

Background: Pin1 expression is regulated by nutrient conditions.
Results: Pin1 binds to γ subunit and suppresses AMPK phosphorylation.
Conclusion: Pin1 expression level affects metabolic regulation.
Significance: Pin1 inhibition is a potential therapy for metabolic syndrome.
Keywords: AMP-activated kinase (AMPK), diabetes, energy metabolism, lipid metabolism, metabolic syndrome, muscle, prolyl isomerase
Abstract
AMP-activated protein kinase (AMPK) plays a critical role in metabolic regulation. In this study, first, it was revealed that Pin1 associates with any isoform of γ, but not with either the α or the β subunit, of AMPK. The association between Pin1 and the AMPK γ1 subunit is mediated by the WW domain of Pin1 and the Thr211-Pro-containing motif located in the CBS domain of the γ1 subunit. Importantly, overexpression of Pin1 suppressed AMPK phosphorylation in response to either 2-deoxyglucose or biguanide stimulation, whereas Pin1 knockdown by siRNAs or treatment with Pin1 inhibitors enhanced it. The experiments using recombinant Pin1, AMPK, LKB1, and PP2C proteins revealed that the protective effect of AMP against PP2C-induced AMPKα subunit dephosphorylation was markedly suppressed by the addition of Pin1. In good agreement with the in vitro data, the level of AMPK phosphorylation as well as the expressions of mitochondria-related genes, such as PGC-1α, which are known to be positively regulated by AMPK, were markedly higher with reduced triglyceride accumulation in the muscles of Pin1 KO mice as compared with controls. These findings suggest that Pin1 plays an important role in the pathogenic mechanisms underlying impaired glucose and lipid metabolism, functioning as a negative regulator of AMPK.
Introduction
To maintain cellular energy homeostasis, starvation triggers cells to inhibit anabolic processes and to promote catabolism. AMP-activated protein kinase (AMPK)2 is a major sensor of cellular energy status and works as a switch for restoration of cellular ATP levels (1–5). AMPK exists as heterotrimers, composed of an α catalytic subunit and β and γ regulatory subunits (1, 6). AMPK activation essentially requires phosphorylation at Thr172 in the α subunit induced by upstream kinase LKB1 or Ca2+/calmodulin-dependent protein kinase II (7–10). Under nutrient-rich conditions, ATP occupies the CBS domain of the γ subunit, and this situation enables phosphatase PP2C to easily access the AMPK α subunit, leading to inactivation of AMPK.
On the contrary, under nutrient-poor conditions, CBS domains are occupied by AMP or ADP, and this binding induces a conformational change in AMPK, thereby protecting the α catalytic subunit from being dephosphorylated by PP2C (1, 11, 12). In addition, upstream kinase LKB1 translocates from the nucleus to the cytosol and phosphorylates the α subunit (10, 13, 14). Thus, LKB1 translocation and occupation of AMP or ADP in the γ subunit are both essential for AMPK activation.
Phosphorylated AMPK transmits signals to numerous downstream substrates, such as acetyl-CoA carboxylase (ACC), TSC2, Raptor, AS160, PGC-1α, etc., the resultant actions of which include lipid oxidation and uptake of glucose (3, 15–20). In fact, deficiency of AMPK subunits in mice reportedly results in glucose intolerance, reduction of exercise capacity, and obesity (21–23). In addition, a line of evidence supports the dysregulation of AMPK in diabetic rodent models (24, 25), and low AMPK activity and high concentrations of malonyl-CoA, the inhibitor of fatty acid oxidation, are observed in muscle from obese and type 2 diabetes patients (26).
AMPK is activated by not only exercise or starvation, but also by numerous secretory factors and anti-diabetic drugs, such as biguanides, DPP4 inhibitors, or GLP-1 (15, 27, 28). In this study, we identified prolyl isomerase Pin1 as a negative regulator of AMPK. Importantly, Pin1 expression was shown to be markedly higher under re-fed than under fasting conditions, or in the high fat-fed state, as compared with feeding of a normal diet (29, 30). Herein, we present evidence showing that increased expression of Pin1 reduces the efficiency of AMPK phosphorylation. This mechanism may be involved the pathogenesis of impaired lipid and glucose metabolism in diabetic and obese subjects.
Experimental Procedures
Materials
Antibodies were purchased from Abcam (Cambridge, UK; S tag and AMPK γ1), Sigma (FLAG), Cell Signaling (Beverly, MA; p/tAMPK α, β, Cyc, PD, and PHB1), and Santa Cruz Biotechnology (Pin1, GAPDH, and tubulin). Anti-rabbit horseradish peroxidase antibodies were obtained from Amersham Biosciences (Buckinghamshire, UK). Dulbecco's modified Eagle's medium (DMEM) was purchased from Nissui (Tokyo, Japan), Metformin was from Wako, and SYBR Green was from Takara (Shiga, Japan). Recombinant LKB1 was purchased from Sigma, and PP2C was from Millipore (Bedford, MA).
Cell Culture
HepG2 and 293T cells were cultured in DMEM containing glutamine, NaHCO3, antibiotics, and 10% fetal calf serum (FCS). One day before the various experiments, cells were cultivated in collagen-coated 24-well plates or 60-mm dishes. To induce overexpression of the subunits of AMPK and Pin1, we prepared expression plasmids. Briefly, cDNAs encoding the α, β, and γ subunits of AMPK and Pin1, with or without S tag or FLAG tag at their N termini, were inserted into pcDNA3. Mutated Trp34 and Lys63 Pin1, lacking the ability to bind the WW protein domain and isomerase activity, respectively, were also prepared. For adenovirus preparation, the GFP or the Pin1 gene was inserted into a pAxCAwtit2 vector, and adenovirus was produced in 293 cells, using an adenovirus dual expression kit (Takara). In plasmid transfection experiments, plasmids were mixed with the transfection reagent X-treme in Opti-MEM for 15 min and then transferred to the cells. For adenovirus experiments, HepG2 cells were infected with adenovirus containing the cDNA encoding GFP or Pin1 in serum-free DMEM for 1 h and then changed to DMEM containing 10% FCS. At 48 h after transfection of the expression plasmid or adenoviral infection, the cells were used for experiments.
Pin1 Inhibitors
Juglone is an agent widely used as a Pin1 inhibitor. However, evidence of tubulin aggregation or the disappearance of BubR1 immunoreactivity in response to Juglone, independently of the presence of Pin1 (31), suggests an additional effect besides Pin1 inhibition. Thus, we employed not only Juglone but also two other Pin1 inhibitor types possibly more highly specific for Pin1, both of which were custom-made by SundiaMediTech Company Ltd. (Shanghai, China). The one termed Pin1 inhibitor 1 is (R)-2-(5-(4-methoxyphenyl)-2-methylfuran-3-caroxamido)-3-(naphthalen-6-yl) propanoic acid (32), and the other, termed Pin1 inhibitor 2, is 4-(N-benzyl-N-phenethylcarbamoyl)-2-(3-chlorophenyl)-1H-imidazole-5-carboxylic acid (33). These inhibitors were added to cells 30 min prior to 2-deoxyglucose (2-DG) treatment.
