

Background: Myocyte enhancer factor 2 (MEF2) proteins are key regulators of cardiac muscle differentiation and hypertrophy, but additional roles in this cell type have not been defined.
Results: MEF2D regulates the cell cycle and survival of post-mitotic cardiomyocytes.
Conclusion: MEF2D is required for proper neonatal cardiomyocyte homeostasis.
Significance: These findings provide opportunities to modulate MEF2D activity in cardiomyocyte proliferation and survival.
Keywords: apoptosis, cardiomyocyte, cell cycle, transcription factor, transcriptomics, survival
Abstract
The cardiomyocyte cell cycle is a poorly understood process. Mammalian cardiomyocytes permanently withdraw from the cell cycle shortly after birth but can re-enter the cell cycle and proliferate when subjected to injury within a brief temporal window in the neonatal period. Thus, investigating the mechanisms of cell cycle regulation in neonatal cardiomyocytes may provide critical insight into the molecular events that prevent adult myocytes from proliferating in response to injury or stress. MEF2D is a key transcriptional mediator of pathological remodeling in the adult heart downstream of various stress-promoting insults. However, the specific gene programs regulated by MEF2D in cardiomyocytes are unknown. By performing genome-wide transcriptome analysis using MEF2D-depleted neonatal cardiomyocytes, we found a significant impairment in the cell cycle, characterized by the up-regulation of numerous positive cell cycle regulators. Expression of Pten, the primary negative regulator of PI3K/Akt, was significantly reduced in MEF2D-deficient cardiomyocytes and found to be a direct target gene of MEF2D. Consistent with these findings mutant cardiomyocytes showed activation of the PI3K/Akt survival pathway. Paradoxically, prolonged deficiency of MEF2D in neonatal cardiomyocytes did not trigger proliferation but instead resulted in programmed cell death, which is likely mediated by the E2F transcription factor. These results demonstrate a critical role for MEF2D in cell cycle regulation of post-mitotic, neonatal cardiomyocytes in vitro.
Introduction
Mammalian cardiomyocytes permanently withdraw from the cell cycle shortly after birth and are maintained in a quiescent state characterized by robust contractile and metabolic activity. Despite the stringent post-mitotic cell cycle control systems in cardiomyocytes, these differentiated cells have the capacity to re-enter the cell cycle and proliferate in response to injury or stress within the first postnatal week (1–5). Beyond this temporal window, cardiomyocytes lose their capacity to proliferate and instead respond to injury or stress through non-proliferative, pathological remodeling pathways that ultimately contribute to cardiac dysfunction (6, 7).
Our understanding of the transcriptional control of the cardiomyocyte cell cycle remains incomplete. Apart from established transcriptional regulators of the cell cycle, such as E2F and c-Myc, few others transcription factors have been shown to regulate this process in cardiomyocytes. Recently, the TALE class homeodomain factor Meis1 was shown to be important in cardiomyocyte cell cycle exit. Deletion of Meis1 promoted cell cycle re-entry in neonatal and adult cardiomyocytes (8). Although known to function in cardiac survival pathways, members of the FOXO transcription factor family also regulate the cell cycle in cardiomyocytes (9, 10).
Previous studies from our laboratory demonstrated that MEF2A (myocyte enhancer factor 2A), but not the related family members MEF2C and MEF2D, regulates the integrity of costameres, specialized focal adhesions in muscle (11, 12). Because costamere structure was not significantly affected by MEF2C or MEF2D deficiency, we were interested in determining the processes dependent on these MEF2 protein isoforms in cardiomyocytes. We have focused on MEF2D because this isoform and MEF2A are the major isoforms expressed in the postnatal heart. Moreover, it has been previously shown that MEF2D is required for pathological cardiac remodeling downstream of pressure overload and chronic adrenergic signaling. Hearts from MEF2D knock-out mice subjected to these stressors displayed significantly less hypertrophy and fibrosis (13). However, the gene programs regulated by MEF2D in cardiac muscle stress or homeostasis are largely unknown.
To gain a better understanding of the cellular processes regulated by MEF2D in cardiac muscle, we depleted MEF2D in neonatal cardiomyocytes, followed by genome-wide expression analysis. Deficiency of MEF2D in neonatal cardiomyocytes stimulated re-entry into the cell cycle, which was characterized by the up-regulation of numerous positive cell cycle regulators and activation of the PI3K/Akt pathway. Despite activation of these cell cycle-promoting pathways, prolonged depletion of MEF2D did not induce proliferation but instead resulted in widespread programmed cell death. Taken together, these results demonstrate an unanticipated critical function for MEF2D in regulating the cell cycle and survival of post-mitotic, neonatal cardiomyocytes.
Experimental Procedures
Cell Culture and Reporter Assays
Neonatal rat ventricular myocytes (NRVMs)2 were isolated from 2-day-old SASCO Sprague-Dawley neonatal rats (Charles River Laboratories). Briefly, ventricles were separated from atria and transferred to 1× Hanks' balanced salt solution, 0.025% trypsin and incubated overnight at 4 °C. The next day, 10 mg/ml collagenase II (Worthington) was added to isolate individual cardiomyocytes and preplated to remove fibroblasts. Cells were plated in growth medium at a density of 4 × 106 cells/10-cm dish on gelatinized dishes. After 24 h in culture, cells were washed with 1× PBS and switched to 0.5× Nutridoma-SP (Roche Applied Sciences) in DMEM.
Plasmids
MEF2D-FLAG (human) was a kind gift of T. Gulick (Sanford Burnham Medical Research Institute, Orlando, FL).
Adenoviruses for Knockdown and Overexpression
Adenoviruses carrying short hairpin RNAs (shRNAs) were generated as described previously (14). The Mef2d and lacZ shRNA adenoviruses were used at a multiplicity of infection of 50 for all assays. MEF2D overexpression (Jeff Molkentin, Children's Hospital, Cincinnati, OH) and β-gal (Ken Walsh, Boston University Medical School) adenoviruses were used at a multiplicity of infection of 25.
siRNA Knockdown
siPTEN was purchased from Dharmacon (ON-TARGETplus PTEN siRNA). siPTEN was transfected in NRVMs using a standard reverse transfection protocol ata final concentration of 100 nm. Briefly, Lipofectamine RNAiMAX transfection reagent (Life Technologies, Inc.) was diluted in Opti-MEM (Life Technologies) and added to the siRNA. Cells were seeded 30 min later.
Microarray
Seventy-two hours post-transduction, total RNA from shlacZ (n = 6) and shMef2d (n = 6) NRVMs was prepared by TRIzol® isolation (Invitrogen). Samples were pooled in sets, for a total of three biological replicates per condition. Samples were hybridized to the Rat GeneChip® Gene 1.0 ST array (Affymetrix) at the Boston University Microarray Facility. Microarray data are available in the GEO (NCBI) database with series ID number GSE72157.
Quantitative RT-PCR
RNA from NRVM MEF2D knockdown experiments (n > 3) was used to synthesize cDNA using reverse transcriptase (Moloney murine leukemia virus) with random hexamers (Promega). Quantitative RT-PCR was performed in triplicate wells using Power SYBR® Green Master Mix (Applied Biosystems) with the 7900HT Sequence Detection System (Applied Biosystems). The primers used are listed in Table 1.
TABLE 1.
Primers used in this study
Top, list of rat quantitative RT-PCR primers used. Bottom, List of oligonucleotides used for EMSA. The wild-type and mutant MEF2 binding sites are underlined.

