germany); and William's Medium E was from Biochrom (Berlin, Germany). The Ntcp antibody (K4) (30) and the Bsep antibody (K24) (53) were the generous gifts from Prof. Dr. B. Stieger (Kantonsspital Zürich, Switzerland). Antibodies recognizing Yes (immunoprecipitation) and Fyn (immunoprecipitation) were from Santa Cruz Biotechnology (Heidelberg, Germany); zona occludens-1 (ZO-1) and c-Src were from Life Technologies, GmbH; and against phosphoserine was from Enzo Life Sciences GmbH (Lörrach, Germany). The antibodies raised against phospho-Src family Tyr-418 were from Cell Signaling Technology, Inc. (Danvers, MA); against Na+/K+-ATPase, Yes (Western blot), Fyn (Western blot), phospho-c-Src, Cy3-conjugated donkey anti-rabbit IgG, and FITC-conjugated donkey anti-mouse IgG were from Merck-Millipore (Darmstadt, Germany); and green fluorescent protein (GFP)-horseradish peroxidase was from Miltenyi (Bergisch Gladbach, Germany). Horseradish peroxidase-conjugated anti-mouse IgG and anti-rabbit IgG were from Bio-Rad (Munich, Germany) and Dako (Hamburg, Germany). All other chemicals were from Merck-Millipore (Darmstadt, Germany) at the highest quality available.
Liver Perfusion
Livers from male Wistar rats (140–160 g) were perfused in a nonrecirculating manner as described previously (54). As a perfusion medium the bicarbonate-buffered Krebs-Henseleit saline plus l-lactate (2.1 mmol/liter) and pyruvate (0.3 mmol/liter) gassed with 5% CO2 and 95% O2 at 37 °C was used (305 mosmol/liter, normo-osmotic). Hyperosmotic (385 mosmol/liter) perfusion was performed by raising the NaCl concentration in the medium. Addition of inhibitors, Bt2cAMP and TUDC to the influent perfusate was made by dissolution into the Krebs-Henseleit buffer. Viability of the perfused livers was assessed by measuring lactate dehydrogenase leakage into the perfusate. The portal pressure, the effluent K+ concentration, and pH were continuously monitored. Livers from male control mice (wild type, 8–10 weeks; on C57BL/6 background) or p47phox knock-out mice (8–10 weeks, C57BL/6J-Ncf1m1J/J, The Jackson Laboratory) were perfused in an adapted version.
Preparation and Culture of Primary Rat Hepatocytes
Hepatocytes were isolated from livers of male Wistar rats (160–180 g) by a collagenase perfusion technique in an adapted version as described previously (55). Animals were fed ad libitum with a standard diet. Aliquots of rat hepatocytes were plated on collagen-coated culture plates and maintained in bicarbonate-buffered Krebs-Henseleit medium (115 mmol/liter NaCl, 25 mmol/liter NaHCO3, 5.9 mmol/liter KCl, 1.18 mmol/liter MgCl2, 1.23 mmol/liter NaH2PO4, 1.2 mmol/liter Na2SO4, 1.25 mmol/liter CaCl2), supplemented with 6 mmol/liter glucose in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. After 2 h, the medium was removed, and the cells were washed twice. Subsequently, the culture was continued for 24 h in William's Medium E, supplemented with 2 mmol/liter glutamine, 100 nmol/liter insulin, 100 units/ml penicillin, 0.1 mg/ml streptomycin, 100 nmol/liter dexamethasone, and 5% FBS. After 24 h, experimental treatments were performed using William's Medium E that contained 2 mmol/liter glutamine and 100 nmol/liter dexamethasone. The viability of hepatocytes was more than 95% as assessed by trypan blue exclusion. To allow comparisons with previous studies, in these cell culture experiments hyperosmolarity of 405 mosmol/liter was used, whereas in rat liver perfusions we used 385 mosmol/liter.
Generation of Stably Expressing FLAG-Ntcp-EGFP, Transfection, Culture, and Immunoprecipitation
The FLAG-Ntcp-EGFP plasmid was cloned as described previously (56). HepG2 cells (ATCC, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium Nutrimix F-12 supplemented with 10% FBS. FLAG-Ntcp-EGFP was transfected into HepG2 cells using Lipofectamine 2000 according to the manufacturer's guidelines. Stable clones were selected by 0.5% G418 geneticin. HepG2 cells stably expressing FLAG-Ntcp-EGFP were cultured until subconfluence, stimulated, and lysed for direct isolation of the GFP-tagged Ntcp using μMACS GFP isolation kit (Miltenyi, Bergisch Gladbach, Germany).
Immunofluorescence Staining
Cryo-sections of liver samples (7 μm) were fixed in methanol (2 min, −20 °C) and blocked with FBS (5% (w/v), 30 min, room temperature). Then, the liver sections were incubated with the primary antibodies against Ntcp (1:200) and Na+/K+-ATPase (1:200) or Bsep (1:200) and ZO-1 (1:500) (overnight, 4 °C), washed three times, and stained with an anti-mouse FITC and an anti-rabbit Cy3-conjugated antibody (1:500 in PBS, 2 h, room temperature, respectively). Following immunofluorescence staining, samples were covered with Fluoromount-G reagent and visualized by confocal laser scanning microscopy (LSM) using LSM510 META or by super resolution structured illumination microscopy (SR-SIM) using the ELYRA microscope (Zeiss, Oberkochen, Germany).
Densitometric Fluorescence Intensity Analysis
Cryosections of perfused rat liver for the analysis at the basolateral membrane were stained for Ntcp and for Na+/K+-ATPase (plasma membrane marker protein). For negative controls, primary antibodies were omitted in each experiment. Na+/K+-ATPase profiles were selected according to apparent integrity and comparability. Acceptable Na+/K+-ATPase intensity profiles have a sufficiently high peak of fluorescence in the central part (corresponding to the basolateral membrane) and a low intracellular fluorescence. For analysis of the canalicular membrane, cryosections were stained for Bsep and the tight junction protein ZO-1, which forms the sealing border between canalicular and sinusoidal membrane. Apparent integrity and comparability of the canaliculi was assumed when the bordering tight junction lines (detected by the immunostained ZO-1) were intact, run in parallel, and showed a width of 1.12 to 1.82 μm. Densitometric analysis was performed as described previously (10, 17). For analysis of digitalized microscopic pictures of the membranes, the software Image-Pro Plus (Media Cybernetics, Rockville, MD) was used. The profile of the fluorescence intensity was measured over a thick line at a right angle to the membrane. The length of the line was always 8 μm. The mean fluorescence intensity to each pixel over the line perpendicular to the length was calculated by Image-Pro Plus. The obtained data (pixel positions with the associated pixel intensities, red and green channel) were transferred to an Excel data sheet (Microsoft Excel for Windows, Redmond, WA). Each measurement was normalized to the sum of all intensities of the respective measurement. Values are given as means ± S.E. Densitometric analysis of protein distribution in immunofluorescence images was performed using Wilcoxon rank sum test. p < 0.05 was considered statistically significant. Data from at least 10 different areas per tissue sample and from at least three independent liver preparations were processed.
Immunoblot Analysis
Liver samples were immediately lysed at 4 °C by using a lysis buffer containing 20 mmol/liter Tris-HCl (pH 7.4), 140 mmol/liter NaCl, 10 mmol/liter NaF, 10 mmol/liter sodium pyrophosphate, 1% (v/v) Triton X-100, 1 mmol/liter EDTA, 1 mmol/liter EGTA, 1 mmol/liter sodium vanadate, 20 mmol/liter β-glycerophosphate, protease inhibitor, and phosphatase inhibitor mixture. The lysates were kept on ice for 10 min and then centrifuged at 8000 rpm for 8 min at 4 °C, and aliquots of the supernatant were taken for protein determination using the protein assay (Bio-Rad, Munich, Germany). Equal amounts of protein were subjected to SDS-PAGE and transferred onto nitrocellulose membranes using a semidry transfer apparatus (GE Healthcare, Freiburg, Germany). Membranes were blocked for 60 min in 5% (w/v) bovine serum albumin containing 20 mmol/liter Tris (pH 7.5), 150 mmol/liter NaCl, and 0.1% Tween 20 (TBS-T) and exposed to primary antibodies overnight at 4 °C. After washing with TBS-T and incubating at room temperature for 2 h with horseradish peroxidase-coupled anti-mouse or anti-rabbit IgG antibody, respectively (all diluted 1:10,000), the immunoblots were washed extensively, and bands were visualized using the FluorChem E detection instrument from ProteinSimple (Santa Clara, CA). Semi-quantitative evaluation was carried out by densitometry using the Alpha View image acquisition and analysis software from ProteinSimple. Protein phosphorylation is given as the ratio of detected phosphoprotein/total protein.
Immunoprecipitation
Liver samples were harvested in lysis buffer containing 136 mmol/liter NaCl, 20 mmol/liter Tris-HCl, 10% (v/v) glycerol, 2 mmol/liter EDTA, 50 mmol/liter β-glycerophosphate, 20 mmol/liter sodium pyrophosphate, 0.2 mmol/liter Pefabloc, 5 mg/liter aprotinin, 5 mg/liter leupeptin, 4 mmol/liter benzamidine, 1 mmol/liter sodium vanadate, supplemented with 1% (v/v) Triton X-100. The protein amount was determined as described above. Samples containing equal protein amounts were incubated for 2 h at 4 °C with the respective antibody to immunoprecipitate the required protein. Then protein A-/G-agarose (Santa Cruz Biotechnology) was added and incubated at 4 °C overnight. Immunoprecipitates were washed three times with lysis buffer supplemented with 0.1% (v/v) Triton X-100 and then transferred to Western blot analysis as described above.
Detection of ROS
Primary hepatocytes were seeded on collagen-coated plates (Falcon, Heidelberg, Germany) and cultured for 24 h. Cells were incubated with PBS containing 5 μmol/liter CM-H2DCFDA for 30 min at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. To detect ROS generation, CM-H2DCFDA-loaded cells were supplemented again with culture medium and then exposed to the respective stimuli for the indicated time period. Thereafter, cells were washed briefly using ice-cold PBS and lysed in 0.1% Triton X-100 (v/v) dissolved in aqua bidest. Lysates were centrifuged immediately (10,000 × g, 4 °C, 1 min), and fluorescence of the supernatant was measured at 515–565 nm using a luminescence spectrometer LS-5B (PerkinElmer Life Sciences, Rodgau, Germany) with an excitation wavelength of 488 nm. Fluorescence intensity of controls was set to 1, and fluorescence found under the respective treatment is given relative to it.
