

Background: Cargo of extracellular vesicles (EVs) may reveal responses to targeted anticancer drugs.
Results: Kinase inhibitors of the oncogenic epidermal growth factor receptor (EGFR) activate emission of exosome-like EVs containing EGFR protein and DNA.
Conclusion: Co-detection of EGFR and DNA in tumor-related EVs reflects the responses to kinase inhibitors.
Significance: EV cargo may serve as a biomarker for targeted therapy.
Keywords: anticancer drug, cancer, caspase, DNA, enzyme inhibitor, epidermal growth factor receptor (EGFR), exosome (vesicle), phosphorylation, protein kinase
Abstract
Cancer cells emit extracellular vesicles (EVs) containing unique molecular signatures. Here, we report that the oncogenic EGF receptor (EGFR) and its inhibitors reprogram phosphoproteomes and cargo of tumor cell-derived EVs. Thus, phosphorylated EGFR (P-EGFR) and several other receptor tyrosine kinases can be detected in EVs purified from plasma of tumor-bearing mice and from conditioned media of cultured cancer cells. Treatment of EGFR-driven tumor cells with second generation EGFR kinase inhibitors (EKIs), including CI-1033 and PF-00299804 but not with anti-EGFR antibody (Cetuximab) or etoposide, triggers a burst in emission of exosome-like EVs containing EGFR, P-EGFR, and genomic DNA (exo-gDNA). The EV release can be attenuated by treatment with inhibitors of exosome biogenesis (GW4869) and caspase pathways (ZVAD). The content of P-EGFR isoforms (Tyr-845, Tyr-1068, and Tyr-1173), ERK, and AKT varies between cells and their corresponding EVs and as a function of EKI treatment. Immunocapture experiments reveal the presence of EGFR and exo-gDNA within the same EV population following EKI treatment. These findings suggest that targeted agents may induce cancer cells to change the EV emission profiles reflective of drug-related therapeutic stress. We suggest that EV-based assays may serve as companion diagnostics for targeted anticancer agents.
Introduction
Agents targeting members of the epidermal growth factor receptor family of kinases (EGFR5/ErbB) represent an informative paradigm as to both the opportunities and challenges facing oncogene-directed, personalized anti-cancer therapy (1). In this regard, experimental evidence supports oncogenic effects associated with the activation, up-regulation, amplification, or mutation of EGFR in several human malignancies (2). The ligand-dependent, or -independent, multi- and heteromerization of ErbB proteins (including EGFR) on the cell surface leads to their activation and generation of transforming intracellular signals. These events involve phosphorylation of critical tyrosine residues within the cytoplasmic domain of EGFR, including Tyr-845, Tyr-1068, and Tyr-1173, to form docking sites for effector and adaptor proteins, such as SRC, GRB2, and SHC, respectively (2, 3). The resulting phosphorylation of MAPK, AKT, STAT, and other signaling modules leads to the expression of genes governing cell proliferation, survival, angiogenesis, and other responses (2).
The constitutive activation of the oncogenic EGFR pathway is thought to be central to the pathogenesis of lung, colorectal, epidermoid, brain (glioblastoma), and other human cancers (2, 4). This realization motivates ongoing efforts to develop EGFR-targeting anticancer therapeutics (5), including neutralizing monoclonal antibodies (cetuximab and panitumumab) and EGFR kinase inhibitors (EKIs), of which several are already in clinical use (gefitinib and erlotinib), whereas others remain at various stages of development (1, 2, 5). The latter category includes second generation, irreversible experimental pan-ErbB inhibitors, such as CI-1033 (canertinib) and PF-00299804 (dacomitinib), thought to exert especially potent and lasting effects on the EGFR transforming activity (5–7).
Although these agents efficiently suppress EGFR signaling in laboratory settings, this is not always predictive of their clinical efficacy (1). This disparity is attributed to numerous mechanisms of therapeutic resistance (8), including tumor cell heterogeneity (9), EGFR mutations, as well as activation of salvage signaling pathways (10), especially in vivo (11, 12). In this regard, the ability to longitudinally monitor the EGFR status, activity, signaling, and cellular responses could provide actionable insights during the course of therapy. However, targeted therapy choices are presently imprecise, being extrapolated mainly from a one-time (static) access to treatment-naive tissues collected at surgery or biopsy (1, 13).
These circumstances underscore the emerging interest in approaches known as “liquid biopsy” designed to gain “remote” access to key molecular characteristics of cancer cells in real time (14). In this setting, blood and body biofluids are thought to be usable for the recovery of circulating tumor cells, soluble factors, tumor-derived cell-free DNA, or extracellular vesicles (EVs) reflective of different aspects of cancer-related molecular repertoire (14–16). EVs are of particular interest, as they are relatively abundant in biofluids and cellular supernatants, and they are known to contain combinatorial mixtures of tumor cell signatures, including intact oncoproteins and nucleic acids. Indeed, previous studies documented the presence of EGFR and its oncogenic mutants (e.g. EGFRvIII) in the cargo of tumor-derived EVs (15–18).
EVs are generated through several still poorly understood biogenetic mechanisms. The resulting structural and molecular diversity of vesicle subtypes (19, 20) includes small EVs known as exosomes (usually 30–100 nm in diameter). Exosomes are thought to originate through an alternative form of endosomal trafficking, and their emission is believed to involve neutral sphingomyelinase (NSMase), Rab proteins (Rab27A/B), and tetraspanins (CD63, CD81, CD82, and CD9), which are often used as exosomal markers (21–23). Cancer cells may also shed larger EVs derived from blebs of the plasma membrane (ectosomes and microvesicles), a process that involves different effectors, such as acidic sphingomyelinase (ASMase) and ARF6 (22, 24). Still larger and more complex particles may exit cells as “large oncosomes” or apoptotic bodies, the latter of which contain genomic DNA (gDNA). However, gDNA was also found in exosome-like particles released from viable cells, adding to the complexity of the EV generation (vesiculation) process (17, 25, 26). Nonetheless, EVs are of great interest due to their role in intercellular communication, content, and exchange of molecular cargo (27, 28).
Oncogenic EGFR is thought to influence EV biogenesis and thereby its own emission and intercellular trafficking (15, 29). This could be explained, at least in part, by the activation of the EGFR recycling mechanisms involving endosomes and exosomes (2). However, the status and changes in the state of EV-associated EGFR, P-EGFR, their effectors (MAPK and AKT) and cellular phosphoproteome in various cancer settings remain poorly studied. This is of special interest in relation to EGFR-targeting therapeutics, which could be expected to impact the EV emission, content, and cargo.
Here, we show that oncogenic EGFR (and EGFRvIII) is detectable in EV isolates from plasma of glioblastoma (GBM) patients, plasma of GBM xenograft-bearing mice, and conditioned medium of EGFRvIII-transfected cancer cells. We also describe detection of phosphorylated RTKs (including P-EGFR) in cargo of EVs circulating in the blood of mice harboring several different human tumor xenografts. However, the pattern of EGFR phosphorylation differs between EVs and their parental cells. The exposure of cancer cells to EKIs stimulates the emission of EVs containing EGFR, P-EGFR, and exo-gDNA, a process that is mitigated by NSMase and caspase inhibitors suggesting a cross-talk between exosomal and apoptotic pathways. Overall, we propose that unique EV characteristics could be explored as biomarkers of targeted therapy responses in cancer.
Experimental Procedures
Cell Cultures and Treatments
Human cancer cell lines, including A431 (epidermal), MDA-MB-231 (breast), BxPC3, PANC-1 (pancreatic), PC-3 (prostate), DLD-1, and CaCo2 (colon), were obtained from American Type Culture Collection (ATCC, Manassas, VA). HCT116 cells and their p53−/− variant (379.2) were a gift from Dr. Bert Vogelstein (Johns Hopkins University), and glioma cell lines U373, U373vIII, and U87vIII were contributed by the late Dr. Ab Guha (University of Toronto). Cell lines were cultured in DMEM (Invitrogen), supplemented with 10% fetal bovine serum (FBS) (Multicell Wisent, Inc., St-Bruno, Quebec, Canada), and penicillin/streptomycin (Invitrogen). The clonal A431-CD63/GFP cell line was prepared by transfecting A431 parental cells with the plasmid pCMV6-AC-GFP (OriGene, Rockville, MD), clone RG 201733, encoding the CD63-GFP fusion protein. Clonal sublines were selected for high GFP expression and one chosen for experimentation. Treatment with the indicated agents was conducted at 80% confluence after serum starvation overnight (0.1% FBS). Cells were washed twice with PBS (1×) and incubated with CI-1033 (Canertinib, 1–5 μm; LC Laboratories, Woburn, MA), PF-00299804 (Dacomitinib 1–5 μm; Selleckchem, Houston, TX), Cetuximab (Erbitux, 50 μg/ml, Bristol-Myers Squibb Co.), and/or TGFα (50 ng/ml; Invitrogen) for 24 h before preparation of EVs.
