

Background: MG53 is a membrane repair gene whose role in wound healing has not been studied.
Results: Topical administration of MG53 protein facilitates wound healing and reduces scar formation.
Conclusion: This study establishes MG53 as facilitator of injury repair and inhibitor of myofibroblast differentiation during wound healing.
Significance: MG53 has therapeutic benefits in treating wounds and fibrotic diseases.
Keywords: cell migration, fibrosis, myofibroblast, TGF-β, wound healing, thermosensitive hydrogel, recombinant human MG53 protein
Abstract
Cell membrane repair is an important aspect of physiology, and disruption of this process can result in pathophysiology in a number of different tissues, including wound healing, chronic ulcer and scarring. We have previously identified a novel tripartite motif family protein, MG53, as an essential component of the cell membrane repair machinery. Here we report the functional role of MG53 in the modulation of wound healing and scarring. Although MG53 is absent from keratinocytes and fibroblasts, remarkable defects in skin architecture and collagen overproduction are observed in mg53−/− mice, and these animals display delayed wound healing and abnormal scarring. Recombinant human MG53 (rhMG53) protein, encapsulated in a hydrogel formulation, facilitates wound healing and prevents scarring in rodent models of dermal injuries. An in vitro study shows that rhMG53 protects against acute injury to keratinocytes and facilitates the migration of fibroblasts in response to scratch wounding. During fibrotic remodeling, rhMG53 interferes with TGF-β-dependent activation of myofibroblast differentiation. The resulting down-regulation of α smooth muscle actin and extracellular matrix proteins contributes to reduced scarring. Overall, these studies establish a trifunctional role for MG53 as a facilitator of rapid injury repair, a mediator of cell migration, and a modulator of myofibroblast differentiation during wound healing. Targeting the functional interaction between MG53 and TGF-β signaling may present a potentially effective means for promoting scarless wound healing.
Introduction
Wound care is a major challenge to public health, and the burden for wound treatment is growing because of an aging population and a sharp rise in the incidence of diseases such as diabetes and obesity that contribute to poor wound healing. In the United States, ∼6.5 million patients are affected by chronic wounds, and more than 72,000 amputations occur each year (1). Inadequate regenerative capacity during wound healing often results in scar formation, caused mainly by the deposition of extracellular matrix (ECM)4 proteins (2–5). Although multiple factors may contribute to wound healing complications, compromised tissue repair capacity represents an underlying cause for wounds that fail to heal. Development of therapeutic approaches that facilitate scarless wound healing represents an important area of biomedical and clinical research.
Several critical steps are involved in wound healing, including the initial inflammation caused by the acute death of cells within the wound area and release of pro-inflammatory factors (2–4). Although recruitment of inflammatory cells to the wound environment is important for prevention of bacterial infection, excessive inflammation is often associated with the death of cells and delayed wound healing. During the proliferative stage of wound healing, the release of cytokines and growth factors, such as TGF-β, stimulates the migration of keratinocytes and fibroblasts toward the wound area to begin the re-epithelialization and tissue rebuilding process (6, 7). The proliferative phase also involves the conversion of fibroblasts into myofibroblasts that secrete ECM proteins, which are required for closure of the wound. It is known that excessive synthesis of ECM components because of uncontrolled differentiation of myofibroblasts can lead to pathologic scarring associated with tissue injuries. Although many studies have been designed to target TGF-β signaling for the treatment of wound healing, these attempts often faced a double-edged sword. For example, blocking TGF-β signaling can reduce scarring but also compromise wound healing and other cellular functions (8–11).
Cell membrane injury repair is an important aspect of normal physiology, and its disruption can result in various pathologies, including insufficient wound healing, chronic ulcers, and scarring (1–3, 12–15). We previously identified a novel tripartite motif family protein, MG53, as an essential component of the cell membrane repair machinery (14). MG53 functions in vesicle trafficking and allows the nucleation of intracellular vesicles at sites of membrane disruption. MG53 knockout mice (mg53−/−) exhibit defective membrane repair in striated muscle that leads to progressive skeletal myopathy and increased vulnerability of cardiomyocytes to ischemia-reperfusion-induced injury (14, 16, 17). Moreover, the recombinant human MG53 (rhMG53) protein can protect various cell types against membrane disruption when applied to the extracellular environment and ameliorate the pathology associated with muscular dystrophy and lung injury (18, 19). We have also shown that rhMG53 is effective in protection against myocardial infarction (20) and acute kidney injury (21).
In this study, we present evidence supporting the biological function of MG53 in the modulation of wound healing and scarring. First, MG53 is capable of nucleating the cell membrane repair machinery, therefore protecting from acute and stress-induced injuries to the epidermis. Such a fast membrane resealing response could reduce the overall inflammatory responses during the early phase of wounding and not only facilitate early closure of the wound but also impact the fibrotic remodeling during later stages of healing. Second, we found that MG53 present in the extracellular solution can facilitate migration of fibroblast cells in response to scratch wounding, which is an important step in efficient wound healing. Third, we found that MG53 can interfere with TGF-β signaling to control the differentiation process of fibroblasts into myofibroblasts. The latter is a major source of secretion of ECM proteinsand the main factor contributing to scarring. We show that treatment of fibroblasts with rhMG53 leads to reduced expression of α-SMA, collagen 1, and fibronectin, which may contribute to the reduction of scarring during wound healing.
Experimental Procedures
Chemicals and Recombinant Human MG53 Protein
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless stated otherwise. rhMG53 protein was purified from Escherichia coli fermentation as described previously (18). rhMG53 was stored as lyophilized powder and dissolved in saline solution or encapsulated in hydrogel before use.
Animal Care and in Vivo Wound Healing Models
All animal care and use followed National Institutes of Health guidelines and Institutional Animal Care and Use Committee approval by Ohio State University. mg53−/− mice and wild-type littermates were bred and generated as described previously (14). Sprague-Dawley rats (250–300 g in weight) were purchased from Charles River Laboratories. During surgical procedures, animals were anesthetized using isoflurane. Surgical excision or incision wounds were created as detailed below. For pain management, all animals received drinking water containing ibuprofen (PrecisionDoseTM, 200 mg/liter in water) for 2 days prior to and 5 more days post-surgery.
Excisional Wound Model
Dorsal hair was shaved using electric clippers, and the area was disinfected by application of betadine solution. Alcohol prep pads were then used to wipe the area clean. Two full-thickness dermal wounds (3 cm apart and 4 cm caudal to the scapulae) were created using sterile 5-mm- (mice) or 6-mm-diameter (rats) biopsy punches (IntegraTM Miltex®) to expose the underlying dorsolateral skeletal muscle fascia. Hemostasis was achieved by even compression with sterile gauze. Wounds were left open with no dressing, and then the mice immediately received a subcutaneous injection of either 200 μl of saline (as a control) or rhMG53 (1 mg/kg) and were treated daily for 3 successive days.
Rat Incision Wound Model
The hind limbs were shaved and disinfected, and marks of 10-mm length were made to guide the incisional wound. Incisional wounds 10 mm long and 3 mm deep were created on both limbs on the gastrocnemius muscle with a sharp blade (World Precision Instruments, Inc.). Wounds were treated subcutaneously with saline or rhMG53 (0.2 mg/wound) in a similar fashion as described above. For the hydrogel experiment, a single dose of hydrogel or rhMG53/hydrogel (containing 0.2 mg of rhMG53) was applied topically. Wounds were left open with no dressing.
Rabbit Incision Wound Model
Four New Zealand rabbits were used for this study. Three incisional wounds of 3 cm (length) × 1 cm (depth) were created on the back of each rabbit. Hydrogel or rhMG53/hydrogel (0.1 mg rhMG53/wound) was applied to the wound immediately after injury, and then the incision was closed with sutures. Of the 12 wounds generated, six received hydrogel and six received rhMG53/hydrogel.
Wound Analysis
A digital camera (Panasonic DMC-ZS3) was used to capture the wound with a metric ruler inside the field of view to set the scale for measurements. The wound bed was measured using digital calipers (CD-6”CSX, Mitutoyo Corp.). Blinded measurement of wound size was performed using ImageJ software. The perimeter of the wound was then traced, and the wound area was recorded. The wound size on day 0 was set to 100 and, on each subsequent day, was reported as a percentage of the initial wound size.
Histopathology and Immunohistochemistry
Skin and muscle samples dissected from experimental animals were fixed in 10% formalin overnight at 4 °C. Skin samples were pinned to Styrofoam rafts to maintain morphology during the fixation. After fixing, samples were washed three times for 5 min with 70% ethanol. Washed samples were processed and embedded in paraffin. 4-μm-thick paraffin sections were cut and stained with H&E and Masson trichrome. Immunohistochemical staining of MG53 in skin tissues was performed using the Ventana Medical Systems Discovery XT automated immunostainer. Deparaffinization and antigen retrieval was performed using cell conditioning solution (CC1, Ventana Medical Systems). Rabbit polyclonal anti-MG53 antibody (14) was applied and incubated at 37 °C for 1 h. Donkey anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories) was applied and incubated for 1 h at 37 °C. The chromogenic detection kit DABMap (Ventana Medical Systems) was used to label the secondary antibody. Slides were counterstained with hematoxylin, dehydrated, and xylene-cleared. The resulting slides showed MG53 protein stained brown and surrounding tissue stained blue.
