

Background: Collectrin is required for membrane expression of the broad neutral amino acid transporters (B0AT1 and -3).
Results: Collectrin activates B0AT1 and B0AT3 in a discrete interaction region of the transporters.
Conclusion: A potential conserved ancillary protein binding region in B0AT1/B0AT3 mediates collectrin interactions.
Significance: This is the first example of a potential common interaction site for multiple solute carrier 6 family ancillary proteins.
Keywords: intestinal epithelium, kidney, membrane transport, protein complex, protein-protein interaction, angiotensin-converting enzyme 2 (ACE2), broad neutral amino acid transporters, collectrin, syntaxin 1A, syntaxin 3
Abstract
Many solute carrier 6 (SLC6) family transporters require ancillary subunits to modify their expression and activity. The main apical membrane neutral amino acid transporters in mouse intestine and kidney, B0AT1 and B0AT3, require the ancillary protein collectrin or ACE2 for plasma membrane expression. Expression and activity of SLC6 neurotransmitter transporters are modulated by interaction with syntaxin 1A. Utilizing monocarboxylate-B0AT1/3 fusion constructs, we discovered that collectrin is also necessary for B0AT1 and B0AT3 catalytic function. Syntaxin 1A and syntaxin 3 inhibit the membrane expression of B0AT1 by competing with collectrin for access. A mutagenesis screening approach identified residues on trans-membrane domains 1α, 5, and 7 on one face of B0AT3 as a key region involved in interaction with collectrin. Mutant analysis established residues that were involved in collectrin-dependent functions as follows: plasma membrane expression of B0AT3, catalytic activation, or both. These results identify a potential binding site for collectrin and other SLC6 ancillary proteins.
Introduction
The SLC6 family includes secondary active amino acid and neurotransmitter transporters, which are involved in a broad variety of physiological functions, most notably neurotransmitter uptake at the synapse and epithelial amino acid transport in kidney and intestine (1). B0AT1 (SLC6A19) catalyzes the secondary active transport of neutral amino acids across the apical membrane in the small intestine and kidney, whereas B0AT3 (SLC6A18) is found only in the kidney (2). Both transporters require collectrin or angiotensin-converting enzyme 2 (ACE2) as an essential subunit for trafficking to the plasma membrane in all in vitro and in vivo expression systems tested to date (3–9). B0AT1, however, retains a small amount of residual activity and membrane expression when expressed alone in Xenopus laevis oocytes (7, 10, 11). Indirect evidence suggests that human B0AT3 is nonfunctional (12); glycine/alanine transport at the human renal brush border is instead mediated by the proton-dependent transporter PAT2 (SLC36A2) (9). The molecular interactions, stoichiometry, and basis of stabilization of B0AT1 and B0AT3 by collectrin/ACE2 have not yet been described in detail. With the exception of SIT1 (SLC6A20) (13), no other transporter in the SLC6 family appears to require heterodimerization to reach the cell surface. Most neurotransmitter transporters in this family rather require homodimerization and/or oligomerization to exit the endoplasmic reticulum (14–16).
Mutations in B0AT1 are the cause of the autosomal recessive Mendelian inherited condition Hartnup disorder (17, 18). Characterized by renal aminoaciduria and intestinal malabsorption, the disorder is normally benign but has been associated with a diverse array of symptoms, including skin rash, cerebellar ataxia, and psychosis (2). B0AT1 knock-out mice replicate human Hartnup disorder and also display a complex metabolic phenotype resulting in enhanced insulin sensitivity (19, 20). Consistent with an essential role of collectrin and ACE2 in trafficking and tissue distribution of B0AT1 and B0AT3, collectrin-deficient mice lack B0AT1 and B0AT3 in the kidney, whereas ACE2-deficient mice lack B0AT1 in the intestine (3, 5). Collectrin has also been associated with glucose-stimulated insulin secretion from pancreatic β-cells in vitro and in vivo (21–23) and aldosterone-independent high sodium-induced hypertension (23–26). Both genes are under the transcriptional control of the transcription factors HNF1α and HNF4α (27). B0AT1 has also been shown to interact with aminopeptidase N in the intestine (28). ACE2 is involved in a number of pathologies due to its role in the degradation of angiotensin II (29–31). With regard to its role in amino acid transporter trafficking, lack of ACE2 has been shown to aggravate intestinal inflammation (32).
Despite these important roles in vital metabolic processes, information on the interaction between collectrin/ACE2 and B0AT1 or B0AT3 is scarce. It is thought to involve a conserved Arg-240 residue in B0AT1 (Arg-225 in B0AT3) (7). Intriguingly, this mutation did not affect B0AT1 when expressed alone but reduced transport severely in the presence of collectrin/ACE2 (2). Several other Hartnup disorder mutations, namely Ala-69 and Pro-265, have also been implicated in ACE2 and collectrin-mediated B0AT1 dysfunction (5).
Although the dependence of B0AT1 and B0AT3 surface expression on collectrin appears unique, protein-protein interactions between SLC6 neurotransmitter sodium symporters and smaller membrane-anchored proteins is widespread (14, 33–37). For instance, syntaxin 1A, a 288-residue type 1 single-pass T-SNARE protein, which regulates vesicular fusion events in the nervous system via formation of coiled-coil bundles with other SNARE proteins (38, 39), has been shown to interact with NET, DAT, SERT, GAT1, and GLYT2 (33–35, 40–43). In particular, the relationship between the neurotransmitter sodium symporters γ-aminobutyric acid (GABA) transporter GAT1 and syntaxin-1A has been well characterized (33). Syntaxin 1A modulates GAT1-mediated GABA uptake, efflux, exchange, and subcellular redistribution (44–49). The net result of these molecular interactions is an ∼75% reduction in the turnover rate of GAT1, although surface expression is increased at the same time (33). Syntaxin 1A paralogs are also involved in membrane protein regulation in epithelial cells (50, 51), and syntaxin isoforms in kidney and small intestinal epithelium localize in a manner that suggests a potential role in apical and basolateral cell polarity (52, 53). Although it is often inferred from evidence of interactions with SNARE proteins that collectrin may mediate vesicular fusion events and formation of SNARE complexes, no direct evidence for this has been shown (23, 54, 55). Even for syntaxin 1A, molecular interactions with other membrane proteins have been shown to be both SNARE complex-dependent and -independent (49, 56).
In this study we undertook a systematic study of molecular interactions between B0AT1/3 and collectrin. We report here that collectrin is essential for catalytic activation, and we identify the molecular interaction site of collectrin with B0AT3. In addition, we identify cross-reactivity between collectrin and syntaxin proteins, which regulate B0AT1 expression.
Experimental Procedures
Molecular Cloning and DNA Manipulation
Molecular cloning of transporters and murine collectrin has been reported previously (6, 7, 17). Monocarboxylate transporter fusion constructs rMCT1-B0AT1 and rMCT1-B0AT3 were generated using overlap extension PCR on cDNA templates. The rMCT1 sense primer was designed to incorporate an XbaI cutting site and Kozak motif, before the start codon. The B0AT1 antisense primer comprised an XbaI site, and the B0AT3 antisense primer comprised an EcoRI site. The overlap primers contained 21 bp from each cDNA in either sense or antisense orientation excluding stop and start codons. Molecular cloning of murine syntaxin 1A was conducted using mRNA isolated from mouse brain as a template (RNeasy Plus, Qiagen). Other syntaxins were cloned using kidney mRNA. Superscript IITM reverse transcriptase (Life Technologies, Inc.) was used to synthesize the cDNA, followed by PCR amplification with gene-specific primers and Pfu polymerase (Agilent). Sequences for all primer pairs are available upon request. For syntaxin 1A and 7, HindIII restriction endonuclease sites were incorporated in both the 5′- and 3′-cloning primers. For syntaxin 3, EcoRI restriction endonuclease sites were added to the 5′ and 3′ ends of the cloning primers. Mouse ACE2 (mACE2) truncation primers were designed based on multiple pairwise alignments of mACE2 and mouse collectrin and used to delete residues 18–580 and 18–500 from mACE2. Δ18–580 retains the collectrin homology consensus sequence, beginning just downstream of the consensus start site (mACE2–591). Δ18–500 was designed to retain an in silico identified tyrosine-based ER sorting signal (ELM accession ELME000120) annotated at mACE2(510–513) which may be important for trafficking to the plasma membrane. The N-terminal signal peptide (residues 2–18) was retained in both constructs. Following amplification, DNA fragment size was verified using agarose gels. DNA was then purified (Wizard® SV, Promega) and digested before ligation (Quick ligase, New England Biolabs) into the pGHJ X. laevis oocyte expression vector.
For CHO cell experiments, syntaxin 1A and syntaxin 3 were subcloned from pGHJ vectors into the pcDNA3.1+ mammalian expression vector using the same HindIII (syntaxin 1A) and EcoRI (syntaxin 3) endonuclease sites as were used for the original cloning. Subcloning was verified by restriction digests. All other cloning products were verified by sequencing (Biomolecular Resource Facility, Australian National University). All pcDNA3.1+ syntaxin constructs were isolated using an endotoxin-free midi DNA preparation (NucleoBond® Xtra EF Plus, Macherey-Nagel) before transfection into mammalian cell cultures.
Preparation of X. laevis Oocytes and Expression of Proteins
X. laevis oocytes were surgically removed and prepared for cRNA injection as described previously (57). Maintenance of animals and preparation of oocytes was approved by the Australian National University animal ethics review board (ANU Protocol A2014/20). Synthetic RNA (cRNA) was prepared for injection and in vitro protein expression in oocytes also as described previously (28) with the following modifications. The pGHJ-based vectors containing transporter and chaperone constructs were linearized using SalI HF (New England Biolabs) for 2 h or NotI (New England Biolabs) overnight. Following in vitro synthesis and purification, cRNA was quantified using a nanodrop spectrophotometer (A260/A280) and diluted to the appropriate concentration.