Western Blot Analysis
Western blots were performed as described previously (14). Antibodies included anti-GAPDH (1:1000; Santa Cruz Biotechnology, Inc.), anti-MEF2D (1:1000; BD Biosciences), anti-proliferating cell nuclear antigen (PCNA) (1:2000; Cell Signaling), PTEN (1:1000; Cell Signaling), Akt (1:1000; Cell Signaling), pAkt Thr-308 (1:1000: Cell Signaling), pAkt Ser-473 (1:1000: Cell Signaling), cyclin D1 (1:1000; Cell Signaling), cyclin D3 (1:1000; Cell Signaling), and CDK2 (1:1000; Cell Signaling). Blots were incubated with horseradish peroxidase-conjugated secondary antibodies (1:10,000; Sigma) and reacted with Western Lightning Chemiluminescent Reagent (PerkinElmer Life Sciences).
PI3K/Akt Inhibition
The PI3Kα/δ inhibitor GDC-0941 (Selleck Chemicals) was added to NRVMs at a final concentration of 10 μm, on the same day as transduction with shRNA adenovirus.
Gel Shift and Luciferase Assays
In vitro translated mouse MEF2D (rabbit reticulocyte lysate; Promega) or nuclear extracts from NRVMs were used for gel shift assays. Supershift assays were performed with anti-MEF2D antibodies (BD Biosciences). Competitions were performed with a 100-fold molar excess of unlabeled probe. Gel shift reactions were fractionated on 5% non-denaturing polyacrylamide gels, dried, and exposed to a phosphorimaging screen (Amersham Biosciences). The oligonucleotides used are listed in Table 1. HEK293T cells were harvested for luciferase activity assay 48 h after transfection and were lysed in 1× passive lysis buffer (Promega). To measure Firefly luciferase activity, 10 μl of cell lysate was mixed with 50 μl of luciferase assay reagent (Promega), and readings were taken on a luminometer.
Immunofluorescence and TUNEL Assay
Cells were cultured on sterilized coverslips coated with Matrigel and transduced with the appropriate shRNA adenoviruses. For immunofluorescence, antibodies included α-actinin (1:500; Sigma), FKHRL-1 (1:200; Millipore), Alexa Fluor 488 donkey anti-mouse H+L (1:200; Invitrogen), and Alexa Fluor 555 donkey anti-rabbit H+L (1:500; Invitrogen).
The TUNEL assay was performed using the DeadEndTM fluorometric TUNEL system (Promega) according to manufacturer's instructions. Fluorescent images were taken using an Olympus DSU spinning disc confocal microscope.
Caspase-3 Activity and Cell TiterBlue Assays
NRVM protein lysates were mixed with the fluorogenic caspase-3 substrate Ac-DEVD-7-amido-4-methylcoumarin (BD Biosciences) to a final 50 μm concentration. Samples were incubated for 1 h at 37 °C. Fluorescence was measured at 440/460 nm using a PerkinElmer Life Sciences Victor3 plate reader. Caspase-3 activity was normalized to total protein level.
NRVMs were cultured in 24-well plates and transduced with either shMef2d or MEF2D overexpression adenovirus, and 10 μl CellTiter-Blue® reagent (Promega) was added to each well 2, 4, or 6 days after transduction. Plates were incubated for 24 h at 37 °C in a tissue culture incubator, and fluorescence was measured at 560/590 nm using a PerkinElmer Life Sciences Victor3 plate reader.
Computational Pathway Analysis
Gene sets sensitive to MEF2D depletion were analyzed using three independent pathway analysis algorithms. Gene ontology term and KEGG pathway analyses were performed through the DAVID bioinformatics database (15, 16). Ingenuity Pathway Analysis® (Ingenuity Systems) was used to determine the canonical cellular pathways associated with MEF2D depletion.
Statistical Analysis
All numerical quantification is representative of the mean ± S.E. of at least three independently performed experiments. Statistically significant differences between two populations of data were determined using Student's t test. p values of ≤0.05 were considered to be statistically significant.
Results
MEF2D Regulates a Cell Cycle Gene Program in Neonatal Cardiomyocytes
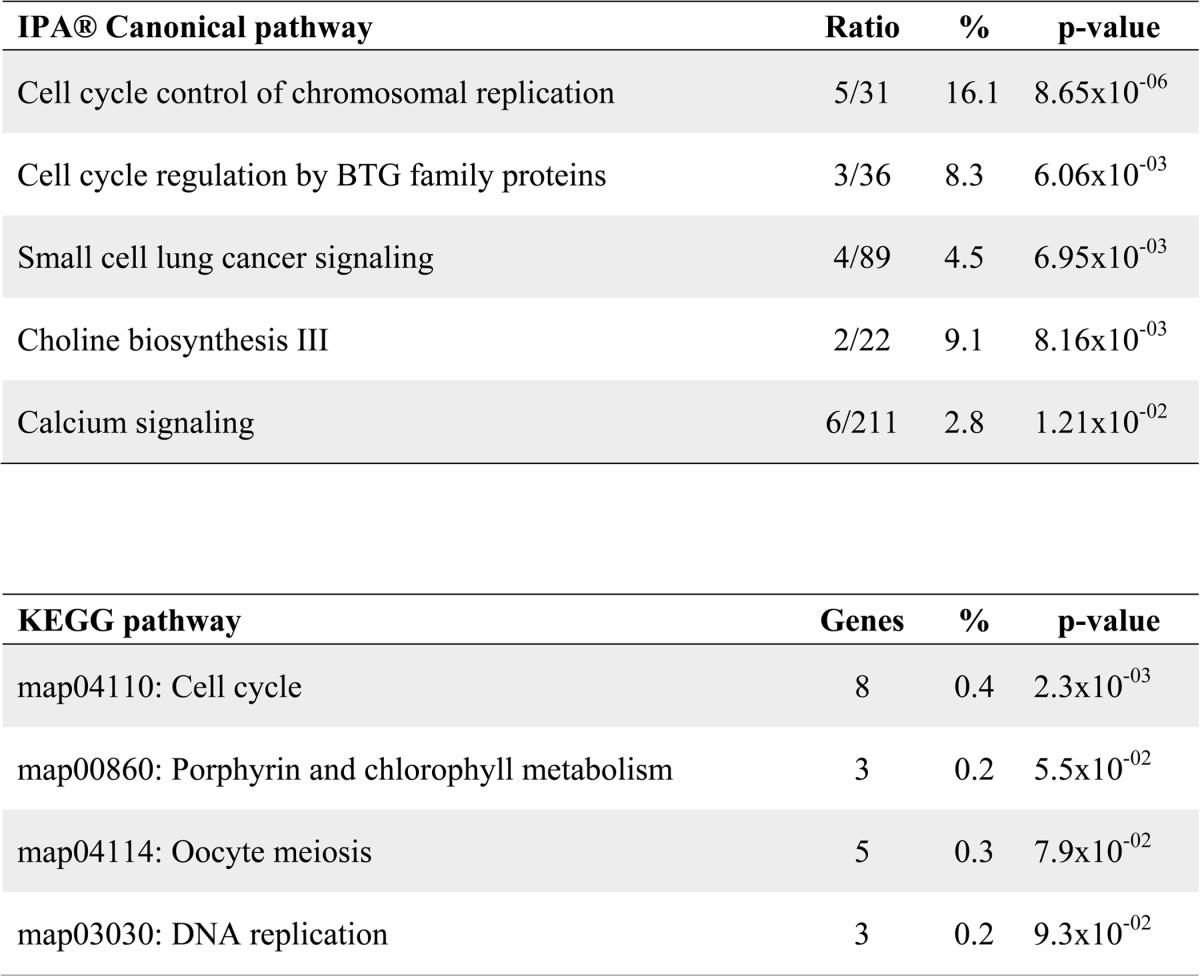
In order to gain a better understanding of the gene programs regulated by MEF2D in cardiac muscle homeostasis, we used RNA interference to deplete this factor in NRVMs. We previously generated a MEF2D-specific shRNA adenovirus that targets the C-terminal region and all alternatively spliced transcripts of Mef2d (14). Transduction of NRVMs with this adenovirus effectively knocked down the expression of endogenous MEF2D (Fig. 1). Subsequently, RNA from MEF2D-depleted NRVMs at day 3 post-transduction was subjected to microarray analysis using the Affymetrix Gene Chip Rat Gene 1.0ST array. Knockdown of MEF2D in neonatal cardiomyocytes resulted in the dysregulation of 279 genes by ±1.5-fold or more. To determine whether a specific cellular process was sensitive to MEF2D depletion, we analyzed these dysregulated genes using Ingenuity Pathway Analysis®. This analysis predicted a significant perturbation of cell cycle and cancer-related processes (Table 2). A complementary analysis using the KEGG algorithm corroborated these results and predicted the cell cycle as the top dysregulated pathway (Table 2).