Hydroxyproline Assay
Hydroxyproline is a major component of collagen that helps stabilize the helical structure of collagen. Therefore, its levels can be used as an indicator of collagen content (22). The hydroxyproline contents from the back skin of WT and mg53−/− mice were extracted and measured using a hydroxyproline assay kit (Sigma-Aldrich) according to the instructions of the manufacturer. The tissue hydroxyproline content (expressed as micrograms per milligram of skin tissue) was analyzed.
Western Blot Analysis and Antibodies
Protein lysates from the indicated tissue and cell sources were separated by SDS-PAGE. Proteins were transferred onto PVDF membranes (Millipore) at 4 °C and then probed with primary antibodies overnight with shaking at 4 °C. The blots were washed with PBST (PBS plus 0.5% Tween 20) and incubated with the appropriate HRP-conjugated secondary antibodies. Immunoblots were detected with an ECL Plus kit (Thermo Scientific/Pierce). The antibodies used in this study were as follows: anti-type I collagen antibody (Novus Bio, Littleton, CO); anti-Smad2/3 and anti-p-Smad2 (Cell Signaling Technologies, Beverly, MA); anti-fibronectin, anti-desmin, and anti-α-SMA (Sigma-Aldrich), anti-myc antibody (clone 9E10, Life Technologies); and anti-GAPDH (Santa Cruz Biotechnology, Dallas, TX).
Plasmids and Cell Cultures
Human and mouse keratinocytes were purchased from Zen-Bio Inc. (Research Triangle Park, NC) and cultured according to the manuals of the manufacturer. 3T3 cells were obtained from the ATCC. Transfection of GFP-MG53 into human keratinocytes was performed using Lipofectamine LTX reagent (Life Technologies) according to the instructions of the manufacturer. 3T3 cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. 3T3 cells were transfected with the pcDNA-myc-MG53 plasmid (23), and cells stably expressing myc-MG53 protein were selected with G148 (Sigma) for 2 weeks and confirmed by Western blotting with anti-MG53 and anti-myc antibodies. All cells were maintained in medium under 5% CO2 culture conditions. 3T3 cells were seeded and cultured in complete medium (DMEM and 10% FBS) in T25 flasks for 16 h until 70% confluence. Cells were washed twice with serum-free DMEM and then subjected to various treatments, including TGF-β (10 ng/ml), rhMG53 (60 μg/ml), or a combination of both TGF-β and rhMG53.
MTT Cell Viability Assay
To evaluate the effect of rhMG53 on the proliferation of 3T3 cells, an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used. 3T3 cells were plated at 1000–1500 cells/well in a 96-well plate and then treated with indicated amounts of rhMG53 for 24 h. 20 μl of MTT (5 mg/ml in PBS) was added to each well and incubated further for 4 h. Absorbance was recorded at 570 nm after adding 150 μl of dimethyl sulfoxide.
In Vitro Cell Membrane Injury Assays
A suspension of primary human keratinocytes (9 × 104 cells/150 μl of PBS/96-well dish) was added to the wells of a 96-well plate along with acid-washed glass microbeads and the indicated amount of rhMG53 (0–200 μg/ml). The plate was shaken at 200 rpm for 6 min to induce cell membrane damage and then centrifuged at 3000 × g for 5 min to collect cells and microbeads at the bottoms of the wells. 50 μl of supernatant was transferred from the well to a new 96-well plate for a lactate dehydrogenase release assay with a lactate dehydrogenase cytotoxicity detection kit (Thermo Scientific). Measurement of the release of lactate dehydrogenase following microglass bead damage was used as an index of membrane injury (18, 23). The released lactate dehydrogenase was calculated by subtracting the basal lactate dehydrogenase release from wells without glass beads (no damage) from the experimental conditions.
In Vitro Cell Migration Assay
3T3 cells or cells stably expressing myc-MG53 were grown to 90% confluence in complete medium in a 6-well plate. Cells were then switched to a medium containing rhMG53 (60 μg/ml) and subjected to a scratch wound assay with 200-μl pipette tips, following the protocol described previously (24). Time-dependent cell migration was recorded using a Zeiss Axiovert 200 phase-contrast microscope 0, 6, and 22 h post-injury.
Encapsulation of rhMG53 in Hydrogel
Hydrogel was synthesized through free radical polymerization of N-isopropylacrylamide, acrylic acid, and hydroxyethyl methacrylate-oligohydroxybutyrate (Sigma) as described previously (25, 26). In brief, N-isopropylacrylamide, acrylic acid, and hydroxyethyl methacrylate-oligohydroxybutyrate at a molar ratio of 86/4/10 were dissolved in dioxane in a three-necked flask. The solution was bubbled in nitrogen gas for 15 min before the degassed benzyl peroxide solution was injected. After 24 h of reaction at 70 °C, the solution was cooled to room temperature and precipitated with hexane. The polymer was purified twice using THF/diethyl ether, and the resulting polymer was vacuum-dried overnight. An equal volume of rhMG53 (2 mg/ml in PBS) was mixed thoroughly with hydrogel (15% (w/v) in PBS) solution at 4 °C overnight to encapsulate rhMG53 in hydrogen (rhMG53/hydrogel) to a final concentration of 1 mg of rhMG53/1 ml of hydrogel.
Immunocytochemistry
3T3 cells grown on glass coverslips were subjected to treatments with the control (PBS), rhMG53, TGF-β, or a combination of both. Cells were then fixed with 4% paraformaldehyde for 30 min at room temperature, permeabilized (0.2% Triton X-100), blocked with BSA, and stained with primary antibodies (anti-phospho-Smad2/3 (p-Smad2/3) or anti-α-SMA) overnight at 4 °C with rocking. Cells were then incubated with fluorophore-conjugated secondary antibodies (Life Technologies) for 2 h at room temperature. F-actin was visualized using Alexa Fluor 647 phalloidin (Life Technologies). Nuclear staining was performed with DAPI (Sigma). Coverslips were washed and mounted with Fluoromount G (Electron Microscopy Sciences, Hatfield, PA). Images were taken and analyzed using a Zeiss 780 confocal microscope. At least 100 stained cells/sample/experiment were analyzed.
Statistical Analysis
All data are expressed as mean ± S.D. Groups were compared by Student's t test and analysis of variance for repeated measures. A value of p < 0.05 was considered significant.
Results
Defective Skin Structure in mg53−/− Mice
To investigate the biological function of MG53 in repairing skin injury, we used Western blot analysis and immunohistochemical staining to determine the expression of MG53 in skin tissues (Fig. 1). Although MG53 was detected in mouse skin, keratinocytes derived either from human or mouse did not express MG53 (Fig. 1a). Immunohistochemical staining confirmed that MG53 is localized to the panniculus carnosus, a subdermal striated muscle layer present in many rodents (Fig. 1b). This result is consistent with our previous observation that MG53 is predominantly expressed in striated muscle tissues (14, 27).
FIGURE 1.