Uptake Experiments
Radiolabeled uptake experiments in oocytes were conducted as described previously (10, 28). For cRNA titration experiments, cRNAs were injected in different ratios as indicated in the experiments. Mouse syntaxin cRNA titrations were fitted to the single competitive binding site Equation 1,
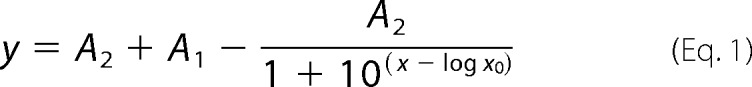 |
where A1 and A2 are the maximum and minimum horizontal (ordinate) asymptotes, respectively.
Electrophysiological Recordings
Electrophysiological recordings were conducted as described previously (10, 57) with some modifications. Briefly, all steady-state recordings were made with an Axon Geneclamp 500B amplifier (Axon Instruments). Voltage clamp was routinely set to −50 mV, and data were sampled at 3 Hz using pClamp 8.2 software (Axon Instruments). Oocytes were chosen for recording when they had a resting membrane potential −25 mV < Vm < −35 mV. Substrate dependence was measured by a stepwise exchange of substrate concentrations using a run-up and run-down series of concentrations for each oocyte. The general oocyte assay buffer 1× ND96, pH 7.4 (96 mm NaCl, 2 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2, 5 mm HEPES, titrated with NaOH), was used as the control solution for all electrophysiological recordings unless otherwise indicated.
For simultaneous measurements of l-[U-14C]lactate uptake and l-alanine-induced currents, oocytes were superfused with ND96, pH 7.4, buffer and clamped to −50 mV. The gravity flow of ND96, pH 7.4, was then stopped, and the bath outflow pump was allowed to evacuate the bath to a volume of ∼100 μl. A 100-μl volume of 10 mm l-alanine and 0.5 mm l-[U-14C]lactate was then added to the bath solution to give a final concentration of ∼5 mm l-alanine and 0.25 mm l-[U-14C]lactate. Subsequently, recordings upon application of both substrates were continued for 10 min to allow for a sufficient quantifiable flux of charge and radiolabeled l-lactate into oocytes. Recordings were stopped and oocytes washed three times with 200 μl ice-cold ND96, pH 7.4, before being transferred individually to scintillation vials and prepared for radioactivity quantification.
Surface Biotinylation, Whole Membrane Preparation and Western Blotting
Surface biotinylation of oocytes has been described previously (28). Whole membrane preparation was used to determine protein expression and degradation in oocytes. Fifteen oocytes per experimental condition were triturated in 1 ml of oocyte homogenization buffer (100 mm NaCl, 50 mm Tris-HCl, pH 7.6, 1 mm EDTA, pH 8.0, Complete EDTA-free protease inhibitor (Roche Applied Science)) until homogenized. The homogenate was cleared by centrifugation (2000 × g, 4 °C, 10 min). Subsequently, total membranes were isolated by centrifugation at 140,000 × g at 4 °C for 30 min (OptimaTM Max, Beckman). The resulting supernatant was aspirated, and the pellet was washed twice with homogenization buffer before being solubilized in 50 μl of homogenization buffer containing 4% (w/v) SDS.
For preparation of CHO cell membranes, cells were washed three times with phosphate buffered saline (PBS) (137 mm NaCl, 2.7 mm KCl, 5 mm Na2HPO4, pH 7.4) supplemented with CaCl2 (0.9 mm) and MgCl2 (0.5 mm) before scraping off cells in the same buffer. Cells were then centrifuged for 5 min at 500 × g, resuspended in hypo-osmotic buffer (10 mm HEPES, pH 8.0, 15 mm KCl, 2 mm MgCl2, 0.1 mm EDTA), and incubated for 5 min on ice. Lysed cells were then homogenized with a hypodermic needle and centrifuged at 600 × g for 10 min to pellet dense cell debris before the supernatant was transferred and centrifuged at 150,000 × g at 4 °C for 60 min. The resulting pellet was dissolved in 5 mm glycine, and a Bradford assay conducted to determine total protein concentration.
Membrane proteins were subjected to SDS-PAGE analysis as described previously (28). Nitrocellulose membranes were incubated with the following primary antibody dilutions: rabbit anti-mouse B0AT1 (1:3000) (custom antibody, Pineda Antibody Service); sheep anti-mouse collectrin (1:2000) (R & D Systems); rabbit anti-mouse B0AT3 (1:200) (custom antibody, Pineda Antibody Service); rabbit anti-human syntaxin-1A (1:1000) (Cell Signaling Technology); rabbit anti-mouse ACE2 (1:4000) (Abcam); and rabbit anti-mouse β-actin (1:2000) (Abcam). For detection, horseradish peroxidase (HRP)-conjugated donkey anti-rabbit or anti-sheep IgG was used at the same dilution as the primary antibody, except for anti-rabbit IgG against mouse B0AT3 (1:1000).
Generation of CHO-cells Stably Expressing SLC6A19 and Collectrin
CHO-SLC6A19-collectrin clone 1 was obtained by a two-step process. CHO-Flip In (Life Technologies, Inc.) cells were transfected with the plasmid pCDNA5/FRT-human SLC6A19 using LTX-Lipofectamine (Life Technologies, Inc.). Cells growing in the presence of hygromycin and expressing SLC6A19, as demonstrated by RT-quantitative PCR, were subsequently transfected with the plasmid pCDNA3.1-collectrin. Double-transfected cells selected by their ability to grow in the presence of hygromycin and G418 were subcloned by limited dilution and analyzed with respect to the expression of both SLC6A19 and collectrin as well as for sodium-dependent uptake of leucine.
CHO Cell Uptake and Transient Transfection of Syntaxin 1A and 3 in CHO-SLC6A19-collectrin
Parental CHO cells were cultured and maintained in Ham's F-12 GlutaMAX media (Life Technologies, Inc.) supplemented with 10% FBS and 100 μg/ml Zeocin (Life Technologies, Inc.). CHO-SLC6A19-collectrin stably transfected cells were cultured and maintained in Ham's F-12 GlutaMAX supplemented with FBS (10% v/v and the antibiotics hygromycin (0.33 μg/ml) (Life Technologies, Inc.)) and G418 (0.275 μg/ml) (Life Technologies, Inc.). Cells were passaged over 7-day intervals with media replaced every 72 h. Experiments were conducted on cells with passage numbers from 3 to 10. For radiolabeled uptake assays, cells were seeded out in 35-mm culture dishes (Nunc) and grown for ∼48 h until >80% confluent. Before uptake, cells were washed three times with Hanks' buffered salt solution (HBSS,2 pH 7.4). Subsequently, uptake of radiolabeled compounds was measured at 37 °C for 6 min in HBSS (pH 7.4). When Na+-independent uptake was measured, NaCl in HBSS was replaced by NMDG-Cl. To terminate transport, cells were washed three times with ice-cold HBSS, pH 7.4. After aspiration, cells were lysed with 0.1 m HCl, scraped together, and homogenized by trituration. Aliquots were used for scintillation counting and protein quantification.
For transient expression of pcDNA3.1+ constructs, cells were seeded into 35-mm dishes and transfected using Lipofectamine LTX® reagent (Life Technologies, Inc.) as recommended by the manufacturer. After incubation for ∼48 h, cells were used for radiolabeled uptake assays.
RNAi Knockdown of Syntaxin 1A and Syntaxin 3
Small interfering RNAs for CHO syntaxin 1A and 3 were selected from mouse MISSION predesigned siRNA (Sigma) binding to sequence regions that are 100% identical to the corresponding Chinese hamster sequences. CHO-SLC6A19-collectrin cells were transfected with the RNAi pairs when 30–50% confluent using 50 pmol of RNAi per target (Lipofectamine® RNAiMAX, Life Technologies, Inc.) and incubated at 37 °C for a further 48 h before experimental use. To verify silencing, total RNA was extracted from transfected cells (Macherey-Nagel, NucleoSpin® RNA), and 1 μg was reverse-transcribed into cDNA (Superscript® II, Life Technologies, Inc.). For semi-quantitative analysis, transcripts were amplified for 25 cycles. Following RT-PCR, samples were loaded onto a 1% (w/v) agarose gel and quantified using UV densitometry after running and staining (SYBR® Safe, Life Technologies, Inc.).
Homology Models of B0AT3 and Creation of Collectrin TM Domain
The PROMALS3D server was used to generate a structure-based alignment of mouse B0AT3 (mB0AT3) using protein sequence of LeuT from Aquifex aeolicus strain V5 (UniProt O67854) or Drosophila melanogaster dopamine transporter DAT (UniProt Q7K4Y6) (58). The following settings were used: identity threshold = 1, secondary structure weight = 0.1, and amino acid weight = 0.5. Minor adjustments were then made to the structure-based sequence alignments to reflect a lack of consensus or hydropathy overlap between them based on a previously established multiple sequence alignment of LeuT to SLC6 neurotransmitter transporters (59). Based on this alignment mouse B0AT3 (mB0AT3) homology models were generated using the Swiss model server with both LeuT from A. aeolicus (PDB 2A65) or the D. melanogaster dopamine transporter DAT (PDB code 4m48) as the nearest homologue with a known x-ray crystal structure as the template for model building. Validation of homology models was conducted as reported previously (28). Collectrin is 222 amino acids in length and has been predicted to contain a single α-helical membrane-spanning domain (Val-142 to Ile-166) (TMHMM version 2.0). To ensure that sufficient residues were included to extend beyond the span of the membrane, residues Met-136 to Arg-171 were modeled as an ideal α-helix using SwissPdbViewer (60).
Docking of Collectrin and Molecular Dynamics Simulations
To investigate the possible interaction interface between mB0AT3 and collectrin, the mB0AT3 and collectrin models were docked using the HADDOCK webserver (61). Based on the experimental predictions for the proposed protein-protein interface between mB0AT3 and collectrin, residues in and around helix 7 of mB0AT3 were defined as the active docking interface, although residues from collectrin were treated as passive. Six docking simulations were performed, in which different subsets of mB0AT3 residues were defined as the active residues, as outlined in Table 1. The docking results were manually analyzed to determine the conformation that best fit a combination of the following criteria: 1) the lowest docking score; 2) the greatest number of interacting residues between mB0AT3 and collectrin; and 3) the conformation in which both mB0AT3 and collectrin adopted a transmembrane orientation. The best fit mB0AT3/collectrin conformation was embedded in a pre-equilibrated 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) bilayer (62) and used as the starting conformation to initiate molecular dynamics (MD) simulations. To avoid the introduction of inappropriate charges, the N and C termini of both mB0AT3 and collectrin were capped by acetylation and amidation, respectively.