Real Time-PCR Analysis
Total RNA was isolated using the RNeasy mini kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. RNA was quantified using NanoDrop1000 System (Thermo Scientific, Wilmington, DE), and first strand cDNA was synthesized from RNA using the RevertAid H Minus First Strand cDNA Synthesis kit (Fermentas, St. Leon-Rot, Germany). Gene expression levels were quantified using SensiMix SYBR No-ROX Kit (Bioline, Luckenwalde, Germany) on a TOptical cycler (Biometra, Göttingen, Germany). Hypoxanthine-guanine phosphoribosyltransferase 1 (HPRT1) was used as reference gene for the normalization of the results obtained by the 2(−ΔΔCt) method. PCR primer sequences are as follows: Ntcp 5′-TCAAGTCCAAAAGGCCACACT-3′ and 5′-AGGGAGGAGGTAGCCAGTAAG-3′, and HPRT1 5′-TGCTCGAGATGTCATGAAGGA-3′ and 5′-CAGAGGGCCACAATGTGATG-3′.
Statistical Analysis
Results from at least three independent experiments are expressed as mean values ± S.E. n refers to the number of independent experiments. For each experimental treatment and time point analyzed, a separate control experiment was carried out. Differences between experimental groups were analyzed by Student's t test or one-way analysis of variance following Dunnett's multiple comparison post hoc test where appropriate (GraphPad Prism; GraphPad, La Jolla, CA; Microsoft Excel for Windows). p < 0.05 was considered statistically significant.
Results
Hyperosmolarity Triggers Ntcp Retrieval from the Basolateral Membrane in the Perfused Rat Liver
To study the subcellular localization of the bile salt transporter Ntcp in hepatocytes, immunofluorescence studies on cryosectioned tissues using confocal laser-scanning microscopy were performed (Fig. 1). For labeling of the plasma membrane, liver sections were stained simultaneously with specific antibodies against the plasma membrane marker protein Na+/K+-ATPase and against Ntcp, respectively. The profile of the fluorescence intensity of Ntcp and Na+/K+-ATPase was measured over a thick line (8 μm) perpendicular to the membranes of adjacent hepatocytes. The mean fluorescence intensity of each pixel along this line was calculated by Image-Pro Plus, and the obtained and normalized data were plotted on a line graph (Fig. 1).
FIGURE 1.
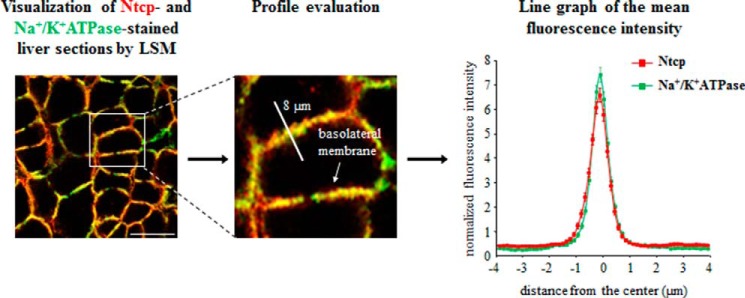
Quantification of Ntcp and Na+/K+-ATPase distribution by fluorescence densitometry. Cryosections from perfused rat liver were immunostained for Ntcp and Na+/K+-ATPase, and fluorescence pictures were recorded by confocal LSM. The profile of the fluorescence intensity was measured over a thick line (8 μm) perpendicular to the plasma membrane of adjacent hepatocytes. The mean fluorescence intensity of each pixel along this line was calculated by Image-Pro Plus, and the obtained data (pixel positions with the associated pixel intensities, red and green channel) were transferred to an MS-Excel data sheet and plotted. The ordinate shows the normalized intensity of Na+/K+-ATPase staining depending on the distance (micrometers) from the center of the basolateral membrane (set as 0) and the normalized intensity of Ntcp-bound fluorescence. The means ± S.E. of 30 measurements in each of three individual experiments are shown.
Subcellular Ntcp distribution in normo-osmotically (305 mosmol/liter) and hyperosmotically (385 mosmol/liter) perfused livers was quantified by densitometric fluorescence intensity analysis as described above. Within 30 min of hyperosmotic perfusion, the fluorescence profile of Ntcp shifted to significantly lower peak values and became more broadened (p < 0.05, Fig. 2A), suggestive for an internalization of the Ntcp in response to hyperosmolarity. In contrast, no changes in Na+/K+-ATPase distribution were observed under these conditions (Fig. 2A). To discriminate Ntcp distribution at a subcellular level, SR-SIM in Ntcp and Na+/K+-ATPase-costained cryosections was performed. In SR-SIM images, obtained from hyperosmotically (385 mosmol/liter) perfused rat livers, intracellular vesicular Ntcp fluorescence intensity was markedly increased within 30 min and maintained for at least 60 min (Fig. 2B). Localization of the basolateral membrane protein Na+/K+-ATPase remained unchanged as compared with normo-osmotic control conditions (Fig. 2B). These results suggest that hyperosmolarity induces within 30 min endocytosis of Ntcp in the intact rat liver, which lasts for at least 60 min. Hyperosmolarity-induced retrieval of Ntcp was not accompanied by changes in Ntcp mRNA expression during the first 60 min of hyperosmotic exposure (0.96 ± 0.07-fold, not significant, n = 4, Fig. 2C). However, a significant decrease of Ntcp mRNA levels was seen after treatment with hyperosmotic medium for 180 min (0.76 ± 0.04-fold, p < 0.05, n = 4) or more (Fig. 2C).
FIGURE 2.

Regulation of Ntcp in hyperosmotically perfused rat liver. A, rat livers were perfused with normo-osmotic (305 mosmol/liter) or hyperosmotic (385 mosmol/liter) Krebs-Henseleit buffer for up to 60 min and immunostained for Ntcp and Na+/K+-ATPase. Densitometric analysis of fluorescence profiles and intensity of Ntcp and Na+/K+-ATPase distribution (visualization by confocal LSM) is shown. Under control conditions (green (0 min) and black (60 min)), Ntcp-bound fluorescence was largely localized in the center of the basolateral membrane. Hyperosmotic perfusion (red (30 min) and blue (60 min)) resulted in a significant lateralization of the Ntcp-bound fluorescence, but no significant changes of Na+/K+-ATPase fluorescence profiles were observed. The fluorescence profiles depicted are statistically significantly (p < 0.05) different from each other with respect to variance and peak height. Means ± S.E. of 30 measurements in each of at least three individual experiments for each condition are shown. B, rat livers were perfused with normo-osmotic (305 mosmol/liter) or hyperosmotic (385 mosmol/liter) Krebs-Henseleit medium for up to 60 min, immunostained for Ntcp and Na+/K+-ATPase, and visualized by SR-SIM. Under normo-osmotic conditions, Ntcp was largely localized in the membrane, and after hyperosmotic exposure Ntcp, but not the Na+/K+-ATPase, was internalized. Representative pictures of at least three independent experiments are depicted. The scale bars correspond to 10 μm in the overviews (upper row) and 1 μm in the four bottom rows with higher magnification of the plasma membrane. C, primary rat hepatocytes were cultured for 24 h and thereafter stimulated with normo-osmotic (305 mosmol/liter) or hyperosmotic (405 mosmol/liter) medium for up to 24 h. RNA was extracted, and Ntcp mRNA expression levels were analyzed by real time PCR. Ntcp mRNA expression levels are given relative to the unstimulated control (0 min). Data represent the mean ± S.E. of four independent experiments *, p < 0.05 statistical significance compared with the unstimulated control. #, p < 0.05 statistical significance between normo-osmotic and hyperosmotic stimulations.
Hyperosmolarity-induced Retrieval of Ntcp from the Basolateral Membrane Is Mediated by Activation of the Src Family Kinase Fyn
Hyperosmolarity was shown to trigger NADPH oxidase-dependent formation of ROS (6, 57) and to activate the Src family kinases Yes and Fyn, although no c-Src activation became detectable within 180 min of hyperosmotic exposure (1). Hyperosmotic Fyn and Yes activations were sensitive to inhibition by NAC, apocynin, and SU6656, whereas PP-2 inhibited hyperosmotic Fyn but not Yes activation (1). Ntcp distribution in hyperosmotically (385 mosmol/liter) perfused livers in the absence and presence of inhibitors of the NADPH oxidase-driven ROS generation (apocynin and NAC) or of Src kinases (PP-2, SU6656) was quantified by densitometric fluorescence intensity analysis (Fig. 3). Fig. 3A depicts the perfusion plans chosen for these experiments. As shown in Fig. 3, B–D, NAC (10 mmol/liter) and apocynin (20 μmol/liter) inhibited the hyperosmolarity (385 mosmol/liter, t = 30 min)-induced Ntcp retrieval from the basolateral membrane, whereas Na+/K+-ATPase distribution remained unchanged. These findings point to a role of NADPH oxidase-driven ROS formation in triggering transporter retrieval from the plasma membrane. Also, the Src kinase inhibitors SU6656 (1 μmol/liter) and PP-2 (250 nmol/liter) prevented hyperosmotic Ntcp retrieval from the basolateral membrane (Fig. 3D). Because PP-2 (58) inhibits Fyn but not Yes activation, and SU6656 (59) inhibits both hyperosmotic Yes and Fyn activation, these findings suggest that Fyn is involved in the hyperosmotic retrieval of Ntcp from the basolateral membrane. The role of NADPH oxidase in triggering hyperosmotic Ntcp retrieval from the plasma membrane was also studied in livers from p47phox knock-out mice. As shown in Fig. 4, hyperosmolarity induced within 30 min the internalization of Ntcp in livers from wild-type mice, whereas no retrieval was detectable in livers from p47phox knock-out mice.
FIGURE 3.