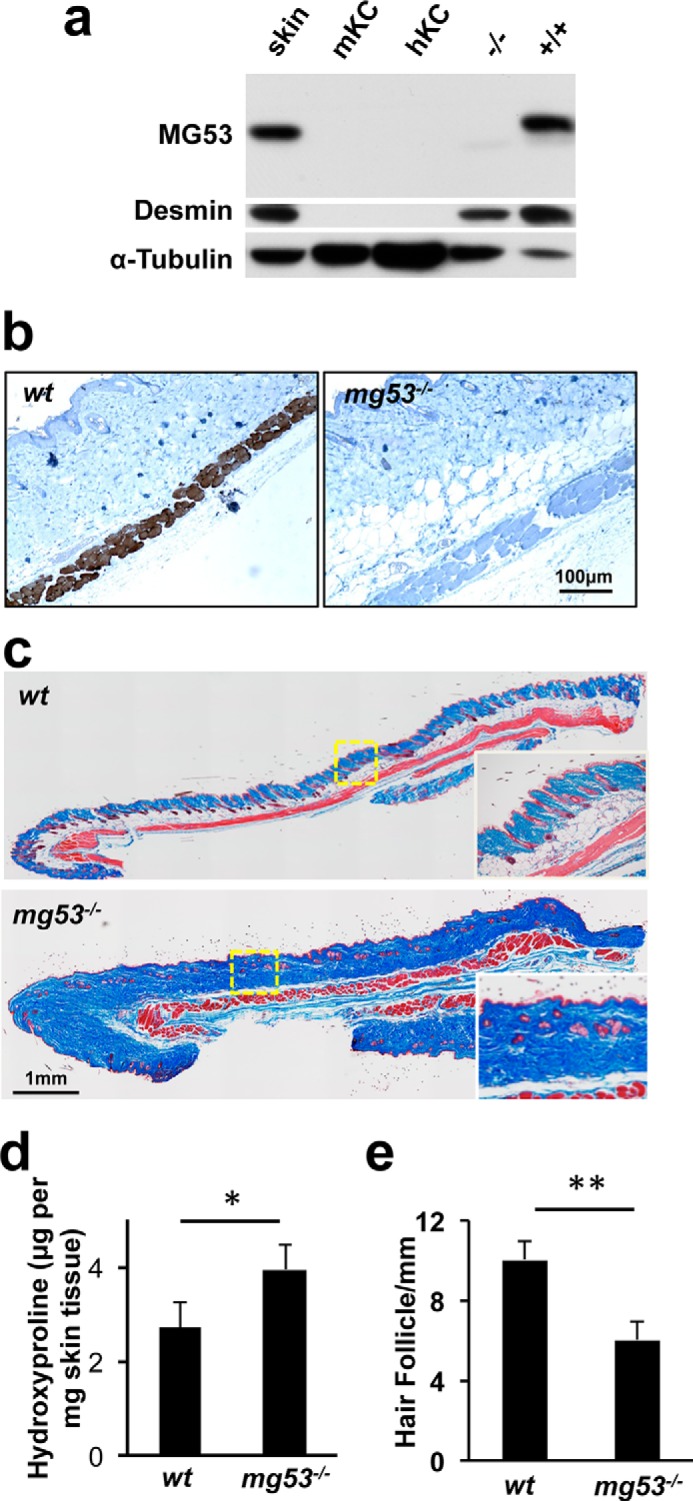
MG53 knockout mice show abnormal skin structure. a, Western blot analysis of MG53 was performed with mouse skin lysates (lane 1, 40 μg), mouse and human keratinocytes (lanes 2 and 3, 40 μg), and skeletal muscle derived from mg53−/− mice (−/−, lane 4, 10 μg) and WT mice (+/+, lane 5, 10 μg). Desmin was used as a marker for skeletal muscle, and tubulin was used as a protein loading control. b, immunohistochemical staining illustrating the presence of MG53 in the panniculus carnosus, a subdermal muscle layer, in WT mice (left panel) but absent in mg53−/− mice (right panel). c, Masson trichrome staining revealed elevated collagen deposition in mg53−/− mouse skin (bottom panel) compared with WT skin (top panel). d, quantification of hydroxyproline content showed higher collagen content in mg53−/− skin compared with WT skin. *, p < 0.05 (n = 6). e, quantification of the number of hair follicles in mouse skin. **, p < 0.01 (n = 12).
Despite the absence of MG53 expression in keratinocytes, remarkable defects in skin architecture were observed in mg53−/− mice. Masson trichrome staining revealed elevated collagen deposition in the mg53−/− dermal layer compared with their WT littermates (Fig. 1c). Measurement of the hydroxyproline content in the skin tissue indicated that mg53−/− skin contained ∼40% more collagen than WT skin (Fig. 1d). In addition, mg53−/− skin possessed roughly 30% fewer hair follicles than WT littermates (Fig. 1e).
Although MG53 is absent from keratinocytes and fibroblasts, circulating MG53 may contribute to the maintenance of normal skin architecture under physiological conditions. Our previous studies have shown that MG53 present in the blood stream plays a protective role against tissue injuries (18–21). The present data show that genetic ablation of MG53 leads to abnormal skin architecture, hair follicle formation, and collagen overproduction in the dermal layer.
Knockout of MG53 Impairs Wound Healing and Increases Scar Formation
Given the abnormal skin phenotype of mg53−/− mice, we evaluated the role of MG53 in wound healing using an established excisional cutaneous wounding model (28–30). Wounds were created in the dorsal region of WT and mg53−/− animals using a biopsy punch to cut through both the epidermal and dermal layers (Fig. 2a). Wound size was then monitored over the subsequent 15 days. In WT mice, wound retraction occurred within the first few days and was significantly greater on day 1 compared with mg53−/− mice. Delayed wound healing in mg53−/− mice was observed consistently on day 7 and beyond (Fig. 2b). On day 11 post-injury, Masson trichrome staining of mg53−/− skin revealed excessive collagen deposition in the interstitial area of the injured muscle fibers (Fig. 2c, bottom panel). The extent of interstitial collagen deposition was significantly lower in WT littermates (Fig. 2c, top panel). Quantification of collagen deposition in WT and mg53−/−mice is summarized in Fig. 2d.
FIGURE 2.
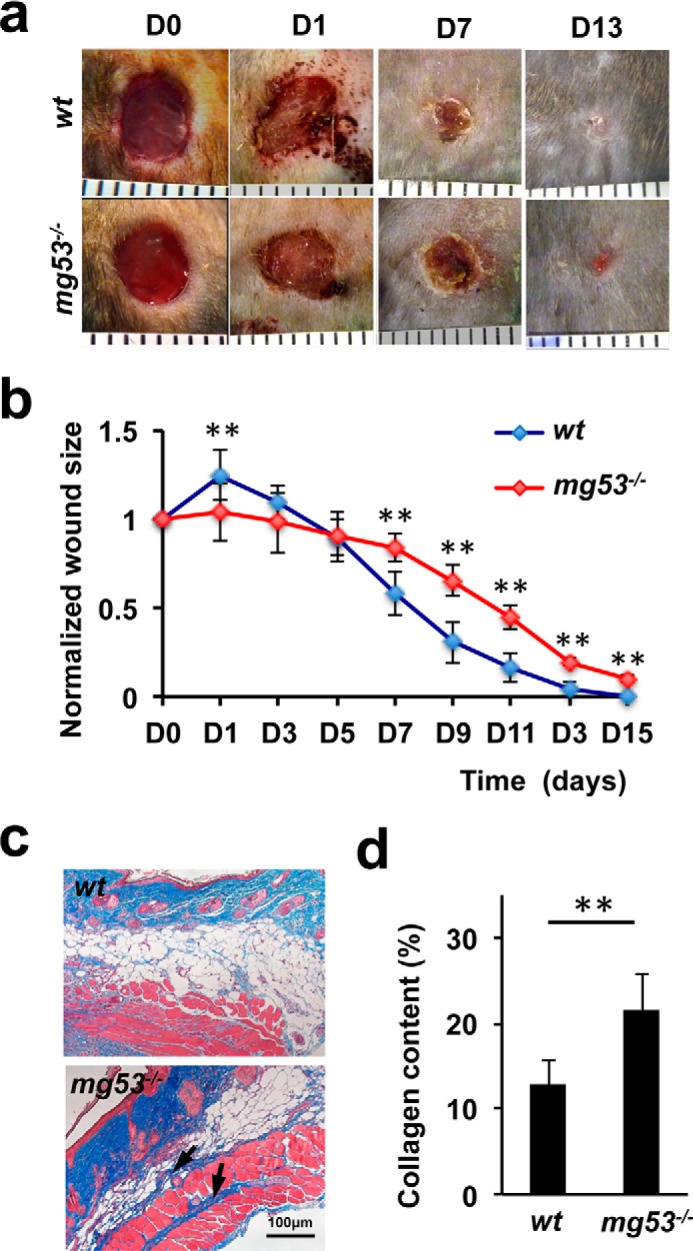
mg53−/− mice display delayed wound healing and excessive collagen deposition following injury. a, representative images of cutaneous wounds from WT (top panel) and mg53−/− (bottom panel) mice at different time points. D, day. b, statistical analyses revealed that mg53−/− mice displayed delayed wound healing compared with their WT littermates. **, p < 0.01 (n = 12). c, Masson trichrome staining of wound sections derived from WT (top panel) and mg53−/− mice (bottom panel) 10 days post-injury. Arrows indicate fibrosis and deposition of collagen. d, quantification of collagen deposition in dorsal skin on day 10 following excisional wounding in WT and mg53−/− mice. **, p < 0.01 (n = 6/group).
MG53 Protects against Keratinocyte Injury
Primary cultured human keratinocytes were transfected with GFP-MG53, a fusion construct with GFP added to the amino-terminal end of MG53, to investigate the extent of MG53 participation in the repair of membrane injury. As shown in Fig. 3a, right panel, in response to injury caused by penetration of a microelectrode into the plasma membrane, GFP-MG53 rapidly translocated toward the acute injury site. This GFP-MG53 translocation to membrane injury sites in keratinocytes is similar to that observed in C2C12, HEK293, and other cell types (14, 18).
FIGURE 3.