Immunoprecipitation
293T cells or mouse tissues were solubilized with lysis buffer containing NaCl, KCl, EGTA, 1% Triton, PMSF, orthovanadate, and NaF. After centrifugation at 15,000 rpm for 30 min, supernatants were transferred to new tubes. Then adequate amounts of antibodies and beads were added, and the lysates were rotated for 2 h at 4 °C. The beads were washed four times with lysis buffer. In the case of using FLAG beads, immunoprecipitates were eluted employing FLAG peptide. Finally, 2× SDS sample buffer was added to each tube, followed by boiling at 95 °C for 5 min.
Immunoblotting
Proteins were electrophoresed on SDS-PAGE and then transferred to PVDF membranes. After blocking with 3% bovine serum albumin in phosphate-buffered saline/Tween for 1 h, the membranes were reacted with the first antibody (1:2000) for 1 h, followed by the secondary antibody for 1 h. The bands were detected using Super Signal West Pico stable peroxidase solution (Thermo).
Gene Silencing of Pin1 Using siRNAs
HepG2 and 293T cells were transfected with either negative siRNA (Qiagen) or Pin1 siRNA (Invitrogen) using RNAiMAX (Invitrogen) according to the manufacturer's protocol: Pin1 siRNA1, CCG UGU UCA CGG AUU CCG GCA UCC A; Pin1 siRNA2, GCC CUG GAG CUG AUC AAC GGC UAC A.
Real-time PCR
RNA extraction was performed using Sepasol according to the manufacturer's protocol. To measure the mRNA levels of PGC-1α, Nrf1, Tfam, and cytochrome c, reverse transcription and real-time PCR were conducted as reported previously (34). Primer sets were as follows: mPGC-1α, gca ctt cgg tca tcc ctg tc (forward) and ggc gac aca tcg aac aat ga (reverse); mNrf1, caa cag gga aga aac gga aa (forward) and gca cca cat tct cca aag gt (reverse); mTfam, aag ctt cca gga ggc aaa gg (forward) and tgt ctc cgg atc gtt tca ca (reverse); mCyt c, caaatctccacggtctgttcg (forward) and ggtctgccctttctcccttc (reverse).
Preparation of Recombinant AMPK Complex
AMPK complexes (α/β/γ) with or without S-tagged Pin1 were obtained from 293T cells. Briefly, the S-tagged α subunit, the β subunit with no tag, and the FLAG-tagged γ1 subunit with or without S-tagged Pin1 were overexpressed in 293T cells by transfection with their respective expression plasmids. The cell lysates were solubilized with the lysis buffer containing NaCl, KCl, EGTA, 1% Triton, PMSF, orthovanadate, and NaF. After centrifugation at 15,000 rpm for 30 min, the solubilized fractions were mixed with FLAG tag antibody-conjugated beads for 2 h. After the beads had been washed with lysis buffer four times, recombinant AMPK and AMPK-Pin1 complexes were prepared by elution with the FLAG peptide.
The in Vitro Dephosphorylation of the AMPK α Subunit at Thr172 by PP2C and Phosphorylation by LKB1
To examine the effect of Pin1 on the dephosphorylation of AMPK by PP2C, AMPK or the AMPK-Pin1 complex, purified employing FLAG tag antibody-conjugated beads, was reacted with 25 ng of recombinant PP2C in the presence or absence of 150 μm AMP or ADP in assay buffer containing Tris-HCl, NaCl, and MgCl2 in a volume of 20 μl for up to 120 min at 30 ºC.
For the LKB1 phosphorylation assay, AMPK or AMPK-Pin1 complexes conjugated with FLAG antibody-conjugated beads were reacted with 25 ng/ml PP2C for 2 h, prior to elution with the FLAG peptide, to reduce the level of AMPK phosphorylation. Then the beads were washed four times, and the AMPK complex was eluted employing FLAG peptide. The largely dephosphorylated AMPK or AMPK-Pin1 complexes were reacted with 50 ng of recombinant LKB1 complex with or without 300 μm AMP in a volume of 20 μl for up to 30 min at 37 °C. The reactions with PP2C or LKB1 were stopped by adding DB buffer, followed by immediate boiling at 95 °C for 5 min. Then the phosphorylation levels at Thr172 in the α subunit were examined by immunoblotting with the antibody recognizing this phosphorylation.
Animals
To generate Pin1 flox mice (accession number CDB1016K; see the Center for Developmental Biology Web site), a genomic sequence including exon2 of Pin1 was inserted into the DT-ApA/conditional KO FW vector (Fig. 7A). The target vector was electroporated into TT2 embryonic stem cells (35), and homologous recombinants were selected by PCR and Southern blotting. To create chimeric mice, positive clones were injected into CD-1 8-cell-stage embryos. The chimeric mice were then mated with C57BL/6J. To create Pin1 null mice, Pin1 flox mice were crossed with CAG-Cre mice. The genotyping primers used were P1 (tct cct tcc act ggg caa ctt cct g) and P2 (tgc tgt ccc cat cgg gac ct) for both the wild and the floxed type allele and P1 and P3 (ctg tgg tgc tct tgg ggg tg) for the KO allele, yielding 180-, 375-, and 419-bp products, respectively. These mice were maintained in a temperature- and light-controlled facility. The animals were handled in accordance with the Animal Experimentation guidelines of Hiroshima University.
FIGURE 7.

AMPK phosphorylation is elevated in Pin1 KO mouse muscle. A, scheme of the genomic construct of Pin1 flox mice. B, loss of Pin1 in muscle promotes AMPK phosphorylation. After the animals had been fasted for 16 h, muscles were harvested from Pin1 KO and wild-type mice, and the indicated protein levels were detected, using the corresponding antibody. C and D, expressions of mitochondria-related genes and proteins were elevated in Pin1 KO mice. E, muscle triglyceride content was lower in Pin1 KO mice. IB, immunoblotting. Error bars, S.E.; *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant.
Statistical Analysis
Data are expressed as means ± S.E., and values of p < 0.05 were considered to be statistically significant.