TABLE 1.
The list of docking sets and the corresponding mB0AT3 active residues
For details see under “Experimental Procedures.”
| Docking set | mB0AT3-active residues |
|---|---|
| 1 | 225, 283, 287, 296, 502 |
| 2 | 210, 268, 283, 286, 272 |
| 3 | 6, 10, 194, 210, 268, 286, 492 |
| 4 | 205, 210, 268, 286, 492 |
| 5 | 6, 10, 194, 210, 218, 280, 286, 492 |
| 6 | 280, 283, 286 |
All MD simulations were performed using the GROMACS (Groningen Machine for Chemical Simulation) package, version 3.3.3, using the GROMOS 54A7 force field (63). The simple point charge water model (64) was used to describe the solvent water. All simulations were performed under periodic boundary conditions in a rectangular box. The dimensions of the box were chosen such that the minimum distance of the protein to the box wall was at least 1.0 nm. A twin-range method was used to evaluate the nonbonded interactions. Interactions within the short-range cutoff of 0.9 nm were updated every step. Interactions within the long-range cutoff of 1.4 nm were updated every three steps together with the pair list. A reaction field correction was applied using a relative dielectric constant of ϵr = 78.5 to minimize the effect of truncating the electrostatic interactions beyond the 1.4-nm long-range cutoff (65). The SHAKE algorithm (66) was used to constrain the lengths of the covalent bonds. The geometry of the water molecules was constrained using the SETTLE algorithm (67). To extend the time scale that could be simulated, explicit hydrogen atoms in the protein were replaced with dummy atoms, the positions of which were calculated each step based on the positions of the heavy atoms to which they were attached. This eliminates high frequency degrees of freedom associated with the bond angle vibrations involving hydrogens, allowing a time step of 4 fs to be used to integrate the equations of motion without affecting thermodynamic properties of the system significantly (68). The simulations were carried out in the NPT-ensemble at T = 300 K, and P = 1 bar. The temperature (T) and pressure (P) were maintained close to the reference values by weakly coupling the system to an external temperature and pressure bath using a relaxation time constant of 0.1 ps (69) and 0.5 ps, respectively. The pressure coupling was semi-isotropic. Data were collected every 40 ps for analysis. Images were produced using VMD (70).
To initiate the simulation, 1000 steps of steepest descent energy minimization were performed. Then the system was equilibrated over a 10-ns period in which the position restraints were gradually lowered from 1000 to 500 to 100 to 50 to 10 kJ/mol/nm2 in successive 2-ns simulations. This gradual lowering of position restraints reduces the backbone fluctuations often associated with homology models in MD simulations (71). After equilibration, all position restraints were removed; new velocities were assigned, and a further 10-ns unrestrained MD simulation was performed to allow the protein-protein interface to adopt an equilibrated conformation. Note, because of the well documented structural limitations of homology models in MD, simulations were not extended beyond 10 ns (71).
Statistical and Data Analysis
A Shapiro-Wilk normality test was used to ensure that experimental data sets in oocyte experiments were drawn from a normally (i.e. Gaussian) distributed population. All experimental data are given as mean ± S.D. The number of biological repeats (experiments) is given as e, and the number of technical repeats is given as n. Multivariant experiments were analyzed for significance using one-way analysis of variance tests. The Bonferroni post hoc test was used to compare between pairs of means for significance. Probability values (p values) of significance thresholds for one-way analysis of variance are indicated in the figure legends. To verify correlation between variables for Eadie-Hofstee and other linear regressions, Pearson's correlation coefficient (r) was determined; adjusted R2 values were hence derived to test for goodness of fit of linear functions. Kinetic parameters K0.5 and Vmax values were first derived by fitting individual current recordings to either the Michaelis-Menten Equation 2
 |
or the Hill Equation 3.
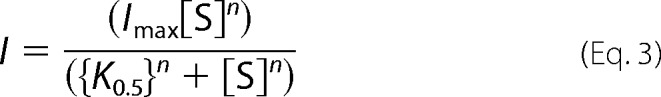 |
Individual curves were averaged and refitted. Nonlinear curve fitting using linear regressions of kinetic data were conducted using the Eadie-Hofstee Equation 4.
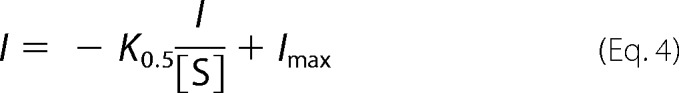 |
All data manipulation and statistical analyses were performed using OriginPro 9.1 (OriginLab) with the exception of Western blot and agarose gel UV densitometry, which was performed using ImageJ version 1.48 (http://imagej.nih.gov/ij/).
Results
Functional Interactions between SLC6 Amino Acid Transporters and Ancillary Proteins Collectrin and ACE2
As reported previously, collectrin and ACE2 are essential to functionally express mouse B0AT1 and B0AT3 (Fig. 1A) (3, 4, 7–9). It is important to note that human B0AT3 is an inactive protein even if coexpressed with ACE2 or collectrin (Fig. 1A). Mouse and human B0AT1 display a small transport activity in the absence of the ancillaries, whereas mouse B0AT3 does not. It appears that the same domains of ACE2 and collectrin interact with B0AT1 and B0AT3 as exemplified by the virtually unchanged transport induction of mouse B0AT1 by a collectrin homologous truncation of ACE2(Δ18–580) compared with full-length ACE2 (Fig. 1B).
FIGURE 1.

Ancillary proteins modify substrate affinity of B0AT1 and B0AT3. X. laevis oocytes were injected with 10 ng of the indicated transporter, 2 ng of collectrin, and 10 ng of ACE2 cRNA in the indicated combinations except where otherwise stipulated. All uptake and electrophysiological measurements were made day 4–6 post-injection. A, uptake of 100 μm l-[U-14C]leucine or 100 μm l-[U-14C]alanine was measured in oocytes expressing B0AT1 or B0AT3 over 30 min. Each bar represents mean ± S.D. (n = 12, e > 3. B, B0AT1 was coexpressed with truncated (18–500del or 18–580del) or wild-type ACE2. Uptake was measured as in A (n = 10−12, e = 3). a and b above the individual bars indicate groupings of conditions whose differences of means are not statistically significant from each other at the p = 0.05 level. C, oocytes were voltage-clamped at −50 mV and then perfused with serial concentrations of l-alanine. Data were transformed according to Eadie-Hofstee and analysed by linear regression, for all regressions 0.99 > R2 > 0.88, and Pearson's r > −0.94 (n = 7). D, oocytes were injected with 6 ng of B0AT3 cRNA, 5 ng of ACE2, and 2 ng of collectrin cRNA. Uptake was measured as in A with the exception that it was challenged by 10 mm unlabeled l-amino acids as indicated. The first pair of bars from the left represents unchallenged 100 μm l-[U-14C]alanine uptake. l-[U-14C]Alanine uptake from uninjected oocytes was subtracted. Each bar represents mean ± S.D. (n = 8−10, *, p < 0.05). E, oocytes were perfused with 10 mm of all the amino acids indicated on the abscissa, and subsequent steady-state Na+ currents were recorded. All currents were normalized to the maximal current induced by 10 mm l-methionine. Each bar represents mean ± S.D. (n = 6, e = 3).
We and others have previously shown that neither collectrin nor ACE2 change the substrate affinity of B0AT1 (3, 7, 28). Mouse B0AT3, by contrast, demonstrated differential substrate affinity for its main substrates l-glycine and l-alanine when coexpressed with either collectrin or ACE2 (Fig. 1C). The apparent affinity (K0.5) for l-alanine was 0.79 ± 0.14 mm in the presence of collectrin, whereas in the presence of ACE2 a value of 0.14 ± 0.02 mm was measured. For glycine, the K0.5 was 0.99 ± 0.05 and 0.27 ± 0.06 mm in the presence of collectrin and ACE2, respectively. To see whether the substrate specificity was also ancillary partner-dependent, we measured uptake of l-[14C]alanine in the presence of 10 mm of a range of amino acids (Fig. 1D). Consistent with the increased apparent affinity of glycine and alanine in the presence of ACE2, a higher degree of inhibition was observed in the presence of unlabeled glycine and alanine. Serine and threonine showed a similar behavior. By contrast, no incremental change of the maximum substrate-induced transport current (Imax) induced by alanine and glycine and other substrates was observed, when currents were normalized to the methionine-induced current in the presence of either ancillary protein (Fig. 1E). These results demonstrate that collectrin and ACE2 are not only trafficking subunits, but they also affect substrate affinity and specificity of the B0AT3 transporter. As a result, these subunits may also be involved in the catalysis of substrate movement by B0-like transporters.
Collectrin Is Necessary for the Catalytic Function of B0AT3 and B0AT1
To differentiate between the trafficking function of collectrin and any potential role in the catalytic process, we created a tandem transporter with the rat monocarboxylate transporter 1 (MCT1) (Fig. 2A). Rat MCT1 is readily expressed at the oocyte surface due to the expression of the Xenopus Basigin protein that is involved in trafficking of MCT1 to the cell surface (72). We first created a MCT1-B0AT3 construct, as B0AT3 has no activity when expressed alone (see Fig. 1A). Surprisingly, we found that MCT1-B0AT3 did not transport l-alanine in the absence of collectrin (Fig. 2B). In contrast, when collectrin was coexpressed with MCT1-B0AT3, full l-alanine transport activity was restored. MCT1-B0AT3 showed active l-lactate transport comparable with MCT1 alone, either with or without collectrin coexpression (Fig. 2C). An MCT1-B0AT1 construct displayed similar dependence on collectrin coexpression as the MCT1-B0AT3 construct (Fig. 2D). To further investigate whether collectrin-induced activity of B0AT1 and B0AT3 was proportional to the surface expression of the tandem construct, we used the natural variability of transporter expression in oocytes. Rat MCT1 is electro-neutral (73) allowing us to compare B0AT3 currents with l-[14C]lactate uptake. These experiments demonstrated a strong correlation between lactate uptake and alanine-induced currents (Fig. 2E). However, the magnitude of l-alanine-induced charge fluxes (Q5 mM L-Ala) was generally lower in MCT1-B0AT3/collectrin coexpressing oocytes than those coexpressing native B0AT3/collectrin.