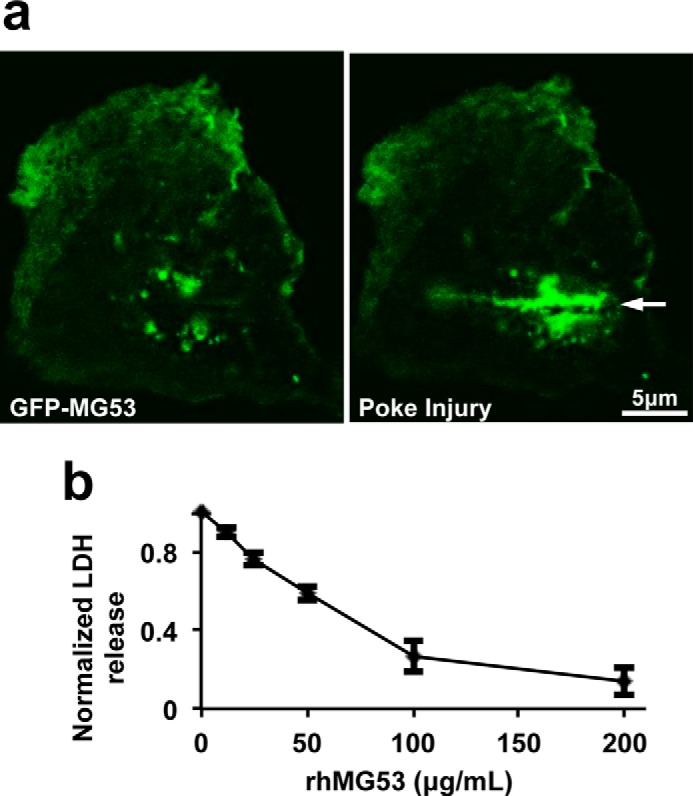
MG53 protects human keratinocyte from membrane injury. a, GFP-MG53 expressed in human keratinocytes rapidly translocated to the acute cell membrane injury site created by penetration of a microelectrode (arrow). A live-cell imaging movie of the cell response to injury can be found in supplemental Movie 1. b, rhMG53 protected human keratinocytes from glass microbead-induced membrane damage in a dose-dependent manner (n = 3 independent experiments). LDH, lactate dehydrogenase.
We have shown previously that rhMG53 protein could protect various cell types against cell membrane disruption when applied to the extracellular environment (18). To quantify the effect of rhMG53 protection against injury to keratinocytes, we first employed an in vitro keratinocyte cell membrane damage assay. In this assay, we measured the amount of lactate dehydrogenase leaked from the cell interior into the extracellular solution following cell membrane injury induced by microglass beads (18, 23). As shown in Fig. 3b, application of rhMG53 to the extracellular solution reduced the release of lactate dehydrogenase from keratinocytes into the culture medium in a dose-dependent manner after microglass bead treatment. This suggests that rhMG53 protects against mechanical injury to keratinocytes.
MG53 Enhances Fibroblast Migration following Scratch Wounding
The wound healing process involves migration of fibroblasts to wound sites and differentiation of fibroblasts into myofibroblasts (2, 4, 5, 8, 11). To test whether MG53 plays a role in the modulation of fibroblast migration during wound healing, we performed an in vitro scratch wound assay with cultured 3T3 fibroblast cells (Fig. 4a). 3T3 fibroblasts grown to ∼95% confluence were subjected to scratch wounding. The cells were treated with phosphate-buffered saline (Fig. 4a, left column), rhMG53 (60 μg/ml, center column), or stably transfected with myc-MG53 (right column). Images were captured 0, 6, and 22 h post-injury. Cell migration was quantified by the distance traveled by cells from the scratch edge toward the center of the scratch (Fig. 4b). Both rhMG53 treatment and myc-MG53 overexpression enhanced the migration of fibroblast cells following scratch wounding.
FIGURE 4.
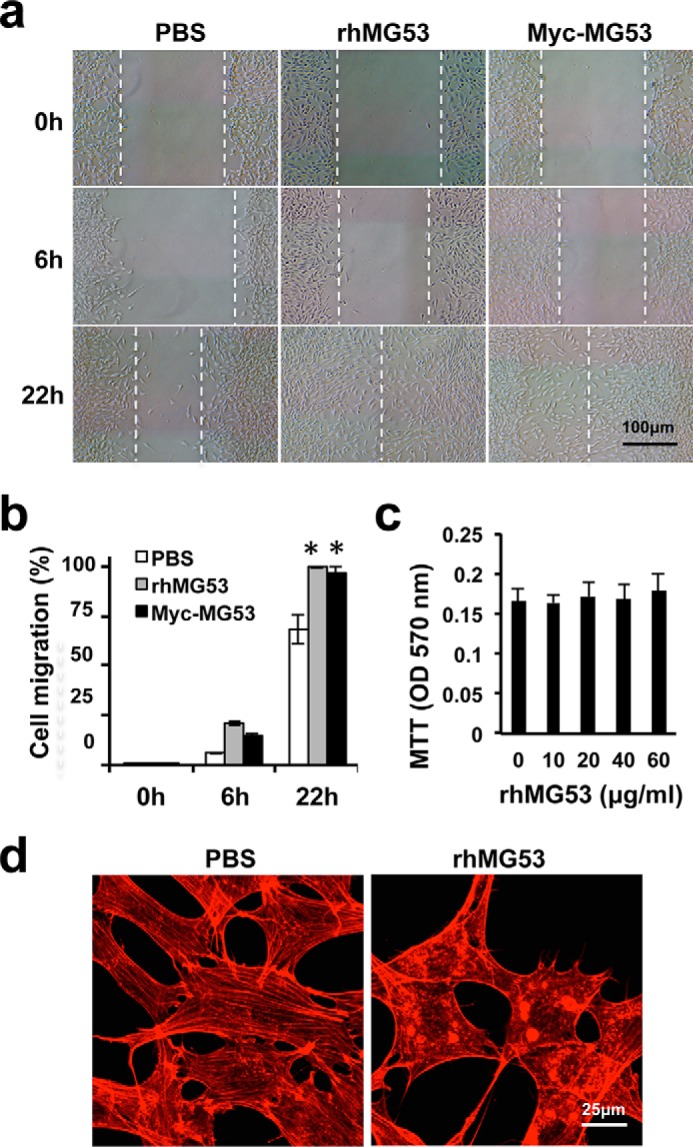
rhMG53 enhances fibroblast migration following scratch wounding. a, monolayers of 3T3 fibroblasts were subjected to scratch wounding. The cells were treated with vehicle (PBS, left column), rhMG53 (center column), or 3T3 cells stably expressing myc-MG53 (right column) in DMEM. Representative images were captured 0, 6, and 22 h post-injury. b, both rhMG53 treatment and myc-MG53 overexpression enhanced the migration of fibroblast cells following scratch wounding (n = 3 independent assays for all groups). **, p < 0.01). c, 3T3 cells were treated with different concentrations (0–60 μg/ml) of rhMG53 for 24 h, and cell growth and viability were analyzed by MTT assay (data represented three independent assays). d, subconfluent 3T3 cells were grown in serum-free medium in the absence of rhMG53 (PBS, left panel) or in the presence of rhMG53 (60 μg/ml, right panel) for 48 h. Cells were fixed and stained with Alexa Fluor 647 phalloidin.
The MG53-mediated enhancement of fibroblast migration was not due to an increase in cell proliferation, as determined by MTT assay because the cell growth rate was not affected by the treatment with rhMG53 (Fig. 4c). Under confocal microscopy, we found that fibroblasts cultured in serum-free medium displayed a formation of intracellular stress fibers that were larger and more contiguous compared with those treated with rhMG53 (these were thinner and dissociated) (Fig. 4d). Other investigators have shown that reduced stress fiber formation is correlated with the enhanced cell migration associated with wound healing (31, 32). Together, these results show that MG53-mediated reduction of stress fiber formation may be a contributing factor for the enhancement of cell migration during wound healing.
Subcutaneous Administration of rhMG53 accelerates Wound Healing
We have shown previously that systemic administration of rhMG53 provided dose-dependent protection against injury to muscles and the heart, kidneys, and lungs under various injurious conditions (18–21). We studied the effect of rhMG53 in protection against dermal injury using established rat models of wound healing (29, 30, 33). Excisional wounds were created on the back of rats with a biopsy punch (diameter, 6 mm) (Fig. 5a). Vehicle or rhMG53 (0.2 mg/wound) was applied to the wound site via subcutaneous injection after injury, and on the following 3 days (once per day). We found that wounds treated with rhMG53 displayed better healing compared with those treated with vehicle (Fig. 5b). On day 1, rhMG53-treated wounds showed improved settlement in the wound bed compared with vehicle-treated wounds despite there being no significant difference in wound closure between the two groups. Improved wound closure was clearly observed starting on day 5 in the rhMG53-treated group. The role of rhMG53 in facilitating wound healing was tested further in an incisional wound model. Consistent with our observations in the excisional wound model, rhMG53 treatment led to significant improvement in healing of the incisional wound starting on day 3 and continuing to wound closure (Fig. 5, c and d). These studies suggest that subcutaneous delivery of rhMG53 has beneficial effects on wound healing.
FIGURE 5.