Results
Pin1 Associates with AMPK γ Subunits
To investigate whether or not Pin1 binds to each of the subunits of AMPK, S-tagged Pin1 was overexpressed with the FLAG-tagged α1, α2, β1, β2, γ1, γ2, or γ3 subunit of AMPK or control LacZ. Immunoblotting of anti-FLAG tag antibody immunoprecipitates with anti-S tag antibody revealed associations of Pin1 with γ1, γ2, and γ3 but not with other isoforms of AMPK (Fig. 1A). With exposure for much longer periods, the bands of the α1, α2, β1, and β2 subunits were detectable in the anti-FLAG tag antibody immunoprecipitates, but this would very probably be due to complex formation with the γ subunit (data not shown). The association between the FLAG-tagged AMPK γ1 subunit and the S-tagged Pin1 was also detected by immunoblotting of anti-S tag immunoprecipitates with anti-FLAG antibody (Fig. 1B). The association with AMPK γ was observed for the wild type and for a Lys63 mutant Pin1 lacking isomerase activity but not for a W34A mutant reportedly lacking the protein binding function to the WW domain (Fig. 1C). In addition, the association between Pin1 and the γ subunit was not affected by either incubation with 10 mm 2-DG for 1 h or by serum starvation for 16 h (Fig. 1D). An endogenous association between Pin1 and the γ1 subunit was demonstrated by immunoprecipitation and subsequent immunoblotting using anti-Pin1 and anti-γ1 subunit antibodies in mouse muscle (Fig. 1E). To examine whether Pin1 binding to the γ subunit affects the association of the γ subunit with the α or the β subunit, the FLAG-tagged γ subunit and S-tagged Pin1 were overexpressed. The amounts of the α1 and β1 subunits co-immunoprecipitated with the FLAG-tagged γ subunit by anti-FLAG tag antibody were not altered by Pin1 overexpression (Fig. 1F). Finally, whether the association between Pin1 and AMPK was affected by either starvation or feeding was examined (Fig. 1, G and H). Pin1 expression in mouse muscle was higher by ∼5-fold in the fed than in the fasted state, whereas expression of the γ1 subunit was unchanged (Fig. 1G). In proportion to the increase in the total amount of Pin1, the amount of Pin1 associated with the γ1 subunit was also markedly elevated in the re-fed state (Fig. 1H).
FIGURE 1.

Pin1 associates with AMPK γ subunit. A and B, Pin1 interacts with the AMPK γ subunit in 293T cells. The 293T cells were transfected with S tag Pin1 (wild type, W34A, and K63A) and the FLAG-tagged α1, α2, β1, β2, γ1, γ2, or γ3 subunit of AMPK. Then cell lysates were immunoprecipitated (IP) with anti-FLAG tag antibody beads, followed by immunoblotting (IB) with anti-S tag or anti-FLAG tag antibody. C, Pin1 associates with the γ1 subunit independently of 2-DG stimulation or serum starvation. The 293T cells overexpressing Pin1 and the γ1 subunit were treated with 2-DG for 1 h or serum-starved overnight. Then cell lysates were immunoprecipitated with anti-FLAG tag antibody, followed by immunoblotting with anti-S tag or anti-FLAG tag antibody. D, Pin1 endogenously binds to the γ1 subunit in muscle. Protein extracts from mouse muscle were immunoprecipitated with anti-Pin1 antibody, followed by immunoblotting with anti-γ1 subunit antibody. E, Pin1 had no effect on formation of the AMPK complex. FLAG-tagged γ1 subunits alone or with S-tagged Pin1 were transfected into 293T cells, followed by immunoprecipitation with anti-FLAG tag antibody and immunoblotting with the indicated antibodies. F, Pin1 expressions changed in response to nutrient conditions. C57BL/6J mice were fasted for 16 h and then re-fed for 4 h. The mice were then sacrificed, and muscles were harvested. The indicated proteins were detected using the corresponding antibody. G, the association between Pin1 and γ1 was increased in the re-fed state. Tissue lysates from muscle were immunoprecipitated with γ1 antibody, followed by immunoblotting. Shown are representative data from three independent experiments. Error bars, S.E.; *, p < 0.05.
Thr211-Pro-containing Motif in γ1 Subunit Is Required for Binding with Pin1
To identify the domain of Pin1 responsible for the association with the γ1 subunit, the pGEX vectors expressing the GST proteins fused with the WW domain, the PPI domain of Pin1, and full-length Pin1 were prepared. GST alone, the GST-WW domain, the GST-PPI domain, and GST-full length Pin1 were expressed in E. coli. BL21 was purified using glutathione beads, as confirmed by Coomassie Blue staining (Fig. 2A, middle). The pull-down experiments, performed by mixing the lysates from the 293 cells overexpressing the FLAG-tagged AMPK γ1 subunit with GST alone, the GST-WW domain, the GST-PPI domain, or the GST-full-length Pin1, revealed that the GST-WW domain and GST-full-length Pin1 but not the GST-PPI domain associated with the γ1 subunit (Fig. 2A). In addition, this interaction was abolished by treating the cell lysates containing the FLAG-tagged AMPK γ1 subunit with calf intestinal alkaline phosphatase, suggesting that phosphorylation of the γ1 subunit is required for this interaction with Pin1 (Fig. 2B). As shown in Fig. 2C, the γ1 subunit possesses four CBS domains and also four Ser/Thr-Pro-containing motifs (Ser10, Ser21, Ser133, and Thr211) necessary for the association with Pin1. To pinpoint the serine or threonine residue(s) in the γ1 subunit interacting with Pin1, mutants of the γ1 subunit in which one of these serine or threonine residues had been replaced by alanine were prepared. Alanine substitution of Thr211 in CBS3 completely abrogated the association with Pin1 (Fig. 2D), whereas those of Ser10, Ser21, and Ser133 did not. Taking these results together, we can reasonably speculate that the Thr211-Pro motif in γ1 of the CBS3 domain is involved in the interaction with Pin1 via its WW domain.
FIGURE 2.

Thr211-Pro motif in γ1 subunit is required for the association with Pin1. A, the AMPK γ1 subunit interacts with the WW domain in Pin1. The cell lysates containing the overexpressed γ1 subunit were incubated with glutathione beads conjugated with GST alone, the GST-WW domain, the GST-PPIase domain, or GST-full-length Pin1. The middle panel shows the Coomassie blue staining of these GST or GST fusion proteins, and the top panel shows the Pin1 associated with each protein. B, phosphorylation of the γ1 subunit is required for the association with Pin1. The cell lysates containing the overexpressed γ1 subunit were incubated with 50 units of calf intestinal alkaline phosphatase for 2 h at 30 °C and subjected to a pull-down assay using GST-Pin1. C, scheme of Ser/Thr-Pro motif in AMPK γ1. D, substitution of Thr211 in the γ1 subunit by alanine abolished the association with Pin1. The 293T cells were transfected with S-tagged Pin1 and the γ1 subunit in which Ser10, Ser21, Ser133, or Thr211 had been substituted with alanine, and their associations were examined employing immunoprecipitation (IP) and immunoblotting (IB). E–H, the AMPK γ2 and γ3 subunits possess 15 and 8 Ser/Thr-Pro motifs, respectively. To identify the Ser/Thr-Pro-containing domains involved in the association with Pin1, the mutant γ2 and γ3 subunits, in which all or all but one of the Ser/Thr-Pro motifs had been generated by substituting Ser/Thr with alanine, were prepared. Their associations with Pin1 were then examined. Next, the 293T cells were transfected with S-tagged Pin1 and mutated AMPK γ2 or γ3, followed by immunoprecipitation with FLAG beads. W, wild type; W+number, only one Ser/Pro or Thr/Pro site is wild type, and other sites have been substituted with alanine; M, all Ser/Pro or Thr/Pro sites substituted with alanine. Shown are representative data from three independent experiments.