FIGURE 1.
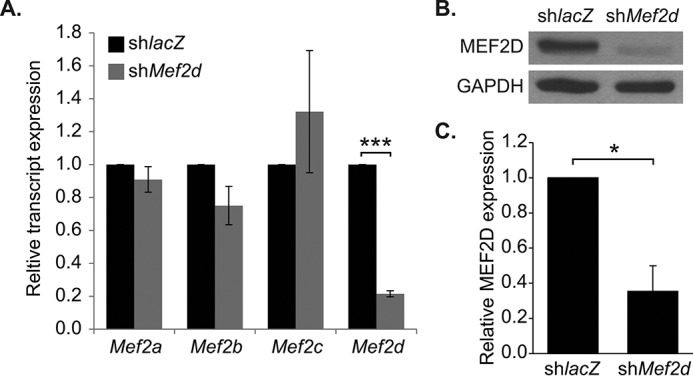
Mef2d shRNA adenovirus efficiently knocks down MEF2D. A, quantitative reverse transcriptase-PCR analysis of endogenous Mef2 knockdown in NRVMs 72 h post-transduction (day 3). The Mef2d shRNA adenovirus specifically knocks down Mef2d, without depleting Mef2a, Mef2b, or Mef2c. B, Western blot analysis of endogenous MEF2D knockdown in NRVMs and densitometry (C). shMef2d adenovirus efficiently knocks down MEF2D at both the transcript and protein level. The data are means ± S.E. (error bars). *, p < 0.05; ***, p < 0.001.
TABLE 2.
Analysis of canonical pathways associated with MEF2D
Dysregulated genes were analyzed using Ingenuity Pathway Analysis (IPA) (Top) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (bottom) prediction software. The top five most significantly dysregulated canonical pathways are provided. These pathways were analyzed for statistically significant association with MEF2D depletion, and a subset was found to be significant (p < 0.05). Ratio values indicate the number of genes dysregulated in the MEF2D knockdown in relation to the accepted number of genes associated with each canonical pathway. Percent values represent the number of dysregulated genes over the total number of genes associated with the indicated pathway. The number of genes dysregulated in the indicated pathway is given. (ratio).
MEF2D Depletion Induces Activation of G1 and S Phase Cell Cycle Genes
Next, we identified specific cell cycle genes dysregulated in MEF2D-depleted NRVMs. Of the total 279 genes dysregulated, 12 have well established roles in the cell cycle. Of the cycle-associated genes identified, seven overlapped with Ingenuity Pathway Analysis® and KEGG pathway analysis predictions and were up-regulated in MEF2D-depleted cardiomyocytes, including the positive cell cycle regulators CDT1 (Cdt1), cyclin E1 (Ccne1), cyclin E2 (Ccne2), Cdc6, Mcm3, Mcm5, and Mcm6 (Fig. 2A). Furthermore, a small number of negative regulators of the cell cycle were downregulated in the absence of MEF2D, but most were largely unaffected (data not shown).
FIGURE 2.

Microarray validation in NRVMs. A, summary of a subset of cell cycle genes dysregulated by 1.5-fold or more in MEF2D depleted NRVMs. The majority of the positive cell cycle genes are up-regulated. Cdt1, chromatin licensing and DNA replication factor 1; Ccne1, cyclin E1; Ccne2, cyclin E2; Mcm3, minichromosome maintenance complex component 3; Mcm5, minichromosome maintenance complex component 5; Mcm5, minichromosome maintenance complex component 6. B, quantitative RT-PCR analysis of cell cycle regulatory genes dysregulated in MEF2D-depleted NRVMs at day 3. Pcna, proliferating cell nuclear antigen. C, Western blot analysis of cell cycle-associated proteins cyclin D1, cyclin D3, and cyclin-dependent kinase 2 (CDK2). D, Western blot densitometry. The data are means ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Among those genes showing an increase in transcript levels on the microarray were genes that function at the G1 or S phase of the cell cycle, such as Mcm3, Mcm5, Mcm6, Ccne1, and Ccne2. PCNA interacts with MCM3, -5, and -6 at the replication fork and is necessary for DNA replication; therefore, although it did not meet the 1.5-fold dysregulation cut-off, we included Pcna in all subsequent analyses. We next focused our analysis on these early cell cycle markers and used quantitative RT-PCR to validate their expression levels. As shown in Fig. 2B, all six positive regulators were significantly up-regulated in MEF2D-depleted NRVMs at day 3. Given the up-regulation of these genes, we extended these results by analyzing the expression of additional positive cell cycle regulators that function at the G1 phase of the cell cycle. The expression of cyclin D1, cyclin D3, and CDK2 were also significantly up-regulated in MEF2D-depleted myocytes (Fig. 2, C and D). The up-regulation of this collection of genes, which function to promote the G1/S phase of the cell cycle, suggests that MEF2D-depleted cardiomyocytes have activated a gene program associated with cell cycle re-entry.
Absence of Proliferation and Hypertrophy but Partial DNA Synthesis in MEF2D-depleted Myocytes
Having confirmed the up-regulation of positive cell cycle regulators that function at the G1/S transition, we reasoned that the activation of these factors would be associated with increased DNA synthesis and proliferation. Initially, we measured DNA replication using EdU incorporation, which stably marks newly synthesized DNA. Surprisingly, there was no significant increase in EdU+ in MEF2D-depleted cardiomyocytes at day 3 post-transduction (Fig. 3A). Subsequently, we examined proliferation using the proliferation marker Ki67. Again, this assay failed to reveal any significant proliferation in MEF2D-depleted myocytes (Fig. 3B). Because these results were inconsistent with our G1/S phase expression analysis, we measured DNA content of individual cardiomyocytes using propidium iodide incorporation followed by flow cytometry. As shown in Fig. 3C, MEF2D-depleted NRVMs had significantly fewer cells with diploid DNA content (2n) and a modest but significant fraction of cells containing between 2n and 4n DNA content (arrow). There was no significant difference in NRVMs containing 4n DNA content. These results suggest that a subset of MEF2D-depleted cells have entered S phase and initiated DNA synthesis but have failed to fully replicate the cardiomyocyte genome.
FIGURE 3.

Analysis of DNA synthesis, proliferation, and cell size in MEF2D-depleted cardiomyocytes. MEF2D-depleted NRVMs were cultured for 3 days post-transduction, followed by morphological analysis. A, MEF2D-depleted NRVMs showed no significant DNA synthesis by EdU incorporation 3 days post-transduction. B, MEF2D depleted NRVMs showed no significant proliferation as examined by Ki67 immunoreactivity. C, flow cytometric measurement of DNA content using propidium iodide (PI) staining shows a significant reduction in the percentage of cells with 2n DNA content and a significant increase in cells with DNA content between 2n and 4n. No significant change was observed in the percentage of cells with 4n DNA content. DNA content was measured 3 and 5 d post-transduction. D, MEF2D-depleted NRVMs showed no significant increase in cell size or markers of hypertrophy (E) 3 days post-transduction. Nppa, natriuretic peptide A (ANF); Nppb, natriuretic peptide B (BNP). The data are means ± S.E. (error bars). *, p < 0.05; **, p < 0.01.