FIGURE 2.

Collectrin is required for catalytic activation of mouse B0AT3 and B0AT1. X. laevis oocytes were injected with 10 ng of the indicated transporter or 18 ng of MCT-B0AT3 fusion cRNA and 2 ng of collectrin cRNA. All measurements were made days 4–6 post-injection of cRNA. A, schematic of B0AT3 expression conditions are as follows: B0AT3 alone cannot traffic to the plasma membrane (panel i) because it requires collectrin (panel ii). We hypothesized it would traffic without collectrin when fused to MCT1 (panel iii). B, uptake of 100 μm l-[U-14C]alanine or 100 μm l-[U-14C]lactate was carried out over 30 min. Each histogram bar represents mean ± S.D. (n = 10−15, e > 3). a–c above the individual bars indicate groupings of conditions whose differences of means are not statistically significant from each other at the p = 0.05 level. C, uptake of 100 μm l-[U-14C]lactate was carried out over 24 min. Each histogram bar represents mean ± S.D. (n = 10, e = 3). a and b above the individual bars indicates groupings of conditions whose differences of means are not statistically significant from each other at the p = 0.05 level. D, uptake of 100 μm l-[U-14C]leucine or 100 μm l-[U-14C]lactate was carried out over 25 min. Each bar represents mean ± S.D. (n = 12−15, e = 3). a–d above the individual bars indicate groupings of conditions whose differences of means are not statistically significant from each other at the p = 0.05 level. E, correlation between l-lactate uptake and l-alanine-induced currents in the MCT-B0AT3 tandem construct. The integral of the alanine-dependent inward charge transfer (Q5 mM L-Ala) was calculated and plotted against the l-[U-14C]lactate uptake in the same oocyte (n = 10 − 13). Pearson's r = 0.92, the adjusted coefficient of determination (R2) is indicated on the graph.
To investigate the behavior of the MCT1-B0AT3 tandem construct further, we measured basic kinetic parameters. Inspection of the original tracings suggested that alanine-induced currents saturated more readily in the tandem construct compared with the conventional transporter (Fig. 3A). Consistent with Fig. 2E, the Vmax of alanine-induced currents was significantly lower in the tandem construct plus collectrin (26.8 ± 2.3 nA) compared with B0AT3 plus collectrin (97.6 ± 13.5 nA) (Fig. 3B). The K0.5 value of alanine-induced currents was also significantly lower in the tandem construct plus collectrin (0.15 ± 0.03 mm) compared with B0AT3 plus collectrin (0.79 ± 0.18 mm). This combination of a change in both steady-state kinetic parameters explains why we observed similar rates of transport in both native B0AT3 and the fusion construct at sub-K0.5 l-alanine concentrations in uptake assays (Fig. 2B). No l-alanine-induced currents were observed in MCT-B0AT3-expressing oocytes in the absence of collectrin (data not shown), confirming the necessity of collectrin for sodium-driven transport activity in MCT-B0AT3. Together these results clearly demonstrate an essential role of collectrin in both trafficking and catalytic activation of B0AT3.
FIGURE 3.

Properties of MCT-B0AT3 activation by collectrin. X. laevis oocytes were injected with 10 ng of the indicated transporter or 18 ng of MCT-B0AT3 fusion cRNA and 2 ng of collectrin cRNA. All measurements were made 4–6 days post-injection. All oocytes were voltage-clamped at −50 mV. A, l-alanine-induced current tracings recorded from single oocytes super-fused with the serial substrate concentrations were used to determine the steady-state kinetics of MCT-B0AT3 compared with B0AT3, both in the presence of collectrin. Steady-state inward currents were recorded for both the descending (shown) and ascending (not shown) order of l-alanine concentrations. B, oocytes were perfused with serial concentrations of l-alanine as in A and averaged data for each concentration fitted to the Michaelis-Menten function. Inset, Eadie-Hofstee linear regression of the Michaelis-Menten data: adjusted R2 = 0.77 for B0AT3 + collectrin and adjusted R2 = 0.88 for MCT-B0AT3 + collectrin (n = 8−10, e = 3).
Identification of a Collectrin Interaction Site in B0AT3
To identify the potential binding site(s) for collectrin in B0AT3 and B0AT1, we used mutagenesis-based approaches and sequence alignments of the SLC6 family. A summary of the results for all mutants is given in Table 2. Mutations were selected by a number of processes as follows: (i) mutations associated with iminoglycinuria in humans (6); (ii) protein sequence differences between mouse B0AT3 (active in oocytes) and human B0AT3 (inactive in oocytes); (iii) potential sites for electrostatic interactions between collectrin and B0AT3; and (iv) molecular dynamics simulation of collectrin binding to B0AT3 in the vicinity of Arg-225 (equivalent to B0AT1 Arg-240). We deliberately excluded mutations of residues lining the translocation pore as these are likely to be involved in substrate and ion binding. The relative activity of single mutations was mapped onto the homology model of B0AT3 (Fig. 4, A and B).
TABLE 2.
Summary of single residue mutations in murine B0AT3, co-expressed with collectrin: selection, position, activity, and expression
| mB0AT3 mutation | Selection categorya | Mutation position | l-[U-14C]Alanine uptake activity (% wild-type ±S.E., e ≥ 3) | Plasma membrane expression (normalized to WT expression) | Relative protein expression (normalized to WT expression) | Orthologous residue in human B0AT1 |
|---|---|---|---|---|---|---|
| Wild type | 100 | 1 | 1 | |||
| D21N | 3 | N terminus | 167.5 ± 9.6b | 1.86 ± 0.25 | 0.96 | Asp-36 |
| Q25V | 4 | TM1α | 32.0 ± 11.0b | 0.29 ± 0.09 | 0.86 | Gln-40 |
| Q25D | 4 | TM1α | 3.9 ± 5.0b | 0.05 ± 0.07 | 0.84 | Gln-40 |
| H50Y | 2 | EL 1 | 141 ± 11.3b | 0.80 ± 0.15 | 1.21 | His-65 |
| Y72H | 2 | TM2 | 79.0 ± 10.3 | 0.89 ± 0.11 | 1.20 | Tyr-87 |
| G78S | 1 | IL 1 | 15.3 ± 1.8b | 0.89 ± 0.19 | 0.88 | Gly-93 |
| K90T | 2 | IL 1 | 72.5 ± 13.4 | 0.86 ± 0.24 | 0.90 | Ser-106 |
| S158D | 3 | EL 2 | 115.2 ± 5.6 | 1.25 ± 0.12 | 1.46 | Asp-173 |
| L213W | 4 | TM5 | 30.3 ± 8.5b | 1.10 ± 0.18 | 1.14 | Thr-228 |
| R225T | 3 | TM5 | 10.2 ± 2.7b | 0.39 ± 0.15 | 1.07 | Arg-240 |
| N283D | 2 | IL 3 | 4.9 ± 1.5b | 0.21 ± 0.03 | 1.32 | Asn-298 |
| D287K | 3 | TM7 | 3.5 ± 2.4b | 0.10 ± 0.12 | 1.03 | Asp-302 |
| D287N | 3 | TM7 | 21.0 ± 12.6b | 0.21 ± 0.06 | 1.08 | Asp-302 |
| S296R | 2 | TM7 | 2.6 ± 0.6b | 0.09 ± 0.07 | 1.01 | Gly-311 |
| M297S | 4 | TM7 | 104.0 ± 5.6 | 0.96 ± 0.10 | 0.77 | Phe-312 |
| M297C | 4 | TM7 | 117.2 ± 4.8 | 1.39 ± 0.42 | 0.84 | Phe-312 |
| I456T | 2 | TM9 | 110.7 ± 6.7 | 1.05 ± 0.23 | 1.12 | Phe-471 |
| G464N | 2 | EL 5 | 90.0 ± 10.8 | 0.94 ± 0.55 | 1.08 | Gln-479 |
| L477P | 1 | TM10 | 120.0 ± 26 | 1.21 ± 0.38 | 0.81 | Ile-492 |
| G495R | 1 | IL 5 | 3.9 ± 3.3b | 0.01 ± 0.02 | 0.76 | Gly-510 |
| D502N | 3 | IL 5 | 9.3 ± 6.8b | 0.12 ± 0.11 | 0.07 | Asp-517 |
| Q568R | 2 | EL 6 | 91.4 ± 9 | 0.92 ± 0.49 | 0.93 | Tyr-583 |
a Mutagenesis selection categories are as follows: 1, iminoglycinuria-associated mutants; 2 = evolved between murine and human B0AT3; 3 = electrostatically mediated interactions in cytosolic and extracellular loops; and 4 = in silico molecular dynamic-associated (for details of mutant selection see under “Experimental Procedures”).
b p < 0.05 one-way analysis of variance;, Bonferroni post hoc test was conducted for all repeats that make up the tabulated data.
FIGURE 4.

Surface localization of B0AT3 mutants displayed on a homology model. Transport activity of all mutants is indicated by color coding and was determined by flux assays in X. laevis oocytes as described in Fig. 1. Data were taken from Table 2. The homology model structure of mouse B0AT3 was built using the D. melanogaster dopamine transporter DAT x-ray crystal structure in the outward open confirmation (PDB 4m48) (A–C) or LeuT from A. aeolicus in an outward occluded confirmation (PDB 2A65) (D) as a template. Mouse B0AT3 is viewed parallel to the membrane. Mutated residues are shown in van der Waals representation. Wild-type activity is 100% (white); lower activity is displayed in red, and higher activity in blue. A, transmembrane helices 1α, 5, and 7 are colored yellow, orange, and green, respectively. B, homology model is rotated 180° on its vertical axis. C, surface representation of B0AT3 in the same orientation as shown in A. The pocket is flanked by TM domains 5 and 7 and partially occupied by TM1α. Only mutants located in, or peripheral to, TM1α, TM5, or TM7 are labeled. D, hypothesized interaction site between collectrin's TM domain (Met-136–Arg-171) and B0-like transporters. The model is based on the end point of a 10-ns MD simulation (see “Experimental Procedures”). The same face of mouse B0AT3 is shown as in other panels. The transporter is viewed parallel to the membrane with the POPC membrane used during MD simulation removed to see the proteins clearly. Mouse B0AT3 is visualized in gray with collectrin Met-136 to Arg-171 in dark green. The same residues are shown as in C.