Xenografts and Plasma Samples
Single cell suspensions of the indicated cells were subcutaneously (s.c.) injected into immunodeficient YFP/SCID mice at 1–5 × 106 cells/animal. Tumor growth in either vehicle- (PBS) or drug (CI-1033, 20 mg/kg/day intraperitoneally)-treated mice was measured using Vernier calipers, and lesion volumes were calculated according to the formula TV = a2 × b/2, where a and b are the shorter and longer perpendicular diameters, respectively (30). Intracranial xenografts of human GBM were generated in the laboratory of Dr. Abhijit Guha (University Health Network) under the Institutional Research Ethics Board approval. The EGFRvIII mutation status was established as described previously (31). Mouse blood was collected by cardiac puncture at the end point, and EVs were isolated as described (15). All animal procedures (Animal Use Protocol) were approved by the Institutional Animal Care Committee according to guidelines of the Canadian Council of Animal Care.
Preparation of Extracellular Vesicles
EV-depleted FBS (centrifuged for 5 h at 150,000 × g) was used to prepare conditioned medium. Conditioned medium or platelet-poor plasma was used to isolate EVs in vitro and in vivo, respectively (15, 22). Briefly, culture supernatants were centrifuged for 10 min at 400 × g to collect floating cells (P1 fraction), followed by 20 min at 2500 × g (P2), 30 min at 10,000 × g (P3), and 1 h at 110,000 × g (P4). Pellets containing P4 fraction were washed in PBS once and centrifuged for 1 h at 110,000 × g. EVs collected for the DNA extraction were filtered through a 0.22-μm filter (or as indicated) prior to the last 110,000 × g centrifugation (17).
Western Blot
RIPA buffer containing protease inhibitor (Roche Applied Science) and phosphatase inhibitor mixtures 2 and 3 (Sigma) was used to isolate total proteins from cells or EVs. Lysates were incubated on ice for 5 min and centrifuged at 13,000 rpm for 10 min at 4 °C, and solubilized proteins were quantified using the Pierce Micro BCATM protein assay (Thermo Scientific, Rockford, IL). Proteins were resolved using SDS-PAGE at 10%, and transferred to polyvinylidene difluoride membranes (PVDF; Bio-Rad). The membranes were probed with indicated primary antibodies and appropriate horseradish peroxidase (HRP)-conjugated secondary anti-mouse (Dako, Mississauga, Ontario, Canada) or anti-rabbit (Cell Signaling, Danvers, MA) antibodies. Chemiluminescence (GE Healthcare) was visualized using ChemiDoc MP system (Bio-Rad). Primary antibodies were from Cell Signaling, including the following: rabbit anti-EGFR; rabbit anti-Tyr-845-P-EGFR; rabbit anti-Tyr-1068-P-EGFR; rabbit anti-Tyr-1173-P-EGFR; rabbit anti-AKT and anti P-AKT; and rabbit anti-ERK and rabbit anti-P-ERK. Antibodies from Sigma and from BD Biosciences were used to probe for either mouse β-actin or for mouse flotillin-1, GAPDH, CD9, GFP, and caspase 3, respectively.
Phosphoprotein Profiling
Indicated cells were grown to 70% confluence following conditioning of the culture medium for 3 days using exosome-depleted FBS. EV pellets (fraction P4) were collected by ultracentrifugation following generation of EV and cell lysates using buffers containing 20 mm MOPS, 2 mm EGTA, 5 mm EDTA, 30 mm sodium fluoride, 40 mm β-glycerophosphate, 20 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride, 3 mm benzamidine, 5 μm pepstatin A, 10 μm leupeptin, and 5% Nonidet P-40. Profiling of phosphoproteins was performed using KPSS1.3 KinexTM phospho-site antibody array (Kinexus Bioinformatics Corp.) and visualized as a heatmap generated by MeV software as described (32).
Phosphoreceptor Tyrosine Kinase (P-RTK) Array
The content of activated human receptors in EVs circulating in blood of xenograft-bearing mice was measured using antibody array (Protein Profiler, R&D Systems, Minneapolis, MN) containing phospho-specific antibodies against 42 human RTKs. Plasma EV preparations were incubated with Profiler membranes, and chemiluminescent signal was quantified by densitometry (ChemiDoc MP, Bio-Rad) and presented as a heatmap (MeV).
Quantification of EV-associated EGFR
Human total EGFR and P-EGFR ELISA kits (R&D Systems, Minneapolis, MN) were used to quantify the respective receptors in cell culture medium. Quantifications were repeated using human phospho-EGFR antibody array (RayBiotech, Norcross, GA; data not shown). Briefly, 80% confluent cultures were serum-starved overnight (0.1% FBS), washed twice with PBS (1×), and pretreated for 20 min with indicated agents in 5% FBS. The media were subsequently replaced, and the treatment was continued for 24 h. Proteins were extracted from adherent cells and EV pellets quantified, and incubated with ELISA reagents as recommended by the manufacturer. Alternatively, array membranes were exposed to equal amounts (5–10 μg) of protein. The signal was visualized using chemiluminescence, quantified (ChemiDoc MP; Bio-Rad), normalized to internal controls, pooled from two experiments, and plotted.
Cell Growth/Survival Assays
MTS assay (Promega, Madison, WI) was used to measure in vitro cell growth/viability in the presence of indicated treatments. In parallel, trypan blue exclusion and changes in cell morphology (200×) were recorded at different time intervals (2, 6, and 24 h). As indicated, the cells were grown to 80% confluence, serum-starved overnight (0.1% FBS), washed, and pretreated for 1 h with the pan-caspase inhibitor Z-VAD-fluoromethyl ketone (ZVAD; 20 μm; Promega, Madison, WI) in EV-depleted media prior to addition of EGFR inhibitors or control compounds (Etoposide, 50 μg/ml; Sigma). After the first 20 min, the medium was changed, and treatment was continued for 24 h, followed by EV isolation.
EV Analysis
The number and size of emitted EVs were analyzed using NS500 nanoparticle tracking analysis system (NTA; Nanosight, Amesbury, UK). Culture media were centrifuged at 400 × g for 10 min to remove cells, and 300-μl aliquots (P2–P4) were loaded into the flow chamber. At least three recordings of 30 s each were obtained under automatic detection and batch processing settings. For inhibitor treatment, cells were grown to 80% confluence, washed, and pretreated for 1 h with sphingomyelinase inhibitors as follows: GW4869, 10 μm or FTY720, 5 μm (Calbiochem, Millipore, Ontario, Canada). After 1 h, the medium was changed, and fresh media containing the EGFR inhibitor (or vehicle) with either GW4869 or FTY720, respectively, were added for additional 24 h, followed by NTA. The results of at least 2–3 independent experiments were combined and plotted. The EVs were also analyzed by floatation in the sucrose gradient (33). Briefly, the combined P2 to P4 fractions were centrifuged twice at 110,000 × g for 70 min. The pellets were resuspended in 2 ml of 20 mm HEPES, 2 m sucrose. The resulting samples were transferred to the bottom of the Beckman SW41 centrifuge tube and slowly layered with a continuous gradient of sucrose, from 2 to 0.25 m. The samples were ultracentrifuged in a swinging bucket rotor for 17 h at 210,000 × g with the brake set on low. After centrifugation, the samples in each tube were separated into 11 fractions of 1 ml each, starting from the top. Each fraction was transferred into a 3-ml tube, and mixed with 2 ml of HEPES (20 mm), centrifuged at 110,000 × g for 1 h, resuspended in 50 μl of PBS (1×), and processed for assays indicated.