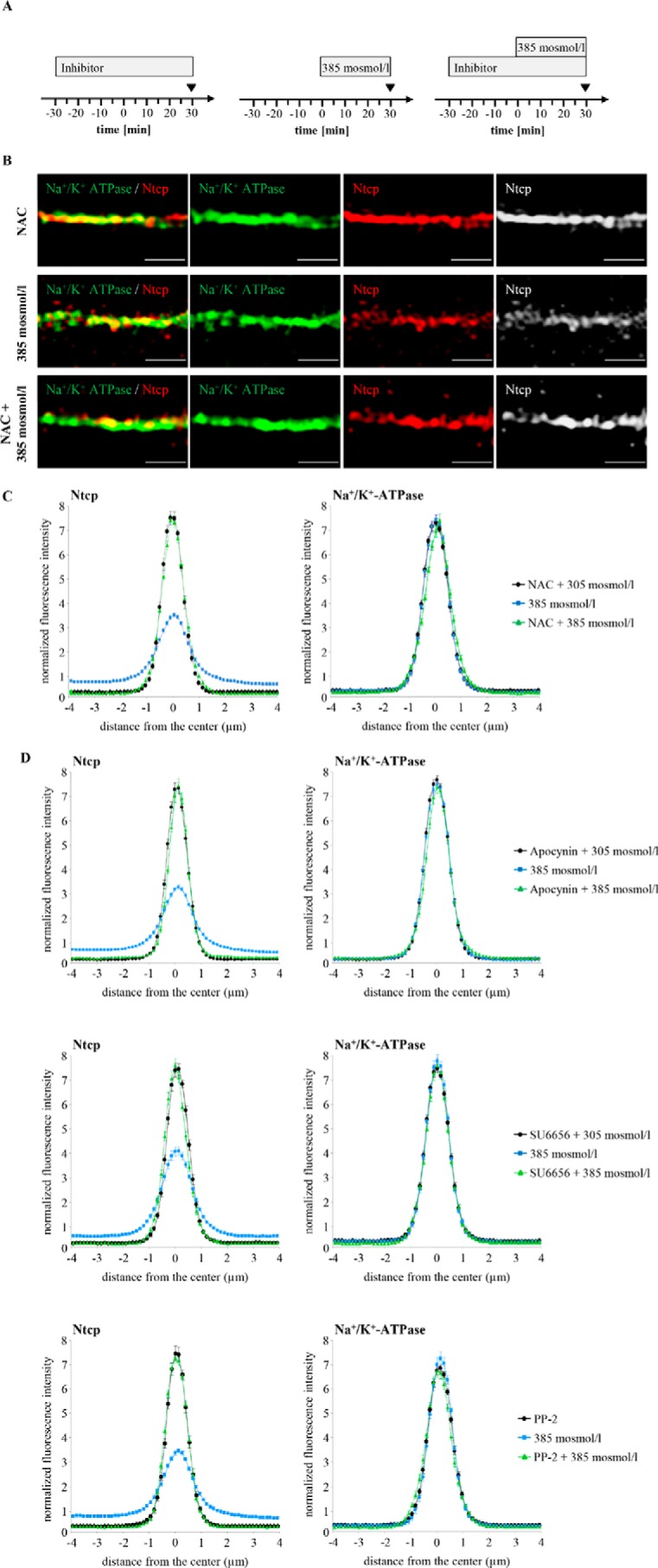
Determination of Ntcp and Na+/K+-ATPase distribution in hyperosmotically perfused rat livers. Rat livers were perfused with either inhibitor-containing Krebs-Henseleit buffer (i.e. apocynin (20 μmol/liter) or NAC (10 mmol/liter) or SU6656 (1 μmol/liter) or PP-2 (250 nmol/liter), black) or hyperosmotic (385 mosmol/liter) Krebs-Henseleit buffer in the absence (blue) or presence of the respective inhibitors (green). B, Ntcp and Na+/K+-ATPase were visualized after perfusion with either NAC, hyperosmotic Krebs-Henseleit buffer, or NAC plus Krebs-Henseleit buffer at t = 30 min (see perfusion plan (A)) by SR-SIM. Representative pictures of three independent experiments are depicted. The scale bar corresponds to 1 μm. C and D, densitometric analysis of fluorescence profiles and intensity of Ntcp and Na+/K+-ATPase staining at t = 30 min (see perfusion plan (A)) is shown. The means ± S.E. of 30 measurements in each of three individual experiments for each condition are shown. NAC (C), apocynin, SU6656, and PP-2 (D) all significantly inhibited hyperosmolarity-induced retrieval of Ntcp from the membrane (p < 0.05), whereas the inhibitors by themselves did not significantly affect Ntcp localization.
FIGURE 4.

Distribution of Ntcp in hyperosmotically perfused wild-type and p47phox knock-out mouse liver. Mouse livers were perfused for 30 min with either normo-osmotic (305 mosmol/liter, black) or hyperosmotic Krebs-Henseleit buffer (385 mosmol/liter, green) and stained for Ntcp and Na+/K+-ATPase. Densitometric analysis of fluorescence profiles and intensity of Ntcp and Na+/K+-ATPase staining is shown. The means ± S.E. of 30 measurements in each of three individual experiments for each condition are shown. As shown by colocalization with Na+/K+-ATPase, Ntcp is largely localized in the membrane under normo-osmotic conditions. After hyperosmotic perfusion, Ntcp is no longer colocalized (p < 0.05) with Na+/K+-ATPase and appears inside the cells in wild-type animals but remained unchanged in p47phox knock-out mice.
TUDC Reverses Hyperosmolarity-induced Internalization of Ntcp via Integrin-dependent Protein Kinase A Activation
The choleretic bile salt TUDC induces a rapid insertion of intracellularly stored Mrp2 and Bsep into the canalicular membrane of the hepatocyte (19, 25), which increases the bile salt excretory capacity in a microtubule-dependent way (60, 61). As shown by SR-SIM, TUDC (20 μmol/liter, 30 min) was able to reverse the hyperosmolarity-induced internalization of Ntcp (Fig. 5). Because β1-integrins were identified as sensors for TUDC in rat liver (19, 48), the integrin-inhibitory peptide GRGDSP, which binds to α5β1-integrin with a KD of 396 nm (62), was used to study the involvement of β1-integrins in TUDC-induced Ntcp insertion into the plasma membrane. GRGDSP (10 μmol/liter) inhibited the counteracting effect of TUDC on hyperosmotic Ntcp retrieval, although the inactive control peptide GRADSP (10 μmol/liter) was ineffective (Fig. 5, A and C). As shown recently, TUDC activates via β1-integrins the PKA (49). When H89 (2 μmol/liter), a PKA inhibitor, was perfused 30 min before and during the experiment, the effect of TUDC on hyperosmolarity-induced Ntcp retrieval was largely abolished, whereas Na+/K+-ATPase immunostaining remained unchanged under all conditions (Fig. 5C). Similar findings were obtained with Bt2cAMP (50 μmol/liter), which is known to increase the uptake capacity of TC by increasing plasma membrane content of Ntcp (36). This effect was also H89 (2 μmol/liter)-sensitive (data not shown). All inhibitors, i.e. GRADSP, GRGDSP, and H89, had per se no effect on Ntcp localization (data not shown).
FIGURE 5.

TUDC-induced re-insertion of Ntcp into the membrane is integrin- and PKA-dependent. A, rat livers were perfused as given in the perfusion plan of the respective experiments, stained for Ntcp and Na+/K+-ATPase, and visualized by SR-SIM. Representative pictures of at least three independent experiments are depicted. Under normo-osmotic (305 mosmol/liter) conditions (t = 0), Ntcp was largely localized in the membrane, whereas after hyperosmotic (385 mosmol/liter) perfusion, Ntcp appears inside the cells and colocalizes no longer with Na+/K+-ATPase (t = 30 min). TUDC (20 μmol/liter) induced the re-insertion of Ntcp in the membrane (t = 60 min). GRGDSP (10 μmol/liter) inhibited the TUDC induced re-insertion in the membrane (p < 0.05), whereas GRADSP (10 μmol/liter) had no effect on Ntcp distribution (t = 60 min). The scale bar corresponds to 1 μm. C, rat livers were perfused as given in the perfusion plan (B), stained for Ntcp and Na+/K+-ATPase, and analyzed densitometrically. Densitometric analysis of fluorescence profiles and intensity of Ntcp and Na+/K+-ATPase at t = 60 min staining is shown. The means ± S.E. of 10 measurements in each of three individual experiments for each condition are shown. Bt2cAMP (Db-cAMP) (50 μmol/liter) and TUDC (20 μmol/liter) significantly inhibited hyperosmolarity-induced retrieval of Ntcp from the membrane (p < 0.05). GRGDSP (10 μmol/liter) and H89 (2 μmol/liter) inhibited the TUDC-induced re-insertion in the membrane (p < 0.05), whereas GRADSP (10 μmol/liter) had no effect on Ntcp distribution. Furthermore, H89 (2 μmol/liter) inhibited the Bt2cAMP-induced re-insertion of Ntcp (p < 0.05).
Hyperosmolarity-induced Fyn Activation Is Inhibited by TUDC via an Integrin- and Protein Kinase A-dependent Mechanism
Rat liver perfusion with hyperosmotic medium (385 mosmol/liter) (Figs. 6 and 7) induced an activating phosphorylation of Fyn and Yes, which was inhibited by TUDC (20 μmol/liter) within 30 min in a PKA-dependent, i.e. H89-sensitive manner (Fig. 6). Furthermore, the β1-integrin antagonistic hexapeptide GRGDSP (10 μmol/liter) largely prevented the TUDC-induced inhibition of Fyn and Yes phosphorylation, however the inactive control peptide GRADSP (10 μmol/liter) had no effect (Fig. 6). No c-Src activation was observed in response to hyperosmolarity (Figs. 6 and 7). Like TUDC, Bt2cAMP (50 μmol/liter, 30 min) inhibited the hyperosmolarity (30 min)-induced activation of Fyn and Yes (Fig. 7). When H89 (2 μmol/liter) was added 30 min before and during the perfusion, the effect of Bt2cAMP on hyperosmolarity-induced Src kinase activation was blunted (Fig. 7).
FIGURE 6.