Because activation of the cell cycle has been associated with hypertrophic growth (1–4), we examined MEF2D-depleted myocytes for increased cell size. Detailed ImageJ analysis failed to reveal a significant increase in area of MEF2D-depleted myocytes (Fig. 3D). Moreover, there was no significant increase in the hypertrophic marker genes ANF and BNP (Fig. 3E) or in the fetal sarcomere gene isoforms Acta1 (skeletal α-actin) and Myh7 (β-myosin heavy chain) (data not shown). Taken together, these results indicate that up-regulation of G1 and S phase positive cell cycle regulators is not sufficient to promote proliferation and is not associated with a hypertrophic response in MEF2D-depleted cardiomyocytes. However, a fraction of these MEF2D-depleted NRVMs is capable of undergoing partial DNA synthesis, consistent with activation of G1/S phase marker genes.
PTEN Is a Direct Target of MEF2D
To understand the mechanism by which cell cycle genes are up-regulated, we initially considered the possibility that MEF2D directly represses these genes to regulate cell cycle exit in cardiomyocytes. MEF2 proteins are known to inhibit gene expression through their interaction with histone deacetylases (17). Examination of the upstream regions of these cell cycle genes, however, failed to find significant enrichment of MEF2 DNA binding sites (data not shown). These results suggested an indirect transcriptional regulation of cell cycle gene expression dependent on MEF2D. We then re-examined our microarray to identify candidate genes whose down-regulation may help explain activation of the cell cycle. Interestingly, the Pten (phosphatase and tensin homolog) gene was found to be downregulated 1.6-fold on the microarray. PTEN is an important negative regulator of the PI3K/Akt pathway, a pathway that promotes cell cycle progression and survival (18, 19). Expression of PTEN was validated by quantitative RT-PCR and Western blot analysis and found to be significantly reduced in MEF2D-deficient myocytes (Fig. 4, A and B).
FIGURE 4.

PTEN expression is down-regulated in MEF2D deficient NRVMs. A, Pten transcripts were down-regulated 2.5-fold in cardiomyocytes depleted of MEF2D. B, Western blot analysis and densitometry confirm decreased levels of PTEN expression, with a 2.5-fold reduction of PTEN protein. C, the MEF2 binding site and flanking sequence located 4596 bases upstream of the putative Pten transcriptional start site is highly conserved between species. D, gel shift assay reveals binding of both in vitro translated and endogenous MEF2D to the wild-type (4596 Mef2) but not mutant (−4596 MUT) Mef2 site identified within the rat Pten regulatory region. Incubation with MEF2D antibodies shift the MEF2 complex bound to the radiolabeled −4596 Mef2 sequence. Binding of MEF2D to the −4596 Mef2 site is competed by the unlabeled wild-type but not the mutant sequence. E, luciferase analysis of the wild-type PTEN promoter fragment containing an evolutionarily conserved MEF2 binding site (−4596 Mef2). HEK293T cells were transfected with mouse MEF2D (n = 5). The data are means ± S.E. (error bars). **, p < 0.01; ***, p < 0.001.
Examination of the rat Pten promoter revealed a candidate MEF2 site, CTATTTATAC, situated ∼4.6 kb upstream of the transcription start site. Alignment of this MEF2 site and flanking sequences showed high conservation among human, mouse, rat, cow, and pig genomic sequences harboring the candidate MEF2 site upstream of the respective Pten genes (Fig. 4C). To determine whether MEF2D binds to the rat MEF2 site, an electrophoretic mobility shift (EMSA) was performed using in vitro translated MEF2D and nuclear extracts from NRVMs. The EMSA demonstrated that in vitro translated MEF2D was able to bind the conserved MEF2 site (−4596 Mef2), and binding was severely diminished when the core A/T sequence within the MEF2 binding site was mutated (−4596 MUT) (Fig. 4D). Furthermore, an antibody specific for MEF2D was able to supershift the protein-DNA complex generated using NRVM extracts, demonstrating that endogenous MEF2D binds to the −4596 Mef2 site (Fig. 4D). Subsequently, a 500-base pair region containing this conserved MEF2 site (wild type) was cloned into pGL3 promoter (−4596 Mef2), and its activity was examined in reporter assays. As shown in Fig. 4E, MEF2D overexpression in HEK293T cells was sufficient to significantly activate this PTEN enhancer region.
MEF2D Modulates Cardiomyocyte Cell Cycle Gene Expression through PI3K/Akt Signaling
Previous studies have shown that activation of Akt correlates with cell cycle re-entry (20, 21). Because MEF2D-depleted cardiomyocytes displayed a decrease in PTEN, we would expect increased activated phospho-Akt. As predicted, Western blot analysis of MEF2D-depleted cardiomyocytes at day 3 exhibited a significant increase in phospho-Akt Thr-308 and Ser-473, suggesting activation of the Akt/PKB pathway (Fig. 5A).
FIGURE 5.

MEF2D depletion results in increased PI3K/Akt activation in NRVMs. A, Western blot and densitometry analysis reveals an increase in Akt phosphorylation at the activating residues threonine 308 and serine 473 in MEF2D-deficient cardiomyocytes. B, PI3K/Akt inhibition by treatment with GDC-0941 blunts up-regulation of positive cell cycle markers at the transcript level. C, FOXO3a protein is enriched in the nuclei of shlacZ-transduced NRVMs, whereas MEF2D-depleted NRVMs display significantly increased levels of cytoplasmic FOXO3a, suggesting FOXO3a inactivation in the absence of MEF2D. White arrows point to FOXO3a enrichment. Mcm3, minichromosome maintenance complex component 3; Mcm5, minichromosome maintenance complex component 5; Mcm5, minichromosome maintenance complex component 6; Pcna, proliferating cell nuclear antigen; Ccne1, cyclin E1; Ccne2, cyclin E2. The data are means ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Given the enhanced Akt activity, we wanted to determine whether this increase in activity was linked to the up-regulation of positive cell cycle regulators. Toward this end, we treated MEF2D-deficient NRVMs with GDC-0941 (Selleck), a potent and specific inhibitor of PI3K that prevents global Akt activation. PI3K inhibition severely attenuated the up-regulation of Mcm5, Pcna, cyclin E1, and cyclin E2 observed in MEF2D-depleted cardiomyocytes (Fig. 5B). Although the addition of GDC-0941 did not prevent up-regulation of Mcm3 or Mcm6, the increase in transcript levels was blunted nearly 2-fold in comparison with MEF2D knockdown alone.