DNA Analysis
As described previously (17), EV aliquots (20 μl) were pelleted at 110,000 × g and mixed with 20 μl of DNA lysis buffer (2× 200 μl of Tris, 20 μl of 0.5 m EDTA, 40 μl of 10% SDS, 80 μl of 5 m NaCl, and proteinase K (1:200)). Following incubation at 37 °C for 2 h, isopropyl alcohol precipitation, and centrifugation at 16,100 × g for 20 min at room temperature, DNA pellets were washed once with 200 μl of 70% ethanol, air-dried, and resuspended in 50 μl of double-distilled H2O. The genomic sequences were PCR-amplified from 5 to 10 μl of DNA samples using a denaturation step set at 95 °C for 10 min followed by 33 or 40 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and synthesis at 72 °C for 1 m. The final elongation step was at 72 °C for 5 min, followed by resolution in 2% agarose gel at 100 V for 20–30 min. The forward (gtgtcctggcacccaagcccatg) and reverse (ccccaccagaccatgaga) primer sequences were designed to amplify the region flanking exon 18 of the EGFR gene.
Capture and DNA Analysis in EGFR-containing EVs
Cells grown in 6-well plates were incubated with CI-1033 or PF-00299804 in 650 μl of EV-depleted media for 24 h. Culture medium was then centrifuged at 400 × g for 10 min to remove cells debris, and 300-μl aliquots were added to EGFR ELISA well (RayBiotech) and incubated overnight at 4 °C. The following day, the media were removed; the wells were washed twice with PBS, and 30 μl of DNA lysis buffer was added to the wells. The solution was removed and transferred to a clean tube for DNA precipitation with isopropyl alcohol. DNA pellets were recovered at 16,100 × g for 20 min at room temperature, washed once with 200 μl of 70% ethanol, air-dried, and resuspended in 50 μl of double distilled H2O, and processed for EGFR PCR (above).
Data Analysis
All experiments were reproduced at least twice with similar results and presented as a number of replicates (n) and their mean value ± S.D. Statistical significance was evaluated using SPSS software, or a computerized two tailed t test, and the differences were considered significant for p < 0.05.
Results
EV-mediated Release of EGFRvIII Oncogene by Glioma Cells
Cells may emit intact EGFR through EV-mediated mechanisms (15, 29, 34). To extend this observation, we examined by immunoblotting the content of mutant EGFRvIII in EVs isolated from plasma samples of patients with GBM and from mice harboring intracranial human GBM xenografts. In both cases, the EGFRvIII status in the tumor mass correlated with the EGFRvIII signal in plasma-derived EVs (Fig. 1, A and B). We also tested the conditioned medium of EGFRvIII-transfected glioma cells (U373vIII) and their parental indolent counterparts (U373; Fig. 1C). Once again, endogenous EGFR and exogenous EGFRvIII were readily and specifically detected in EVs shed by the former cell line, although U373 released only small amounts of endogenous EGFR. As expected, we also detected increased levels of P-EGFR (Tyr-1068) in transformed U373vIII cells and their EVs, relative to parental controls (Fig. 1C). Indeed, EGFRvIII induces widespread changes in the cellular and EV phosphoproteome, as indicated by profiling of U373 and U373vIII cells and their EVs using KinexTM array platform (data not shown). Collectively, these results highlight the ability of EVs to mediate the release of the oncogenic EGFR and P-EGFR from cancer cells.
FIGURE 1.

Evidence for EV-mediated extracellular release of the oncogenic EGFRvIII by human GBM cells in vivo and in vitro. A, analysis of GBM tissue samples for EGFRvIII mRNA. Hypoxanthine phosphoribosyltransferase 1 (HGPRT1) was used as a loading control, as described previously (31). Lower lane, EGFRvIII protein signal was detected by Western blotting in EV preparations from plasma of patients with EGFRvIII-expressing tumors where the EGFRvIII band is apparent, despite the fact that the quality of the archival plasma samples was relatively poor and negatively impacted protein integrity in exosomal isolates and the image quality. B, detection of EGFRvIII in plasma of SCID mice harboring human GBM intracranial xenografts positive (EGFRvIII+) or negative (EGFRvIII−) for EGFRvIII expression. C, detection of EGFR and P-EGFR in cell lysates and EV preparations of conditioned medium from control U373 indolent glioma cells and their aggressive variant (U373vIII) transfected with EGFRvIII expression vector (31).
Tumor-related Emission of Phosphorylated RTKs into the Circulating Blood
We asked whether EV-mediated emission of phosphorylated RTKs (P-RTKs) extends beyond EGFR in glioma. To address this question, plasma was collected from mice harboring xenografts of human brain (U87vIII and U373vIII), epidermal (A431), breast (MDA-MB-231), lung (A549), prostate (PC3), pancreatic (BxPC3 and PANC-1), and colorectal (379.2, HCT116, CaCo2, and DLD-1) cancers, and purified EVs were profiled using the antibody array containing probes for 42 different P-RTKs. Among several detectable P-RTKs, only some were relatively ubiquitous, including P-EGFR and macrophage-stimulating protein receptor (MSPR/RON, member of the MET family) (Fig. 2). These experiments document the ability of multiple human cancer types to emit P-RTKs, including P-EGFR, in vivo.
FIGURE 2.
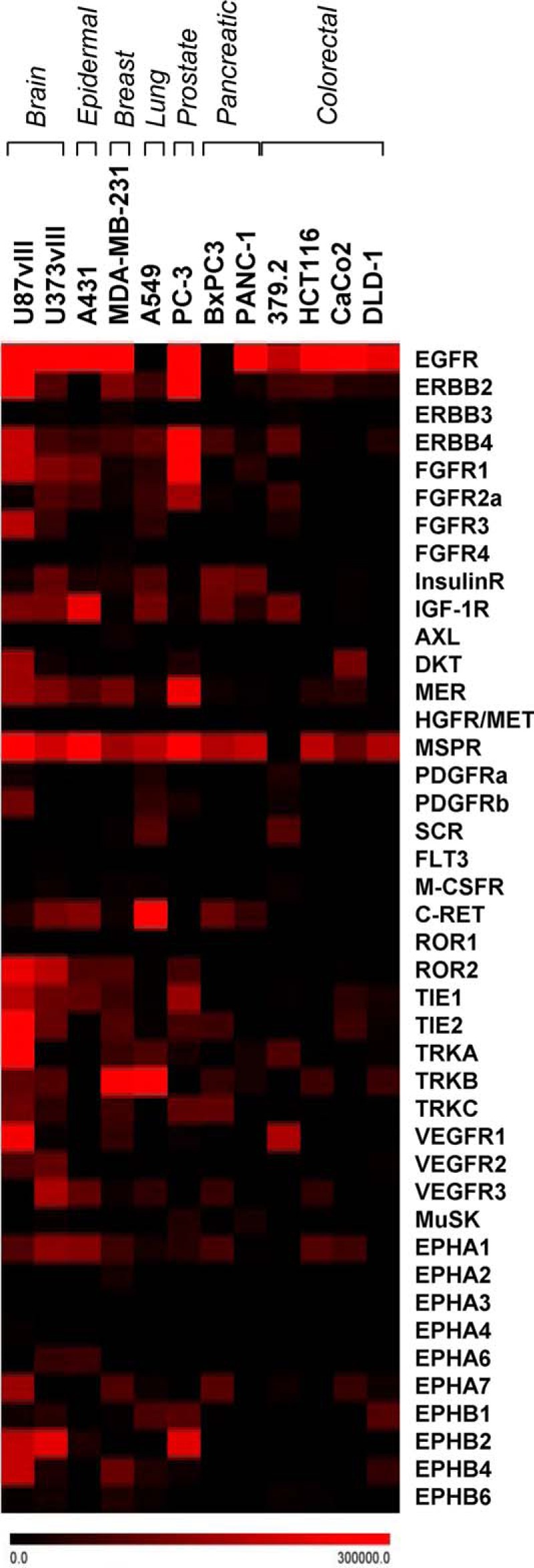
Detection of phosphorylated receptor tyrosine kinases in EVs released to mouse blood from human cancer cell xenografts. Indicated cell lines are representative of brain, epidermal, breast, lung, prostate, pancreatic, and colorectal cancers. Subcutaneous tumors were grown in SCID mice until subjected to terminal blood collection. Plasma EVs were isolated by ultracentrifugation, lysed, and tested for 42 different human phospho-RTKs using the indicated antibody array platform. Notably, several human RTKs, including EGFR, were detected in the circulating blood in the intact, phosphorylated, and EV-associated form (see text for details).