TUDC triggers integrin- and PKA-dependent inhibition of hyperosmolarity-induced Fyn and Yes activation. A, rat livers were perfused as mentioned in the perfusion plans. B, liver samples were analyzed for activating phosphorylation of Src kinase family members Fyn, Yes, and c-Src. C, blots were analyzed densitometrically. t = 60 min was compared with the respective control (t = 0 min, set as 1), i.e. sample without institution of either hyperosmolarity (385 mosmol/liter) or TUDC (20 μmol/liter), or with the respective inhibitor (i.e. H89 (2 μmol/liter), GRADSP and GRGDSP (each 10 μmol/liter). Densitometric analysis (means ± S.E.) of at least three independent perfusion experiments is shown. In line with a previous study (1), hyperosmolarity induced within 30 min a significant phosphorylation of Fyn and Yes (*, p < 0.05), however no c-Src activation was observed. Phosphorylation of Fyn and Yes was inhibited by TUDC (#, p < 0.05). Inhibition of Fyn and Yes phosphorylation by TUDC was sensitive to H89 and GRGDSP ($, p < 0.05), however GRADSP had no effect.
FIGURE 7.

Bt2cAMP triggers PKA-dependent inhibition of hyperosmolarity-induced Src kinase family activation. Rat livers were perfused as described under “Experimental Procedures” and as mentioned in the perfusion plans (A) of the respective experiments. B, liver samples were analyzed for activation of Src kinase family members Fyn, Yes, and c-Src. Blots were analyzed densitometrically. t = 60 min was compared with the respective control (t = 0 min, set as 1), i.e. sample without institution of either hyperosmolarity (385 mosmol/liter) or Bt2cAMP (50 μmol/liter) or with H89 (2 μmol/liter). Representative blots and densitometric analysis (means ± S.E.) of at least three independent perfusion experiments are shown. In line with Fig. 5, hyperosmolarity induced within 30 min a significant phosphorylation of Fyn and Yes (*, p < 0.05), although no c-Src activation was observed. Phosphorylation of Fyn and Yes was inhibited by Bt2cAMP (#, p < 0.05). Inhibition of Fyn and Yes phosphorylation by Bt2cAMP was sensitive to H89 ($, p < 0.05).
A decrease in Ntcp serine phosphorylation was shown to be associated with Ntcp insertion into the membrane (34, 37). This was confirmed in our study in Ntcp-transfected HepG2 cells (Fig. 8). Bt2cAMP (10 μmol/liter) decreased serine phosphorylation of the Ntcp within 15 min to 83.0 ± 2.1% of control (p < 0.05, n = 5, Fig. 8). In line with this, TUDC also lowered Ntcp serine phosphorylation under normo-osmotic conditions to 82.5 ± 5.2% of control (p < 0.05, n = 5, Fig. 8). Hyperosmolarity (405 mosmol/liter) increased Ntcp serine phosphorylation to 107.9 ± 4.2% (p < 0.05, n = 5) of control (305 mosmol/liter), and addition of TUDC lowered Ntcp serine phosphorylation to 95.9 ± 3.1% (p < 0.05, n = 5) (Fig. 8).
FIGURE 8.
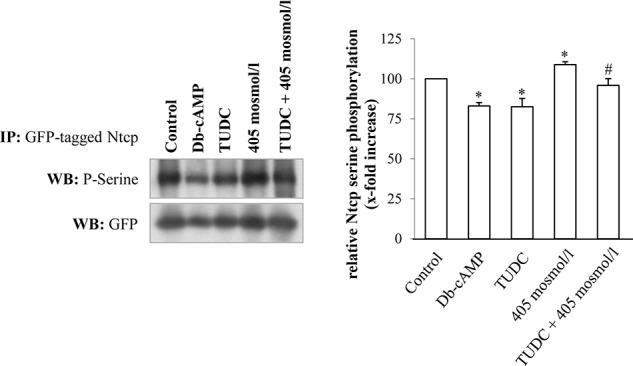
TUDC and cAMP induce serine dephosphorylation of Ntcp in HepG2-Ntcp cells. Ntcp-transfected HepG2 cells were stimulated with either TUDC (100 μmol/liter), Bt2cAMP (10 μmol/liter) under normo-osmotic (305 mosmol/liter) conditions, or with hyperosmotic medium (405 mosmol/liter) without and with TUDC. GFP-tagged Ntcp was analyzed with regard to serine phosphorylation. Treatment with TUDC as well as treatment with Bt2cAMP resulted in decreased serine phosphorylation, although hyperosmolarity triggered an increase in serine phosphorylation of the Ntcp. Ntcp serine phosphorylation was analyzed by Western blot (WB) and subsequent densitometric analysis. GFP served as a loading control. Ntcp serine phosphorylation under control condition was set to 100%. Data represent means ± S.E. of at least five independent experiments. IP, immunoprecipitation. *, p < 0.05 statistically significant compared with the unstimulated control. #, p < 0.05 statistical significance between hyperosmolarity and hyperosmolarity plus TUDC.
TUDC Reverses the Hyperosmolarity-induced Internalization of the Bile Salt Transporter Bsep
In line with a TUDC-induced reversal of hyperosmotic Fyn activation, TUDC not only prevented the hyperosmolarity-induced Ntcp retrieval from the plasma membrane, but also the hyperosmolarity-induced Bsep retrieval from the canalicular membrane (Fig. 9), which was previously shown to be Fyn-dependent, too (1). Fig. 9A shows the experimental approach, which was also used in previous studies (1, 9, 10, 17). Immunofluorescence stainings of the canalicular bile salt transporter Bsep and of ZO-1 (a protein of the tight junction complex) were investigated by confocal laser scanning microscopy and densitometric analysis. ZO-1 delineates the bile canaliculi and a linear staining pattern of ZO-1 was found under all conditions. During normo-osmotic perfusion (305 mosmol/liter), Bsep was located predominantly in the canalicular membrane, i.e. between the two rows of ZO-1 fluorescence (Fig. 9, A and B). Hyperosmotic perfusion (385 mosmol/liter) led to an increase of Bsep fluorescence intensity inside the hepatocytes at the expense of its canalicular localization, suggesting Bsep internalization (Fig. 9B). Subsequent addition of TUDC (20 μmol/liter) to the same liver preparation led to an almost complete disappearance of intracellular Bsep, indicating re-insertion of Bsep into the canalicular membrane. Under all these conditions, ZO-1 fluorescence distribution remained unaffected (Fig. 9B).
FIGURE 9.
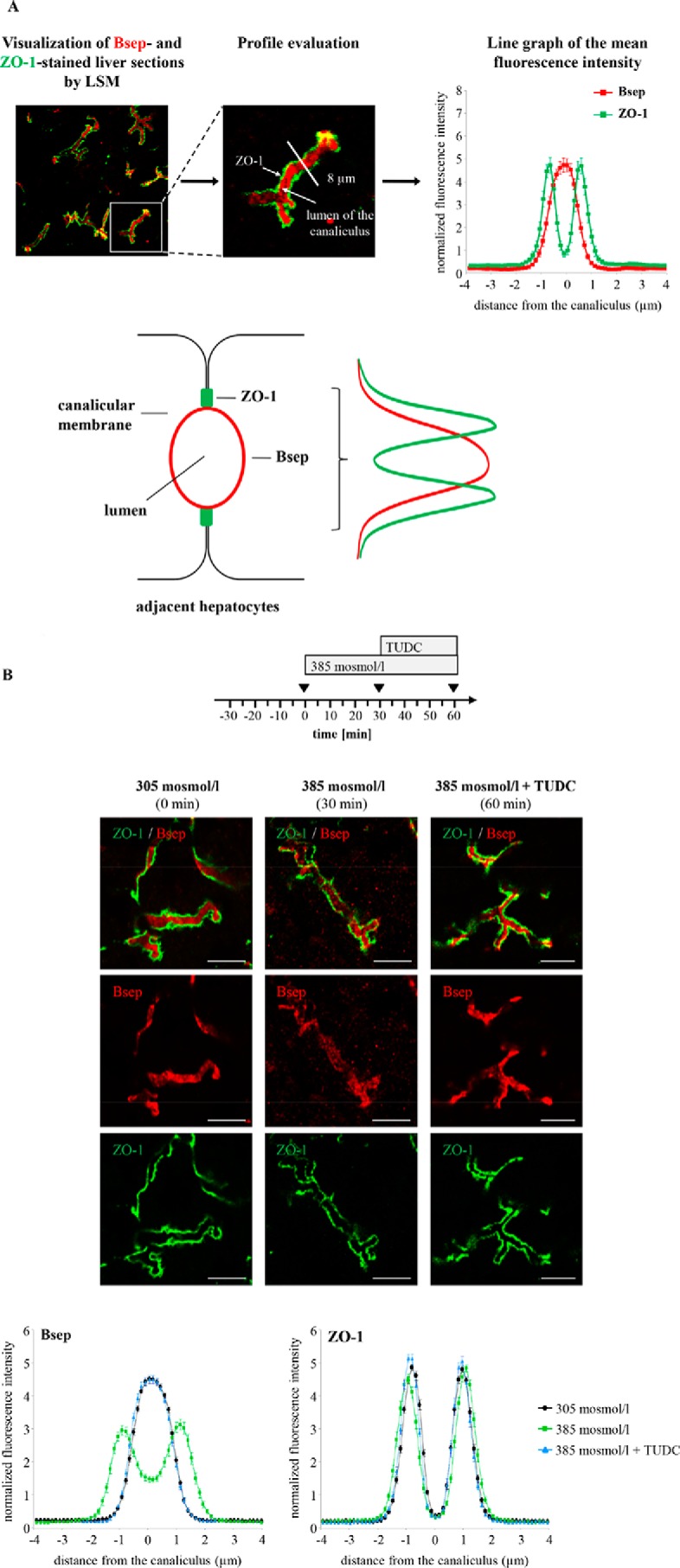
Distribution of the hepatobiliary transporter Bsep. A, cryosections from perfused rat liver were immunostained for Bsep and ZO-1, and fluorescence images were recorded by confocal LSM. The profile of the fluorescence intensity was measured over a thick line (8 μm) perpendicular to the canaliculus. The mean fluorescence intensity of each pixel along the line was calculated by Image-Pro Plus, and the obtained data (pixel positions with the associated pixel intensities, red and green channel) were transferred to an MS-Excel data sheet and plotted. The ordinate shows the normalized intensity of ZO-1 staining depending on the distance (micrometers) from the center of the canaliculus (set as 0) and the normalized intensity of Bsep-bound fluorescence. The means ± S.E. of 30 measurements in each of three individual experiments are shown. Under control conditions, ZO-1 fluorescence profiles show two peaks, whereas Bsep-bound fluorescence is largely localized in the canalicular membrane between the ZO-1 fluorescence. B, rat livers were perfused as given in the perfusion plan, stained for Bsep and ZO-1, and visualized by confocal laser scanning microscopy. Representative pictures of at least three independent experiments are depicted. The scale bar corresponds to 5 μm. Under normo-osmotic conditions (t = 0), Bsep was largely localized in the canalicular membrane, whereas after hyperosmotic perfusion Bsep appears inside the cells (t = 30 min). TUDC (20 μmol/liter) induced re-insertion in the membrane (t = 60 min). Under control conditions (black, t = 0 min), Bsep-bound fluorescence was largely localized in the canalicular membrane. Hyperosmotic perfusion (green, t = 30 min) resulted in a significant lateralization of the Bsep-bound fluorescence. TUDC (20 μmol/liter) induced re-insertion in the membrane (blue, t = 60 min). The fluorescence profiles depicted are statistically significantly (p < 0.05) different from each other with respect to variance and peak height. Under control conditions (305 mosmol/liter, black), ZO-1 fluorescence profiles show two peaks. Liver perfusion experiments with hyperosmolarity (green) and hyperosmolarity plus TUDC (blue) resulted in no significant changes of ZO-1 fluorescence profiles with respect to the distance of the peaks and to the variance of fluorescence profiles. Means ± S.E. of 30 measurements in each of at least three individual experiments for each condition are shown.