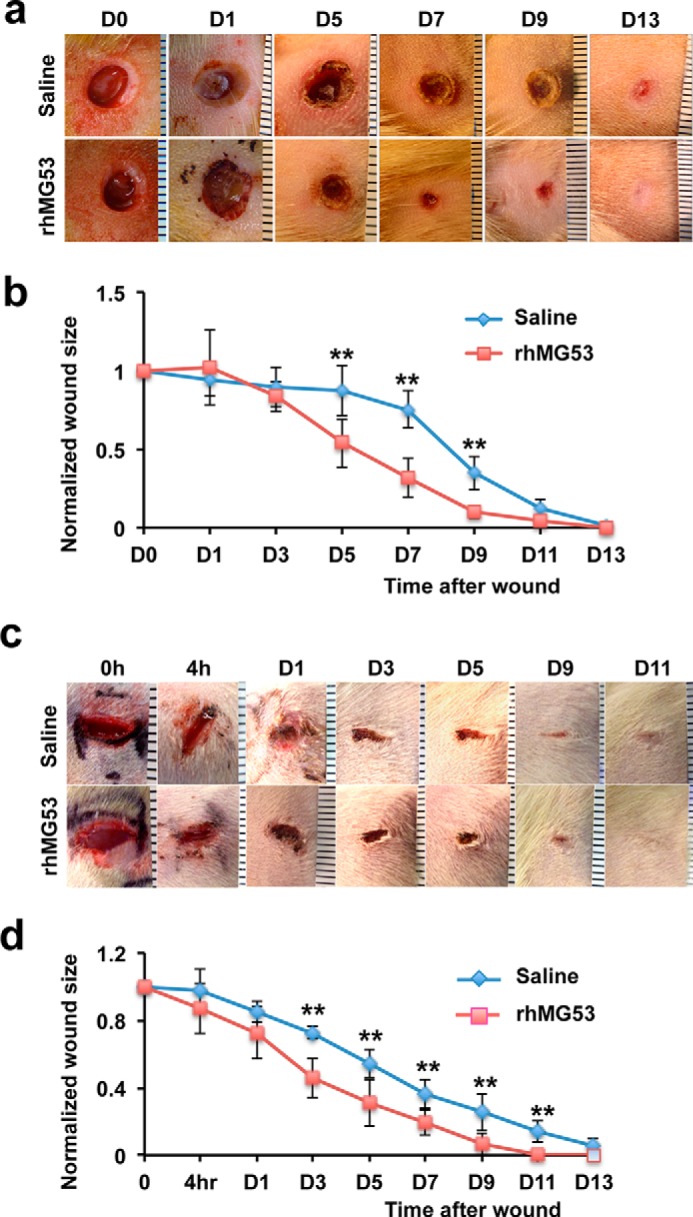
rhMG53 improves dermal wound healing in rats. a, representative micrograph images of rat excisional wounds were taken at different time points. Wounds were created on the back skin of rats and treated with saline (top row) or rhMG53 (0.2 mg/wound, subcutaneous injection, bottom row) and three additional daily treatments. D, day. b, quantification of wound healing of rhMG53-treated rats compared with saline-treated control rats. **, p < 0.01 (n = 8/group). c, incisional wound images at different time points in rats receiving saline as a control (top row) or a subcutaneous injection of rhMG53 (bottom row). d, quantification of incisional wound healing in rats treated with rhMG53 compared with controls. **, p < 0.01 (n = 8/group).
Hydrogel Encapsulation Provides Efficient Delivery of rhMG53 to Dermal Wounds
Pharmacokinetic studies have shown that rhMG53 has a short half-life of ∼1.5 h in the blood circulation after intravenous or subcutaneous administration (18, 21). Therefore, treatment of open dermal wounds required repetitive subcutaneous administration of rhMG53. Our group has previously developed a thermosensitive hydrogel drug delivery system that is biodegradable and safe for topical application (25). rhMG53 encapsulated in hydrogel exists in liquid gel format at room temperature and quickly solidifies to form a flexible solid gel when at 37 °C (body temperature). This hydrogel encapsulation allows better retention and time-controlled release of rhMG53 at the wound site to facilitate wound healing.
We investigated the therapeutic efficacy of the rhMG53/hydrogel formulation in the rat incisional wound model. As shown in Fig. 6a, rat skin incisions that received a local administration of rhMG53/hydrogel (0.2 mg rhMG53/wound) healed significantly faster than those treated with hydrogel alone. Statistical analyses with multiple animals indicate that rhMG53 treatment both improves wound healing (Fig. 6b) and reduces scar formation (Fig. 6c). On day 18 post-injury, scars were clearly diminished in rats treated with hydrogel/rhMG53 compared with animals treated with hydrogel alone (Fig. 6c, left panels). Histological analysis was used to examine collagen deposition in sections of gastrocnemius muscle by Masson trichrome staining (Fig. 6c, right panels). The results showed that rhMG53/hydrogel treatment markedly reduced collagen content (blue staining) on day 8 post-injury (summarized in Fig. 6d). Similar results have been reported in our previous studies, where systemic application of rhMG53 reduced the fibrotic content in skeletal (18) and cardiac muscle (20).
FIGURE 6.

Hydrogel delivery of rhMG53 enhances wound healing and reduces scar formation. a, incisional wounds in the hind limbs of rats were treated with hydrogel (top row) or hydrogel encapsulating rhMG53 (bottom row). 200 μl of the hydrogel or rhMG53/hydrogel (1 mg/ml) was applied immediately after wounding (0 h, n = 8/group). b, time-dependent wound closure compared with the original wound sizes. Hydrogel/rhMG53 treatment enhanced healing of the incisional wound. **, p < 0.01 (n = 8/group). c, representative macroscopic (left panels) and microscopic (right panels) images of skin treated with hydrogel (top panels) or rhMG53/hydrogel (bottom panels). The macroscopic images were taken from day 18 post-injury to observe scar formation (left panels). Masson trichrome staining of gastrocnemius muscle was performed on day 8 post-injury (right panels). Insets, decreased collagen deposition in muscle sections receiving rhMG53/hydrogel treatment (bottom panels) compared with those receiving hydrogel alone (top panels). d, quantification of collagen deposition of the gastrocnemius muscle from animals treated with hydrogel or rhMG53/hydrogel on day 8 after incisional wounding. **, p < 0.01 (n = 8/group). e, macroscopic images of wounds in rabbits were taken from day 14 after the incisional wound injuries. f, Masson trichrome staining of rabbit skeletal muscle taken from the wound sites as indicated above on day 14 post-injury. Muscle sections derived from the rabbit receiving rhMG53/hydrogel treatment show less fibrosis (collagen deposition, blue) compared with those of the control.
To further assess the effect of rhMG53 in reducing fibrosis during wound healing, we used a rabbit model of dermal incisional wounding. An incisional wound 2 cm in length and 0.5 cm in depth was created on the back of the rabbit (Fig. 6e). Animals received either hydrogel or rhMG53/hydrogel on the wound sites immediately after injury, followed by another administration on day 1 post-wounding. Rabbits treated with rhMG53/hydrogel healed better than those receiving hydrogel alone. Masson trichrome staining of the skeletal muscle layer was conducted 14 days post injury. Sections derived from rabbits receiving rhMG53/hydrogel treatment showed significantly less fibrosis compared with controls (Fig. 6f). Together, our in vivo studies with mouse, rat, and rabbit models of dermal injury support the therapeutic effect of rhMG53 in facilitating wound healing and inhibiting scar formation.
MG53 Modulates Fibroblast-to-Myofibroblast Differentiation to Reduce α-SMA Expression
TGF-β is a growth factor that regulates wound healing and fibrotic remodeling associated with the healing process (7, 34–36). Many studies have suggested that TGF-β contributes to wound healing and scarring by controlling the differentiation of myofibroblasts from fibroblasts (5, 8, 34). The expression of α-SMA is considered a marker of myofibroblasts differentiation (8, 35). We therefore examined whether MG53 affects the expression level of α-SMA associated with fibroblast differentiation. Western blotting showed an elevation of α-SMA in skin tissues derived from mg53−/− mice compared with WT mice (Fig. 7, a and b), suggesting the possibility that MG53 in the circulation may contribute to fibroblast differentiation.
FIGURE 7.