EGFR Inhibitors Impact Growth and Vesiculation of Cancer Cells
Among cancer models found to emit EVs containing P-EGFR, A431 cells represent an interesting paradigm due to their exquisite and well documented dependence on the endogenous EGFR activity (29). Therefore, we chose these cells to interrogate in more detail whether EGFR and EV emission may change in the presence of clinically relevant EGFR-targeting agents. Indeed, the exposure of A431 cells to two different irreversible pan-ErbB/EKIs, CI-1033 and PF-00299804, produced a marked and dose-dependent decline of metabolic activity (MTS assay), especially above the 1 μm threshold. In contrast, the growth inhibitory effect of the anti-EGFR-neutralizing antibody (Cetuximab) with no direct anti-kinase activity was relatively mild (Fig. 3, A–C), as described earlier (4).
FIGURE 3.

Responses of EGFR-driven cancer cells to EGFR inhibitors. A–C, A431 cells harboring genomic amplification of the EGFR gene were grown in monolayer cultures and exposed for 24 h to the indicated irreversible EGFR kinase inhibitors (EKIs: CI-1033 and PF-00299804) or neutralizing anti-EGFR antibody (Cetuximab) at increasing concentrations. The cells were tested for metabolic activity using the MTS assay. EKIs, but not Cetuximab, triggered marked and dose-dependent reduction in metabolic activity. D, effects of drug treatment on EGFR phosphorylation (P-EGFR; Western blotting); numerical values represent mean ± S.D. of several independent experiments; *, p > 0.05.
Intriguingly, the EV-mediated emission of EGFR into the conditioned medium of cancer cells increased dramatically in the presence of EKIs but not Cetuximab (Fig. 4, A–C). Moreover, additional stimulation of EGFR with transforming growth factor α (TGFα) did not change the ability of TKIs (CI-1033) to drive the EV-mediated release of EGFR (Fig. 4, B–D). Collectively, these observations suggest that the efficient blockade of the EGFR kinase activity facilitates (rather than inhibits) extracellular emission of EVs containing this RTK.
FIGURE 4.

Marked increase in the EV-mediated EGFR emission from EKI-treated cancer cells. A, unchanged levels of EGFR protein in lysates of A431 cells cultured for 24 h in the presence of control medium (control), CI-1033 (5 μm), PF-00299804 (5 μm), or Cetuximab (Cetux., 50 μg/ml), as measured by ELISA. B, unchanged levels of EGFR protein in lysates of A431 cells cultured in the presence of CI-1033 (5 μm), TGFα (50 ng/ml), or both (EGFR ELISA). C, dramatic increase in EGFR signal in the EV (P4) fraction of the conditioned medium corresponding to CI-1033 and PF-00299804 treatment; pooled data were from two independent experiments. D, increase in EV-mediated emission of EGFR upon treatment with CI-1033 and TGFα/CI-1033. The combined treatment highlights the ability of EGFR kinase inhibitor to trigger EV release; p values were as indicated. The results were independently reproduced using an antibody array (data not shown). NS, not significant.
EKI Treatment Impacts the Profile of EV-associated P-EGFR
EGFR signaling is a multifactorial process involving phosphorylation of several tyrosine residues (including Tyr-845, Tyr-1068, and Tyr-1173) and activation of downstream transduction modules, especially AKT and ERK. These events would be expected to subside in the presence of EKIs (5, 11, 35, 36), but their representation in the EV cargo has not been studied in detail. Thus, we compared the expression of the respective phosphorylated EGFR isoforms, AKT and ERK, in EKI-treated A431 cells and their EVs. Of note, the constitutive P-EGFR levels were considerably lower in EVs than in corresponding cells, especially the Tyr-1173 phosphorylation. As expected, the exposure to PF-00299804 (Fig. 5) and CI-1033 (Fig. 6) markedly reduced cellular EGFR phosphorylation levels on all tyrosines tested. Somewhat surprisingly, however, the content of P-EGFR in EVs released from EKI-treated cells was visibly and selectively increased, especially on Tyr-845 and Tyr-1068. P-EGFR was also elevated in cells and EVs following stimulation with TGFα, including the Tyr-1173 residue. Addition of EKI (CI-1033) completely reversed this effect in cells, but only partially in the case of their derived EVs, in which Tyr-845 and Tyr-1068 remained phosphorylated. These results may suggest that certain P-EGFR isoforms may be preferentially positioned at sites of EV biogenesis, especially when EGFR signaling is perturbed in the presence of EKIs (Figs. 4–6).
FIGURE 5.

Differential EGFR phosphorylation profiles of cancer cells and their EVs in the presence or absence of PF-00299804 treatment. A431 cells and corresponding EV preparations were subjected to Western blotting for EGFR and for three different EGFR phosphosites (Tyr-845, Tyr-1068, and Tyr-1173). EVs differ in their EGFR phosphorylation profiles from their parental cells, including preponderance of 2 out of 3 phosphoisoforms of EGFR following drug treatment.
FIGURE 6.

Differential phosphorylation profiles of EGFR, AKT, and ERK in cancer cells and their EVs following treatment with pan-Erb kinase inhibitor CI-1033. A431 cells were treated with the EGFR agonist (TGFα, 50 ng/ml), irreversible ErbB kinase inhibitor (CI-1033, 5 μm), or both. The cells and EVs were collected 24 h later and immunoblotted for total EGFR, three different EGFR phosphosites (P-EGFR Tyr-845, Tyr-1068, and Tyr-1173), AKT, P-AKT (Ser-473), ERK, and P-ERK (Thr-202/Tyr-Y204). As loading controls, β-actin and Ponceau red were used for cell and EV lysates, respectively. EVs differ in their EGFR phosphorylation profile from their parental cells. P-EGFR is less abundant in EVs than in corresponding cells. EVs accumulate Tyr-845 and Tyr-1068 P-EGFR isoforms, but not Tyr-1173, following treatment with CI-1033. EV-associated Tyr-1173 is increased following stimulation with TGFα. EVs contain no detectable P-AKT but are enriched for ERK and P-ERK following CI-1033 treatment (see text for details).
It is also noteworthy that EVs emitted from A431 cells contained AKT, but no detectable P-AKT, regardless of treatment. In contrast, although the levels of ERK and P-ERK in EVs of intact cells were exceedingly low, EKI treatment markedly increased the packaging of these proteins into the EV cargo, especially following prestimulation of cells with TGFα. Thus, EVs do not mimic the cellular status of crucial proteins involved in oncogenic EGFR signaling, but instead they may reflect the cross-talk between EGFR signaling and vesiculation pathways.
EVs Released Following EGFR Blockade Exhibit Exosome-like Properties
Exosomes represent the major subset of EVs implicated in EGFR emission (29, 34). To explore whether EKI-induced vesiculation leads to the exaggerated emission of exosomes, or triggers formation of other types of EVs (e.g. larger microvesicles or apoptotic bodies), we employed the variant of A431 cells stably expressing CD63-GFP fusion protein. Because CD63 is present in exosomes, this modification serves to visualize production of these EVs by tracking the GFP signal. Thus, conditioned medium of A431-CD63/GFP cells was subjected to differential ultracentrifugation under either low (P2 fraction) or high centrifugal force (P4 fraction), to preferentially sediment microvesicles and exosomes, respectively (22). As expected, EGFR was markedly enriched in the exosomal (P4) fraction, especially after EKI (CI-1033) treatment (Fig. 7A). Next, EVs were resolved in the sucrose gradient, followed by NTA for the number and size distribution in the respective fractions. This placed the majority of particles between 1.11 and 1.21 g/ml density and 50–250 nm in diameter, which corresponds to properties of exosomes. It should be noted that exosomal fractions contained a range of EV sizes, suggesting a level heterogeneity. After EKI treatment, the abundance and size range of EVs markedly increased (Fig. 7, B and C). We also probed individual sucrose fractions for EGFR and exosomal markers (CD63/GFP and CD9), all of which co-localized within the exosomal density range, between 1.10 and 1.21 g/ml (33). Notably, CI-1033 treatment led to a marked increase in the EGFR, CD63/GFP, and CD9 content of these fractions, further suggesting that EKI treatment stimulates emission of EVs with exosome-like characteristics.
FIGURE 7.