PKC-dependent Inhibition of Hyperosmolarity-induced Generation of Reactive Oxygen Species and Activation of Fyn
In line with previous data (57), hyperosmolarity (405 mosmol/liter) increased CM-H2-DCFDA fluorescence significantly 3.03 ± 0.44-fold (p < 0.05, n = 6) within 15 min (Fig. 10). Administration of TUDC (100 μmol/liter) together with hyperosmotic medium (405 mosmol/liter) inhibited the hyperosmolarity-induced oxidative stress response (1.54 ± 0.39-fold, p < 0.05, n = 6, Fig. 10). As shown in Fig. 10, the specific PKCζ inhibitor (PKCζ pseudosubstrate, 100 μmol/liter) and the PKC inhibitors chelerythrine (20 μmol/liter) and Gö 6850 (10 μmol/liter) (63) also reversed the hyperosmolarity (385 mosmol/liter, 15 min)-induced ROS generation (PKCζ pseudosubstrate 1.03 ± 0.14-fold; chelerythrine 1.21 ± 0.25-fold, and Gö 6850 1.17 ± 0.33-fold, each p < 0.05, n = 3), whereas Gö 6976, a potent inhibitor of Ca2+-dependent PKCs, had no significant inhibitory effect, and hyperosmolarity increased ROS formation under these conditions 2.37 ± 0.29-fold (not significant, n = 3).
FIGURE 10.
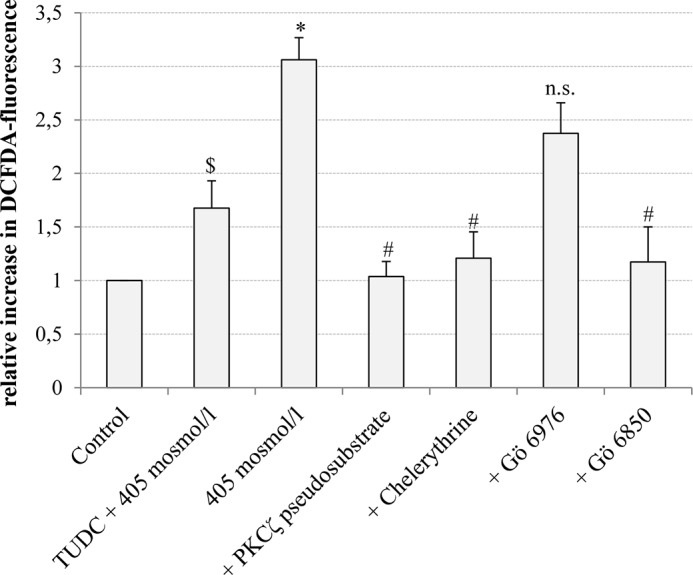
Inhibition of hyperosmolarity-induced generation of reactive oxygen species. Primary rat hepatocytes were cultured for 24 h, loaded with 5 μmol/liter CM-H2DCFDA as described under “Experimental Procedures,” and thereafter stimulated with hyperosmotic medium (405 mosmol/liter) or hyperosmotic medium plus TUDC (100 μmol/liter) for 15 min. When indicated, PKCζ pseudosubstrate (100 μmol/liter), chelerythrine (20 μmol/liter), Gö 6976 (200 nmol/liter), or Gö 6850 (10 μmol/liter) were preincubated for 30 min. ROS levels of at least three independent experiments were expressed as the mean-fold increase over control (set as 1) ± S.E. * indicates the statistical significance compared with the control (p < 0.05); $ indicates the statistical significance of the TUDC effect (p < 0.05), and # indicates the statistical significance of the inhibitor effect (p < 0.05). n.s., not significant.
Paralleling the inhibition of ROS formation, the hyperosmolarity-induced Fyn phosphorylation was sensitive to inhibition of PKCζ by the specific PKCζ pseudosubstrate and by the broad spectrum PKC inhibitors chelerythrine and Gö 6850 (Fig. 11A). Gö 6850 also prevented the hyperosmolarity-induced retrieval of Ntcp (Fig. 11B). Gö 6976, a selective inhibitor of Ca2+-dependent PKC isoforms, had no significant effect on hyperosmotic Fyn phosphorylation (Fig. 11A). These findings suggest that activation of PKCζ in response to hyperosmolarity may play a role in triggering hyperosmotic Fyn phosphorylation (Fig. 11A) and Ntcp retrieval (Fig. 11B).
FIGURE 11.

Effect of PKC inhibitors on hyperosmotic Fyn phosphorylation and inhibition of hyperosmotic Ntcp internalization by the broad spectrum PKC inhibitor Gö 6850. A, primary rat hepatocytes were cultured for 24 h and thereafter stimulated with hyperosmotic medium (405 mosmol/liter) for 30 min. When indicated, PKCζ pseudosubstrate (100 μmol/liter), chelerythrine (20 μmol/liter), Gö 6976 (200 nmol/liter), or Gö 6850 (10 μmol/liter) were preincubated for 30 min. Fyn was immunoprecipitated and detected for phosphorylation by Western blotting. Total Fyn served as a loading control. Hyperosmolarity-induced Fyn phosphorylation was sensitive to inhibition of PKCζ (PKCζ pseudosubstrate, chelerythrine, and Gö 6850). Fyn phosphorylation under control condition was set to 1. Data represent means ± S.E. of three independent experiments. *, p < 0.05 statistically significant compared with the unstimulated control. #, p < 0.05 statistical significance between hyperosmolarity and hyperosmolarity plus PKC inhibitor. B, rat livers were perfused with either Gö 6850 (1 μmol/liter)-containing normo-osmotic (305 mosmol/liter) buffer (black) or hyperosmotic (385 mosmol/liter) buffer in the absence (blue) or presence of the inhibitor (green) (see perfusion plan in Fig. 3A) and immunostained for Ntcp and Na+/K+-ATPase. Densitometric analysis of fluorescence profiles and intensity of Ntcp and Na+/K+-ATPase staining at t = 30 min (see perfusion plan) is shown. The means ± S.E. of 10 measurements in each of three individual experiments for each condition are shown. Gö 6850 significantly inhibited hyperosmolarity-induced retrieval of Ntcp from the membrane (p < 0.05), whereas the inhibitor by itself did not significantly affect Ntcp localization.
Discussion
Hyperosmolarity was shown to stimulate the retrieval of Bsep and Mrp2 from the canalicular membrane (1, 10, 17), whereas the hydrophilic bile acid TUDC stimulates the insertion of these transporters into the canalicular membrane (25, 29). However, the effects of hyperosmolarity and TUDC on the short term regulation of the basolateral transporter Ntcp remained unexplored. This study shows the following: (i) hyperosmolarity induces an oxidative stress- and Fyn-dependent retrieval of Ntcp from the membrane, and (ii) TUDC is able to stimulate re-insertion of Ntcp into the plasma membrane by a β1-integrin- and PKA-dependent mechanism. To what extent TUDC-induced PI3K activation (64) contributes to this effect remains to be established. Fig. 12 schematically summarizes our current view on short term regulation of the basolateral bile salt transporter Ntcp.
FIGURE 12.
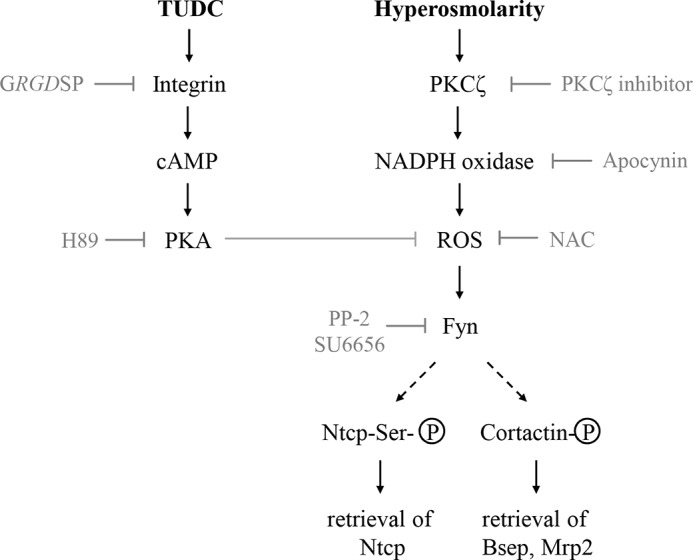
Short term regulation of bile salt transporters in rat liver. Schematic illustration of signaling pathways involved in the short term regulation of basolateral Ntcp and canalicular Bsep and Mrp2. Hyperosmolarity-induced and NADPH-dependent ROS formation triggers activation of the Src family kinase Fyn and induces cholestasis by endocytosis not only of canalicular Bsep and Mrp2 but also of basolateral Ntcp. TUDC induces re-insertion of Ntcp by activation of β1-integrin signaling.