Several mutations causing functional changes of B0AT3 activity, D21N, Q25V, L213W, R225T, N283D, D287N, and S296R, were found on one face of the B0AT3 homology model in transmembrane (TM) domains 1α, 5, 7 and adjacent cytosolic and extracellular regions. Mutants Y72H, K90T, S158D, M297S, I456T, L477P, and Q568R, by contrast, had no significant effects on B0AT3 transport activity and, with the exception of M297S, were facing the side of the transporter opposite TM1α, -5, and -7. A space-filled rendering of B0AT3 shows TM domains 5 and 7 are separated by a groove in the side of the transporter, with the mutated residues D21N, Q25V, L213W, R225T, N283D, D297N, and S296R surrounding this groove (Fig. 4C). TM1α is proposed to move during the transport cycle through this gap between TM5 and TM7 (74). All mutated B0AT3 variants with the exception of D502N were expressed in oocytes at comparable protein levels to wild-type B0AT3 (Table 2). With the exception of mutants H50Y, G78S, L213W, R225T, and N283D, transport activity was highly correlated with surface expression (Table 2). This is illustrated by the correlation coefficient (r), which increased from 0.87 to 0.99 when the five mutants were excluded. Mutants H50Y and G78S were not investigated further as they are not located at the cell surface. As there seems to be a preponderance of functional and surface expression mutants in B0AT3 TMs 5 and 7, we hypothesized that the collectrin's TM domain may occupy an interaction site in the groove flanked by these TM domains.
To test this notion, a series of six docking simulations between collectrin TM domain and B0AT3 were performed, using different sets of residues from TM5 and TM7 as docking constraints (Table 1). The lowest scoring docking conformer was used to initiate a 10-ns molecular dynamics simulation (see under “Experimental Procedures”). This simulation identified Met-297 in TM7, Leu-213 in TM5, and Gln-25 in TM1α as key interacting residues with collectrin's TM domain (Fig. 4D). Fig. 4, C and D, appears different in the loop regions as they are based on Drosophila DAT and LeuT crystal structures, respectively. The TM5/TM7 groove is present in both homology models and the experimental structures on which they were based.
Next, we sought to differentiate whether these residues in TM1α, -5 and -7 interact directly with collectrin or affect the transporter independently of it. To do so, we hypothesized the existence of three different types of mutations that could cause loss of function in B0AT3. The first are catalytic mutations that interfere directly with the transport mechanism, independent of collectrin. A second type of mutation interferes with the interaction between collectrin and B0AT3 resulting in a trafficking modification. These should be rescued when expressed in the MCT1-B0AT3 tandem construct. A third type of mutation affects the catalytic role of collectrin and should not be rescued when incorporated into the tandem construct. Although it is difficult to discriminate between nonfunctional type 1 and type 3 mutants, a true type 3 mutant is likely to be expressed at the cell surface. A possible type 3 mutant is L213W (located in TM5) as it is neither active in B0AT3 nor in the auto-trafficked MCT1-B0AT3 when both are coexpressed with collectrin (Fig. 5A). Furthermore, it is localized and fully expressed at the plasma membrane (Fig. 5B). N283D can be classified as a type 2 mutant; it lacks surface expression and activity when coexpressed with collectrin, but it is re-activated in MCT-B0AT3 by the presence of collectrin (Fig. 5, B and C). The TM1α mutant B0AT3(D21N) is also type 2 but with an opposite effect. It consistently shows higher surface expression when coexpressed with collectrin, but it shows wild-type-like activity when expressed in the tandem construct in the presence of collectrin. Thus, both D21N and N283D change trafficking but display wild-type-like activity in the tandem construct. Both of these mutants are likely to interact with collectrin.
FIGURE 5.

Analysis of mutants in a MCT-B0AT3 tandem construct supports a bi-functional role for collectrin. A–E, X. laevis oocytes were injected with the indicated constructs and uptake of 100 μm l-[U-14C]alanine measured over 30 min. Each bar represents the mean ± S.D. activity of n = 10−15 (e = 3). a–d above the individual bars indicate groupings of conditions whose differences of means are not statistically significant from each other at the p = 0.05 level. B, surface biotinylation and detection of collectrin and B0AT3 was carried out as indicated under “Experimental Procedures.” Molecular mass markers (in kDa) are indicated on the left, and detected proteins are indicated on the right. No TP = no transporter. F, uptake of 100 μm l-[U-14C]lactate by wild-type and mutated MCT-B0AT3 tandem constructs. Lactate uptake for all conditions was normalized to lactate uptake of wild-type MCT-B0AT3-expressing oocytes from the same batch (i.e. either with or without collectrin). Each bar represents mean ± S.D. No difference at the p < 0.05 was detected for lactate uptake between MCT-B0AT3 WT and any MCT-B0AT3 mutant (n = 10−15, e = 3).
A mix between type 2 and 3 is represented by R225T; the mutant has an almost complete loss of surface expression, but it regains limited activity in the auto-trafficking MCT-B0AT3 construct in the presence of collectrin (Fig. 5, B and D). Another type 2/3 mutant is B0AT3(Q25V), which is partially active in the presence of collectrin despite having a strong trafficking defect, as shown by Western blotting after surface biotinylation (Fig. 5, B and E). However, transport activity of MCT-B0AT3(Q25V) was even lower in the presence of collectrin, suggesting that Gln-25 is also important in the catalytic activation by collectrin. In most transporters of the SLC6 family, Gln-25 is an acidic residue (Asp or Glu). Replication of the negative charge (B0AT3(Q25D)) caused a total loss-of-function in both B0AT3 and the tandem construct when coexpressed with collectrin (Fig. 5E). Membrane expression was consistent across all mutants in the tandem construct as ascertained by l-[U-14C]lactate uptake (Fig. 5F).
Interactions with Syntaxin-1A and Syntaxin 3
Protein-protein interactions with single-pass trans-membrane helical chaperones or ancillary proteins is a common feature in the SLC6 family (33). Syntaxin 1A, in particular, has been shown to interact with a number of neurotransmitter transporters (33). We therefore wondered whether the collectrin binding region identified here represents a common interaction site. The GABA transporter GAT1 (SLC6A1) has been shown to interact with syntaxin 1A at aspartate residues in the N terminus of the transporter (44, 46, 48), which is part of our identified collectrin binding region in B0AT3 and B0AT1. For syntaxin and collectrin interaction experiments, we utilized mouse B0AT1, as it has a larger transport signal than mouse B0AT3, and small but resolvable levels of collectrin-independent transport activity and cell surface expression. In confirmation of published results, we found that the presence of murine syntaxin 1A significantly reduced rat GAT1-mediated GABA transport (Fig. 6A) (46). Coexpression of collectrin and syntaxin 1A with rat GAT1 recovered GABA transport to a similar rate observed with GAT1 expressed alone. When expressed with GAT1 alone, collectrin slightly, but significantly, increased transport activity. Similarly, B0AT1 transport activity, measured in the absence of collectrin, was virtually abolished when it was coexpressed with syntaxin 1A. Coexpression with collectrin resulted in reconstitution of ∼80% of transport observed in the absence of syntaxin 1A. A similar inhibition of l-alanine transport activity was observed when syntaxin 1A was coexpressed with B0AT3 and collectrin (data not shown). To determine the mode of action of syntaxin 1A, we titrated syntaxin 1A cRNA against B0AT1 and collectrin cRNA (Fig. 6B). Surface expression of B0AT1in the presence of collectrin was very sensitive to expression of small amounts of syntaxin 1A. This effect could be reversed by increasing collectrin cRNA (Fig. 6C). However, this reversal required large amounts of collectrin cRNA (10–20 ng/oocyte). Notably, increasing titration of collectrin reduced the apparent molecular mass of B0AT1 from about 80 to 65 kDa (Fig. 6C). l-Leucine uptake rates of B0AT1 confirmed the suppression of B0AT1-mediated activity by syntaxin 1A in the presence of collectrin (Fig. 6D). Functional recovery was observed when the amount of collectrin cRNA was increased, confirming competition between both ancillary proteins. The inhibition of B0AT1-mediated transport activity fitted well to a single competitive binding site equation (Fig. 6D, inset).
FIGURE 6.

Interactions between B0AT1, collectrin, and syntaxin 1A. Uptake measurements in X. laevis were performed as outlined in Fig. 2. A, cRNA was injected as follows: transporter = 10 ng/oocyte; collectrin = 2 ng/oocyte; syntaxin-1A = 5 ng/oocyte. Uptake of 100 μm l-[U-14C]alanine was measured over 30 min. Each bar represents the mean ± S.D. (n = 10−15, e = 3). a–c (GAT1) and d–f (B0AT1) above the individual bars indicate groupings of conditions whose differences of means are not statistically significant from each other at the p = 0.05 level. B, collectrin cRNA = 2 ng/oocyte B0AT1 cRNA = 10 ng/oocyte. Syntaxin-1A cRNA was titrated as indicated. Surface biotinylation was carried out with 15 oocytes as described under “Experimental Procedures.” Molecular mass markers (in kDa) are added to the left, and the detected proteins are indicated on the right. C, syntaxin-1A cRNA = 5 ng/oocyte B0AT1 cRNA = 10 ng/oocyte. Collectrin cRNA was titrated as indicated. 15 oocytes per sample were treated as in B. D, uptake of 100 μm l-[U-14C]leucine was carried out over 30 min. For each condition, one ancillary protein cRNA was injected at a constant amount, and the other ancillary cRNA was titrated as indicated in the legend. Each data point represents mean ± S.D. (n = 10−15, e = 3). E, cRNA was injected as follows: B0AT1 = 10 ng/oocyte; collectrin = 2 ng/oocyte; all syntaxins = 5 ng/oocyte. Uptake of 100 μm l-[U-14C]leucine was carried out over 30 min. Each bar represents mean ± S.D. (n = 15, e = 3). a–d above the individual bars indicate groupings of conditions whose differences of means are not statistically significant from each other at the p = 0.05 level.