Pharmacological blockade of the oncogenic EGFR triggers emission of EVs with exosomal characteristics. A431-CD63/GFP cells were cultured for 24 h in the presence of control media or EGFR blocking concentrations of CI-1033 (5 μm). A, EVs were collected by ultracentrifugation at either 2500 × g (EV-P2) or 110,000 × g (EV-P4) and immunoblotted for EGFR or GFP, the latter to reveal the exogenous GFP/CD63 chimeric protein marker of exosomes. P4 fraction containing exosome-sized EVs was enriched in EGFR and GFP, especially after the CI-1033 treatment. B and C, A431-derived EVs were floated on the sucrose gradient, and the respective fractions were profiled for size and numerical EV distribution using nanoparticle tracking analysis system (NTA, Nanosight). Of note is the fact that CI-1033 treatment stimulated production of mainly small EVs (exosome-like), with sizes ranging between 51 and 150 nm and density between 1.11 and 1.21 g/ml, which corresponded to sucrose density fractions 3–8. NTA of individual fractions containing cancer cell-derived EVs suggests a highly heterogeneous size distribution. D and E, exosomal fractions (1.12–1.17 g/ml) of EVs purified from conditioned medium of control A431-CD63/GFP cells contain both EGFR and GFP (CD63) markers (immunoblotting). CI-1033 treatment triggered the increase in the EGFR and exosomal marker content (GFP-CD63 and CD9) across exosomal fractions 1.10–1.21 g/ml of A431-GFP/CD63-derived EVs. Data are representative of three independent experiments (see text for details).
We also examined the role of EV biogenesis pathways in vesiculation triggered by EKIs. To accomplish this, A431 cells were treated with inhibitors of either NSMase (GW4869) or ASMase (FTY720), implicated in formation of exosomes and microvesicles, respectively (Fig. 8) (22). As expected, GW4869 diminished the emission of exosome-sized EVs at baseline (Fig. 8A, peak), while shifting the NTA spectrum to larger particles. Interestingly, this treatment also significantly reduced the emission of exosome-sized EVs from CI-1033-treated cancer cells. In contrast, FTY720 treatment not only did not suppress, but instead exacerbated the EKI-triggered EV emission. Collectively these results suggest that EKIs trigger formation of exosome-like EVs, as judged by their size, markers, density, and dependence on the NSMase activity.
FIGURE 8.

EKI-induced cellular vesiculation depends on the neutral sphingomyelinase pathway of exosomal biogenesis. A, NTA analysis of EVs emitted from A431 cells treated with EKI (CI-1033 (CI)), inhibitor of neutral sphingomyelinase (GW4869), inhibitor of acidic sphingomyelinase (FTY720), or their indicated combinations. Of note is a shift to the right in the median size peak of EVs in presence of GW4869 indicative of reduced numbers of exosome-seized vesicles and the preponderance of larger EV sizes. B, EV output into the A431 conditioned medium as measured within the median exosome size (range 150–200 nm) of vesicles. CI-1033-induced increase in EV numbers is attenuated by GW4869 pretreatment but not by FTY720 pretreatment, which increases the EV production; compilation is of three independent experiments, and p value is as indicated. NS, nonsignificant.
Emission of Exosome-like EVs from EGFR-inhibited Cancer Cells Involves the Caspase Pathway
It is conceivable that cellular stress may lead to a cross-talk between vesiculation and apoptotic pathways. To test whether this may occur in the context of EKI exposure, A431 cells were pretreated with the pan-caspase inhibitor (ZVAD) and assayed for vesiculation. Indeed, EKI treatment triggered the cleavage of caspase 3 in conjunction with increased emission of EVs (Fig. 9), both mitigated by ZVAD. Cleaved caspase 3 was detected in cells exposed to both relatively inert (1 μm) and strong growth inhibitory (5 μm) concentrations of CI-1033 (Fig. 9) and PF-00299804 (data not shown). Likewise, etoposide, which induces apoptosis in an EGFR-unrelated manner, predictably triggered caspase activation. Cleaved caspase 3 was mainly observed in floating cells (P1), whereas their adherent counterparts remained unaffected, and no caspase 3 was detected in the EV cargo (P2 and P4 fractions). It should be noted that although all aforementioned treatments activated caspase 3, only strong inhibitory concentrations of EKIs (CI-1033) stimulated the increased EV emission in a ZVAD-sensitive manner (Fig. 9, B–E). The ZVAD effect was especially apparent within the exosomal size range (100–200 nm) of EVs. Thus, EKI responses differ from programmed cell death resulting in formation of large apoptotic bodies (>1000 nm) (27), especially because etoposide did not trigger any measurable release of exosome-like EVs (Fig. 9, D and E). These observations suggest that EKI-induced cellular death pathways are associated with a distinct profile of cellular vesiculation.
FIGURE 9.

EKI-triggered vesiculation involves caspase activity. A, activation of caspase 3 in A431 cells treated with CI-1033 and etoposide. Floating (P1) cells reveal the cleaved caspase 3 (lower) band. Cell cultures were treated with growth inhibitory (5 μm) and subthreshold (1 μm) concentrations of CI-1033, and with pro-apoptotic concentrations of etoposide. In all cases caspase 3 cleavage was inhibited by ZVAD peptide. No cleaved caspase was detectable in adherent cells. Caspase 3 was undetectable in microvesicle-like (P2) and exosome-like (P4) EV fractions. B–E, NTA profile reveals that growth inhibitory dosing of CI-1033 (5 μm), but not of etoposide, evoked vesicular emissions from A431 cells, despite caspase 3 activation in both cases. CI-1033-induced EV release was selectively inhibited by ZVAD pretreatment, as demonstrated by NTA of total (C), and exosome-like (150–200 nm (D)) vesicle populations, but this effect was not observed in larger microvesicle-like EV subsets (350–400 nm; E); data are representative of three independent experiments, p value is as indicated. NS, nonsignificant.
EVs Released from EGFR Inhibited Cancer Cells Contain Genomic DNA
We and others have recently described the ability of cancer cells to emit exosome-like EVs containing genomic DNA (exo-gDNA). The underlying processes remain unclear and may involve both viable and dying cells (17, 25, 26). To test whether EKI treatment alters formation of DNA-containing exosomal EVs, their preparation from culture medium was resolved on the sucrose density gradient, and fractions were analyzed for exo-gDNA. This included total DNA (QC Analyzer) and genomic EGFR (exon 18; PCR) (Fig. 10, A and B). Although exosomal fraction 6 isolated from media of untreated cells contained little or no DNA, the exposure to CI-1033 led to a dramatic increase in both total DNA and genomic EGFR signal. It should be noted that EVs with higher density contained DNA regardless of treatment, probably due to the presence of larger vesicles and cell fragments (Fig. 10B). Nonetheless, these results suggest that EKI-induced cellular stress provokes an increase in the emission of exo-gDNA contained in exosome-like EVs.
FIGURE 10.

EKI treatment leads to cellular emission of genomic DNA within exosome-like fraction of EVs. A, A431 cells were cultured and treated with CI-1033 (5 μm), and sucrose fractions of their EVs were tested for extracellular genomic DNA using QC Analyzer (A) or PCR (B) assays. EV fractions are as follows: 3 (below exosomal density), 6 (exosomal like), and 9 (above exosomal density) were analyzed for DNA content. DNA was detected (arrowhead) only in fractions 6 and 9, and upon treatment with CI-1033. B, PCR amplification of the EGFR (exon 18) genomic sequence in A431-derived EV fractions obtained with and without CI-1033 treatment. These profiles suggest the emission of DNA within exosome-like fractions of EVs following the EGFR blockade. EVs at a higher density contain gDNA regardless of treatment (see text for details).
EV-mediated Co-emission of EGFR and Exo-gDNA under Therapeutic Stress
Because EV populations and biogenetic pathways are inherently heterogeneous, we asked whether EKI treatment triggers the co-emission of exo-gDNA and EGFR by the same or different EV subsets. To accomplish this, we designed an assay predicated on a selective capture of EGFR-expressing EVs on dishes coated with the anti-EGFR antibody, followed by detection of DNA content using PCR (Fig. 11A). We also used two different filtration protocols (0.45 and 0.8 μm) to exclude cells, large EVs, and apoptotic bodies (>1 μm). These experiments documented no EGFR DNA amplification from EGFR-exposing EVs collected under control conditions. However, exo-gDNA content was readily detected when donor cells were treated with growth-suppressing concentrations of either CI-1033 or PF-00299804 (Fig. 11B). Thus, EKI treatment causes co-emission of EGFR and exo-gDNA, at least in part, within the same population of exosome-like EVs.