Under normo-osmotic conditions, Ntcp was found mainly in the basolateral membrane and also in transporter-containing vesicles below the plasma membrane (Figs. 2 and 5). Hyperosmolarity increased the fraction of Ntcp, which is located inside the hepatocytes (Figs. 2 and 5). This shift in Ntcp localization occurred within 30 min, persisted for at least 60 min, and was reversible in response to TUDC (Fig. 5). Activation of the Src kinase Fyn was shown to occur in response to hyperosmotic cell shrinkage and to trigger the retrieval of Bsep and Mrp2 from the canalicular membrane (1). As shown in this study, hyperosmotic Fyn activation also triggers Ntcp retrieval from the basolateral membrane (Fig. 3). PP-2, which blocks the hyperosmolarity-induced phosphorylation of Fyn (1), abolished internalization of Ntcp (Fig. 3). Likewise, inhibitors of the upstream events leading to Fyn activation, such as the Nox inhibitor apocynin or the antioxidant NAC, prevented transporter internalization (Fig. 3). Furthermore, Ntcp retrieval was inhibited in p47phox knock-out mice (Fig. 4), in which the hyperosmolarity-induced activation of Fyn is abolished (1). These data strongly suggest that Fyn mediates not only Bsep and Mrp2 retrieval from the canalicular membrane, but simultaneously it also triggers the retrieval of Ntcp from the basolateral membrane. This suggests a coordinated regulation of sinusoidal bile salt uptake and canalicular export systems, which may help to avoid accumulation of potentially toxic bile acids in the hepatocyte. This view is also corroborated by the finding that TUDC and cAMP could reverse hyperosmolarity-induced Fyn activation (Figs. 6 and 7), Ntcp retrieval from the plasma membrane (Fig. 5), and Bsep retrieval from the canalicular membrane (Fig. 9). Thus, TUDC exerts its choleretic effect by a coordinated insertion of both Ntcp and Bsep into plasma and canalicular membrane, respectively. A recent study showed that vesicle motility and ATP production is transiently reduced in a variety of cell types, including hepatocytes after treatment with hyperosmotic medium (65). Movement of dextran-loaded endosomes was tracked by live-cell imaging. Because dextran is distributed to early, late, and recycling endosomes, this approach, however, will not distinguish between differences in motility among these compartments. Whether the motility is restricted also in newly endocytosed vesicles therefore remains unclear (65).
Hyperosmotic perfusion of rat liver induced NADPH oxidase-dependent oxidative stress response as an upstream signal for Fyn activation. Cell hydration and oxidative stress are apparently closely interlinked. On the one hand cell shrinkage induces oxidative stress (66), and on the other hand oxidative stress leads to hepatocyte shrinkage (67). Inflammation is frequently accompanied by ROS formation and can induce cholestasis. It is likely that inflammation also triggers Ntcp retrieval from the plasma membrane. Indeed, proapoptotic bile salts such as taurolithocholic acid 3-sulfate or glycochenodeoxycholate, which induce a NOX2-dependent ROS formation and cell shrinkage (68), also trigger Fyn activation and Ntcp internalization.
Hyperosmolarity-induced retrieval of Ntcp was not accompanied by a down-regulation of Ntcp mRNA levels within 60 min, whereas LPS triggered both Ntcp retrieval and a significant decrease of Ntcp expression at the mRNA and protein level (69). This difference between hyperosmotic and LPS-induced Ntcp retrieval may be explained by cytokine effects.
PKCs belong to a family of serine/threonine kinases, which consists of at least 12 isoforms (70). These kinases are involved in the regulation of diverse cellular functions (70), including bile formation (35, 71). It was suggested that choleretic and cholestatic effects may involve different PKC isoforms; however, the picture is not entirely consistent. The toxic bile acid TCDC inhibits bile salt transport across the sinusoidal membrane by PKC- and PP2B-mediated retrieval of Ntcp from the plasma membrane (33). Phorbol ester-induced endocytosis of Ntcp was shown to be mediated in part by Ca2+-dependent PKCs (56). Furthermore, it was shown recently that PKCα can directly interact with Ntcp in stably Ntcp-transfected HepG2 cells (72). PKCζ (73, 74) was suggested to mediate a choleretic effect by inserting Ntcp into the membrane, whereas PKCϵ (75, 76) may trigger cholestasis by retrieving Mrp2 from the canalicular membrane. However, PKCζ was also shown to trigger p47phox phosphorylation and to induce oxidative stress (57) as an upstream event for Bsep and Mrp2 retrieval from the canalicular membrane (1). This study shows that PKCζ is involved in the Fyn-regulated retrieval of the Ntcp from the membrane (Fig. 11), whereas PKCζ inhibitors were reported to prevent the cAMP-induced Ntcp translocation to the plasma membrane (73). These discrepant findings strongly suggest that regulation of Ntcp retrieval/insertion by PKCζ may be dependent on the signaling context and the underlying stimulus.
As part of its choleretic effect, TUDC stimulates insertion of the canalicular transporter Bsep and Mrp2 by activation of Erks and p38MAPK (25, 50) and counteracts internalization in taurolithocholate-induced cholestasis (29). In this study, the choleretic effect of TUDC may reside in the prevention of hyperosmotic Fyn activation and cAMP formation. Recently, β1-integrins were identified as sensors for TUDC in the liver (19, 48), and molecular dynamics simulation studies showed that TUDC directly interacts with α5β1-integrins resulting in an activation of β1-integrin and the initiation of the downstream integrin signaling (19, 48). This also explains the TUDC-induced activation of c-Src as observed in Fig. 6C. Translocation of Ntcp to the plasma membrane by TUDC was sensitive to GRGDSP and H89 (Fig. 5), which suggests that also Ntcp insertion by TUDC involves a β1-integrin-mediated activation of PKA. Similarly to TUDC, cAMP significantly increased Ntcp translocation to the basolateral membrane in a PKA-dependent way (data not shown). The effect of cAMP is known to be mediated via two major pathways. Activation of PI3K, probably via PKA (77), leads to activation of Akt (78) and PKC isoforms (73, 74, 79), which in turn induce the translocation of Ntcp from an intracellular compartment to the membrane.
Ntcp is a serine/threonine-phosphorylated protein (34), and dephosphorylation at Ser-226 (37) is associated with translocation to the plasma membrane. cAMP activates the serine and threonine protein phosphatase PP2B by increasing intracellular Ca2+. Activated PP2B directly dephosphorylates Ntcp, allowing translocation to the basolateral membrane (80). It was shown recently that TUDC by itself can induce a β1-integrin-dependent rapid generation of cAMP (49), which may explain Ntcp dephosphorylation in response to TUDC.
Taken together, this study for the first time shows that hyperosmolarity induces the retrieval of Ntcp from the plasma membrane and that this effect is mediated by the Src family kinase Fyn, which is activated by a PKCζ-dependent and NADPH oxidase-driven ROS formation. TUDC counteracts the effect of hyperosmolarity through activation of a β1-integrin/PKA signaling pathway, which prevents hyperosmotic ROS formation and Fyn activation and is associated with dephosphorylation of the Ntcp on serine residues.
Author Contributions
D. H. and A. S. designed and planned the research and wrote the paper. A. S., P. G. K. M., and M. C. performed and analyzed the transporter localization experiments. A. S. and M. C. performed and analyzed Western blot data. A. S. performed the SR-SIM pictures and analyzed ROS and real time-PCR data. All authors approved the final version of the manuscript.
Acknowledgments
We thank Nicole Eichhorst, Lisa Knopp, and Janina Thies for their expert technical assistance.
This work was supported by Deutsche Forschungsgemeinschaft through Collaborative Research Center 974 (Düsseldorf) “Communication and System Relevance in Liver Injury and Regeneration.” The authors declare that they have no conflicts of interest with the contents of this article.
- Bsep
- bile salt export pump
- CM-H2DCFDA
- 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
- Bt2cAMP
- dibutyryl-cAMP
- EGFP
- enhanced green fluorescent protein
- LSM
- laser scanning microscopy
- NAC
- N-acetylcysteine
- Ntcp
- Na+-taurocholate cotransporting polypeptide
- ROS
- reactive oxygen species
- SR-SIM
- super resolution structured illumination microscopy
- TC
- taurocholate
- TUDC
- tauroursodeoxycholate
- ZO-1
- zona occludens-1.