On the other hand, the AMPK γ2 and γ3 subunits possess 19 and 8 Ser/Thr-Pro motifs, respectively (Fig. 2, E and G). To identify the Ser/Thr-Pro-containing domains involved in the association with Pin1, mutant γ2 and γ3 subunits, in which all or all but one of the Ser/Thr-Pro motifs had been mutated by substituting Ser/Thr with alanine, were prepared. Their associations with Pin1 were then examined. The mutant γ2 and γ3 subunits lacking a Ser/Thr-Pro motif did not associate with Pin1 (Fig. 2, F and H). The mutant γ2 subunits having a Thr165-Pro or Ser365-Pro motif (W12 and W17, respectively) were able to associate with Pin1 (Fig. 2F). Similarly, the mutant γ3 subunits with Thr12-Pro, Ser39-Pro, Thr109-Pro, Ser162-Pro, or Ser288-Pro (W1, W2, W4, W5, and W7, respectively) associated with Pin1 (Fig. 2H). Taken together, these observations indicate a very high likelihood of the γ2 subunit having two binding domains containing Thr165 and Ser365, whereas the γ3 subunit has five domains containing Thr12, Ser39, Thr109, Ser162, and Ser288. Ser365 in the γ2 isoform and Ser288 in the γ3 isoform are located in their CBS2 domains.
Pin1 Is a Negative Regulator of AMPK Phosphorylation
Thr172 phosphorylation in the α subunit is essential for AMPK activation. To examine whether or not association of Pin1 with the γ subunit affects the phosphorylation level of the α subunit, S-tagged Pin1 was overexpressed in 293T cells, and these cells were then stimulated with 2-DG. As shown in Fig. 3A, Pin1 overexpression markedly suppressed the AMPK α subunit phosphorylation induced by 2-DG without changing the protein amount of the α subunit, whereas overexpression of a K63A Pin1 mutant maintaining the binding ability but lacking PPIase activity did not (Fig. 3A, left). In addition, overexpression of W34A mutant Pin1, lacking the ability to bind to its target proteins, did not affect AMPK phosphorylation (Fig. 3A, right). These results indicate that proline isomerization via PPIase activity, besides the binding to AMPK, of Pin1 is necessary for its suppressive effect on AMPK phosphorylation.
FIGURE 3.
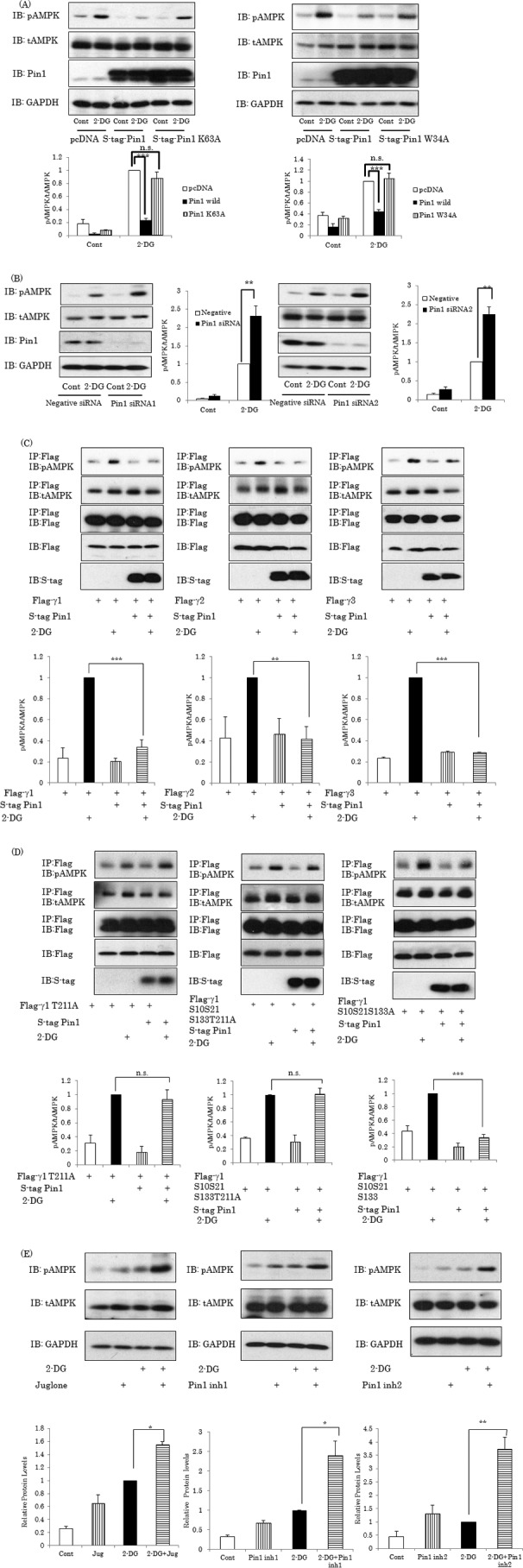
Pin1 suppresses AMPK α subunit phosphorylation by 2-DG. A, Pin1 overexpression suppresses AMPK α subunit phosphorylation. 293T cells were transfected with S-tagged wild-type, W34A, or W63A Pin1 and stimulated with or without 10 mm 2-DG for 1 h. B, knockdown of Pin1 enhances AMPK phosphorylation. The 293T cells were treated with each of the two Pin1 siRNAs. After 72 h, the protein expression levels of Pin1 and the α subunit were determined. After stimulation with or without 2-DG for 1 h, phosphorylation levels of the α subunit were examined. C, Pin1 suppresses α subunit phosphorylation, regardless of whether the γ subunit is γ1, γ2, or γ3. The FLAG-tagged γ1, γ2, or γ3 subunit and S-tagged Pin1 were overexpressed, followed by stimulation with or without 2-DG. The phosphorylation levels and amount of the α subunit in the FLAG tag antibody immunoprecipitates were examined. D, the α subunit phosphorylation level of the AMPK complex containing the γ1 subunit with T211A replacement, unable to associate with Pin1, was not affected by Pin1 overexpression. E, treatment with Pin1 inhibitors promotes AMPK phosphorylation. Three Pin1 inhibitors, including juglone (30 μm), were independently added to the cells, which were then treated with 2-DG. The bar graphs indicate the relative pAMPK levels normalized by tAMPK. Shown are representative data from three independent experiments. IB, immunoblotting; IP, immunoprecipitation. Error bars, S.E. **, p < 0.01; ***, p < 0.001; n.s., not significant.