FIGURE 11.

EV-mediated extracellular co-emission of EGFR and DNA following cancer cell exposure to EKIs. A, experimental design: ImmunoCapture of pre-filtered, EGFR-containing EVs produced by A431 cells either in the absence of in the presence of EKIs, followed by detection of extracellular genomic DNA (exo-gDNA; PCR). B, analysis of EGFR genomic sequences in EV fractions captured on plates coated with anti-EGFR antibody. This assay reveals that the same population of small EGFR-positive exosome-like EVs, which are able to pass through both 0.8- and 0.45-μm filters, is enriched in exo-gDNA, but only if the cells were pretreated with growth inhibitory doses of EKIs, CI-1033 or PF-00299804 (see text for details). UC, ultracentrifugation; -, possible but not obligatory step in the procedure.
Discussion
Our report focuses on EGFR as a paradigm of oncogene-directed targeted anti-cancer therapy. In this regard, we explore how EV biogenesis and cargo may reflect cellular consequences of targeting EGFR kinase activity, and we report several novel findings related to this question. First, we document the ability of multiple types of human cancer cells to emit phosphorylated (intact/activated) RTKs as cargo of their EVs, both in vitro and in vivo. This includes oncogenic EGFR and EGFRvIII. Second, we report differences in molecular profiles between EGFR-driven cancer cells and their derived EVs. We suggest that subsets of EGFR phospho-isoforms, AKT and ERK, are unevenly distributed between cellular and EV compartments. This is significant from both a biological and biomarker perspective, because EVs are often perceived as entities 'representative' of their parental tumors, a notion that we wish to qualify here. Although further studies are required to fully understand this molecular “asymmetry,” we propose that EV-mediated EGFR emission may be a part of an alternative EGFR recycling process sensitive to specific post-translational modifications of this potent oncogenic receptor. Third, we describe the previously unrecognized (and surprising) ability of targeted kinase inhibitors (EKIs) to trigger the emission of exosome-like EVs containing EGFR and exo-gDNA. This process is mitigated by inhibitors of both exosome biogenesis pathways (GW4869) and caspase cascade (ZVAD), an observation suggestive of a possible cross-talk between these respective mechanisms. Fourth, we describe a new capture assay that enables a sensitive detection of combined extracellular release of EGFR (RTK) and exo-gDNA. This is relevant, as EGFR/exo-gDNA co-expression in our hands signifies the effective anti-tumor EKI treatment, which may be of interest from the therapeutic and biomarker perspective.
In particular, this study extends the link between pathways governing oncogenic transformation and cellular vesiculation. Thus, we and others have previously described the quantitative and qualitative influences exerted by oncogenic signals on the EV release from cancer cells (15, 17, 28, 37, 38). These earlier studies also revealed intriguing aspects of EV-mediated intercellular trafficking of oncogenic and bioactive proteins and their corresponding nucleic acids (15, 29, 39, 40). EGFR is especially well studied in this regard and presents itself as an informative (albeit not exclusive) paradigm of the horizontal molecular exchange in cancer (16, 18, 41, 42). Although there is an understandable interest in biological impact of EVs in the cell-to-cell communication (15, 18, 26, 34, 43–45), diagnostic implications of the EV release from cancer cells in their various functional states also deserve more thorough exploration.
Different mechanisms of cellular vesiculation may enable cancer-specific molecular cargo to exit tumor cells and enter biofluid spaces serving as unique “liquid biopsy” portals (14, 42, 46). Unlike circulating cell-free molecules (e.g. cell-free DNA), individual EV carry multiple molecular entities, the combination of which could be informative as to complex properties and responses of EV donor cells. Despite considerable EV profiling efforts currently underway (19, 47–49), the analysis of molecular combinatorial co-emission patterns is scarcely available, especially given the remarkable heterogeneity both between and within the specific and still poorly characterized EV subpopulations (exosomes, ectosomes, and apoptotic bodies) (48). The EV-mediated co-emission of EGFR and exo-gDNA by cancer cells targeted with EKI, as described in our study, is but one example of such biologically meaningful combinatorial characterization.
Diagnostic potential may also lie in a better factual documentation, characterization, and understanding of protein sorting mechanisms between various cellular and extracellular (EV) compartments. We suggest that post-translational modifications, especially phosphorylation, may contribute to these processes, including with respect to EGFR (and beyond) (15, 49, 50, 51). Indeed, recent findings suggest that HER2 may provoke widespread changes in the EV phosphoproteome (52), which is similar to the effects we observed in the case of EGFRvIII. Interestingly, phosphorylation patterns of specific tyrosine residues within the EGFR itself differ between EVs and their parental cancer cells. In particular, the levels of P-EGFR are generally lower (per overall protein content) in EVs relative to parental cancer cells, suggesting that only a fraction of activated EGFR is normally released to the extracellular space. However, the P-EGFR content of EVs is markedly increased in cells subjected to either maximal stimulation (TGFα) or irreversible blockade (EKIs) of the EGFR kinase activity. In addition, we observed that not all P-EGFR isoforms were equally distributed to EVs. For example, we noticed a greater representation of EGFR phosphorylated on Tyr-845 and Tyr-1068 residues versus Tyr-1173, in the EV compartment of A431 cells under several different conditions. The mechanisms regulating this P-EGFR partitioning remain presently unknown.
Phosphorylated residues Tyr-845, Tyr-1068, and Tyr-1173 are thought to mediate contacts between, the cytoplasmic domain of P-EGFR and its key molecular interactors, such as SRC, GRB2 and SHC, respectively (3). The nonrandom association of P-EGFR subsets with the EV compartment could suggest a role of the vesiculation process in modulating the corresponding signaling patterns within the EGFR pathway, including in response to ligands and EKIs (53, 54). For example, the Src homology 2 domain of SHC is thought to interact mainly with P-EGFR on the Tyr-1173 residue, which is under-represented in EVs, although the Tyr-1068 site interacts with GRB2 and is readily detected in EVs of EKI-treated cells. Because GRB2 interacts with a number of EGFR effectors (SOS, GAB1, and RhoU), it is interesting to consider the possibility that their content could undergo a selective exosomal export (“dumping”) resulting in regulatory biases in cellular signaling (34, 55, 56).
We observed that the nearly complete inhibition of EGFR was coupled with dramatically up-regulated global emission of exosome-like EVs. This effect was attenuated following pharmacological inhibition of NSMase (GW4869), which was previously implicated in exosome biogenesis. In contrast, ASMase inhibitor (FTY720) was devoid of this activity and instead provoked increased vesiculation. ASMase is often linked to formation of membrane blebs and microvesicles, rather than exosomes (22) and implicated in ceramide-dependent apoptosis of endothelial cells (57). In keeping with these reports, we have previously documented unperturbed release of small exosome-like EVs containing ras oncogene from ASMase−/− brain tumor cells (17). Our present analysis of EKI-related emission of small EVs containing EGFR also suggests no obvious requirement for ASMase in this process.
The possible link between vesiculation and apoptotic pathways is intriguing. Indeed, EKI-induced vesiculation of cancer cells was associated with caspase 3 cleavage, cell detachment, reduced metabolic activity, sensitivity to ZVAD, and incorporation of exo-gDNA into the EV cargo. However, these events were also distinct from those known to accompany the apoptotic cell death. For example, EVs emitted by EKI-treated cells exhibited many characteristics of exosomes, and their sizes were orders of magnitude smaller that those described for apoptotic bodies (27). Moreover, low concentrations of EKIs triggered caspase 3 cleavage without cell death or increased vesiculation. Conversely, no burst in exosome-like EV emission was observed in the presence of EGFR-independent pro-apoptotic effects of etoposide. We have not detected caspase 3 in the cargo of tumor-related EVs, at variance with some prior reports (55). However, our observations are reminiscent of caspase-dependent exosomal export of MHCII complexes from macrophages, involving regulation of this process by purinergic P2X7 receptor agonists (58). It is possible that cancer and inflammatory cells share some nodes of the caspase and vesiculation signaling networks. It is also possible that in EGFR “addicted” cancer cells (59), the acute blockade of oncogenic signals may lead to cellular stress that engulfs both exosomal and apoptotic pathways leading to unique changes in vesicular emission.