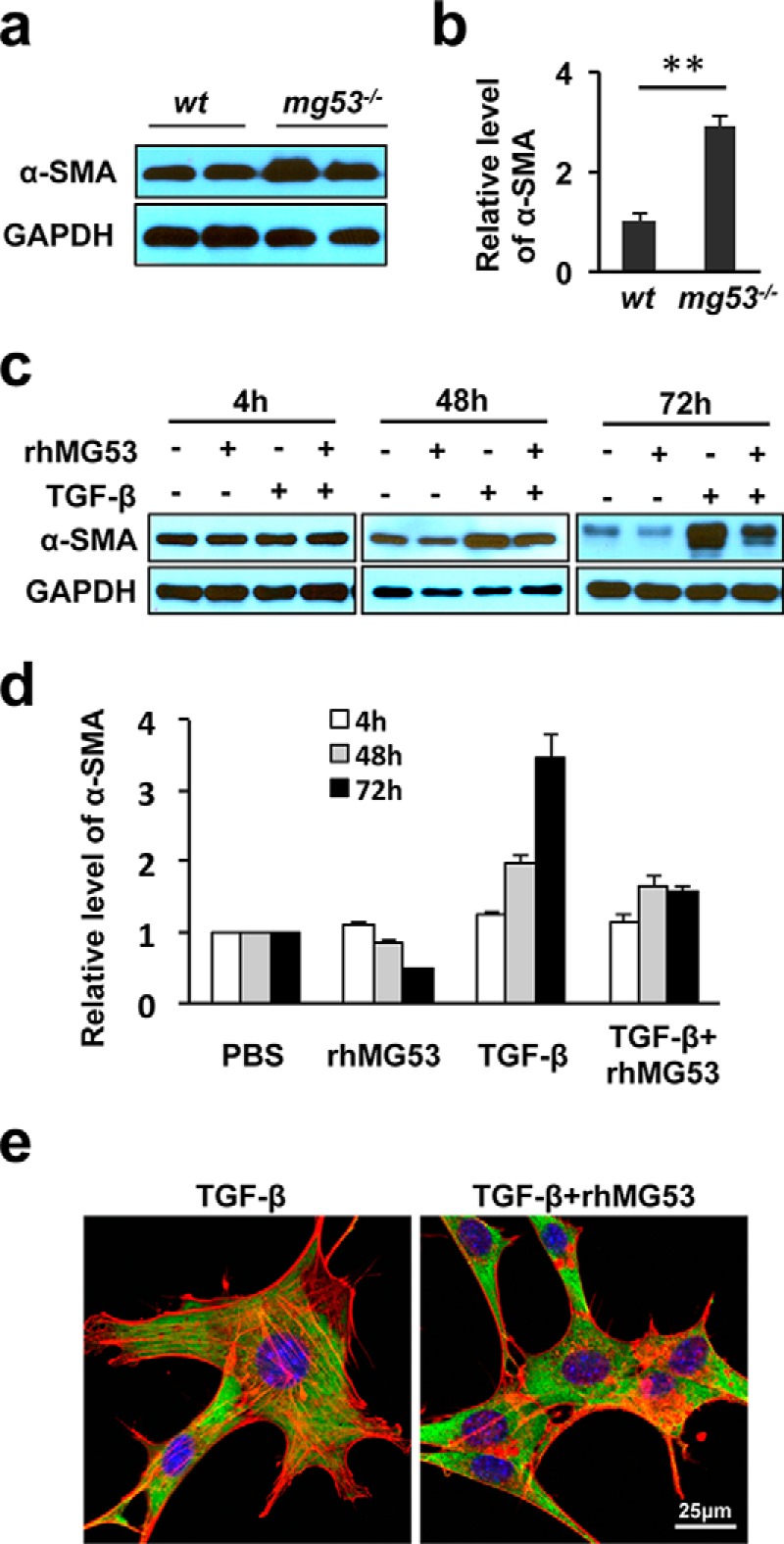
rhMG53 treatment suppresses TGFβ-mediated α-SMA activation in fibroblasts. a, Western blotting shows elevated levels of α-SMA in skin tissues derived from mg53−/− mice compared with those from WT littermates. GAPDH was used as a loading control. b, summary data from multiple animals. **, p < 0.01. (n = 6/group). c, immunoblots of α-SMA expression in cultured 3T3 fibroblasts grown in serum-free medium in the presence of TGF-β (10 ng/ml), rhMG53 (60 μg/ml), or both TGF-β and rhMG53. Western blotting shows that rhMG53 suppressed TGF-β-mediated activation of α-SMA, with a maximal effect at the 72-h time point. d, the relative expression of α-SMA at different time points. Data are the average of three independent experiments. **, p < 0.01. e, immunofluorescence staining of α-SMA (green) and F-actin (phalloidin staining, red) reveals the reduction of stress fibers in cells treated with TGF-β plus rhMG53 (right panel) compared with cells treated with TGF-β alone (left panel) (n = 3 independent experiments).
Changes in the expression of α-SMA were assessed in cultured 3T3 fibroblasts with or without rhMG53 (60 μg/ml) or TGF-β (10 ng/ml) treatment a or combination of TGF-β and rhMG53 in serum-free medium for 4, 48, or 72 h (Fig. 7c). Although TGF-β stimulation led to a more than 3-fold increase in the expression of α-SMA, rhMG53 significantly suppressed TGF-β induced α-SMA expression in 3T3 cells (Fig. 7d). Immunofluorescence staining of α-SMA and phalloidin revealed the diminished appearance of F-actin stress fibers in cells treated with both TGF-β and rhMG53 (Fig. 7e), suggesting that rhMG53 treatment prevents the differentiation of myofibroblasts.
MG53 Affects TGF-β Signaling to Control Fibrotic Remodeling during Wound Healing
Many studies have suggested that the TGF-β/Smad axis is a principal signaling pathway for controlling the transcriptional activation of α-SMA, fibroblast-myofibroblast differentiation, and secretion of ECM proteins during the fibrogenic response of wound healing (8, 10, 37–39). This TGF-β-mediated signaling involves the nuclear translocation of p-Smad2/3 to stimulate the expression of downstream substrates and the ECM proteins, such as fibronectin and collagen 1. We examined the activation of Smad2/3 signaling with biochemical and immunocytochemical staining approaches on 3T3 cells undergoing similar treatments as described above. Although TGF-β-dependent activation of p-Smad2/3 was observed in 3T3 cells, cells treated with both rhMG53 and TGF-β clearly showed a reduced activation of p-Smad2/3 (Fig. 8, a and b).
FIGURE 8.
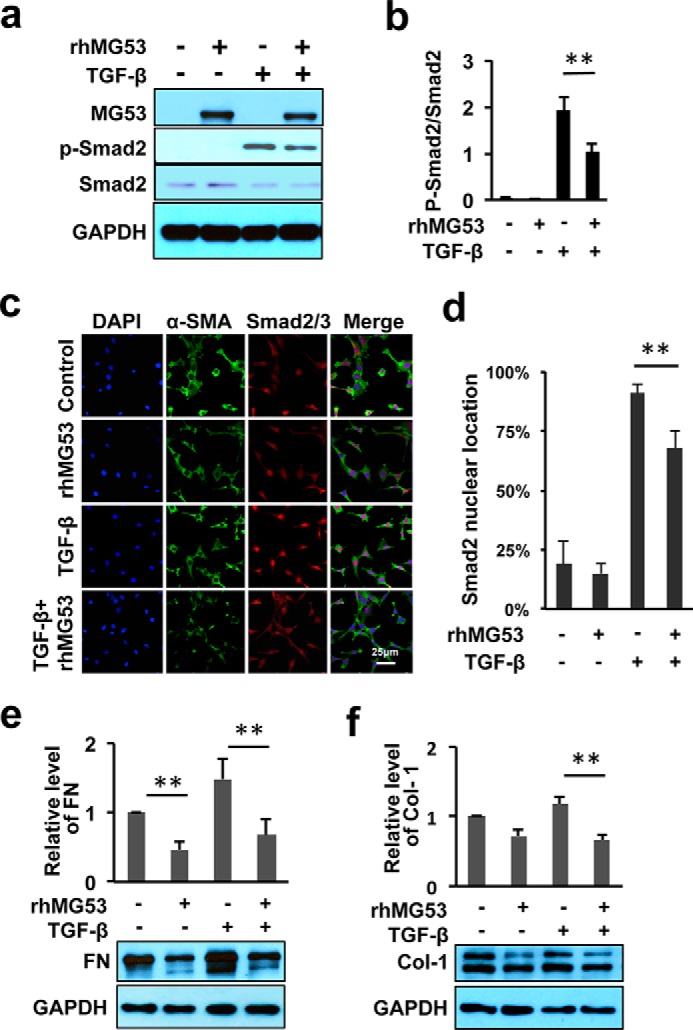
rhMG53 negatively regulates TGF-β/Smad-dependent signaling and suppresses the synthesis of ECM proteins. a, immunoblots of MG53, p-Smad2/3, total Smad 2/3, and GAPDH expression were derived from 3T3 cells treated for 4 h under various culture conditions as denoted at the top of the blots. b, data represent the relative level of p-Smad2/3 as normalized to the expression of total Smad2/3. c, immunofluorescence staining of α-SMA (green), p-Smad2/3 (red), and nuclei (DAPI, blue) derived from 48-h cultures of 3T3 cells under the indicated conditions. Cells were treated with serum-free medium (control), rhMG53 (60 μg/ml), TGF-β (10 ng/ml), or both. d, quantification of p-Smad2 nuclear translocation (n > 120 cells for each of the three independent experiments). **, p < 0.01 for comparison between the indicated groups. e and f, rhMG53 suppresses TGFβ-mediated expression of FN (e) and collagen 1 (Col-1) in 3T3 cells treated for 48 h. (f). The relative expression of collagen-1 and fibronectin was calculated from four independent experiments. **, p < 0.01.