In contrast, knockdown of Pin1 by each of the two siRNAs enhanced the AMPK α subunit phosphorylation by 2-DG (Fig. 3B). These results clearly indicate Pin1 to be a negative regulator of AMPK. To examine whether the effect of Pin1 emerges irrespective of the γ subunit isoform, the FLAG-tagged γ1, γ2, or γ3 subunit with or without S-tagged Pin1 was overexpressed in 293T cells, and the phosphorylation levels of the α subunit co-immunoprecipitated with the γ1, γ2, or γ3 subunit by anti-FLAG tag antibody were examined (Fig. 3C). Overexpression of Pin1 did not alter the amount of the α subunit co-immunoprecipitated with the FLAG-tagged γ subunit but strongly suppressed the 2-DG-induced α subunit phosphorylations, regardless of whether the γ subunit was γ1, γ2, or γ3. Furthermore, it was shown that the α subunit phosphorylation level of the AMPK complex containing the γ1 subunit with T211A replacement or S10A/S21A/S133A/T211A replacement (γ1 T211A or γ1 S10S21S133T211A, respectively), unable to associate with Pin1, was not affected by Pin1 overexpression (Fig. 3D, left and middle panels). In contrast, the α subunit phosphorylation level of the AMPK complex containing the γ1 subunit with mutation of Ser/Pro motifs (γ1 S10S21S133A) besides the Pin1-associated T211-contained motif was reduced by Pin1 overexpression (Fig. 3D, right), observations similar to those for the wild-type γ1 subunit (Fig. 3C, right). These results indicate that AMPK inhibition by Pin1 requires a direct interaction between Pin1 and the γ1 subunit of AMPK.
To investigate whether PPIase activity is involved in the Pin1-induced reduction of α subunit phosphorylation, the effects of three Pin1 inhibitors were examined. These experiments revealed that Juglone, Pin1 inhibitor 1 (Pin1 inh1), and Pin1 inhibitor 2 (Pin1 inh2), administered independently, markedly enhanced AMPK phosphorylation under both basal and 2-DG-stimulated conditions (Fig. 3E).
The inhibitory effects of Pin1 on AMPK phosphorylations were also observed when HepG2 cells were stimulated with metformin after Pin1 overexpression or knockdown had been induced (Fig. 4). Overexpression of Pin1 by adenovirus gene transfer markedly reduced the phosphorylation level of Thr172 in the α subunit under basal as well as metformin-stimulated conditions (Fig. 4A). Conversely, in the cells treated with each of the two Pin1 siRNAs, the enhanced phosphorylation levels of Thr172 in response to metformin stimulation were more marked than with control siRNA (Fig. 4B).
FIGURE 4.

Pin1 suppresses AMPK α subunit phosphorylation by metformin. A, FLAG-tagged Pin1 or control GFP was overexpressed in HepG2 cells employing adenoviral gene transfer. B, HepG2 cells were treated with each of the two Pin1 siRNAs. Then the cells were treated with or without 1 mm metformin for 1 h, and phosphorylation of the α subunit was examined. The bar graph indicates the relative pAMPK levels normalized by tAMPK. Shown are representative data from three independent experiments. Error bars, S.E.; *, p < 0.05; **, p < 0.01.
Pin1 Had No Effect on AMPK γ1 Stability
To elucidate the molecular mechanism underlying Pin1-induced suppression of AMPK α subunit phosphorylation, we investigated whether or not the protein expression level of γ1 subunit is affected by Pin1. As shown in Fig. 5A, Pin1 overexpression or knockdown did not alter the protein amount of the γ1 subunit.
FIGURE 5.

Pin1 does not affect AMPK γ1 stability but inhibits the enhancing effect of AMP on LKB1-induced AMPK α subunit phosphorylation. A, expression of AMPK γ1 was not affected by Pin1 overexpression or Pin1 gene silencing. B and C, Pin1 inhibited the enhancing effect of AMP on LKB1-induced AMPK phosphorylation in vitro. AMPK complexes were reacted with recombinant LKB1 in the presence or absence of 300 μm AMP for 20 or 30 min. Then the phosphorylation level of the α subunit was determined by immunoblotting (IB). Shown are representative data from three independent experiments. Error bars, S.E. n.s., not significant; *, p < 0.05.
Pin1 Blocks the Enhancing Effect of AMP on LKB1-induced AMPK Phosphorylation
CBS domains in the γ subunit are reportedly involved in AMP-mediated phosphorylation of the α subunit by LKB1 (1, 46). To examine the effect of Pin1 on the LKB1-induced phosphorylation of AMPK γ1, AMPK and AMPK-Pin1 complexes were prepared from the cell lysates overexpressing S-tagged α and β subunits and the FLAG-tagged γ1 subunit with or without Pin1 employing FLAG tag antibody-conjugated beads, followed by FLAG peptide elution. The incubation of the obtained AMPK or AMPK-Pin1 complexes with recombinant LKB1 resulted in time-dependent phosphorylation of the α subunit, and the addition of AMP enhanced its LKB1-induced phosphorylation (Fig. 5B, left). However, the presence of Pin1 was found to significantly inhibit the enhancing effect of AMP on LKB1-induced AMPK α subunit phosphorylation (Fig. 5B, right).
Pin1 Abolishes the Protective Effect of AMP against AMPK α Subunit Dephosphorylation by PP2C
In the ATP-depleted state, CBS domains in the γ subunit are reportedly occupied by AMP or ADP, thereby preventing dephosphorylation of the α subunit by PP2C, a phosphatase (1). Because Pin1 binds to one of the CBS domains in the γ subunit, we examined whether or not Pin1 affects this function of AMP. When an AMPK complex consisting of α, β, and γ (AMPKαβγ) was reacted with recombinant PP2C, phosphorylation of the α subunit was time-dependently decreased (Fig. 6). The addition of AMP or ADP to this reaction mixture reduced the PP2C-induced dephosphorylation of the α subunit (left panels of Fig. 6, A and B). The AMPK complex containing Pin1 (AMPKαβγ-Pin1) was dephosphorylated by PP2C, much like the action of AMPKαβγ in the absence of AMP or ADP, and interestingly, this dephosphorylation of the α subunit was not suppressed by the addition of AMP or ADP (middle panels of Fig. 6, A and B). In addition, if the γ1 subunit was mutated so as to be unable to associate with Pin1 (T211A), the dephosphorylation of AMPKαβγ(T211A)-Pin1 by PP2C was prevented by the addition of AMP or ADP (right panels of Fig. 6, A and B), results similar to those obtained with AMPKαβγ (left panels of Fig. 6, A and B). These results indicate that Pin1 abolished the protective effect of AMP or ADP against the dephosphorylation of AMPK by PP2C.
FIGURE 6.

Pin1 abolishes the protective effect of AMP or ADP against PP2C. A and B, the AMPK complex with or without Pin1 was reacted with recombinant PP2C in the presence or the absence of AMP or ADP for the indicated periods, and the phosphorylation level and amount of each subunit were then determined by immunoblotting (IB). The bar graphs indicate the relative protein levels of pAMPK normalized by tAMPK. Shown are representative data from three independent experiments. Error bars, S.E.; *, p < 0.05; **, p < 0.01; n.s., not significant.