References
- 1. Cantore M., Reinehr R., Sommerfeld A., Becker M., Häussinger D. (2011) The Src family kinase Fyn mediates hyperosmolarity-induced Mrp2 and Bsep retrieval from canalicular membrane. J. Biol. Chem. 286, 45014–45029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Elferink R. O., Groen A. K. (2002) Genetic defects in hepatobiliary transport. Biochim. Biophys. Acta 1586, 129–145 [DOI] [PubMed] [Google Scholar]
- 3. Reinehr R., Graf D., Häussinger D. (2003) Bile salt-induced hepatocyte apoptosis involves epidermal growth factor receptor-dependent CD95 tyrosine phosphorylation. Gastroenterology 125, 839–853 [DOI] [PubMed] [Google Scholar]
- 4. Watanabe M., Houten S. M., Mataki C., Christoffolete M. A., Kim B. W., Sato H., Messaddeq N., Harney J. W., Ezaki O., Kodama T., Schoonjans K., Bianco A. C., Auwerx J. (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439, 484–489 [DOI] [PubMed] [Google Scholar]
- 5. Graf D., Kohlmann C., Haselow K., Gehrmann T., Bode J. G., Häussinger D. (2006) Bile acids inhibit interleukin-6 signaling via gp130 receptor-dependent and -independent pathways in rat liver. Hepatology 44, 1206–1217 [DOI] [PubMed] [Google Scholar]
- 6. Becker S., Reinehr R., Graf D., vom Dahl S., Häussinger D. (2007) Hydrophobic bile salts induce hepatocyte shrinkage via NADPH oxidase activation. Cell Physiol. Biochem. 19, 89–98 [DOI] [PubMed] [Google Scholar]
- 7. Gerloff T., Stieger B., Hagenbuch B., Madon J., Landmann L., Roth J., Hofmann A. F., Meier P. J. (1998) The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J. Biol. Chem. 273, 10046–10050 [DOI] [PubMed] [Google Scholar]
- 8. Akita H., Suzuki H., Ito K., Kinoshita S., Sato N., Takikawa H., Sugiyama Y. (2001) Characterization of bile acid transport mediated by multidrug resistance associated protein 2 and bile salt export pump. Biochim. Biophys. Acta 1511, 7–16 [DOI] [PubMed] [Google Scholar]
- 9. Schmitt M., Kubitz R., Wettstein M., vom Dahl S., Häussinger D. (2000) Retrieval of the mrp2 gene encoded conjugate export pump from the canalicular membrane contributes to cholestasis induced by tert-butyl hydroperoxide and chloro-dinitrobenzene. Biol. Chem. 381, 487–495 [DOI] [PubMed] [Google Scholar]
- 10. Schmitt M., Kubitz R., Lizun S., Wettstein M., Häussinger D. (2001) Regulation of the dynamic localization of the rat Bsep gene-encoded bile salt export pump by anisoosmolarity. Hepatology 33, 509–518 [DOI] [PubMed] [Google Scholar]
- 11. Pérez L. M., Milkiewicz P., Elias E., Coleman R., Sánchez Pozzi E. J., Roma M. G. (2006) Oxidative stress induces internalization of the bile salt export pump, Bsep, and bile salt secretory failure in isolated rat hepatocyte couplets: a role for protein kinase C and prevention by protein kinase A. Toxicol. Sci. 91, 150–158 [DOI] [PubMed] [Google Scholar]
- 12. Vos T. A., Hooiveld G. J., Koning H., Childs S., Meijer D. K., Moshage H., Jansen P. L., Müller M. (1998) Up-regulation of the multidrug resistance genes, Mrp1 and Mdr1b, and down-regulation of the organic anion transporter, Mrp2, and the bile salt transporter, Spgp, in endotoxemic rat liver. Hepatology 28, 1637–1644 [DOI] [PubMed] [Google Scholar]
- 13. Trauner M., Arrese M., Lee H., Boyer J. L., Karpen S. J. (1998) Endotoxin downregulates rat hepatic ntcp gene expression via decreased activity of critical transcription factors. J. Clin. Invest. 101, 2092–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kubitz R., Wettstein M., Warskulat U., Häussinger D. (1999) Regulation of the multidrug resistance protein 2 in the rat liver by lipopolysaccharide and dexamethasone. Gastroenterology 116, 401–410 [DOI] [PubMed] [Google Scholar]
- 15. Koopen N. R., Wolters H., Havinga R., Vonk R. J., Jansen P. L., Müller M., Kuipers F. (1998) Impaired activity of the bile canalicular organic anion transporter (Mrp2/cmoat) is not the main cause of ethinylestradiol-induced cholestasis in the rat. Hepatology 27, 537–545 [DOI] [PubMed] [Google Scholar]
- 16. Román I. D., Fernández-Moreno M. D., Fueyo J. A., Roma M. G., Coleman R. (2003) Cyclosporin A induced internalization of the bile salt export pump in isolated rat hepatocyte couplets. Toxicol. Sci. 71, 276–281 [DOI] [PubMed] [Google Scholar]
- 17. Kubitz R., D'urso D., Keppler D., Häussinger D. (1997) Osmodependent dynamic localization of the multidrug resistance protein 2 in the rat hepatocyte canalicular membrane. Gastroenterology 113, 1438–1442 [DOI] [PubMed] [Google Scholar]
- 18. Warskulat U., Kubitz R., Wettstein M., Stieger B., Meier P. J., Häussinger D. (1999) Regulation of bile salt export pump mRNA levels by dexamethasone and osmolarity in cultured rat hepatocytes. Biol. Chem. 380, 1273–1279 [DOI] [PubMed] [Google Scholar]
- 19. Häussinger D., Kurz A. K., Wettstein M., Graf D., Vom Dahl S., Schliess F. (2003) Involvement of integrins and Src in tauroursodeoxycholate-induced and swelling-induced choleresis. Gastroenterology 124, 1476–1487 [DOI] [PubMed] [Google Scholar]
- 20. Häussinger D. (1996) Regulation and functional significance of liver cell volume. Prog. Liver Dis. 14, 29–53 [PubMed] [Google Scholar]
- 21. Lang F., Busch G. L., Ritter M., Völkl H., Waldegger S., Gulbins E., Häussinger D. (1998) Functional significance of cell volume regulatory mechanisms. Physiol. Rev. 78, 247–306 [DOI] [PubMed] [Google Scholar]
- 22. Schliess F., Reinehr R., Häussinger D. (2007) Osmosensing and signaling in the regulation of mammalian cell function. FEBS J. 274, 5799–5803 [DOI] [PubMed] [Google Scholar]
- 23. Häussinger D., Saha N., Hallbrucker C., Lang F., Gerok W. (1993) Involvement of microtubules in the swelling-induced stimulation of transcellular taurocholate transport in perfused rat liver. Biochem. J. 291, 355–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Noé B., Schliess F., Wettstein M., Heinrich S., Häussinger D. (1996) Regulation of taurocholate excretion by a hypo-osmolarity-activated signal transduction pathway in rat liver. Gastroenterology 110, 858–865 [DOI] [PubMed] [Google Scholar]
- 25. Kurz A. K., Graf D., Schmitt M., Vom Dahl S., Häussinger D. (2001) Tauroursodesoxycholate-induced choleresis involves p38(MAPK) activation and translocation of the bile salt export pump in rats. Gastroenterology 121, 407–419 [DOI] [PubMed] [Google Scholar]
- 26. Roelofsen H., Soroka C. J., Keppler D., Boyer J. L. (1998) Cyclic AMP stimulates sorting of the canalicular organic anion transporter (Mrp2/cMoat) to the apical domain in hepatocyte couplets. J. Cell Sci. 111, 1137–1145 [DOI] [PubMed] [Google Scholar]
- 27. Misra S., Varticovski L., Arias I. M. (2003) Mechanisms by which cAMP increases bile acid secretion in rat liver and canalicular membrane vesicles. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G316–G324 [DOI] [PubMed] [Google Scholar]
- 28. Schonhoff C. M., Thankey K., Webster C. R., Wakabayashi Y., Wolkoff A. W., Anwer M. S. (2008) Rab4 facilitates cyclic adenosine monophosphate-stimulated bile acid uptake and Na+-taurocholate cotransporting polypeptide translocation. Hepatology 48, 1665–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beuers U., Bilzer M., Chittattu A., Kullak-Ublick G. A., Keppler D., Paumgartner G., Dombrowski F. (2001) Tauroursodeoxycholic acid inserts the apical conjugate export pump, Mrp2, into canalicular membranes and stimulates organic anion secretion by protein kinase C-dependent mechanisms in cholestatic rat liver. Hepatology 33, 1206–1216 [DOI] [PubMed] [Google Scholar]
- 30. Stieger B., Hagenbuch B., Landmann L., Höchli M., Schroeder A., Meier P. J. (1994) In situ localization of the hepatocytic Na+/Taurocholate cotransporting polypeptide in rat liver. Gastroenterology 107, 1781–1787 [DOI] [PubMed] [Google Scholar]
- 31. Vaz F. M., Paulusma C. C., Huidekoper H., de Ru M., Lim C., Koster J., Ho-Mok K., Bootsma A. H., Groen A. K., Schaap F. G., Oude Elferink R. P., Waterham H. R., Wanders R. J. (2015) Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: conjugated hypercholanemia without a clear clinical phenotype. Hepatology 61, 260–267 [DOI] [PubMed] [Google Scholar]
- 32. Schonhoff C. M., Yamazaki A., Hohenester S., Webster C. R., Bouscarel B., Anwer M. S. (2009) PKCϵ-dependent and -independent effects of taurolithocholate on PI3K/PKB pathway and taurocholate uptake in HuH-NTCP cell line. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G1259–G1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mühlfeld S., Domanova O., Berlage T., Stross C., Helmer A., Keitel V., Häussinger D., Kubitz R. (2012) Short-term feedback regulation of bile salt uptake by bile salts in rodent liver. Hepatology 56, 2387–2397 [DOI] [PubMed] [Google Scholar]
- 34. Mukhopadhyay S., Ananthanarayanan M., Stieger B., Meier P. J., Suchy F. J., Anwer M. S. (1998) Sodium taurocholate cotransporting polypeptide is a serine, threonine phosphoprotein and is dephosphorylated by cyclic adenosine monophosphate. Hepatology 28, 1629–1636 [DOI] [PubMed] [Google Scholar]
- 35. Anwer M. S., Stieger B. (2014) Sodium-dependent bile salt transporters of the SLC10A transporter family: more than solute transporters. Pflugers Arch. 466, 77–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mukhopadhayay S., Ananthanarayanan M., Stieger B., Meier P. J., Suchy F. J., Anwer M. S. (1997) cAMP increases liver Na+-taurocholate cotransport by translocating transporter to plasma membranes. Am. J. Physiol. 273, G842–G848 [DOI] [PubMed] [Google Scholar]
- 37. Anwer M. S., Gillin H., Mukhopadhyay S., Balasubramaniyan N., Suchy F. J., Ananthanarayanan M. (2005) Dephosphorylation of Ser-226 facilitates plasma membrane retention of Ntcp. J. Biol. Chem. 280, 33687–33692 [DOI] [PubMed] [Google Scholar]
- 38. Webster C. R., Blanch C. J., Phillips J., Anwer M. S. (2000) Cell swelling-induced translocation of rat liver Na+/taurocholate cotransport polypeptide is mediated via the phosphoinositide 3-kinase signaling pathway. J. Biol. Chem. 275, 29754–29760 [DOI] [PubMed] [Google Scholar]