We (17) and others (25, 26) have recently described the release of exosome-like EVs containing double-stranded oncogenic gDNA from viable cancer cells. Balaj et al. (41) have earlier described single-stranded DNA sequences containing oncogenic Myc amplicon contained in EVs emitted from intact medulloblastoma cells. These findings suggest that the vesicular release of exo-gDNA may not necessarily signify cellular demise (17, 41). Indeed, we observed detectable amounts of histones and chromatin complexes in EVs isolated from cultures of viable cancer cells ((17) data not shown). However, in this study, the burst in emission of EVs containing both exo-gDNA and EGFR coincides with a dramatic reduction in cellular P-EGFR levels and diminished cell viability. Whether such a combined extracellular export (protein target and gDNA) could be used to gauge the lethality of targeted kinase inhibitors remains to be investigated, including through the use of receptor capture assays similar to the one we employed in this study.
Overall, our study describes a new process of EV emission under the influence of cellular stress resulting from the acute blockade of the oncogenic EGFR driver. Although biological underpinnings of these effects remain to be elucidated, our results could be regarded as a proof-of-principle, and a starting point of further quantitative, qualitative, and molecular characterization of targeted drug-induced EV emission. This avenue could be informative as to therapeutic responses in a wider spectrum of human cancers driven by other oncogenes. We suggest that EV analysis could help personalize the use of targeted anticancer therapeutics in the clinic.
Author Contributions
L. M. and B. M. designed and conducted the majority of experiments (Figs. 2–11), analyzed the data, and wrote the manuscript; D. G., W. J. L., and T. H. L. conducted the experiments, analyzed the data, and provided intellectual input, especially the NTA analyses, PCR assays, and cell culture; A. G. (deceased, last valid e-mail was entered) provided cell lines, human samples, and the glioma xenograft models shown in Fig. 1, inspired and contributed intellectually to the earlier phase of the project; K. A. N. initiated the early stage of the project, designed and conducted experiments shown in Figs. 1 and 2, and analyzed the data; J. R. designed the project, supervised the study, analyzed the data, and wrote the manuscript.
Acknowledgment
We are indebted to our families and colleagues for their support.
This work was supported by operating grants from the Canadian Institutes of Health Research (to J. R.), and infrastructure support was provided by Fonds de Recherche en Santé du Québec. The authors declare that they have no conflicts of interest with the contents of this article.
- EGFR
- epidermal growth factor receptor
- P-EGFR
- phosphorylated EGFR
- P-AKT
- phosphorylated AKT
- P-ERK
- phosphorylated ERK
- EV
- extracellular vesicle
- EKI
- EGFR kinase inhibitor
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- NSMase
- neutral sphingomyelinase
- ASMase
- acidic sphingomyelinase
- NTA
- nanoparticle tracking analysis
- GBM
- glioblastoma
- Z
- benzyloxycarbonyl.
References
- 1. Wistuba I. I., Gelovani J. G., Jacoby J. J., Davis S. E., Herbst R. S. (2011) Methodological and practical challenges for personalized cancer therapies. Nat. Rev. Clin. Oncol. 8, 135–141 [DOI] [PubMed] [Google Scholar]
- 2. Avraham R., Yarden Y. (2011) Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 12, 104–117 [DOI] [PubMed] [Google Scholar]
- 3. Batzer A. G., Rotin D., Ureña J. M., Skolnik E. Y., Schlessinger J. (1994) Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 14, 5192–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Viloria-Petit A. M., Rak J., Hung M.-C., Rockwell P., Goldstein N., Fendly B., Kerbel R. S. (1997) Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors. Am. J. Pathol. 151, 1523–1530 [PMC free article] [PubMed] [Google Scholar]
- 5. Wheeler D. L., Dunn E. F., Harari P. M. (2010) Understanding resistance to EGFR inhibitors–impact on future treatment strategies. Nat. Rev. Clin. Oncol. 7, 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roskoski R., Jr. (2014) ErbB/HER protein-tyrosine kinases: structures and small molecule inhibitors. Pharmacol. Res. 87, 42–59 [DOI] [PubMed] [Google Scholar]
- 7. Brzezniak C., Carter C. A., Giaccone G. (2013) Dacomitinib, a new therapy for the treatment of non-small cell lung cancer. Expert Opin. Pharmacother. 14, 247–253 [DOI] [PubMed] [Google Scholar]
- 8. Viloria-Petit A., Crombet T., Jothy S., Hicklin D., Bohlen P., Schlaeppi J. M., Rak J., Kerbel R. S. (2001) Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 61, 5090–5101 [PubMed] [Google Scholar]
- 9. Inda M. M., Bonavia R., Mukasa A., Narita Y., Sah D. W., Vandenberg S., Brennan C., Johns T. G., Bachoo R., Hadwiger P., Tan P., Depinho R. A., Cavenee W., Furnari F. (2010) Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 24, 1731–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang P. H., Mukasa A., Bonavia R., Flynn R. A., Brewer Z. E., Cavenee W. K., Furnari F. B., White F. M. (2007) Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. U.S.A. 104, 12867–12872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hegi M. E., Diserens A. C., Bady P., Kamoshima Y., Kouwenhoven M. C., Delorenzi M., Lambiv W. L., Hamou M. F., Matter M. S., Koch A., Heppner F. L., Yonekawa Y., Merlo A., Frei K., Mariani L., Hofer S. (2011) Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib–a phase II trial. Mol. Cancer Ther. 10, 1102–1112 [DOI] [PubMed] [Google Scholar]
- 12. Karapetis C. S., Khambata-Ford S., Jonker D. J., O'Callaghan C. J., Tu D., Tebbutt N. C., Simes R. J., Chalchal H., Shapiro J. D., Robitaille S., Price T. J., Shepherd L., Au H. J., Langer C., Moore M. J., Zalcberg J. R. (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 359, 1757–1765 [DOI] [PubMed] [Google Scholar]
- 13. Gerlinger M., Rowan A. J., Horswell S., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A., Tarpey P., Varela I., Phillimore B., Begum S., McDonald N. Q., Butler A., et al. (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crowley E., Di Nicolantonio F., Loupakis F., Bardelli A. (2013) Liquid biopsy: monitoring cancer–genetics in the blood. Nat. Rev. Clin. Oncol. 10, 472–484 [DOI] [PubMed] [Google Scholar]
- 15. Al-Nedawi K., Meehan B., Micallef J., Lhotak V., May L., Guha A., Rak J. (2008) Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumor cells. Nat. Cell Biol. 10, 619–624 [DOI] [PubMed] [Google Scholar]
- 16. Skog J., Würdinger T., van Rijn S., Meijer D. H., Gainche L., Sena-Esteves M., Curry W. T. Jr., Carter B. S., Krichevsky A. M., Breakefield X. O. (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumor growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee T. H., Chennakrishnaiah S., Audemard E., Montermini L., Meehan B., Rak J. (2014) Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys. Res. Commun. 451, 295–301 [DOI] [PubMed] [Google Scholar]
- 18. Bergsmedh A., Szeles A., Henriksson M., Bratt A., Folkman M. J., Spetz A. L., Holmgren L. (2001) Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. U.S.A. 98, 6407–6411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nawaz M., Camussi G., Valadi H., Nazarenko I., Ekström K., Wang X., Principe S., Shah N., Ashraf N. M., Fatima F., Neder L., Kislinger T. (2014) The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat. Rev. Urol. 11, 688–701 [DOI] [PubMed] [Google Scholar]
- 20. Bobrie A., Théry C. (2013) Exosomes and communication between tumors and the immune system: are all exosomes equal? Biochem. Soc. Trans. 41, 263–267 [DOI] [PubMed] [Google Scholar]
- 21. Trajkovic K., Hsu C., Chiantia S., Rajendran L., Wenzel D., Wieland F., Schwille P., Brügger B., Simons M. (2008) Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247 [DOI] [PubMed] [Google Scholar]
- 22. Bianco F., Perrotta C., Novellino L., Francolini M., Riganti L., Menna E., Saglietti L., Schuchman E. H., Furlan R., Clementi E., Matteoli M., Verderio C. (2009) Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 28, 1043–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Colombo M., Raposo G., Théry C. (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 30, 255–289 [DOI] [PubMed] [Google Scholar]