Deficiency of Pin1 in Muscle Enhances AMPK Phosphorylation and Mitochondria Biogenesis
To determine the role of Pin1 in AMPK phosphorylation and its associated downstream events in murine muscle, we compared muscles from Pin1 KO and wild-type mice. Consistent with the in vitro data, Pin1 deficiency in muscle resulted in marked enhancement of AMPK α subunit phosphorylation with no change in the protein amount (Fig. 7B). AMPK activation reportedly induces expressions of mitochondria-related genes, such as PGC-1α and Nrf1, via coordination with Sirt1 (36). In fact, mRNA levels of PGC-1α, Nrf1, Tfam, and cytochrome c were up-regulated in the muscles of Pin1 KO mice (Fig. 7C). The amounts of cytochrome c, PHB1, and PD proteins were also markedly increased in Pin1 KO mice (Fig. 7D). In addition, muscles from Pin1 KO mice were shown to have significantly lower triglyceride levels (Fig. 7E).
Discussion
Prolyl isomerase Pin1 is a unique enzyme that changes the conformations of target proteins by associating with Ser(P)/Thr(P)-Pro containing motifs and thereby modifying protein conformations (37–42). Because the expression level of Pin1 was increased by severalfold in muscle, liver, and adipose tissues by high-fat diet feeding or under conditions of re-feeding (29, 30), we endeavored to elucidate the role of Pin1 in metabolic regulation. Pin1 reportedly enhances the insulin-induced phosphorylation of insulin receptor substrate1 and suppresses the translocation of CRTC2 into the nucleus, thereby reducing CRE transcriptional activity (29, 30). Interestingly, Pin1 KO mice were shown to be resistant to high-fat diet-induced obesity and to the non-alcoholic steatohepatitis development induced by a methionine choline-deficient diet (29, 34).
We demonstrated herein that Pin1 associates with the CBS domain in the γ subunit and functions as a negative regulator of AMPK (Fig. 8). Despite careful and repeated experiments, we could not reproduce the association between Pin1 and the α2 subunit, described in a previous report (43). Although the WW domain of Pin1 is sufficient for association with the γ subunit, suppression of α subunit phosphorylation by Pin1 requires not only its binding ability but also its PPIase activity because the K63A mutant Pin1 lacking PPIase activity exerted no effects (Fig. 3A). The importance of PPIase activity was also confirmed by the experiments showing treatment with Pin1 inhibitors to enhance AMPK phosphorylation (Fig. 3E).
FIGURE 8.
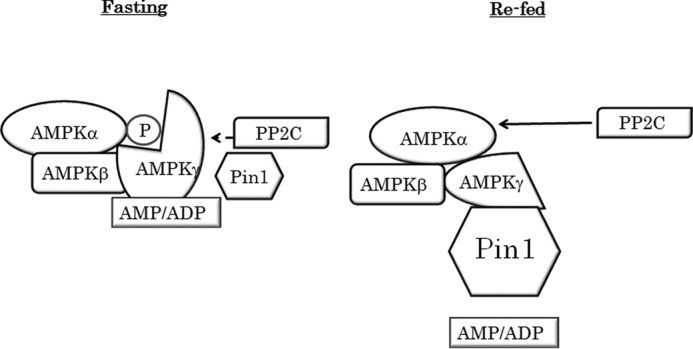
Proposed mechanism of AMPK regulation by Pin1. As the Pin1 expression level is low in the fasted state, AMP can protect the α subunit from dephosphorylation. In the re-fed state, however, Pin1 impairs AMP binding to the g subunit and thereby abolishes the protective effect of AMP.
Our results also provide further insights into the detailed molecular mechanisms underlying Pin1 functions. The observations made in this study strongly suggest that reduced α subunit phosphorylation by Pin1 is mediated via abolishing the protective effect of AMP or ADP against dephosphorylation by PP2C. This interpretation is reasonable, taking into consideration that the CBS domain of the γ subunit, reportedly responsible for the association with AMP or ADP, is the Pin1 binding portion. Although the only domain of the γ1 subunit responsible for the association with Pin1 is in the CBS3 domain, the γ2 and γ3 subunits possess multiple sequences that can bind to Pin1. Nevertheless, one of their Pin1-binding domains is located in the CBS2 domain, and Pin1-induced suppression of α subunit phosphorylation was observed regardless of whether the isoform of the γ subunit was γ1, γ2, or γ3. In addition, whereas it was confirmed that application of AMP increased α subunit phosphorylation by LKB1, in good agreement with the results of previous reports (44–46), Pin1 blocked the promoting effect of AMP on LKB1-induced α subunit phosphorylation.
The AMPK γ subunit has four CBS domains with different roles. CBS2 is empty, and the other three sites are occupied by an adenosine nucleotide. Only AMP binds to CBS4, whereas nucleotides (AMP, ADP, or ATP) binding at CBS1 and CBS3 are interchangeable. AMP binding at CBS1 is required for allosteric activation, and the presence of AMP or ADP at CBS3 protects the α subunit from dephosphorylation and promotes phosphorylation by LKB1 (1, 47–49). Taking these observations together, we can reasonably speculate that the CBS domain associated with Pin1 is no longer capable of binding to AMP or ADP. Alternatively, the conformational change toward protection from PP2C would not occur in Pin1-associated AMPK, even if the CBS domain maintains its ability to bind to AMP or ADP.
On the other hand, treating cells with kinase inhibitors, including Go7874 (PKC inhibitor), wortmannin (PI3K inhibitor), SB202190 (p38 kinase inhibitor), U-0126 (MEK inhibitor), SP600125 (JNK inhibitor), BMS-345541 (IKK inhibitor), 1-azakenpaullone (glycogen synthase kinase inhibitor), TG003 (ClK inhibitor), TBB (CK inhibitor), SB218078 (ChK), and olomoucine (cyclin-dependent kinase inhibitor), did not significantly affect the association between Pin1 and the γ subunit (data not shown). Thus, at present, the kinase phosphorylating the Thr211 in the γ1 subunit remains unknown, and further study is required.
In good agreement with these in vitro results, muscle tissues in Pin1 KO mice showed markedly higher levels of AMPK phosphorylation than those in wild mice. AMPK activation reportedly induces mitochondria-related genes via coordination with Sirt1 in muscle (35, 50, 51). Consistent with these reports, the gene expressions involved in mitochondria biogenesis and lipid oxidation were revealed to be elevated in Pin1 KO mice, which could be one of the mechanisms contributing to the observed resistance to high-fat diet-induced obesity in Pin1 KO mice. The interaction between Pin1 and the γ subunit is low in the fasted state, whereas it is high under re-fed conditions, indicating a physiological role in metabolic regulation via effects on the efficiency of AMPK phosphorylation, according to nutritional intake. Finally, it should be noted that despite the marked difference in muscles, Pin1 KO and control mouse livers did not differ in this respect. Further study is necessary to elucidate the mechanism underlying this difference.