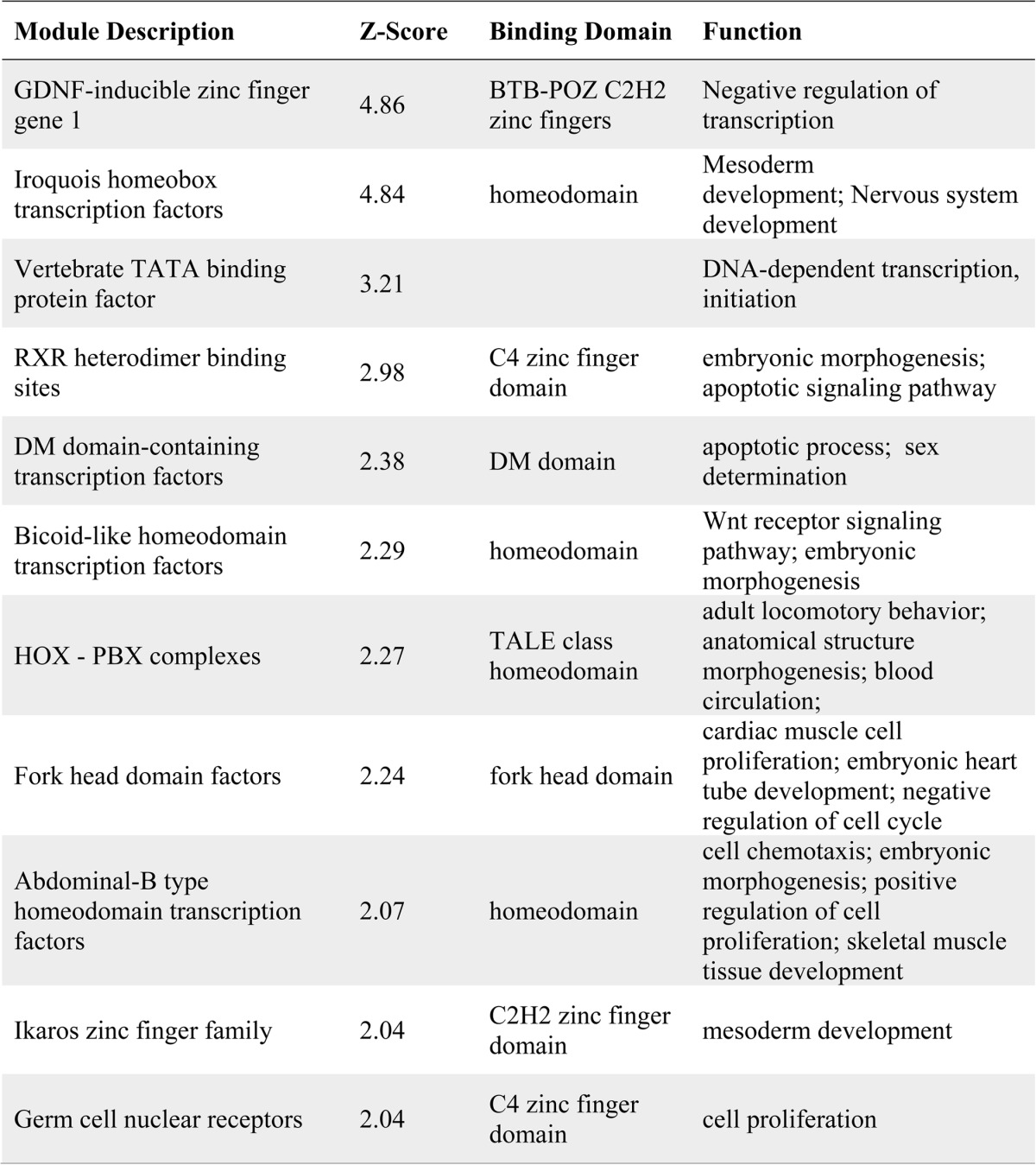
We next wanted to dissect the mechanism by which these cell cycle genes were up-regulated. For these studies, we performed transcription factor binding site enrichment analysis (Genomatix) on the up-regulated cell cycle gene set. Eleven transcription factor binding site motifs were significantly enriched, with a calculated Z score greater than 2 (Table 3). Interestingly, we noted that the binding motif for the Forkhead box (FOX) transcription factors was among the significantly enriched motifs. In this transcription factor superfamily, the FOXO subgroup of Forkhead factors is modulated by Akt signaling (22). Active Akt induces the cytoplasmic retention of FOXO proteins, thereby preventing regulation of their target genes. FOXOs have established roles in the heart and have been shown to regulate cardiomyocyte size (23, 24); therefore, we examined FOXO activity by analyzing the subcellular localization of the FOXO3 isoform in MEF2D-depleted NRVMs. Immunofluorescence analysis of FOXO3a revealed that MEF2D-deficient cardiomyocytes had significantly increased levels of cytoplasmic FOXO3a compared with the shlacZ control (Fig. 5C). These findings suggest that MEF2D depletion results in FOXO3a cytoplasmic retention and, through an unknown mechanism, induces the expression of positive cell cycle regulators.
TABLE 3.
Overrepresented transcription factor binding sites associated with positive cell cycle genes
Binding site enrichment analysis was performed on cell cycle genes sensitive to the loss of MEF2D using the Genomatix software suite. Eleven motifs were enriched with a calculated Z score greater than 2. The known binding factors and their relevant binding domains are included. Finally, a summary of the function of each of these binding factors is provided.
MEF2D Is Cardioprotective and Its Depletion Results in Reduced Viability
Because we failed to observe significantly increased proliferation in MEF2D-depleted NRVMs at day 3 post-transduction, we reasoned that these mutant myocytes were not given sufficient time to display a proliferative phenotype. Therefore, NRVMs were depleted of MEF2D for 5 or 7 days. To our surprise, beginning at day 5, we noted a number of detached, rounded cells in the supernatant, which was highly suggestive of cell death rather than proliferation. To confirm these observations, we assayed MEF2D depleted NRVMs for cell viability. Consistent with our observations, after 5 days of MEF2D depletion, there was a significant reduction in cardiomyocyte viability (Fig. 6A). By day 7, MEF2D-depleted NRVMs showed a widespread decline in viability (Fig. 6B), suggesting that aberrant cell cycle gene expression at early time points eventually perturbs neonatal cardiomyocyte survival.
FIGURE 6.

Reduced cardiomyocyte viability and increased programmed cell death in MEF2D-depleted cardiomyocytes. MEF2D-depleted NRVMs were cultured for 5 and 7 days, and viability was measured with CellTiter Blue cell viability assay. Five (A) and 7 days (B) of MEF2D depletion resulted in decreased cardiomyocyte viability and significantly increased caspase-3 activity (C and D). E, MEF2D-deficient cardiomyocytes have 3.5-fold more TUNEL-positive cells than the control, indicating that apoptosis is occurring in these cells. The data are means ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
In order to determine whether apoptosis was being induced in MEF2D-depleted NRVMs at days 5 and 7, we performed a caspase-3 activity assay on these cells. Consistent with the cell viability assays, we noted a significant increase in caspase activity at days 5 and 7 post-transduction (Fig. 6, C and D), suggesting that reduced cardiomyocyte survival resulted from apoptotic cell death. In a complementary set of experiments, we performed a TUNEL assay to measure apoptotic cell death. As shown in Fig. 6E, the TUNEL assay revealed a significant increase in apoptotic cell death in MEF2D-depleted NRVMs. It is important to note that there was no significant difference in either cell viability or caspase-3 activation at day 3 (data not shown).
To demonstrate that PTEN down-regulation contributes to reduced viability in MEF2D-depleted cells, we silenced PTEN alone in NRVMs. PTEN-depleted NRVMs showed significantly reduced viability at day 5 and day 7 (Fig. 7, A and B). Moreover, TUNEL assay showed a significant increase in cell death (Fig. 7C). These results are consistent with the reduced PTEN expression in MEF2D-depleted NRVMs, reinforcing that notion that it is a physiologically relevant target in this pathway.
FIGURE 7.

PTEN knockdown in NRVMs results in increased cell death. NRVMs were transfected with siPTEN and cultured for 5 and 7 days, and viability was measured with the CellTiter Blue cell viability assay. Five (A) and 7 days (B) of knockdown of PTEN resulted in decreased cardiomyocyte viability. C, PTEN knockdown NRVMs have 1.6-fold more TUNEL-positive cells than the control on day 5, indicating apoptosis is occurring in these cells. The data are means ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Given the decreased viability upon knockdown of MEF2D, we asked whether overexpressing MEF2D in wild type NRVMs could prolong cardiomyocyte survival. It is firmly established that survival of primary neonatal cardiomyocytes begins to decline as they approach 1 week in culture. MEF2D was overexpressed in NRVMs and analyzed for cell viability at days 3, 5, and 7 post-transduction. MEF2D overexpression did not significantly affect viability at day 3 (data not shown) but resulted in a significant increase in viability in NRVMs at days 5 and 7 (Fig. 8, A and B). Although there was no difference in caspase-3 activity at day 5, by 7 days after transduction, there was a marked decrease in caspase-3 activity in MEF2D-overexpressing cells (Fig. 8, C and D). In addition, the increased viability in MEF2D overexpression resulted from enhanced survival of NRVMs and not from increased cell numbers. Finally, consistent with increased viability, we found significantly decreased phospho-Akt and increased PTEN expression in NRVMs overexpressing MEF2D (Fig. 8, E and F). Taken together, these results clearly demonstrate that MEF2D is cardioprotective.