- 39. Hofmann A. F. (1994) Pharmacology of ursodeoxycholic acid, an enterohepatic drug. Scand. J. Gastroenterol. Suppl. 204, 1–15 [DOI] [PubMed] [Google Scholar]
- 40. Beuers U., Spengler U., Kruis W., Aydemir U., Wiebecke B., Heldwein W., Weinzierl M., Pape G. R., Sauerbruch T., Paumgartner G. (1992) Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: a placebo-controlled trial. Hepatology 16, 707–714 [DOI] [PubMed] [Google Scholar]
- 41. Leuschner U., Fischer H., Kurtz W., Güldütuna S., Hübner K., Hellstern A., Gatzen M., Leuschner M. (1989) Ursodeoxycholic acid in primary biliary cirrhosis: results of a controlled double-blind trial. Gastroenterology 97, 1268–1274 [DOI] [PubMed] [Google Scholar]
- 42. Poupon R. E., Balkau B., Eschwège E., Poupon R. (1991) A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group. N. Engl. J. Med. 324, 1548–1554 [DOI] [PubMed] [Google Scholar]
- 43. Poupon R. (2012) Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: an overview of their mechanisms of action. Clin. Res. Hepatol. Gastroenterol. 36, S3–S12 [DOI] [PubMed] [Google Scholar]
- 44. Beuers U., Trauner M., Jansen P., Poupon R. (2015) New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J. Hepatol. 62, S25–37 [DOI] [PubMed] [Google Scholar]
- 45. Faubion W. A., Guicciardi M. E., Miyoshi H., Bronk S. F., Roberts P. J., Svingen P. A., Kaufmann S. H., Gores G. J. (1999) Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J. Clin. Invest. 103, 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Graf D., Kurz A. K., Fischer R., Reinehr R., Häussinger D. (2002) Taurolithocholic acid-3 sulfate induces CD95 trafficking and apoptosis in a c-Jun N-terminal kinase-dependent manner. Gastroenterology 122, 1411–1427 [DOI] [PubMed] [Google Scholar]
- 47. Paumgartner G., Beuers U. (2002) Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 36, 525–531 [DOI] [PubMed] [Google Scholar]
- 48. Gohlke H., Schmitz B., Sommerfeld A., Reinehr R., Häussinger D. (2013) α5β1-integrins are sensors for tauroursodeoxycholic acid in hepatocytes. Hepatology 57, 1117–1129 [DOI] [PubMed] [Google Scholar]
- 49. Sommerfeld A., Reinehr R., Häussinger D. (2015) Tauroursodeoxycholate protects rat hepatocytes from bile acid-induced apoptosis via β1-integrin- and protein kinase A-dependent mechanisms. Cell Physiol. Biochem. 36, 866–883 [DOI] [PubMed] [Google Scholar]
- 50. Schliess F., Kurz A. K., vom Dahl S., Häussinger D. (1997) Mitogen-activated protein kinases mediate the stimulation of bile acid secretion by tauroursodeoxycholate in rat liver. Gastroenterology 113, 1306–1314 [DOI] [PubMed] [Google Scholar]
- 51. Yan H., Zhong G., Xu G., He W., Jing Z., Gao Z., Huang Y., Qi Y., Peng B., Wang H., Fu L., Song M., Chen P., Gao W., Ren B., et al. (2014) Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012 13;3. [DOI] [PubMed] [Google Scholar]
- 52. Bouezzedine F., Fardel O., Gripon P. (2015) Interleukin 6 inhibits HBV entry through NTCP down regulation. Virology 481, 34–42 [DOI] [PubMed] [Google Scholar]
- 53. Noé J., Stieger B., Meier P. J. (2002) Functional expression of the canalicular bile salt export pump of human liver. Gastroenterology 123, 1659–1666 [DOI] [PubMed] [Google Scholar]
- 54. Sies H. (1978) The use of perfusion of liver and other organs for the study of microsomal electron-transport and cytochrome P-450 systems. Methods Enzymol. 52, 48–59 [DOI] [PubMed] [Google Scholar]
- 55. Meijer A. J., Gimpel J. A., Deleeuw G. A., Tager J. M., Williamson J. R. (1975) Role of anion translocation across the mitochondrial membrane in the regulation of urea synthesis from ammonia by isolated rat hepatocytes. J. Biol. Chem. 250, 7728–7738 [PubMed] [Google Scholar]
- 56. Stross C., Helmer A., Weissenberger K., Görg B., Keitel V., Häussinger D., Kubitz R. (2010) Protein kinase C induces endocytosis of the sodium taurocholate cotransporting polypeptide. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G320–G328 [DOI] [PubMed] [Google Scholar]
- 57. Reinehr R., Becker S., Braun J., Eberle A., Grether-Beck S., Haüssinger D. (2006) Endosomal acidification and activation of NADPH oxidase isoforms are upstream events in hyperosmolarity-induced hepatocyte apoptosis. J. Biol. Chem. 281, 23150–23166 [DOI] [PubMed] [Google Scholar]
- 58. Hanke J. H., Gardner J. P., Dow R. L., Changelian P. S., Brissette W. H., Weringer E. J., Pollok B. A., Connelly P. A. (1996) Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem. 271, 695–701 [DOI] [PubMed] [Google Scholar]
- 59. Blake R. A., Broome M. A., Liu X., Wu J., Gishizky M., Sun L., Courtneidge S. A. (2000) SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol. Cell. Biol. 20, 9018–9027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Häussinger D., Hallbrucker C., Saha N., Lang F., Gerok W. (1992) Cell volume and bile acid excretion. Biochem. J. 288, 681–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Busch G. L., Schreiber R., Dartsch P. C., Völkl H., Vom Dahl S., Häussinger D., Lang F. (1994) Involvement of microtubules in the link between cell volume and pH of acidic cellular compartments in rat and human hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 91, 9165–9169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xia W., Springer T. A. (2014) Metal ion and ligand binding of integrin α5β1. Proc. Natl. Acad. Sci. U.S.A. 111, 17863–17868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Toullec D., Pianetti P., Coste H., Bellevergue P., Grand-Perret T., Ajakane M., Baudet V., Boissin P., Boursier E., Loriolle F. (1991) The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 266, 15771–15781 [PubMed] [Google Scholar]
- 64. Kurz A. K., Block C., Graf D., Dahl S. V., Schliess F., Häussinger D. (2000) Phosphoinositide 3-kinase-dependent Ras activation by tauroursodesoxycholate in rat liver. Biochem. J. 350, 207–213 [PMC free article] [PubMed] [Google Scholar]
- 65. Nunes P., Roth I., Meda P., Féraille E., Brown D., Hasler U. (2015) Ionic imbalance, in addition to molecular crowding, abates cytoskeletal dynamics and vesicle motility during hypertonic stress. Proc. Natl. Acad. Sci. U.S.A. 112, E3104–E3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Reinehr R., Schliess F., Häussinger D. (2003) Hyperosmolarity and CD95L trigger CD95/EGF receptor association and tyrosine phosphorylation of CD95 as prerequisites for CD95 membrane trafficking and DISC formation. FASEB J. 17, 731–733 [DOI] [PubMed] [Google Scholar]
- 67. Saha N., Schreiber R., vom Dahl S., Lang F., Gerok W., Häussinger D. (1993) Endogenous hydroperoxide formation, cell volume and cellular K+ balance in perfused rat liver. Biochem. J. 296, 701–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Reinehr R., Becker S., Keitel V., Eberle A., Grether-Beck S., Häussinger D. (2005) Bile salt-induced apoptosis involves NADPH oxidase isoform activation. Gastroenterology 129, 2009–2031 [DOI] [PubMed] [Google Scholar]
- 69. Donner M. G., Schumacher S., Warskulat U., Heinemann J., Häussinger D. (2007) Obstructive cholestasis induces TNF-α- and IL-1-mediated periportal downregulation of Bsep and zonal regulation of Ntcp, Oatp1a4, and Oatp1b2. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G1134–G1146 [DOI] [PubMed] [Google Scholar]
- 70. Reyland M. E. (2009) Protein kinase C isoforms: Multi-functional regulators of cell life and death. Front. Biosci. 14, 2386–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Anwer M. S. (2014) Role of protein kinase C isoforms in bile formation and cholestasis. Hepatology 60, 1090–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Stross C., Kluge S., Weissenberger K., Winands E., Häussinger D., Kubitz R. (2013) A dileucine motif is involved in plasma membrane expression and endocytosis of rat sodium taurocholate cotransporting polypeptide (Ntcp). Am. J. Physiol. Gastrointest. Liver Physiol. 305, G722–G730 [DOI] [PubMed] [Google Scholar]
- 73. McConkey M., Gillin H., Webster C. R., Anwer M. S. (2004) Cross-talk between protein kinases Cζ and B in cyclic AMP-mediated sodium taurocholate co-transporting polypeptide translocation in hepatocytes. J. Biol. Chem. 279, 20882–20888 [DOI] [PubMed] [Google Scholar]
- 74. Sarkar S., Bananis E., Nath S., Anwer M. S., Wolkoff A. W., Murray J. W. (2006) PKCζ is required for microtubule-based motility of vesicles containing the ntcp transporter. Traffic 7, 1078–1091 [DOI] [PubMed] [Google Scholar]
- 75. Beuers U., Probst I., Soroka C., Boyer J. L., Kullak-Ublick G. A., Paumgartner G. (1999) Modulation of protein kinase C by taurolithocholic acid in isolated rat hepatocytes. Hepatology 29, 477–482 [DOI] [PubMed] [Google Scholar]
- 76. Schonhoff C. M., Webster C. R., Anwer M. S. (2013) Taurolithocholate-induced MRP2 retrieval involves MARCKS phosphorylation by protein kinase C in HUH-NTCP cells. Hepatology 58, 284–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Grüne S., Engelking L. R., Anwer M. S. (1993) Role of intracellular calcium and protein kinases in the activation of hepatic Na+/taurocholate cotransport by cyclic AMP. J. Biol. Chem. 268, 17734–17741 [PubMed] [Google Scholar]
- 78. Webster C. R., Srinivasulu U., Ananthanarayanan M., Suchy F. J., Anwer M. S. (2002) Protein kinase B/Akt mediates cAMP- and cell swelling-stimulated Na+/taurocholate cotransport and Ntcp translocation. J. Biol. Chem. 277, 28578–28583 [DOI] [PubMed] [Google Scholar]
- 79. Park S. W., Schonhoff C. M., Webster C. R., Anwer M. S. (2012) Protein kinase Cdelta differentially regulates cAMP-dependent translocation of NTCP and MRP2 to the plasma membrane. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G657–G665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Webster C. R., Blanch C., Anwer M. S. (2002) Role of PP2B in cAMP-induced dephosphorylation and translocation of NTCP. Am. J. Physiol. Gastrointest. Liver Physiol. 283, G44–G50 [DOI] [PubMed] [Google Scholar]