In conclusion, our results have clearly demonstrated Pin1 to be a negative regulator of AMPK, and this may be one of the mechanisms underlying the phenotype of Pin1 KO mice that are resistant to high-fat diet-induced obesity (29). Thus, the interaction between Pin1 and AMPK complexes is a potential treatment target for obesity and diabetes.
Author Contributions
Y. N. designed, performed, and analyzed the experiments. M. I., K. N., and Y. M. performed the experiments. H. S., H. O., T. F., M. F., A. K., H. Kumata, S.-I. T., H. Katagiri, H. H., H. Kiyonari, and T. U. provided technical assistance. T. A. designed experiments and wrote the paper. All authors approved the final version of the manuscript.
This study was supported in part by a Grant-in-Aid for Scientific Research (C) (to N. Y.) and a Grant-in-Aid for Scientific Research (B) (to T. A.) from the Ministry of Education, Science, Sports, and Culture, Japan, and grants from the Ono Medical Research Foundation, the Tsuchiya Medical Foundation, and the 2012 Hiroshima University Fujii Research Promotion Fund. The authors declare that they have no conflicts of interest with the contents of this article.
- AMPK
- AMP-activated kinase
- CBS
- cystathionine β-synthase
- PP2C
- protein phosphatase 2C
- ACC
- acetyl-CoA carboxylase
- 2-DG
- 2-deoxyglucose
- PPIase
- prolyl isomerase
- IKK
- IkκB kinase
- ClK
- Cdc2-like kinase
- CK
- casein kinase
- ChK
- checkpoint kinase.
References
- 1. Hardie D. G., Ross F. A., Hawley S. A. (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kahn B. B., Alquier T., Carling D., Hardie D. G. (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25 [DOI] [PubMed] [Google Scholar]
- 3. Hardie D. G. (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785 [DOI] [PubMed] [Google Scholar]
- 4. Hardie D. G., Carling D. (1997) The AMP-activated protein kinase: fuel gauge of the mammalian cell? Eur. J. Biochem. 246, 259–273 [DOI] [PubMed] [Google Scholar]
- 5. Andersson U., Filipsson K., Abbott C. R., Woods A., Smith K., Bloom S. R., Carling D., Small C. J. (2004) AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 279, 12005–12008 [DOI] [PubMed] [Google Scholar]
- 6. Carling D. (2004) The AMP-activated protein kinase cascade: a unifying system for energy control. Trends Biochem. Sci. 29, 18–24 [DOI] [PubMed] [Google Scholar]
- 7. Woods A., Johnstone S. R., Dickerson K., Leiper F. C., Fryer L. G., Neumann D., Schlattner U., Wallimann T., Carlson M., Carling D. (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 [DOI] [PubMed] [Google Scholar]
- 8. Hawley S. A., Pan D. A., Mustard K. J., Ross L., Bain J., Edelman A. M., Frenguelli B. G., Hardie D. G. (2005) Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2, 9–19 [DOI] [PubMed] [Google Scholar]
- 9. Shaw R. J., Kosmatka M., Bardeesy N., Hurley R. L., Witters L. A., DePinho R. A., Cantley L. C. (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. U.S.A. 101, 3329–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lizcano J. M., Göransson O., Toth R., Deak M., Morrice N. A., Boudeau J., Hawley S. A., Udd L., Mäkelä T. P., Hardie D. G., Alessi D. R. (2004) LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23, 833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davies S. P., Helps N. R., Cohen P. T., Hardie D. G. (1995) 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase: studies using bacterially expressed human protein phosphatase-2Cα and native bovine protein phosphatase-2A(c). FEBS Lett. 377, 421–425 [DOI] [PubMed] [Google Scholar]
- 12. Sanders M. J., Grondin P. O., Hegarty B. D., Snowden M. A., Carling D. (2007) Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 403, 139–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dorfman J., Macara I. G. (2008) STRAD α regulates LKB1 localization by blocking access to importin-α, and by association with Crm1 and exportin-7. Mol. Biol. Cell 19, 1614–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boudeau J., Baas A. F., Deak M., Morrice N. A., Kieloch A., Schutkowski M., Prescott A. R., Clevers H. C., Alessi D. R. (2003) MO25 α/β interact with STRAD α/β enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 22, 5102–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., Musi N., Hirshman M. F., Goodyear L. J., Moller D. E. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Minokoshi Y., Kim Y. B., Peroni O. D., Fryer L. G., Müller C., Carling D., Kahn B. B. (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415, 339–343 [DOI] [PubMed] [Google Scholar]
- 17. Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corradetti M. N., Inoki K., Bardeesy N., DePinho R. A., Guan K. L. (2004) Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 18, 1533–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Treebak J. T., Glund S., Deshmukh A., Klein D. K., Long Y. C., Jensen T. E., Jørgensen S. B., Viollet B., Andersson L., Neumann D., Wallimann T., Richter E. A., Chibalin A. V., Zierath J. R., Wojtaszewski J. F. (2006) AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes 55, 2051–2058 [DOI] [PubMed] [Google Scholar]
- 20. Jäger S., Handschin C., St-Pierre J., Spiegelman B. M. (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1 alpha. Proc. Natl. Acad. Sci. U.S.A. 104, 12017–12022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Steinberg G. R., O'Neill H. M., Dzamko N. L., Galic S., Naim T., Koopman R., Jørgensen S. B., Honeyman J., Hewitt K., Chen Z. P., Schertzer J. D., Scott J. W., Koentgen F., Lynch G. S., Watt M. J., van Denderen B. J., Campbell D. J., Kemp B. E. (2010) Whole body deletion of AMP-activated protein kinase β2 reduces muscle AMPK activity and exercise capacity. J. Biol. Chem. 285, 37198–37209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Neill H. M., Maarbjerg S. J., Crane J. D., Jeppesen J., Jørgensen S. B., Schertzer J. D., Shyroka O., Kiens B., van Denderen B. J., Tarnopolsky M. A., Kemp B. E., Richter E. A., Steinberg G. R., (2011) AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. U.S.A. 108, 16092–16097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lessard S. J., Rivas D. A., Chen Z. P., van Denderen B. J., Watt M. J., Koch L. G., Britton S. L., Kemp B. E., Hawley J. A. (2009) Impaired skeletal muscle β-adrenergic activation and lipolysis are associated with whole-body insulin resistance in rats bred for low intrinsic exercise capacity. Endocrinology 150, 4883–4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin T. L., Alquier T., Asakura K., Furukawa N., Preitner F., Kahn B. B. (2006) Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J. Biol. Chem. 281, 18933–18941 [DOI] [PubMed] [Google Scholar]