FIGURE 8.

MEF2D overexpression in NRVMs prolongs survival. MEF2D overexpression for 5 d (A) and 7 d (B) resulted in increased cell viability of cultured cardiomyocytes. C, there was no change in caspase-3 activity 5 d after transduction in MEF2D-overexpressing cells. D, 7 d post-transduction, MEF2D-overexpressing NRVMs displayed a significant decrease in caspase-3 activity. E, Western blot analysis and quantification reveal a significant decrease in Akt phosphorylation at the activating residues threonine 308 and serine 473 in MEF2D-overexpressing cardiomyocytes. F, Western blot analysis and quantification revealed a significant increase in PTEN in MEF2D-overexpressing cardiomyocytes. OE, overexpression. The data are means ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Inhibition of Cell Cycle Activation Blocks Apoptosis in MEF2D-deficient NRVMs
Because cell cycle activation was associated with cell death and not proliferation, we asked whether these two phenotypes were linked by a common regulatory pathway. Initially, we examined our microarray data and found a number of dysregulated genes associated with cell death (Fig. 9A), reinforcing the notion that an apoptotic gene program has been activated in MEF2D-depleted NRVMs. Next, we considered the E2F transcription factor family as a prime candidate in molecularly integrating the two phenotypes because some members of this family have been shown to trigger apoptosis in response to aberrant cell cycle regulation (25, 26). We examined transcript levels of E2F1, -2, and -3 and two of their pro-apoptotic targets, Apaf1 and Casp8. MEF2D-depleted cardiomyocytes demonstrated a 1.8-, 4-, and 2.6-fold increase in E2f1, -2, and -3 mRNA levels, respectively (Fig. 9B). Furthermore, APAF1 (Apaf1) and caspase-8 (Casp8) transcripts were up-regulated 1.7- and 3-fold in MEF2D-depleted cardiomyocytes, respectively (Fig. 9C).
FIGURE 9.

PI3K/Akt inhibition rescues apoptotic cell death associated with MEF2D depletion. A, summary of apoptosis-associated genes dysregulated by 1.5-fold or more in MEF2D-depleted NRVMs. B, quantitative RT-PCR analysis of the activating E2Fs, E2f1, E2f2, and E2f3, revealed that MEF2D deficiency results in their increased expression. C, two pro-apoptotic transcriptional targets of E2F, Apaf1 and Casp8, are significantly up-regulated in MEF2D depleted NRVMs. Up-regulation of E2f1, E2f2, and E2f3 transcript levels (D) and the E2F pro-apoptotic targets Apaf1 and Casp8 (E) is abolished when PI3K/Akt activation is inhibited by the addition of GDC-0941 in MEF2D-depleted NRVMs. F, TUNEL assay of NRVMs treated with GDC-0941 demonstrates an abrogation of apoptosis observed in MEF2D-deficient NRVMs 5 days post-transduction. G, post-natal cardiomyocytes are terminally differentiated and are unable to re-enter the cell cycle. MEF2D depletion results in an up-regulation of Mcm3, Mcm5, Mcm6, Pcna, Ccne1, and Ccne2 and aberrant cell cycle re-entry. E2F transcription factors likely sense unprogrammed cell cycle activation and mediate cardiomyocyte apoptosis by transcriptionally activating pro-apoptotic genes Apaf1 and Casp8. Apaf1, apoptotic peptidase-activating factor 1; Casp8, caspase-8. The data are means ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We next wanted to determine whether the up-regulation of E2F and its downstream target genes was related to activation of the Akt pathway. As shown in Fig. 9, D and E, treatment of NRVMs with GDC-0941 blocked the up-regulation of E2f1–3 as well as Apf1 and Casp8. Furthermore, MEF2D-depleted cardiomyocytes treated with GDC-0941 attenuated the cell death observed in the absence of the PI3K inhibitor (Fig. 9F). Collectively, these results suggest that cell cycle induction and programmed cell death in MEF2D-depleted cardiomyocytes are both regulated by the PI3K/Akt signaling pathway. We propose a model whereby cell cycle exit in neonatal cardiomyocytes is critically dependent on the ability of MEF2D to depress Akt signaling (Fig. 9G).
Discussion
The transcriptional control of cell cycle arrest in differentiated cardiomyocytes remains largely unexplored. Here, we demonstrate that MEF2D regulates the cell cycle in post-mitotic, neonatal cardiomyocytes. Acute depletion of MEF2D in primary neonatal cardiomyocytes triggered cell cycle re-entry through activation of the Akt signaling pathway and the up-regulation of genes that function at the G1 and S phases of the cell cycle. MEF2D-depleted cardiomyocytes, however, did not display increased proliferation and only partial DNA synthesis. Instead, prolonged depletion of MEF2D in neonatal cardiomyocytes resulted in significant programmed cell death, possibly mediated via the activation of the E2F transcription factor. Taken together, our results identify MEF2D as an essential transcription factor in cell cycle regulation and survival in postnatal cardiac muscle.
Numerous studies have focused on the role of MEF2 in mediating stress and remodeling pathways in the heart downstream of pathological insults (27–29). Although these investigations have firmly established the central and important role of this core cardiac transcription factor in adult cardiac disease pathways, the function of this factor in cardiomyocyte homeostasis, particularly in neonatal myocytes that undergo dramatic changes in growth and metabolism within the first week after birth, is largely unknown. Our studies have revealed an essential, non-stress-related function for MEF2D in neonatal cardiomyocyte cell cycle regulation and survival. This function differs from that described for MEF2D in adult hearts in which global MEF2D deletion did not affect cardiac viability, structure, or function in the absence of stress (13). A physiological role for MEF2D in the heart only emerged when subjected to various stress signals. Perhaps the most notable difference between the two MEF2D-deficient models relates to the requirement of MEF2D in cardiomyocyte survival. Adult MEF2D knock-out mice subjected to pressure overload displayed significantly less fibrosis in the heart, an indicator of the extent of cardiomyocyte cell death, suggesting a pro-apoptotic role for this transcription factor in adult myocytes. By contrast, MEF2D deficiency in unstressed neonatal cardiomyocytes triggered significant cell death, pointing to an anti-apoptotic function in this temporal context. Based on the present and previously reported results, it appears that MEF2D has dual and opposing roles in cardiomyocyte survival. Not only are these opposing functions influenced by the relative maturation of cardiomyocytes (i.e. neonatal versus adult), but also by whether or not these cells are exposed to stress. Finally, because cell cycle gene expression was not examined in MEF2D knock-out hearts in either basal or stressed conditions or in neonates, it is unknown whether this gene program is dependent on MEF2D in vivo.
MEF2 proteins are involved in proliferation, survival, and apoptotic pathways in a variety of specialized cell types, such as neurons, immune cells, and vascular smooth muscle and endothelial cells (30, 31). Until now, it was not known whether MEF2 regulated any of these aforementioned processes in cardiac muscle. Cardiomyocyte survival is dependent on MEF2D, and one of its primary targets is PTEN. It has long been known that PTEN is involved in the progression of heart failure and maladaptive cardiac remodeling (32–34). However, it was recently shown that the miR-17–92 cluster promotes cardiomyocyte proliferation, most likely through the inhibition of PTEN expression (35). These observations suggest that PTEN is involved in neonatal cardiomyocyte proliferation and lend support to our findings regarding the up-regulation of positive cell cycle regulators. Curiously, the phenotype of PTEN knock-out mice resembles that of MEF2D knock-out in response to stress. Given the similarity of the cardiac phenotypes in PTEN and MEF2D knock-out mice, it is tempting to speculate that PTEN also functions downstream of MEF2D to modulate pathologic cardiac remodeling in stressed, adult hearts.