Although our data suggest competition for a common binding site between collectrin and syntaxin-1A, the cellular expression of both proteins does not overlap (38). Database searches comparing tissue-specific E.S.T. expression data (www.ncbi.nlm.nih.gov) and literature searches (52, 53), however, identified several syntaxin isoforms (3, 4, 7 and 17) with overlapping expression to B0AT1 and/or collectrin. Coexpression experiments showed strong inhibition of B0AT1 + collectrin activity by syntaxin 3, whereas syntaxin 7 had no effect (Fig. 6E).
To confirm syntaxin 1A and 3 inhibition of B0AT1-mediated leucine uptake in a different expression system, we generated stably transfected CHO cells expressing human B0AT1 and human collectrin. CHO-SLC6A19-collectrin cells showed a significant increase in the sodium-dependent uptake of leucine above CHO parental cells (Fig. 7A). Expression of both B0AT1 and collectrin was confirmed by Western blotting and immunodetection in whole membrane preparation (Fig. 7B). CHO parental cells exhibited no B0AT1 or collectrin expression (Fig. 7B). As observed in oocytes, transient expression of either syntaxin 1A or syntaxin 3 in CHO-SLC6A19-collectrin cells significantly reduced sodium-dependent uptake of leucine compared with pcDNA3.1+ vector-only controls (Fig. 7C, left panel). We also discovered that both syntaxin 1A and 3 are endogenously expressed in CHO-SLC6A19-collectrin cells (Fig. 7D). If competition with collectrin and syntaxin occurs, we reasoned that silencing of endogenous syntaxin would increase B0AT1-mediated leucine uptake. Accordingly, RNAi against both syntaxin 1A and syntaxin 3 led to an increase in sodium-dependent leucine uptake (Fig. 7C, right panel). Simultaneous RNAi of both mRNAs had a small cumulative effect on the inhibition of sodium-dependent uptake. RT-PCR analysis confirmed significant reduction of syntaxin 1A and syntaxin 3 mRNA after siRNA transfection (Fig. 7D).
FIGURE 7.

Syntaxin 1A and syntaxin 3 inhibit B0AT1 activity. A, uptake of 100 μm l-[U-14C]leucine was measured over 6 min in CHO parental and CHO-SLC6A19-collectrin cells in either Hanks' buffered saline (Na+) or with NMDG replacing sodium (NMDG). Each bar represents mean transport activity ± S.D. (e = 3, n = 3, ***, p = 0.005 level). p values are calculated as difference from Na+-independent uptake condition. B, immunodetection of B0AT1 and collectrin in whole membrane preparations of CHO parental and CHO-SLC6A19-collectrin cells; 20 μg of total protein was loaded into each well. Molecular mass markers (in kDa) are indicated on the left, and detected proteins are indicated on the right. C, CHO-SLC6A19-collectrin cells were transfected with 3 μg of pcDNA3.1+ vector DNA encoding the indicated murine syntaxin gene (overexpression, left panel) or 50 pmol of siRNA against the CHO endogenous syntaxins (RNAi, right panel). 100 μm l-[U-14C]leucine uptake in CHO-SLC6A19-collectrin cells was measured over 6 min in either Hanks' buffered saline (Na+) or with NMDG replacing sodium (NMDG). The ratio of net sodium-dependent/sodium-independent uptake for each uptake condition was normalized to the pcDNA3.1+ vector-only control (overexpression) or scrambled siRNA (RNAi). Each bar represents mean ± S.E. (n = 3, e = 3, ***, p 0.005 level; **, p 0.01 level; *, p 0.05 level). p values are calculated as difference from control conditions, vector only (overexpression) or scramble (RNAi). D, levels of syntaxin 1A and syntaxin 3 mRNA transcripts were estimated by RT-PCR with 25 cycles of amplification. Closed bars represent control samples using scrambled RNAi, and open bars show the effect of specific RNAi. Samples were run on agarose gel, and quantification was made relative to clathrin mRNA expression in the same sample. Each bar represents mean ± S.E. (e = 3, ***, p 0.005 level). p values are calculated as difference from scramble conditions.
Discussion
In this paper we report for the first time that collectrin is not only required for trafficking of B0AT1 and B0AT3 to the plasma membrane, but also for catalytic activation. We also note here for the first time that human B0AT3 is an inactive protein, which is consistent with evolution from B0AT3-mediated glycine/alanine uptake in mice to PAT2-mediated glycine/alanine uptake in humans (9) and the absence of glycinuria in humans with homozygous nonsense mutations in B0AT3 (6). Moreover, we report the localization of a collectrin interaction region in B0AT3, positioned in a groove between TM5 and -7 of the transporter, which also contains TM1α in all experimentally isolated structural conformations of the SLC6 ancestors LeuT and Drosophila DAT (74–77). Interestingly, the groove suggested here to harbor collectrin is occupied by a cholesterol molecule in the DAT outward open structure (75). The authors suggest that cholesterol may stabilize the transporter in the outward open conformation, which is consistent with functional studies of the human serotonin transporter (78). We would thus propose that collectrin and syntaxin isoforms can occupy this binding site, increasing or inhibiting transport activity, respectively. In the occluded structure of LeuT, TM1α and the cytoplasmic end of TM5 are in contact but become separated during the transition to the cytoplasmic-facing conformation (74–76, 79, 80). If collectrin would reduce the activation energy of the transporter's rate-limiting step, it could speed up the transport cycle. Syntaxin isoforms by contrast could stabilize transporter conformations other than the rate-limiting transition state, thereby slowing down transport. Syntaxin 3 could have a physiological role in regulating the transporter by displacing collectrin and thus inhibiting B0AT1.
Our results clearly demonstrate a dual role of collectrin. It is required for trafficking of the transporter to the cell surface and is required for the catalytic activity. Both functions can be separated. L213W is involved in the catalytic activation of the transporter, although it is located >10 Å from the ionic radius of the second sodium-binding site in the DAT-based outward open homology model; and it is even further away from all other predicted ion/substrate-binding sites. In contrast, type 2 mutants N283D and D21N selectively affect trafficking to the cell surface. The remaining nonfunctional mouse B0AT3 mutants, the previously identified R225T (R240Q in B0AT1) (7) and Q25V, were classified as type 2/3 and appear to be involved in both collectrin-mediated functions. Alternatively, the small but significant reduction of the B0AT1 molecular weight, when coexpressed with collectrin (see, for example, Fig. 6C) maybe associated with its activation. The mechanism of this activation remains to be elucidated, but it may involve recruitment of additional proteins. We cannot rule out the possibility that neither collectrin nor syntaxin proteins directly bind to B0-like transporters. They may interact indirectly through other proteins, although there is evidence for direct interaction and colocalization between B0AT1, collectrin, and ACE2 from mouse kidney and intestinal epithelial tissue (3, 28).
In contrast to collectrin, syntaxin 1A mediates an opposing bi-functional effect (33). It enhances trafficking of GAT1 to the surface, but at the same time reduces its activity. Several lines of evidence are converging to establish the N-terminal domain and TM1α of SLC6 transporters as a key domain in establishing conformational equilibrium and intracellular gating (81–83). This region also represents a binding/interaction region for syntaxin 1A to the SLC6 serotonin transporter SERT (44, 48, 84), the norepinephrine transporter NET (42), and a Caenorhabditis elegans dopamine transporter DAT-1 (41). Although the N-terminal domain has been implicated in syntaxin binding, our results, for the first time, suggest binding of syntaxin to a groove between helix 5 and 7. This interaction would most likely be mediated by the membrane anchored domain of syntaxin, a region shown to be crucial to the syntaxin 1A-mediated regulation of GAT1 (44). The intracellular loop 4 (IL 4) region of GAT1 has also been shown to inhibit GABA transport via an arginine-rich domain that competes with syntaxin 1A for binding to the transporter's N-terminal domain (44). Only Arg-420 in human GAT1 (Lys-455 in human B0AT1) is conserved by chemical identity across solute carrier transporters in the IL 4 region, and we did not investigate the role of the IL 4 region in the B0-like transport-collectrin interaction.
The interactions between collectrin and the B0-like transporters cannot involve canonical coiled-coil domains as these are absent in collectrin. It may, however, involve different stoichiometric combinations with collectrin and/or associations between SNARE proteins as has been described in pancreatic β-islet cells (23, 54, 55). To date, the only structural analysis of collectrin comes from islet cells and islet cell cultures, where disulfide-mediated dimerization, N-linked glycosylation, and partial proteolysis by an extracellular protease regulate collectrin-associated insulin secretion and cell mass (22, 85). One unexplained aspect of the effect of catalytic activation by collectrin on B0AT3 is the significant increase in substrate affinity when the transporter is auto-trafficked to the plasma membrane as part of the MCT-B0AT3 fusion construct. This construct could have an effect on the overall conformation of the transporter resulting in a slight change of the on- or off-rates of transporter substrates, which in turn would alter the affinity.
We believe that in light of its dual role and identified binding region collectrin should now be viewed as an essential subunit of a heteromultimeric secondary active transporter, rather than simply as an ancillary protein or chaperone.
Author Contributions
S. B. and S. J. F. conceived and coordinated the study and wrote the paper. S. J. F. designed, performed, and analyzed experiments shown in Figs. 1–6. A. B., D. S., and Q. C. designed, performed, and analyzed the experiments shown in Figs. 1 and 7. M. O. M. and N. S. performed MD simulations. E. T. provided technical assistance and contributed to the preparation of figures. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Thuvaraka Thavyogarajah for technical support.
This work was supported by a sponsored research agreement with Sanofi and National Health and Medical Research Council Grant 525415 (to S. B.). The authors declare that they have no conflicts of interest with the contents of this article.
- HBSS
- Hanks' buffered salt solution
- TM
- transmembrane
- NMDG
- N-methyl-d-glucamine
- PDB
- Protein Data Bank
- POPC
- 1-palmitoyl-2-oleoylphosphatidylcholine
- MD
- molecular dynamics.