Taking advantage of the monoclonal antibody that recognizes the phosphorylated form of Smad2/3, we examined the nuclear translocation of Smad2/3 in 3T3 cells following stimulation with TGF-β with or without cotreatment of rhMG53 (Fig. 8, c and d). Localization of Smad2/3 in the cell nuclei was observed infrequently in 3T3 cells maintained under basal serum deprivation conditions (Fig. 8c). Treatment with TGF-β (10 ng/ml) for 4 h led to a significant increase in nuclear localization of Smad2/3 because more than 90% of 3T3 cells were positive for immunofluorescence labeling with p-Smad2/3. However, cotreatment of rhMG53 diminished the TGF-β-induced nuclear localization of Smad2/3 because only 68% of cells were observed with pSmad2/3 nuclear translocation (Fig. 8d).
We next investigated the expression levels of ECM substrates such as collagen type 1 and fibronectin (FN). We found that treatment of 3T3 cells with rhMG53 led to a significant reduction in FN under serum-deprived conditions (Fig. 8e). Although the level of FN was increased significantly following stimulation with TGF-β, TGF-β-mediated elevation of FN expression was largely suppressed following cotreatment with rhMG53. Similarly, the expression of collagen type 1 from cells receiving cotreatment of TGF-β and rhMG53 showed a significantly lower expression compared with those receiving TGF-β treatment alone (Fig. 8f).
Discussion
Our group has demonstrated previously that MG53 acts as an essential component in the cell membrane repair machinery (14) and that the rhMG53 protein can protect against injury in various cell and tissue types when applied to the extracellular environment or administered systemically (18–21). In this study, we show that MG53 has therapeutic benefits in treating dermal wounds. We provide evidence that topical administration of rhMG53 not only facilitates wound healing but also suppresses scar formation. These studies reveal a role for MG53 in the modulation of TGF-β-dependent fibroblast-myofibroblast differentiation in addition to its conventional cell membrane repair function.
Because MG53 is predominantly expressed in striated muscle tissues, most published studies have focused on characterizing the skeletal muscle and cardiac phenotype of mg53−/− mice (14, 16). Here we show that, despite the absence of MG53 expression in keratinocytes and fibroblasts, genetic ablation of MG53 leads to prominent defects in skin structure and its wound healing property. Excessive collagen deposition in the dermal layer of mg53−/− mice may reveal a compensatory response of the animal to the chronic absence of MG53 in the blood circulation. The delayed wound healing response of mg53−/− skin further suggests that MG53 in circulation may function to protect the dermal layer under injurious conditions.
The therapeutic effect of rhMG53 on the facilitation of wound healing was evaluated in three animal models, including incisional and excisional wounds in rats and incisional wounds in rabbits. Although subcutaneous delivery of rhMG53 was effective in accelerating skin wound healing, the short half-life of rhMG53 in the circulation necessitated repeated administration (18–21). We encapsulated rhMG53 in a thermosensitive hydrogel to control its release at the wound site. The thermosensitive hydrogel can be mixed readily with rhMG53 in liquid formulation at 4 °C to room temperature, facilitating convenient topical administration. Upon delivery to the wound site at body temperature (∼37 °C), rhMG53/hydrogel solidifies quickly, allowing the retention of rhMG53 at the injury site. This approach enhanced the effective dosage of rhMG53 at the wound site, achieving improved therapeutic efficacy of the protein in wound healing compared with the conventional approach of repeated subcutaneous delivery. Hydrogel-mediated delivery of rhMG53 represents a potentially novel and effective means to treat dermal injuries.
Through these studies, we identified the exciting phenomenon that rhMG53 suppresses scar formation during wound healing. The role of MG53 in the modulation of scarring was first observed in mg53−/− mice. These animals displayed excessive fibrotic remodeling following dermal injury. With hydrogel-mediated delivery of rhMG53, reduced scar formation was observed consistently in both rat and rabbit models of dermal injuries. The promotion of scarless wound healing is a major challenge because the natural biological wound healing process is often accompanied by fibrotic remodeling because of the limited regenerative capacity of the dermis. Our finding that rhMG53 facilitates wound healing and reduces scarring has potentially broad applications in treating tissue injuries where excessive scarring can be detrimental to the biological function of the organ.
We conducted biochemical and cellular studies to understand the mechanisms that underlie the biological function of MG53 in facilitating wound healing and suppressing scar formation (Fig. 9). First, MG53, as a membrane repair molecule, can protect against acute injuries to keratinocytes at the dermal injury site. Such a fast membrane resealing response could reduce the level of inflammatory cytokines released to the wound milieu, significantly affecting immunogenic and fibrotic remodeling during later stages of healing. Second, MG53 present in the extracellular solution can facilitate the migration of fibroblast cells in response to dermal wounding, which is an important step in efficient wound healing. Confocal microscopic imaging revealed that fibroblast cells exposed to rhMG53 show reduced formation of stress fibers, which can lead to increased mobility of fibroblasts during wound healing. Third, MG53 can interfere with TGF-β signaling to control the differentiation process of fibroblasts into myofibroblasts. The latter is a major source of secretion of ECM proteins and the main factor contributing to scarring. We have data showing that treatment of fibroblasts with rhMG53 leads to reduced expression of α-SMA and ECM components, which underlie reduced scar formation during wound healing.
FIGURE 9.

Schematic illustrating the role of MG53 in wound healing and scar formation. a, MG53 nucleates the cell membrane repair machinery and protects against acute injury to keratinocytes and fibroblasts in response to dermal injury. b, MG53 promotes the migration of fibroblasts to the wound sites by modulating stress fiber formation. c, MG53 regulates TGF-β/Smad signaling to control the differentiation of fibroblasts into myofibroblasts, leading to down-regulation of ECM proteins during the tissue remodeling process of wound healing.
Fibroblasts play important roles in wound repair, and their uncontrolled differentiation into myofibroblasts is responsible for the excessive synthesis of extracellular matrix proteins as part of scar formation (8, 11, 39–41). TGF-β is an important cytokine that controls the conversion of fibroblasts into myofibroblasts. Although many studies have been designed to target TGF-β signaling for the treatment of wound healing, these attempts often faced a double-edged sword. For example, blocking TGF-β signaling can reduce scarring but can also compromise wound healing and other immune functions (5, 10, 39, 40, 42). Our identification of a functional link between MG53 and TGF-β signaling has important therapeutic implications regarding wound healing. The trifunctional role of MG53 in wound healing, e.g. protection against acute injury to cell membrane, promotion of fibroblast migration, and control of myofibroblast differentiation, has advantages over many of the current approaches for the treatment of wound healing. Although the biological function of MG53 in cell membrane repair has received much attention, the cytokine function of MG53 in controlling fibroblast-myofibroblast differentiation opens a new avenue for reducing fibrotic remodeling in response to recovery from traumatic injuries not only to the dermal layer but also to many vital organs, such as myocardial infarction, acute lung injury, and acute kidney injury, where prevention of scarring is of prominent importance for restoring normal organ function (18–21). One avenue for future study may include elucidating the molecular interaction between MG53 and TGF-β signaling cascades, information that will be instrumental for developing effective therapies to facilitate scarless wound healing.
Author Contributions
H. L. and J. M. developed the concept for the study. H. L., P. D., D. H., H. Z., S. M. S., E. J., and C. Z. performed the animal model studies with wound healing. H. L., P. L., L. Z., X. Z., M. S., M. F., M. O., and M. S. conducted biochemical studies, the in vitro cell assay, and imaging. Z. F. and J. G. performed hydrogel studies. T. T. and H. M. performed toxicological studies of rhMG53. J. M. oversaw the entire project. H. L., S. S., R. H., J. G., and J. M. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Novoprotein Scientific, Inc., for the rhMG53 protein.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-AR061385 and R01-HL069000 (to J. M.). This work was also supported by National Science Foundation grants 1006734 and 1160122 (to J. G.) and the Ohio State University Lockwood Early Career Development Award (to P. L. and H. L.). J. M. and T. T. have an equity interest in TRIM-edicine, which develops MG53 for the treatment of human disease. Patents on the use of MG53 are held by Rutgers University Robert Wood Johnson Medical School.

This article contains supplemental Movie 1.
- ECM
- extracellular matrix
- rh
- recombinant human
- α-SMA
- α smooth muscle actin
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- FN
- fibronectin.