Downstream of PTEN, Akt signaling plays a critical role in cell cycle progression and cell survival in the heart (36–39). Akt activity has been shown to be cardioprotective in myocardial injury, by its ability to promote cell survival and block cell death (37, 38). It is intriguing that activation of Akt by other pathways, namely the Hippo-YAP pathway, in cardiomyocytes is associated with proliferation and survival (40, 41). Our data clearly reveal involvement of the Akt pathway, but activation of this pathway in MEF2D-deficient myocytes is not cardioprotective; nor does it stimulate proliferation. These observations suggest that MEF2D regulates other gene programs that cross-talk with PI3K/Akt to counteract the pro-survival and proliferation effects often mediated by this pathway.
The relationship between cell cycle re-entry and programmed cell death has been well studied in neurons (42), whereas in cardiomyocytes, these mechanisms are poorly understood. In neurodegenerative diseases, in an attempt to replenish damaged neurons, populations of neurons will re-enter the cell cycle, but these neurons fail to complete the cell cycle and instead undergo cell death. Unlike post-mitotic neurons, differentiated cardiomyocytes in the newborn mammalian heart have the ability to re-enter the cell cycle in response to injury, thereby affording the neonatal mammalian heart the capacity to regenerate (43). Despite the up-regulation of positive cell cycle markers and partial DNA synthesis in MEF2D-depleted neonatal cardiomyocytes, these cells fail to progress through the cell cycle and ultimately die. It is worth noting that expression of selected cell cycle inhibitors was not significantly decreased (data not shown), which may, in part, prevent full execution of cell cycle progression. The apoptotic phenotype of MEF2D-depleted neonatal cardiomyocytes is reminiscent of the role of MEF2 in post-mitotic neurons. Degradation of MEF2D is associated with neuronal cell death induced by neurotoxic stimuli (44, 45). Conversely, enhancing MEF2 activity downstream of neuronal insults, such as DNA damage, is required for the survival of neurons (46). It remains to be determined whether certain types of insults, such as oxidative stress or doxorubicin, stressors that cause DNA damage and cell death but not hypertrophy, modulate the activity of MEF2D in cardiomyocytes.
The up-regulation of E2F gene expression appears to be the most plausible mechanistic link to the cell death phenotype in MEF2D-deficient NRVMs. Although commonly known as regulators of proliferation, the E2F family of transcription factors also regulate cell death, primarily the E2F1 and -3 protein isoforms (25, 26). Interestingly, the role of E2F has been investigated in the heart. Overexpression of E2F1 in the adult heart promotes widespread myocardial apoptosis (47). Consistent with these observations, overexpression of E2F1 or E2F3 in neonatal cardiomyocytes promoted cell death (48). Whereas MEF2D knockdown activates the Akt pathway, the simultaneous induction of E2F expression seems to counteract and override any effects of this pro-survival and proliferative signaling cascade.
Genome-wide expression analysis of genes sensitive to the loss of MEF2D demonstrated that this protein isoform regulates gene programs required for neonatal cardiomyocyte cell cycle withdrawal and survival. Thus, exploiting gene programs regulated by MEF2D in cardiomyocytes is likely to provide us with avenues in which to promote the survival and possibly proliferation of cardiomyocytes in cardiac disease.
Author Contributions
N. L. E. and F. J. N. designed the study and wrote the paper. N. L. E. performed and analyzed the experiments in Figs. 1–6, 8, and 9. A. L. C. performed and analyzed the experiments in Figs. 2, 5, 7, and 8. C. A. D. performed and analyzed the experiments in Fig. 3. S. E. N. provided technical assistance and contributed to the experiments described in Fig. 4. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank members of the Naya laboratory for critical reading of the manuscript. We are grateful to Jeff Molkentin (Cincinnati Children's Hospital) for providing the MEF2D overexpression adenovirus and to Tod Gulick (Sanford Burnham Medical Research Institute) for the MEF2D-FLAG expression plasmid. We also thank Geof Cooper (Boston University) for insightful comments regarding the relationship between cell cycle dysregulation and cell death and the potential role of E2F in this process and Ulla Hansen (Boston University) for critical analysis and interpretation of the cell cycle data. Finally, we thank Todd Blute for technical assistance with flow cytometry and Tiffany Dill for quantification of FoxO3 subcellular localization.
This work was supported, in whole or in part, by National Heart, Lung, and Blood Institute, National Institutes of Health Grant HL73304 (to F. J. N.) and a diversity supplement (parent Grant HL73304) (to N. L. E.). This work was also supported by the Beckman Scholars Program (to S. E. N.) and Boston University Bioinformatics CTSI Grant U54-TR001012. The authors declare that they have no conflicts of interest with the contents of this article.
- NRVM
- neonatal rat ventricular myocyte
- PCNA
- proliferating cell nuclear antigen.
References
- 1. Pasumarthi K. B., Field L. J. (2002) Cardiomyocyte cell cycle regulation. Circ. Res. 90, 1044–1054 [DOI] [PubMed] [Google Scholar]
- 2. Ahuja P., Sdek P., MacLellan W. R. (2007) Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 87, 521–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zebrowski D. C., Engel F. B. (2013) The cardiomyocyte cell cycle in hypertrophy, tissue homeostasis, and regeneration. Rev. Physiol. Biochem. Pharmacol. 165, 67–96 [DOI] [PubMed] [Google Scholar]
- 4. Zacchigna S., Giacca M. (2014) Extra- and intracellular factors regulating cardiomyocyte proliferation in postnatal life. Cardiovasc. Res. 102, 312–320 [DOI] [PubMed] [Google Scholar]
- 5. Rumyantsev P. P. (1977) Interrelations of the proliferation and differentiation processes during cardiac myogenesis and regeneration. Int. Rev. Cytol. 51, 186–273 [PubMed] [Google Scholar]
- 6. Hill J. A., Olson E. N. (2008) Cardiac plasticity. N. Engl. J. Med. 358, 1370–1380 [DOI] [PubMed] [Google Scholar]
- 7. Heineke J., Molkentin J. D. (2006) Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 7, 589–600 [DOI] [PubMed] [Google Scholar]
- 8. Mahmoud A. I., Kocabas F., Muralidhar S. A., Kimura W., Koura A. S., Thet S., Porrello E. R., Sadek H. A. (2013) Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 497, 249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Evans-Anderson H. J., Alfieri C. M., Yutzey K. E. (2008) Regulation of cardiomyocyte proliferation and myocardial growth during development by FOXO transcription factors. Circ. Res. 102, 686–694 [DOI] [PubMed] [Google Scholar]
- 10. Sengupta A., Kalinichenko V. V., Yutzey K. E. (2013) FoxO1 and FoxM1 transcription factors have antagonistic functions in neonatal cardiomyocyte cell-cycle withdrawal and IGF1 gene regulation. Circ. Res. 112, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ewen E. P., Snyder C. M., Wilson M., Desjardins D., Naya F. J. (2011) The Mef2A transcription factor coordinately regulates a costamere gene program in cardiac muscle. J. Biol. Chem. 286, 29644–29653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Estrella N. L., Naya F. J. (2014) Transcriptional networks regulating the costamere, sarcomere, and other cytoskeletal structures in striated muscle. Cell Mol. Life Sci. 71, 1641–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim Y., Phan D., van Rooij E., Wang D. Z., McAnally J., Qi X., Richardson J. A., Hill J. A., Bassel-Duby R., Olson E. N. (2008) The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J. Clin. Invest. 118, 124–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Estrella N. L., Desjardins C. A., Nocco S. E., Clark A. L., Maksimenko Y., Naya F. J. (2015) MEF2 transcription factors regulate distinct gene programs in mammalian skeletal muscle differentiation. J. Biol. Chem. 290, 1256–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 16. Huang da W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McKinsey T. A., Zhang C. L., Olson E. N. (2001) Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev. 11, 497–504 [DOI] [PubMed] [Google Scholar]