References
- 1. Bröer S., Gether U. (2012) The solute carrier 6 family of transporters. Br. J. Pharmacol. 167, 256–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bröer S. (2013) Diseases associated with general amino acid transporters of the solute carrier 6 family (SLC6). Curr. Mol. Pharmacol. 6, 74–87 [DOI] [PubMed] [Google Scholar]
- 3. Danilczyk U., Sarao R., Remy C., Benabbas C., Stange G., Richter A., Arya S., Pospisilik J. A., Singer D., Camargo S. M., Makrides V., Ramadan T., Verrey F., Wagner C. A., Penninger J. M. (2006) Essential role for collectrin in renal amino acid transport. Nature 444, 1088–1091 [DOI] [PubMed] [Google Scholar]
- 4. Singer D., Camargo S. M., Huggel K., Romeo E., Danilczyk U., Kuba K., Chesnov S., Caron M. G., Penninger J. M., Verrey F. (2009) Orphan transporter SLC6A18 is renal neutral amino acid transporter B0AT3. J. Biol. Chem. 284, 19953–19960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Camargo S. M., Singer D., Makrides V., Huggel K., Pos K. M., Wagner C. A., Kuba K., Danilczyk U., Skovby F., Kleta R., Penninger J. M., Verrey F. (2009) Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with Hartnup mutations. Gastroenterology 136, 872–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bröer S., Bailey C. G., Kowalczuk S., Ng C., Vanslambrouck J. M., Rodgers H., Auray-Blais C., Cavanaugh J. A., Bröer A., Rasko J. E. (2008) Iminoglycinuria and hyperglycinuria are discrete human phenotypes resulting from complex mutations in proline and glycine transporters. J. Clin. Invest. 118, 3881–3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kowalczuk S., Bröer A., Tietze N., Vanslambrouck J. M., Rasko J. E., Bröer S. (2008) A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 22, 2880–2887 [DOI] [PubMed] [Google Scholar]
- 8. Malakauskas S. M., Quan H., Fields T. A., McCall S. J., Yu M. J., Kourany W. M., Frey C. W., Le T. H. (2007) Aminoaciduria and altered renal expression of luminal amino acid transporters in mice lacking novel gene collectrin. Am. J. Physiol. Renal Physiol. 292, F533–F544 [DOI] [PubMed] [Google Scholar]
- 9. Vanslambrouck J. M., Bröer A., Thavyogarajah T., Holst J., Bailey C. G., Bröer S., Rasko J. E. (2010) Renal imino acid and glycine transport system ontogeny and involvement in developmental iminoglycinuria. Biochem. J. 428, 397–407 [DOI] [PubMed] [Google Scholar]
- 10. Böhmer C., Bröer A., Munzinger M., Kowalczuk S., Rasko J. E., Lang F., Bröer S. (2005) Characterization of mouse amino acid transporter B0AT1 (slc6a19). Biochem. J. 389, 745–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Camargo S. M., Makrides V., Virkki L. V., Forster I. C., Verrey F. (2005) Steady-state kinetic characterization of the mouse B(0)AT1 sodium-dependent neutral amino acid transporter. Pflugers Arch. 451, 338–348 [DOI] [PubMed] [Google Scholar]
- 12. Bröer S. (2013) Epithelial neutral amino acid transporters: lessons from mouse models. Curr. Opin. Nephrol. Hypertens. 22, 539–544 [DOI] [PubMed] [Google Scholar]
- 13. Vuille-dit-Bille R. N., Camargo S. M., Emmenegger L., Sasse T., Kummer E., Jando J., Hamie Q. M., Meier C. F., Hunziker S., Forras-Kaufmann Z., Kuyumcu S., Fox M., Schwizer W., Fried M., Lindenmeyer M., et al. (2015) Human intestine luminal ACE2 and amino acid transporter expression increased by ACE-inhibitors. Amino Acids 47, 693–705 [DOI] [PubMed] [Google Scholar]
- 14. Eriksen J., Jørgensen T. N., Gether U. (2010) Regulation of dopamine transporter function by protein-protein interactions: new discoveries and methodological challenges. J. Neurochem. 113, 27–41 [DOI] [PubMed] [Google Scholar]
- 15. Farhan H., Freissmuth M., Sitte H. H. (2006) Oligomerization of neurotransmitter transporters: a ticket from the endoplasmic reticulum to the plasma membrane. Handb. Exp. Pharmacol. 175, 233–249 [DOI] [PubMed] [Google Scholar]
- 16. Sitte H. H., Farhan H., Javitch J. A. (2004) Sodium-dependent neurotransmitter transporters: oligomerization as a determinant of transporter function and trafficking. Mol. Interv. 4, 38–47 [DOI] [PubMed] [Google Scholar]
- 17. Seow H. F., Bröer S., Bröer A., Bailey C. G., Potter S. J., Cavanaugh J. A., Rasko J. E. (2004) Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat. Genet. 36, 1003–1007 [DOI] [PubMed] [Google Scholar]
- 18. Kleta R., Romeo E., Ristic Z., Ohura T., Stuart C., Arcos-Burgos M., Dave M. H., Wagner C. A., Camargo S. R., Inoue S., Matsuura N., Helip-Wooley A., Bockenhauer D., Warth R., Bernardini I., et al. (2004) Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat. Genet. 36, 999–1002 [DOI] [PubMed] [Google Scholar]
- 19. Jiang Y., Rose A. J., Sijmonsma T. P., Bröer A., Pfenninger A., Herzig S., Schmoll D., Bröer S. (2015) Mice lacking neutral amino acid transporter B(0)AT1 (Slc6a19) have elevated levels of FGF21 and GLP-1 and improved glycaemic control. Mol. Metab. 4, 406–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bröer A., Juelich T., Vanslambrouck J. M., Tietze N., Solomon P. S., Holst J., Bailey C. G., Rasko J. E., Bröer S. (2011) Impaired nutrient signaling and body weight control in a Na+ neutral amino acid cotransporter (Slc6a19)-deficient mouse. J. Biol. Chem. 286, 26638–26651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saisho K., Fukuhara A., Yasuda T., Sato Y., Fukui K., Iwahashi H., Imagawa A., Hatta M., Shimomura I., Yamagata K. (2009) Glucose enhances collectrin protein expression in insulin-producing MIN6 beta cells. Biochem. Biophys. Res. Commun. 389, 133–137 [DOI] [PubMed] [Google Scholar]
- 22. Akpinar P., Kuwajima S., Krützfeldt J., Stoffel M. (2005) Tmem27: a cleaved and shed plasma membrane protein that stimulates pancreatic beta cell proliferation. Cell Metab. 2, 385–397 [DOI] [PubMed] [Google Scholar]
- 23. Yasuhara A., Wada J., Malakauskas S. M., Zhang Y., Eguchi J., Nakatsuka A., Murakami K., Kanzaki M., Teshigawara S., Yamagata K., Le T. H., Makino H. (2008) Collectrin is involved in the development of salt-sensitive hypertension by facilitating the membrane trafficking of apical membrane proteins via interaction with soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex. Circulation 118, 2146–2155 [DOI] [PubMed] [Google Scholar]
- 24. Bril A., Feletou M. (2014) Collectrin: a new component of the renin-angiotensin system? Med. Sci. 30, 136–139 [DOI] [PubMed] [Google Scholar]
- 25. Egan B. M. (2013) Collectrin, an X-linked, angiotensin converting enzyme 2 homolog, causes hypertension in a rat strain through gene-gene and gene-environment interactions: relevance to human hypertension. Circulation 128, 1727–1728 [DOI] [PubMed] [Google Scholar]
- 26. Cechova S., Zeng Q., Billaud M., Mutchler S., Rudy C. K., Straub A. C., Chi L., Chan F. R., Hu J., Griffiths R., Howell N. L., Madsen K., Jensen B. L., Palmer L. A., Carey R. M., et al. (2013) Loss of collectrin, an angiotensin-converting enzyme 2 homolog, uncouples endothelial nitric oxide synthase and causes hypertension and vascular dysfunction. Circulation 128, 1770–1780 [DOI] [PubMed] [Google Scholar]
- 27. Tümer E., Bröer A., Balkrishna S., Jülich T., Bröer S. (2013) Enterocyte-specific regulation of the apical nutrient transporter SLC6A19 (B(0)AT1) by transcriptional and epigenetic networks. J. Biol. Chem. 288, 33813–33823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fairweather S. J., Bröer A., O'Mara M. L., Bröer S. (2012) Intestinal peptidases form functional complexes with the neutral amino acid transporter B(0)AT1. Biochem. J. 446, 135–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Mello W. C., Frohlich E. D. (2014) Clinical perspectives and fundamental aspects of local cardiovascular and renal renin-angiotensin systems. Front. Endocrinol. 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Patel S. K., Velkoska E., Freeman M., Wai B., Lancefield T. F., Burrell L. M. (2014) From gene to protein-experimental and clinical studies of ACE2 in blood pressure control and arterial hypertension. Front. Physiol. 5, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moreira de Macêdo S., Guimarães T. A., Feltenberger J. D., Sousa Santos S. H. (2014) The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides 62, 189–196 [DOI] [PubMed] [Google Scholar]
- 32. Hashimoto T., Perlot T., Rehman A., Trichereau J., Ishiguro H., Paolino M., Sigl V., Hanada T., Hanada R., Lipinski S., Wild B., Camargo S. M., Singer D., Richter A., Kuba K., et al. (2012) ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 487, 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quick M. W. (2006) The role of SNARE proteins in trafficking and function of neurotransmitter transporters. Handb. Exp. Pharmacol. 175, 181–196 [DOI] [PubMed] [Google Scholar]
- 34. López-Corcuera B., Aragón C., Geerlings A. (2001) Regulation of glycine transporters. Biochem. Soc. Trans. 29, 742–745 [DOI] [PubMed] [Google Scholar]
- 35. Blakely R. D., Sung U. (2000) SNARE-ing neurotransmitter transporters. Nat. Neurosci. 3, 969–971 [DOI] [PubMed] [Google Scholar]
- 36. Oaks A. W., Sidhu A. (2011) Synuclein modulation of monoamine transporters. FEBS Lett. 585, 1001–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhong H., Sánchez C., Caron M. G. (2012) Consideration of allosterism and interacting proteins in the physiological functions of the serotonin transporter. Biochem. Pharmacol. 83, 435–442 [DOI] [PubMed] [Google Scholar]