- 25. Liu Y., Wan Q., Guan Q., Gao L., Zhao J. (2006) High-fat diet feeding impairs both the expression and activity of AMPKa in rats' skeletal muscle. Biochem. Biophys. Res. Commun. 339, 701–707 [DOI] [PubMed] [Google Scholar]
- 26. Bandyopadhyay G. K., Yu J. G., Ofrecio J., Olefsky J. M. (2006) Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes 55, 2277–2285 [DOI] [PubMed] [Google Scholar]
- 27. Ben-Shlomo S., Zvibel I., Shnell M., Shlomai A., Chepurko E., Halpern Z., Barzilai N., Oren R., Fishman S. (2011) Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J. Hepatol. 54, 1214–1223 [DOI] [PubMed] [Google Scholar]
- 28. Mu J., Brozinick J. T. Jr., Valladares O., Bucan M., Birnbaum M. J. (2001) A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol. Cell 7, 1085–1094 [DOI] [PubMed] [Google Scholar]
- 29. Nakatsu Y., Sakoda H., Kushiyama A., Zhang J., Ono H., Fujishiro M., Kikuchi T., Fukushima T., Yoneda M., Ohno H., Horike N., Kanna M., Tsuchiya Y., Kamata H., Nishimura F., Isobe T., Ogihara T., Katagiri H., Oka Y., Takahashi S., Kurihara H., Uchida T., Asano T. (2011) Peptidyl-prolyl cis/trans isomerase NIMA-interacting 1 associates with insulin receptor substrate-1 and enhances insulin actions and adipogenesis. J. Biol. Chem. 286, 20812–20822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakatsu Y., Sakoda H., Kushiyama A., Ono H., Fujishiro M., Horike N., Yoneda M., Ohno H., Tsuchiya Y., Kamata H., Tahara H., Isobe T., Nishimura F., Katagiri H., Oka Y., Fukushima T., Takahashi S., Kurihara H., Uchida T., Asano T. (2010) Pin1 associates with and induces translocation of CRTC2 to the cytosol, thereby suppressing cAMP-responsive element transcriptional activity. J. Biol. Chem. 285, 33018–33027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fila C., Metz C., van der Sluijs P. (2008) Juglone inactivates cysteine-rich proteins required for progression through mitosis. J. Biol. Chem. 283, 21714–21724 [DOI] [PubMed] [Google Scholar]
- 32. Potter A. J., Ray S., Gueritz L., Nunns C. L., Bryant C. J., Scrace S. F., Matassova N., Baker L., Dokurno P., Robinson D. A., Surgenor A. E., Davis B., Murray J. B., Richardson C. M., Moore J. D. (2010) Structure-guided design of α-amino acid-derived Pin1 inhibitors. Bioorg. Med. Chem. Lett. 20, 586–590 [DOI] [PubMed] [Google Scholar]
- 33. Potter A., Oldfield V., Nunns C., Fromont C., Ray S., Northfield C. J., Bryant C. J., Scrace S. F., Robinson D., Matossova N., Baker L., Dokurno P., Surgenor A. E., Davis B., Richardson C. M., Murray J. B., Moore J. D. (2010) Discovery of cell-active phenyl-imidazole Pin1 inhibitors by structure-guided fragment evolution. Bioorg. Med. Chem. Lett. 20, 6483–6488 [DOI] [PubMed] [Google Scholar]
- 34. Nakatsu Y., Otani Y., Sakoda H., Zhang J., Guo Y., Okubo H., Kushiyama A., Fujishiro M., Kikuch T., Fukushima T., Ohno H., Tsuchiya Y., Kamata H., Nagamachi A., Inaba T., Nishimura F., Katagiri H., Takahashi S., Kurihara H., Uchida T., Asano T. (2012) Role of Pin1 protein in the pathogenesis of nonalcoholic steatohepatitis in a rodent model. J. Biol. Chem. 287, 44526–44535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yagi T., Tokunaga T., Furuta Y., Nada S., Yoshida M., Tsukada T., Saga Y., Takeda N., Ikawa Y., Aizawa S. (1993) A novel ES cell line, TT2, with high germline-differentiating potency. Anal. Biochem. 214, 70–76 [DOI] [PubMed] [Google Scholar]
- 36. Cantó C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu K. P., Zhou X. Z. (2007) The prolyl isomerase PIN 1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8, 904–916 [DOI] [PubMed] [Google Scholar]
- 38. Lu K. P., Finn G., Lee T. H., Nicholson L. K.(2007) Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 3, 619–629 [DOI] [PubMed] [Google Scholar]
- 39. Butterfield D. A., Abdul H. M., Opii W., Newman S. F., Joshi G., Ansari M. A., Sultana R. (2006) Pin1 in Alzheimer's disease. J. Neurochem. 98, 1697–1706 [DOI] [PubMed] [Google Scholar]
- 40. Liou Y. C., Zhou X. Z., Lu K. P. (2011) Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 36, 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lu K. P., Hanes S. D., Hunter T. (1996) A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380, 544–547 [DOI] [PubMed] [Google Scholar]
- 42. Lu P. J., Wulf G., Zhou X. Z., Davies P., Lu K. P. (1999) The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated Tau protein. Nature 399, 784–788 [DOI] [PubMed] [Google Scholar]
- 43. Khanal P., Kim G., Yun H. J., Cho H. G., Choi H. S. (2013) The prolyl isomerase Pin1 interacts with and downregulates the activity of AMPK leading to induction of tumorigenicity of hepatocarcinoma cells. Mol. Carcinog. 52, 813–823 [DOI] [PubMed] [Google Scholar]
- 44. Xiao B., Sanders M. J., Underwood E., Heath R., Mayer F. V., Carmena D., Jing C., Walker P. A., Eccleston J. F., Haire L. F., Saiu P., Howell S. A., Aasland R., Martin S. R., Carling D., Gamblin S. J. (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oakhill J. S., Steel R., Chen Z. P., Scott J. W., Ling N., Tam S., Kemp B. E. (2011) AMPK is a direct adenylate charge-regulated protein kinase. Science 332, 1433–1435 [DOI] [PubMed] [Google Scholar]
- 46. Gowans G. J., Hawley S. A., Ross F. A., Hardie D. G. (2013) AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 18, 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adams J., Chen Z. P., Van Denderen B. J., Morton C. J., Parker M. W., Witters L. A., Stapleton D., Kemp B. E. (2004) Intrasteric control of AMPK via the γ1 subunit AMP allosteric regulatory site. Protein Sci. 13, 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hardie D. G. (2004) The AMP-activated protein kinase pathway: new players upstream and downstream. J. Cell Sci. 117, 5479–5487 [DOI] [PubMed] [Google Scholar]
- 49. Towler M. C., Hardie D. G. (2007) AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 100, 328–341 [DOI] [PubMed] [Google Scholar]
- 50. Hou X., Xu S., Maitland-Toolan K. A., Sato K., Jiang B., Ido Y., Lan F., Walsh K., Wierzbicki M., Verbeuren T. J., Cohen R. A., Zang M. (2008) SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 283, 20015–20026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cantó C., Auwerx J. (2009) PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 20, 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]