References
- 1. Sen C. K. (2009) Wound healing essentials: let there be oxygen. Wound Repair Regen. 17, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martin P. (1997) Wound healing: aiming for perfect skin regeneration. Science 276, 75–81 [DOI] [PubMed] [Google Scholar]
- 3. Singer A. J., Clark R. A. (1999) Cutaneous wound healing. N. Engl. J. Med. 341, 738–746 [DOI] [PubMed] [Google Scholar]
- 4. Gurtner G. C., Werner S., Barrandon Y., Longaker M. T. (2008) Wound repair and regeneration. Nature 453, 314–321 [DOI] [PubMed] [Google Scholar]
- 5. Greaves N. S., Ashcroft K. J., Baguneid M., Bayat A. (2013) Current understanding of molecular and cellular mechanisms in fibroplasia and angiogenesis during acute wound healing. J. Dermatol. Sci. 72, 206–217 [DOI] [PubMed] [Google Scholar]
- 6. Darby I. A., Laverdet B., Bonté F., Desmoulière A. (2014) Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 7, 301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leask A., Abraham D. J. (2004) TGF-β signaling and the fibrotic response. FASEB J. 18, 816–827 [DOI] [PubMed] [Google Scholar]
- 8. Hinz B. (2007) Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 127, 526–537 [DOI] [PubMed] [Google Scholar]
- 9. Darby I. A., Hewitson T. D. (2007) Fibroblast differentiation in wound healing and fibrosis. Int. Rev. Cytology 257, 143–179 [DOI] [PubMed] [Google Scholar]
- 10. Wynn T. A., Ramalingam T. R. (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gabbiani G. (2003) The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200, 500–503 [DOI] [PubMed] [Google Scholar]
- 12. Bansal D., Miyake K., Vogel S. S., Groh S., Chen C. C., Williamson R., McNeil P. L., Campbell K. P. (2003) Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423, 168–172 [DOI] [PubMed] [Google Scholar]
- 13. McNeil P. L., Kirchhausen T. (2005) An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 6, 499–505 [DOI] [PubMed] [Google Scholar]
- 14. Cai C., Masumiya H., Weisleder N., Matsuda N., Nishi M., Hwang M., Ko J. K., Lin P., Thornton A., Zhao X., Pan Z., Komazaki S., Brotto M., Takeshima H., Ma J. (2009) MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 11, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Howard A. C., McNeil A. K., Xiong F., Xiong W. C., McNeil P. L. (2011) A novel cellular defect in diabetes: membrane repair failure. Diabetes 60, 3034–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cao C. M., Zhang Y., Weisleder N., Ferrante C., Wang X., Lv F., Zhang Y., Song R., Hwang M., Jin L., Guo J., Peng W., Li G., Nishi M., Takeshima H., Ma J., Xiao R. P. (2010) MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation 121, 2565–2574 [DOI] [PubMed] [Google Scholar]
- 17. Wang X., Xie W., Zhang Y., Lin P., Han L., Han P., Wang Y., Chen Z., Ji G., Zheng M., Weisleder N., Xiao R. P., Takeshima H., Ma J., Cheng H. (2010) Cardioprotection of ischemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ. Res. 107, 76–83 [DOI] [PubMed] [Google Scholar]
- 18. Weisleder N., Takizawa N., Lin P., Wang X., Cao C., Zhang Y., Tan T., Ferrante C., Zhu H., Chen P. J., Yan R., Sterling M., Zhao X., Hwang M., Takeshima M., Cai C., Cheng H., Takeshima H., Xiao R. P., Ma J. (2012) Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 4, 139ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jia Y., Chen K., Lin P., Lieber G., Nishi M., Yan R., Wang Z., Yao Y., Li Y., Whitson B. A., Duann P., Li H., Zhou X., Zhu H., Takeshima H., Hunter J. C., McLeod R. L., Weisleder N., Zeng C., Ma J. (2014) Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat. Commun. 5, 4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu J., Zhu H., Zheng Y., Xu Z., Li L., Tan T., Park K. H., Hou J., Zhang C., Li D., Li R., Liu Z., Weisleder N., Zhu D., Lin P., Ma J. (2014) Cardioprotection of recombinant human MG53 protein in a porcine model of ischemia and reperfusion injury. J. Mol. Cell. Cardiol. 80, 10–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Duann P., Li H., Lin P., Tan T., Wang Z., Chen K., Zhou X., Gumpper K., Zhu H., Ludwig T., Mohler P. J., Rovin B., Abraham W., Zeng C., Ma J. (2015) MG53-mediated cell membrane repair protects against acute kidney injury. Sci. Transl. Med. 7, 279ra236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reddy G. K., Enwemeka C. S. (1996) A simplified method for the analysis of hydroxyproline in biological tissues. Clin. Biochem. 29, 225–229 [DOI] [PubMed] [Google Scholar]
- 23. Lin P., Zhu H., Cai C., Wang X., Cao C., Xiao R., Pan Z., Weisleder N., Takeshima H., Ma J. (2012) Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 26, 1875–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liang C. C., Park A. Y., Guan J. L. (2007) In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2, 329–333 [DOI] [PubMed] [Google Scholar]
- 25. Li Z., Guan J. (2011) Thermosensitive hydrogels for drug delivery. Expert Opin. Drug Deliv. 8, 991–1007 [DOI] [PubMed] [Google Scholar]
- 26. Li Z., Xu Y., Li H., Guan J. (2014) Immobilization of insulin-like growth factor-1 onto thermosensitive hydrogels to enhance cardiac progenitor cell survival and differentiation under ischemic conditions. Science China 4, 568–578 [Google Scholar]
- 27. Yi J. S., Park J. S., Ham Y. M., Nguyen N., Lee N. R., Hong J., Kim B. W., Lee H., Lee C. S., Jeong B. C., Song H. K., Cho H., Kim Y. K., Lee J. S., Park K. S., Shin H., Choi I., Lee S. H., Park W. J., Park S. Y., Choi C. S., Lin P., Karunasiri M., Tan T., Duann P., Zhu H., Ma J., Ko Y. G. (2013) MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat. Commun. 4, 2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frank S., Kämpfer H. (2003) Excisional wound healing. An experimental approach. Methods Mol. Med. 78, 3–15 [DOI] [PubMed] [Google Scholar]
- 29. Galiano R. D., Michaels J. 5th, Dobryansky M., Levine J. P., Gurtner G. C. (2004) Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen. 12, 485–492 [DOI] [PubMed] [Google Scholar]
- 30. Wong V. W., Sorkin M., Glotzbach J. P., Longaker M. T., Gurtner G. C. (2011) Surgical approaches to create murine models of human wound healing. J. Biomed. Biotechnol. 2011, 969618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pellegrin S., Mellor H. (2007) Actin stress fibres. J. Cell Sci. 120, 3491–3499 [DOI] [PubMed] [Google Scholar]
- 32. Tojkander S., Gateva G., Lappalainen P. (2012) Actin stress fibers: assembly, dynamics and biological roles. J. Cell Sci. 125, 1855–1864 [DOI] [PubMed] [Google Scholar]
- 33. Gamelli R. L., He L. K. (2003) Incisional wound healing. Model and analysis of wound breaking strength. Methods Mol. Med. 78, 37–54 [DOI] [PubMed] [Google Scholar]
- 34. Mustoe T. A., Pierce G. F., Thomason A., Gramates P., Sporn M. B., Deuel T. F. (1987) Accelerated healing of incisional wounds in rats induced by transforming growth factor-β. Science 237, 1333–1336 [DOI] [PubMed] [Google Scholar]
- 35. Kane C. J., Hebda P. A., Mansbridge J. N., Hanawalt P. C. (1991) Direct evidence for spatial and temporal regulation of transforming growth factor beta 1 expression during cutaneous wound healing. J. Cell. Physiol. 148, 157–173 [DOI] [PubMed] [Google Scholar]
- 36. Schmid P., Itin P., Cherry G., Bi C., Cox D. A. (1998) Enhanced expression of transforming growth factor-β type I and type II receptors in wound granulation tissue and hypertrophic scar. Am. J. Pathol. 152, 485–493 [PMC free article] [PubMed] [Google Scholar]
- 37. Desmoulière A., Geinoz A., Gabbiani F., Gabbiani G. (1993) Transforming growth factor-β 1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Evans R. A., Tian Y. C., Steadman R., Phillips A. O. (2003) TGF-β1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp. Cell Res. 282, 90–100 [DOI] [PubMed] [Google Scholar]
- 39. Grotendorst G. R., Rahmanie H., Duncan M. R. (2004) Combinatorial signaling pathways determine fibroblast proliferation and myofibroblast differentiation. FASEB J. 18, 469–479 [DOI] [PubMed] [Google Scholar]
- 40. Leask A. (2010) Potential therapeutic targets for cardiac fibrosis: TGFβ, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 106, 1675–1680 [DOI] [PubMed] [Google Scholar]
- 41. Wynn T. A. (2008) Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zeisberg M., Kalluri R. (2013) Cellular mechanisms of tissue fibrosis: 1: common and organ-specific mechanisms associated with tissue fibrosis. Am. J. Physiol. Cell Physiol. 304, C216–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.