- 38. Lang T., Jahn R. (2008) Core proteins of the secretory machinery. Handb. Exp. Pharmacol. 184, 107–127 [DOI] [PubMed] [Google Scholar]
- 39. Sørensen J. B. (2005) SNARE complexes prepare for membrane fusion. Trends Neurosci. 28, 453–455 [DOI] [PubMed] [Google Scholar]
- 40. de Wit H. (2010) Molecular mechanism of secretory vesicle docking. Biochem. Soc. Trans. 38, 192–198 [DOI] [PubMed] [Google Scholar]
- 41. Carvelli L., Blakely R. D., DeFelice L. J. (2008) Dopamine transporter/syntaxin 1A interactions regulate transporter channel activity and dopaminergic synaptic transmission. Proc. Natl. Acad. Sci. U.S.A. 105, 14192–14197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Binda F., Lute B. J., Dipace C., Blakely R. D., Galli A. (2006) The N terminus of the norepinephrine transporter regulates the magnitude and selectivity of the transporter-associated leak current. Neuropharmacology 50, 354–361 [DOI] [PubMed] [Google Scholar]
- 43. Sung U., Apparsundaram S., Galli A., Kahlig K. M., Savchenko V., Schroeter S., Quick M. W., Blakely R. D. (2003) A regulated interaction of syntaxin 1A with the antidepressant-sensitive norepinephrine transporter establishes catecholamine clearance capacity. J. Neurosci. 23, 1697–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hansra N., Arya S., Quick M. W. (2004) Intracellular domains of a rat brain GABA transporter that govern transport. J. Neurosci. 24, 4082–4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang D., Deken S. L., Whitworth T. L., Quick M. W. (2003) Syntaxin 1A inhibits GABA flux, efflux, and exchange mediated by the rat brain GABA transporter GAT1. Mol. Pharmacol. 64, 905–913 [DOI] [PubMed] [Google Scholar]
- 46. Quick M. W. (2002) Substrates regulate gamma-aminobutyric acid transporters in a syntaxin 1A-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 99, 5686–5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Horton N., Quick M. W. (2001) Syntaxin 1A up-regulates GABA transporter expression by subcellular redistribution. Mol. Membr. Biol. 18, 39–44 [PubMed] [Google Scholar]
- 48. Deken S. L., Beckman M. L., Boos L., Quick M. W. (2000) Transport rates of GABA transporters: regulation by the N-terminal domain and syntaxin 1A. Nat. Neurosci. 3, 998–1003 [DOI] [PubMed] [Google Scholar]
- 49. Fan H. P., Fan F. J., Bao L., Pei G. (2006) SNAP-25/syntaxin 1A complex functionally modulates neurotransmitter γ-aminobutyric acid reuptake. J. Biol. Chem. 281, 28174–28184 [DOI] [PubMed] [Google Scholar]
- 50. Saxena S., Quick M. W., Warnock D. G. (2000) Interaction of syntaxins with epithelial ion channels. Curr. Opin. Nephrol. Hypertens. 9, 523–527 [DOI] [PubMed] [Google Scholar]
- 51. Schwartz J. H., Li G., Yang Q., Suri V., Ross J. J., Alexander E. A. (2007) Role of SNAREs and H+-ATPase in the targeting of proton pump-coated vesicles to collecting duct cell apical membrane. Kidney Int. 72, 1310–1315 [DOI] [PubMed] [Google Scholar]
- 52. Delgrossi M. H., Breuza L., Mirre C., Chavrier P., Le Bivic A. (1997) Human syntaxin 3 is localized apically in human intestinal cells. J. Cell Sci. 110, 2207–2214 [DOI] [PubMed] [Google Scholar]
- 53. Li X., Low S. H., Miura M., Weimbs T. (2002) SNARE expression and localization in renal epithelial cells suggest mechanism for variability of trafficking phenotypes. Am. J. Physiol. Renal Physiol. 283, F1111–F1122 [DOI] [PubMed] [Google Scholar]
- 54. Yamagata K., Nammo T., Sato Y., Saisho K., Shoda H., Fukui K. (2007) The HNF-1α-SNARE connection. Diabetes Obes. Metab. 9, 40–45 [DOI] [PubMed] [Google Scholar]
- 55. Fukui K., Yang Q., Cao Y., Takahashi N., Hatakeyama H., Wang H., Wada J., Zhang Y., Marselli L., Nammo T., Yoneda K., Onishi M., Higashiyama S., Matsuzawa Y., Gonzalez F. J., et al. (2005) The HNF-1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab. 2, 373–384 [DOI] [PubMed] [Google Scholar]
- 56. Ganeshan R., Di A., Nelson D. J., Quick M. W., Kirk K. L. (2003) The interaction between syntaxin 1A and cystic fibrosis transmembrane conductance regulator Cl− channels is mechanistically distinct from syntaxin 1A-SNARE interactions. J. Biol. Chem. 278, 2876–2885 [DOI] [PubMed] [Google Scholar]
- 57. Bröer S. (2003) Xenopus laevis oocytes. Methods Mol. Biol. 227, 245–258 [DOI] [PubMed] [Google Scholar]
- 58. Pei J., Kim B. H., Grishin N. V. (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Beuming T., Shi L., Javitch J. A., Weinstein H. (2006) A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 70, 1630–1642 [DOI] [PubMed] [Google Scholar]
- 60. Guex N., Peitsch M. C. (1997) SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 61. Dominguez C., Boelens R., Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 62. Poger D., Mark A. E. (2010) On the validation of molecular dynamics simulations of saturated and cis-monounsaturated phosphatidylcholine lipid bilayers: a comparison with experiment. J. Chem. Theory Comput. 6, 325–336 [DOI] [PubMed] [Google Scholar]
- 63. Schmid N., Eichenberger A. P., Choutko A., Riniker S., Winger M., Mark A. E., van Gunsteren W. F. (2011) Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 40, 843–856 [DOI] [PubMed] [Google Scholar]
- 64. Berendsen H. J., Postma J. P., van Gunsteren W. F., Hermans J. (1981) Interaction models for water in relation to protein hydration. Intermol. Forces 11, 331–338 [Google Scholar]
- 65. Tironi I. G., Sperb R., Smith P. E., van Gunsteren W. F. (1995) A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys. 102, 5451–5459 [Google Scholar]
- 66. Hess B., Bekker H., Berendsen H. J., Fraaije J. G. (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [Google Scholar]
- 67. Miyamoto S., Kollman P. A. (1992) Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 [Google Scholar]
- 68. Feenstra K. A., Hess B., Berendsen H. J. (1999) Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 20, 786–798 [DOI] [PubMed] [Google Scholar]
- 69. Berendsen H. J., Postma J. P., van Gunsteren W. F., Dinola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J Chem. Phys. 81, 3684–3690 [Google Scholar]
- 70. Humphrey W., Dalke A., Schulten K. (1996) VMD–visual molecular dynamics. J. Mol. Graph. Model. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 71. Fan H., Mark A. E. (2004) Refinement of homology-based protein structures by molecular dynamics simulation techniques. Protein Sci. 13, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wilson M. C., Meredith D., Fox J. E., Manoharan C., Davies A. J., Halestrap A. P. (2005) Basigin (CD147) is the target for organomercurial inhibition of monocarboxylate transporter isoforms 1 and 4: the ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J. Biol. Chem. 280, 27213–27221 [DOI] [PubMed] [Google Scholar]
- 73. Bröer S., Schneider H. P., Bröer A., Rahman B., Hamprecht B., Deitmer J. W. (1998) Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem. J. 333, 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Krishnamurthy H., Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Penmatsa A., Wang K. H., Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gouaux E., Mackinnon R. (2005) Principles of selective ion transport in channels and pumps. Science 310, 1461–1465 [DOI] [PubMed] [Google Scholar]
- 77. Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 78. Hong W. C., Amara S. G. (2010) Membrane cholesterol modulates the outward facing conformation of the dopamine transporter and alters cocaine binding. J. Biol. Chem. 285, 32616–32626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Radestock S., Forrest L. R. (2011) The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J. Mol. Biol. 407, 698–715 [DOI] [PubMed] [Google Scholar]
- 80. Forrest L. R., Rudnick G. (2009) The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters. Physiology 24, 377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fraser R., Chen Y., Guptaroy B., Luderman K. D., Stokes S. L., Beg A., DeFelice L. J., Gnegy M. E. (2014) An N-terminal threonine mutation produces an efflux-favorable, sodium-primed conformation of the human dopamine transporter. Mol. Pharmacol. 86, 76–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Guptaroy B., Fraser R., Desai A., Zhang M., Gnegy M. E. (2011) Site-directed mutations near transmembrane domain 1 alter conformation and function of norepinephrine and dopamine transporters. Mol. Pharmacol. 79, 520–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Guptaroy B., Zhang M., Bowton E., Binda F., Shi L., Weinstein H., Galli A., Javitch J. A., Neubig R. R., Gnegy M. E. (2009) A juxtamembrane mutation in the N terminus of the dopamine transporter induces preference for an inward-facing conformation. Mol. Pharmacol. 75, 514–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fenollar-Ferrer C., Stockner T., Schwarz T. C., Pal A., Gotovina J., Hofmaier T., Jayaraman K., Adhikary S., Kudlacek O., Mehdipour A. R., Tavoulari S., Rudnick G., Singh S. K., Konrat R., Sitte H. H., Forrest L. R. (2014) Structure and regulatory interactions of the cytoplasmic terminal domains of serotonin transporter. Biochemistry 53, 5444–5460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Esterházy D., Akpinar P., Stoffel M. (2012) Tmem27 dimerization, deglycosylation, plasma membrane depletion, and the extracellular Phe-Phe motif are negative regulators of cleavage by Bace2. Biol. Chem. 393, 473–484 [DOI] [PubMed] [Google Scholar]