- 18. Song M. S., Salmena L., Pandolfi P. P. (2012) The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296 [DOI] [PubMed] [Google Scholar]
- 19. Worby C. A., Dixon J. E. (2014) PTEN. Annu. Rev. Biochem. 83, 641–669 [DOI] [PubMed] [Google Scholar]
- 20. Liang J., Slingerland J. M. (2003) Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2, 339–345 [PubMed] [Google Scholar]
- 21. Brazil D. P., Yang Z. Z., Hemmings B. A. (2004) Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem. Sci. 29, 233–242 [DOI] [PubMed] [Google Scholar]
- 22. Huang H., Tindall D. J. (2007) Dynamic FoxO transcription factors. J. Cell Sci. 120, 2479–2487 [DOI] [PubMed] [Google Scholar]
- 23. Skurk C., Izumiya Y., Maatz H., Razeghi P., Shiojima I., Sandri M., Sato K., Zeng L., Schiekofer S., Pimentel D., Lecker S., Taegtmeyer H., Goldberg A. L., Walsh K. (2005) The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J. Biol. Chem. 280, 20814–20823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ni Y. G., Berenji K., Wang N., Oh M., Sachan N., Dey A., Cheng J., Lu G., Morris D. J., Castrillon D. H., Gerard R. D., Rothermel B. A., Hill J. A. (2006) Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 114, 1159–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iaquinta P. J., Lees J. A. (2007) Life and death decisions by the E2F transcription factors. Curr. Opin. Cell Biol. 19, 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Polager S., Ginsberg D. (2008) E2F: at the crossroads of life and death. Trends Cell Biol. 18, 528–535 [DOI] [PubMed] [Google Scholar]
- 27. Passier R., Zeng H., Frey N., Naya F. J., Nicol R. L., McKinsey T. A., Overbeek P., Richardson J. A., Grant S. R., Olson E. N. (2000) CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Invest. 105, 1395–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. el Azzouzi H., van Oort R. J., van der Nagel R., Sluiter W., Bergmann M. W., De Windt L. J. (2010) MEF2 transcriptional activity maintains mitochondrial adaptation in cardiac pressure overload. Eur. J. Heart Fail. 12, 4–12 [DOI] [PubMed] [Google Scholar]
- 29. Konno T., Chen D., Wang L., Wakimoto H., Teekakirikul P., Nayor M., Kawana M., Eminaga S., Gorham J. M., Pandya K., Smithies O., Naya F. J., Olson E. N., Seidman J. G., Seidman C. E. (2010) Heterogeneous myocyte enhancer factor-2 (Mef2) activation in myocytes predicts focal scarring in hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 107, 18097–18102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McKinsey T. A., Zhang C. L., Olson E. N. (2002) MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 27, 40–47 [DOI] [PubMed] [Google Scholar]
- 31. Potthoff M. J., Olson E. N. (2007) MEF2: a central regulator of diverse developmental programs. Development 134, 4131–4140 [DOI] [PubMed] [Google Scholar]
- 32. Crackower M. A., Oudit G. Y., Kozieradzki I., Sarao R., Sun H., Sasaki T., Hirsch E., Suzuki A., Shioi T., Irie-Sasaki J., Sah R., Cheng H. Y., Rybin V. O., Lembo G., Fratta L., Oliveira-dos-Santos A. J., Benovic J. L., Kahn C. R., Izumo S., Steinberg S. F., Wymann M. P., Backx P. H., Penninger J. M. (2002) Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 110, 737–749 [DOI] [PubMed] [Google Scholar]
- 33. Oudit G. Y., Sun H., Kerfant B. G., Crackower M. A., Penninger J. M., Backx P. H. (2004) The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J. Mol. Cell Cardiol. 37, 449–471 [DOI] [PubMed] [Google Scholar]
- 34. Oudit G. Y., Kassiri Z., Zhou J., Liu Q. C., Liu P. P., Backx P. H., Dawood F., Crackower M. A., Scholey J. W., Penninger J. M. (2008) Loss of PTEN attenuates the development of pathological hypertrophy and heart failure in response to biomechanical stress. Cardiovasc. Res. 78, 505–514 [DOI] [PubMed] [Google Scholar]
- 35. Chen J., Huang Z. P., Seok H. Y., Ding J., Kataoka M., Zhang Z., Hu X., Wang G., Lin Z., Wang S., Pu W. T., Liao R., Wang D. Z. (2013) mir-17–92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ. Res. 112, 1557–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsui T., Li L., del Monte F., Fukui Y., Franke T. F., Hajjar R. J., Rosenzweig A. (1999) Adenoviral gene transfer of activated phosphatidylinositol 3′-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation 100, 2373–2379 [DOI] [PubMed] [Google Scholar]
- 37. Fujio Y., Nguyen T., Wencker D., Kitsis R. N., Walsh K. (2000) Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 101, 660–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matsui T., Tao J., del Monte F., Lee K. H., Li L., Picard M., Force T. L., Franke T. F., Hajjar R. J., Rosenzweig A. (2001) Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 104, 330–335 [DOI] [PubMed] [Google Scholar]
- 39. Sussman M. A., Völkers M., Fischer K., Bailey B., Cottage C. T., Din S., Gude N., Avitabile D., Alvarez R., Sundararaman B., Quijada P., Mason M., Konstandin M. H., Malhowski A., Cheng Z., Khan M., McGregor M. (2011) Myocardial AKT: the omnipresent nexus. Physiol. Rev. 91, 1023–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xin M., Kim Y., Sutherland L. B., Qi X., McAnally J., Schwartz R. J., Richardson J. A., Bassel-Duby R., Olson E. N. (2011) Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 4, ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin Z., Zhou P., von Gise A., Gu F., Ma Q., Chen J., Guo H., van Gorp P. R., Wang D. Z., Pu W. T. (2015) Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ. Res. 116, 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herrup K., Yang Y. (2007) Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci. 8, 368–378 [DOI] [PubMed] [Google Scholar]
- 43. Porrello E. R., Mahmoud A. I., Simpson E., Hill J. A., Richardson J. A., Olson E. N., Sadek H. A. (2011) Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gong X., Tang X., Wiedmann M., Wang X., Peng J., Zheng D., Blair L. A., Marshall J., Mao Z. (2003) Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron 38, 33–46 [DOI] [PubMed] [Google Scholar]
- 45. Tang X., Wang X., Gong X., Tong M., Park D., Xia Z., Mao Z. (2005) Cyclin-dependent kinase 5 mediates neurotoxin-induced degradation of the transcription factor myocyte enhancer factor 2. J. Neurosci. 25, 4823–4834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chan S. F., Sances S., Brill L. M., Okamoto S., Zaidi R., McKercher S. R., Akhtar M. W., Nakanishi N., Lipton S. A. (2014) ATM-dependent phosphorylation of MEF2D promotes neuronal survival after DNA damage. J. Neurosci. 34, 4640–4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Agah R., Kirshenbaum L. A., Abdellatif M., Truong L. D., Chakraborty S., Michael L. H., Schneider M. D. (1997) Adenoviral delivery of E2F-1 directs cell cycle reentry and p53-independent apoptosis in postmitotic adult myocardium in vivo. J. Clin. Invest. 100, 2722–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ebelt H., Hufnagel N., Neuhaus P., Neuhaus H., Gajawada P., Simm A., Müller-Werdan U., Werdan K., Braun T. (2005) Divergent siblings: E2F2 and E2F4 but not E2F1 and E2F3 induce DNA synthesis in cardiomyocytes without activation of apoptosis. Circ. Res. 96, 509–517 [DOI] [PubMed] [Google Scholar]