- 24. Muralidharan-Chari V., Clancy J., Plou C., Romao M., Chavrier P., Raposo G., D'Souza-Schorey C. (2009) ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 19, 1875–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thakur B. K., Zhang H., Becker A., Matei I., Huang Y., Costa-Silva B., Zheng Y., Hoshino A., Brazier H., Xiang J., Williams C., Rodriguez-Barrueco R., Silva J. M., Zhang W., Hearn S., et al. (2014) Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 24, 766–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kahlert C., Melo S. A., Protopopov A., Tang J., Seth S., Koch M., Zhang J., Weitz J., Chin L., Futreal A., Kalluri R. (2014) Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 289, 3869–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. György B., Szabó T. G., Pásztói M., Pál Z., Misják P., Aradi B., László V., Pállinger E., Pap E., Kittel A., Nagy G., Falus A., Buzás E. I. (2011) Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell. Mol. Life Sci. 68, 2667–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Di Vizio D., Kim J., Hager M. H., Morello M., Yang W., Lafargue C. J., True L. D., Rubin M. A., Adam R. M., Beroukhim R., Demichelis F., Freeman M. R. (2009) Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 69, 5601–5609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Al-Nedawi K., Meehan B., Kerbel R. S., Allison A. C., Rak J. (2009) Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. U.S.A. 106, 3794–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu J. L., May L., Lhotak V., Shahrzad S., Shirasawa S., Weitz J. I., Coomber B. L., Mackman N., Rak J. W. (2005) Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood 105, 1734–1741 [DOI] [PubMed] [Google Scholar]
- 31. Micallef J., Taccone M., Mukherjee J., Croul S., Busby J., Moran M. F., Guha A. (2009) Epidermal growth factor receptor variant III-induced glioma invasion is mediated through myristoylated alanine-rich protein kinase C substrate overexpression. Cancer Res. 69, 7548–7556 [DOI] [PubMed] [Google Scholar]
- 32. D'Asti E., Kool M., Pfister S. M., Rak J. (2014) Coagulation and angiogenic gene expression profiles are defined by molecular subgroups of medulloblastoma: evidence for growth factor-thrombin cross-talk. J. Thromb. Haemost. 12, 1838–1849 [DOI] [PubMed] [Google Scholar]
- 33. Thery C., Clayton A., Amigorena S., Raposo G. (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. Chapter 3, unit 3.22 [DOI] [PubMed] [Google Scholar]
- 34. Garnier D., Magnus N., Lee T. H., Bentley V., Meehan B., Milsom C., Montermini L., Kislinger T., Rak J. (2012) Cancer cells induced to express mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue factor. J. Biol. Chem. 287, 43565–43572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morandell S., Stasyk T., Skvortsov S., Ascher S., Huber L. A. (2008) Quantitative proteomics and phosphoproteomics reveal novel insights into complexity and dynamics of the EGFR signaling network. Proteomics 8, 4383–4401 [DOI] [PubMed] [Google Scholar]
- 36. Huang P. H., Miraldi E. R., Xu A. M., Kundukulam V. A., Del Rosario A. M., Flynn R. A., Cavenee W. K., Furnari F. B., White F. M. (2010) Phosphotyrosine signaling analysis of site-specific mutations on EGFRvIII identifies determinants governing glioblastoma cell growth. Mol. Biosyst. 6, 1227–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu X., Harris S. L., Levine A. J. (2006) The regulation of exosome secretion: a novel function of the p53 protein. Cancer Res. 66, 4795–4801 [DOI] [PubMed] [Google Scholar]
- 38. Putz U., Howitt J., Doan A., Goh C. P., Low L. H., Silke J., Tan S. S. (2012) The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci. Signal. 5, ra70. [DOI] [PubMed] [Google Scholar]
- 39. Peinado H., Aleckovic M., Lavotshkin S., Matei I., Costa-Silva B., Moreno-Bueno G., Hergueta-Redondo M., Williams C., Garcia-Santos G., Ghajar C. M., Nitadori-Hoshino A., Hoffman C., Badal K., Garcia B. A., Callahan M. K., et al. (2012) Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 18, 833–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Demory Beckler M., Higginbotham J. N., Franklin J. L., Ham A. J., Halvey P. J., Imasuen I. E., Whitwell C., Li M., Liebler D. C., Coffey R. J. (2013) Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol. Cell. Proteomics 12, 343–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Balaj L., Lessard R., Dai L., Cho Y. J., Pomeroy S. L., Breakefield X. O., Skog J. (2011) Tumor microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. EL Andaloussi S., Mäger I., Breakefield X. O., Wood M. J. (2013) Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 12, 347–357 [DOI] [PubMed] [Google Scholar]
- 43. Antonyak M. A., Li B., Boroughs L. K., Johnson J. L., Druso J. E., Bryant K. L., Holowka D. A., Cerione R. A. (2011) Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. U.S.A. 108, 4852–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luga V., Zhang L., Viloria-Petit A. M., Ogunjimi A. A., Inanlou M. R., Chiu E., Buchanan M., Hosein A. N., Basik M., Wrana J. L. (2012) Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 151, 1542–1556 [DOI] [PubMed] [Google Scholar]
- 45. Wolfers J., Lozier A., Raposo G., Regnault A., Théry C., Masurier C., Flament C., Pouzieux S., Faure F., Tursz T., Angevin E., Amigorena S., Zitvogel L. (2001) Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 7, 297–303 [DOI] [PubMed] [Google Scholar]
- 46. Rak J. (2013) Extracellular vesicles–biomarkers and effectors of the cellular interactome in cancer. Front. Pharmacol. 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tauro B. J., Mathias R. A., Greening D. W., Gopal S. K., Ji H., Kapp E. A., Coleman B. M., Hill A. F., Kusebauch U., Hallows J. L., Shteynberg D., Moritz R. L., Zhu H. J., Simpson R. J. (2013) Oncogenic H-ras reprograms Madin-Darby canine kidney (MDCK) cell-derived exosomal proteins following epithelial-mesenchymal transition. Mol. Cell. Proteomics 12, 2148–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Garnier D., Magnus N., Meehan B., Kislinger T., Rak J. (2013) Qualitative changes in the proteome of extracellular vesicles accompanying cancer cell transition to mesenchymal state. Exp. Cell Res. 319, 2747–2757 [DOI] [PubMed] [Google Scholar]
- 49. van Dommelen S. M., Van der Meel R., Coimbra M., Vader P., Schiffelers R. M. (2015) Cetuximab treatment alters effects of tumor cell-derived extracellular. J. Extracell. Vesicles 4, 60, P-VI-17 [Google Scholar]
- 50. Myers M. V., Manning H. C., Coffey R. J., Liebler D. C. (2012) Protein expression signatures for inhibition of epidermal growth factor receptor-mediated signaling. Mol. Cell. Proteomics 11, M111.015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moreno-Gonzalo O., Villarroya-Beltri C., Sánchez-Madrid F. (2014) Post-translational modifications of exosomal proteins. Front. Immunol. 5, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Amorim M., Fernandes G., Oliveira P., Martins-de-Souza D., Dias-Neto E., Nunes D. (2014) The overexpression of a single oncogene (ERBB2/HER2) alters the proteomic landscape of extracellular vesicles. Proteomics 14, 1472–1479 [DOI] [PubMed] [Google Scholar]
- 53. Citri A., Alroy I., Lavi S., Rubin C., Xu W., Grammatikakis N., Patterson C., Neckers L., Fry D. W., Yarden Y. (2002) Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J. 21, 2407–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tomas A., Futter C. E., Eden E. R. (2014) EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 24, 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chairoungdua A., Smith D. L., Pochard P., Hull M., Caplan M. J. (2010) Exosome release of β-catenin: a novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 190, 1079–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Verweij F. J., Middeldorp J. M., Pegtel D. M. (2012) Intracellular signaling controlled by the endosomal-exosomal pathway. Commun. Integr. Biol. 5, 88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Garcia-Barros M., Paris F., Cordon-Cardo C., Lyden D., Rafii S., Haimovitz-Friedman A., Fuks Z., Kolesnick R. (2003) Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 300, 1155–1159 [DOI] [PubMed] [Google Scholar]
- 58. Qu Y., Ramachandra L., Mohr S., Franchi L., Harding C. V., Nunez G., Dubyak G. R. (2009) P2X7 receptor-stimulated secretion of MHC class II-containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase-1. J. Immunol. 182, 5052–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Weinstein I. B., Joe A. K. (2006) Mechanisms of disease: Oncogene addiction–a rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 3, 448–457 [DOI] [PubMed] [Google Scholar]